
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-directed spin labeling of proteins for distance measurements in vitro and in cells
P.
Roser†
a,
M. J.
Schmidt†
a,
M.
Drescher
*a and
D.
Summerer
*b
aDepartment of Chemistry, Zukunftskolleg, and Konstanz Research School Chemical Biology, University of Konstanz, Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: malte.drescher@uni-konstanz.de
bDepartment of Chemistry and Chemical Biology, Technical University of Dortmund, Otto-Hahn-Str. 6, 44227 Dortmund, Germany. E-mail: daniel.summerer@tu-dortmund.de
First published on 5th May 2016
Abstract
Site-directed spin labeling (SDSL) in combination with electron paramagnetic resonance (EPR) spectroscopy allows studying the structure, dynamics, and interactions of proteins via distance measurements in the nanometer range. We here give an overview of available spin labels, the strategies for their introduction into proteins, and the associated potentials for protein structural studies in vitro and in the context of living cells.
Introduction
Site-directed spin labeling (SDSL) is the concept of introducing paramagnetic centers into a biomacromolecule of interest at user-defined sites. Pioneered by Hubbell and co-workers more than 25 years ago,1,2 this technique is widely used for studying the structure and function of macromolecules by EPR spectroscopy.3 This includes proteins, but also synthetic polymers,4,5 nucleic acids6 and lipids.7 Though natural paramagnetic centers such as metal ions are present as cofactors in certain classes of proteins and can be used as probes for EPR spectroscopy, most proteins are diamagnetic and thus EPR silent. Hence, SDSL provides insights into protein structure and dynamics at sites of interest virtually without background. Four main sources of information are typically exploited:8 Label dynamics, solvent accessibility, polarity of the microenvironment, and distance distributions between two spin labels in the nanometer range on which we will particularly focus in the following. A variety of spin labels can be used for such studies, with nitroxides being the most popular class.9EPR spectroscopy
SDSL enables detailed studies of a large variety of protein properties. In this review, we will give only a short overview of most of these properties and will focus on distance measurements as main application. For a detailed discussion of all properties, we refer to references cited in the corresponding paragraphs.Rotational motions of spin labels, often characterized by one or several rotational correlation times result in characteristic spectral shapes. Three contributions to the overall dynamics can be distinguished: dynamics of the spin label linker itself, dynamics of the proteins secondary structure element at the incorporation site, and rotation of the entire protein or protein complex. The spectra represent a superposition of all three dynamic processes.10–12
Since the linker dynamics of the spin label can be restricted depending on the steric properties of the molecular environment,10 changes in this environment can be monitored. These can arise from e.g. conformational changes, ligand binding, or differences in the exposure to membrane lipids or intra/extracellular environments in the case of transmembrane proteins. Spectral characteristics can also be assigned to different types of secondary structure elements.13,14
The solvent accessibility of a labeled site can be measured via Heisenberg exchange. When exposed to paramagnetic quenchers like molecular oxygen or water soluble nickel complexes (e.g. Ni-EDDA), Heisenberg exchange processes result in a significant increase of spin–lattice relaxation.15
Since this interaction is highly dependent on a close interaction with the quencher, the choice of a suitable reagent polarity can visualize e.g. membrane bound areas of a protein when nonpolar molecular oxygen is added to the system. In contrast, polar quenchers like Ni-EDDA complexes will only interact with parts of the target molecule that are exposed to the solvent.16
Analysis of EPR spectra reveals the polarity of the microenvironment. Via SDSL, subsequent labeling and thus mapping of the local polarity of e.g. a protein chain is possible.17–20
Distance distributions in the nanometer range
The magnetic dipolar interaction between two spin labels is proportional to the inverse cube of the distance and can thus be employed for distance measurements.21 Additionally, the orientation of the labels with respect to each other can be determined. EPR does not only allow to determine a mean distance, but gives access to a fully quantitative distance distribution representing the complete conformational ensemble. The obtained distance distributions are quantitatively derived without the need for assumptions. Intermolecular distances can be measured between singly spin labeled proteins for analyzing supramolecular aspects such as oligomerization, formation of fibrils or complex formation. SDSL of a protein at two sites can be used to determine intramolecular distance constraints for protein structure determination (Fig. 1). As different secondary structures will lead to a predictable change in the spin–spin distance, for instance folding and unfolding processes can be observed. | ||
| Fig. 1 EPR distance measurements between spin labels for obtaining not only distance constraints but quantitative distance distributions. | ||
EPR distance measurements are complementary to Förster resonance energy transfer (FRET) where distances between a donor and an acceptor fluorophore can be detected when these labels are positioned close to the Förster radius. FRET allows single molecule detection, however, precise distances or distance distributions are usually not measured, and the FRET pairs have to be carefully selected based on expected distances. EPR distance measurements, on the other hand, allow for precise determination of distance constraints, distance distributions and their shape over a wide range of distances. Spin labels are significantly smaller than typical fluorophores, and have been shown to not perturb the structure of proteins in various cases.22 Moreover, the possibility to use two identical spin labels instead of a dedicated pair of fluorescence donor and acceptor significantly simplifies labeling approaches.
For EPR distance measurements, the sample is usually shock-frozen in liquid nitrogen and the subsequent measurements are carried out at cryogenic temperature. By shock-freezing, a snapshot of the complete conformational ensemble in solution-like environment is generated. Low temperatures facilitate distance measurements, as they result in reduced spin-relaxation and slow down the reorientation of the spin–spin vector between the labels, which deteriorates the signal. Current developments aim for measurements at physiological temperatures using either nitroxides23–25 or triarylmethyl (trityl or TAM) spin labels.26
By EPR, a large range of distances between around 1.2 and 10 nm can be measured. Suitable methods have to be chosen carefully depending on the occurring distances and the choice of spin labels as each method has limiting factors.27 Techniques for distance measurements are e.g. double-quantum coherence (DQC)28,29 or T1 relaxation measurements,30–32 but the most commonly applied method in the field is double electron electron resonance (DEER), also called pulsed electron–electron double resonance (PELDOR).33–38 When the distance between spin labels is less than approximately 2.5 nm, the resulting dipole–dipole coupling significantly broadens the EPR spectrum, which can be directly resolved. For distances smaller than approximately 1.2 nm, a lower limit for precise distance determination is reached because of large exchange contributions. For distances larger than 2.5 nm, the dipole–dipole interaction cannot be directly resolved in the EPR spectrum and thus has to be separated from all other contributions.39 Therefore, pulsed EPR methods are applied, which have been extensively reviewed.40 For a summary of distance determination methods and feasible ranges, we refer to recent literature.39 DEER allows distance determinations of usually up to 8 nm, in perdeuterated samples with an improved transverse relaxation time T2 more than 10 nm are feasible.41,42 Initially started as an X-Band technique, Q-Band measurements with increased sensitivity are on the rise.43 The experimental raw data shows a distance-dependent modulation, but is folded with a background function that can be determined individually. Data analysis after removal of the background leads to the desired distance distributions. A key advantage of DEER measurements is that they can be carried out virtually background-free even in complex environments as encountered e.g. in membranes or in the cytoplasm of cells.
Site directed spin labeling
A variety of protein classes, such as flavoproteins and photosystems, are intrinsically paramagnetic and can thus be directly monitored via EPR techniques.44,45 However, most proteins are intrinsically diamagnetic and become accessible for DEER distance measurements only by SDSL. The two central aspects of SDSL are the chemical and spectroscopic properties of the spin label itself, and the strategy used for its introduction into the protein under study. A large number of requirements apply to both aspects, and currently there is no general strategy that satisfactorily fulfills all of them. This makes it necessary to customize the SDSL strategy to the specific demands of the envisaged experiment.46–48A critical property of spin labels is the conformational flexibility of both the label scaffold itself and of the linker between this scaffold and the protein backbone. This contributes to the overall dynamics of the paramagnetic center, which complicates the analysis of protein dynamics.
A second important property of spin labels is the chemical stability of the label itself and of the linker, most importantly in the context of reducing and biological environments such as the cytoplasm of living cells. Therefore, nitroxide radicals (that are typically prone to e.g. reduction) are kinetically stabilized by steric shielding via bulky, quaternary carbon centres in α-position to the nitrogen atom with four methyl, ethyl or even larger alkyl substituents. However, larger spin labels expectedly have an increased potential to perturb the structure of the labeled protein. Spin labels based on chelated metal cations feature a high stability, but are often both bulky and charged.49–51
Besides these chemical and spectroscopic properties of the spin label, the strategies for its introduction into the target protein strongly differ in the scope of labels and target proteins that can be used, the applicability for in vitro or in cell studies, and last but no least the technical ease as well as labeling efficiency and selectivity.
For example, peptide synthesis (Fig. 2A) offers a wide scope of labels that can be introduced, including labels with excellent spectroscopic properties that cannot be introduced by other methods.46,47 However, the scope of applicable proteins is limited by the inherent difficulties of solid phase synthesis in the production of large proteins, and by the lack of natural folding processes and posttranslational modifications that may be required for natural function. Ligation of peptides to expressed proteins (Fig. 2B)52 offers access to larger proteins that can be at least in part naturally folded and modified, but still have limitations in respect to the possible incorporation sites and technical ease.
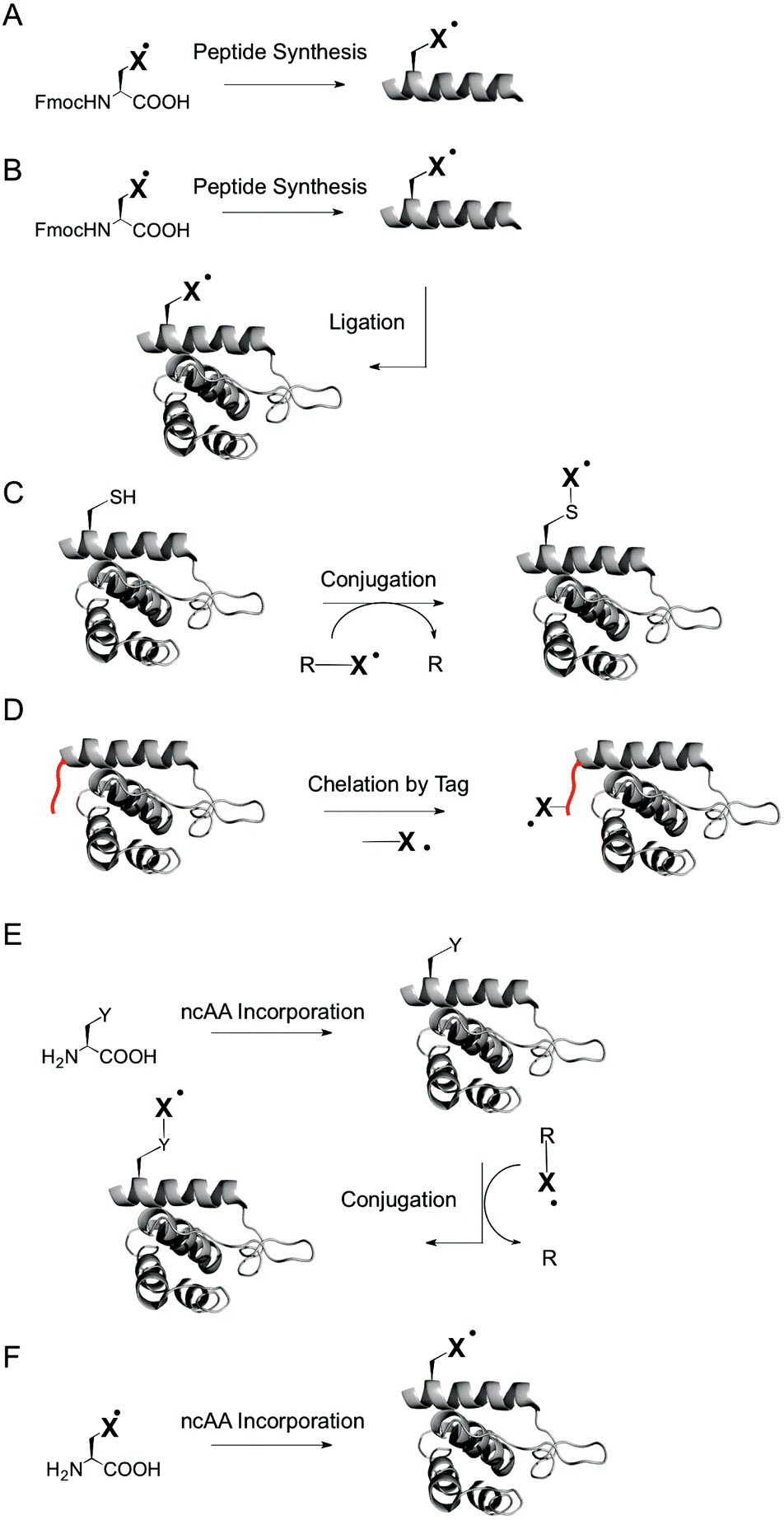 | ||
| Fig. 2 Protein SDSL strategies. (A) Solid phase peptide synthesis. (B) Peptide–protein ligation. (C) Conjugation reactions with canonical amino acids. (D) Chelation by genetically encoded peptide tags. (E) Conjugation reactions with genetically encoded ncAA. (F) Direct genetic encoding of spin labeled ncAA. | ||
Labeling of expressed proteins by chemoselective conjugation reactions at specific amino acids (Fig. 2C) offers the modification of fully natural, endogenous proteins with great technical ease.46–48 On the other hand, the scope of applicable spin labels for chemoselective labeling is reduced compared to approaches based on peptide synthesis. In particular, paramagnetic centers introduced via chemoselective labeling have a minimum flexibility that is dictated by the side chain of the targeted amino acid. Moreover, labeling canonical amino acids – most importantly cysteines – requires mutations to introduce cysteines at user-defined sites and is thus not applicable to proteins that rely on cysteines for their natural function, e.g. as part of catalytic centers or of disulfide bridges for structural stability. Targeting amino acids other than cysteine (such as lysine) suffers from analogous limitations. A solution to this problem are peptide tags (such as His-tags) for noncovalent introduction of spin labels via metal chelation (Fig. 2D),53,54 but for the cost of introducing comparably large changes into the protein, and with limitations in respect to the applicable sites.
The incorporation of noncanonical amino acids (ncAA) with unique reactivity by nonsense suppression, i.e. by translation with an expanded genetic code,55,56 offers chemoselective labeling of endogenous proteins irrespective of the presence of cysteines, and with introducing only minimal changes into the protein (Fig. 2E).57,58 Moreover, spin labels can be directly genetically encoded as ncAA during translation in vivo59–61 (and using microinjection with chemically aminoacylated tRNAs with lower protein yields in vitro62,63), without the need for conjugation reactions at the ncAA (Fig. 2F). Besides technical ease, a direct genetic encoding in vivo conceptually offers a high potential for intracellular studies, since proteins are biosynthesized directly in the cell, whereas chemical labelling procedures require the transformation or microinjection of labeled proteins into cells. This expectedly does not lead to a state that resembles the one of endogenous proteins that are e.g. processed and transported in and between specific sub-cellular compartments.
Nitroxide spin labels
The most commonly employed spin labels are nitroxides9 that exist in various designs and differ in overall ring structure, substituents in α-position to the nitrogen atom, and linkers (Fig. 3A shows a general structure that covers most nitroxides currently in use). Besides the mobility of the paramagnetic center, in particular the chemical stability of nitroxides differs widely, and is controlled by ring size and α-substituents as two major factors. Here, nitroxides with five-membered rings tend to be more stable than ones with six-membered rings. Moreover, increased steric shielding by larger alkyl α-substituents (e.g. ethyl, propyl) can contribute to stability.64–70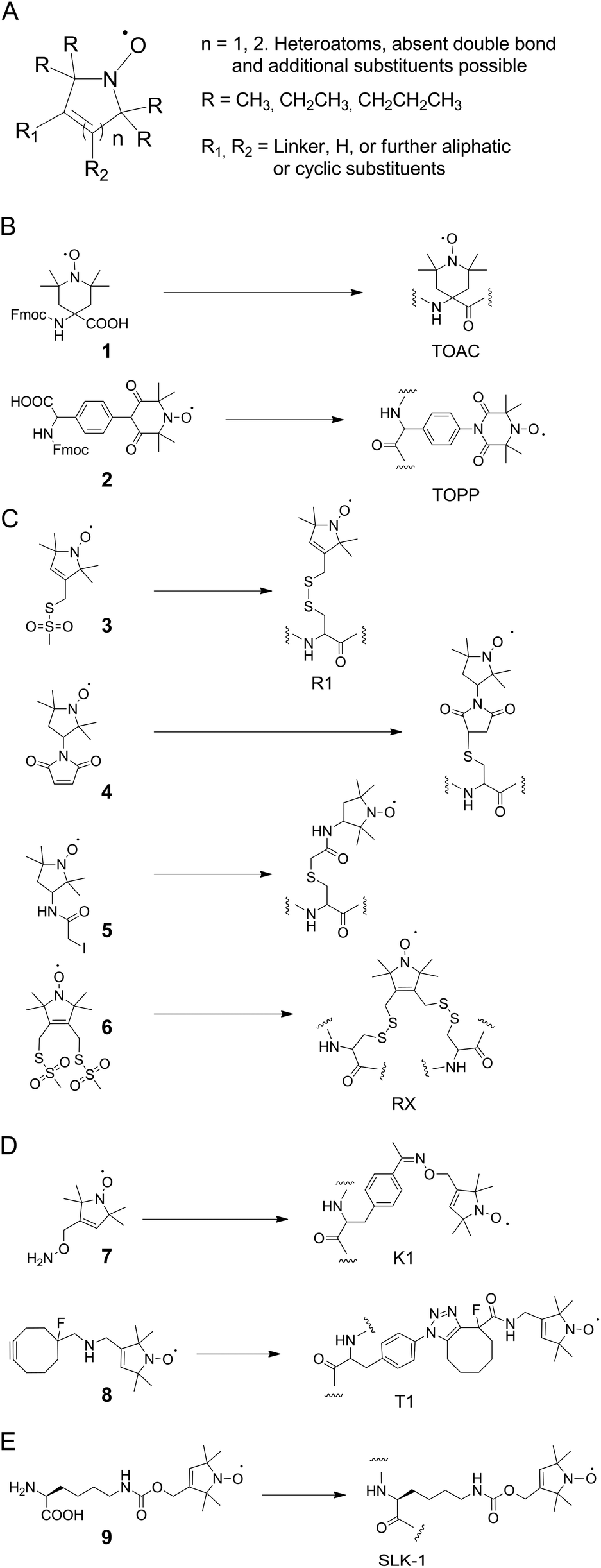 | ||
| Fig. 3 Nitroxide spin labels. (A) General structure of commonly employed nitroxide spin labels. (B) Nitroxides for peptide labeling by solid phase synthesis. (C) Sulfhydryl-reactive nitroxides for protein labeling by cysteine conjugation reactions. (D) Nitroxides for protein labeling by conjugation with genetically encoded ncAA. (E) Nitroxide ncAA for protein labeling by direct genetic encoding. | ||
Among the large variety of nitroxides that can be introduced into synthetic peptides by direct solid phase synthesis or post-synthetic labeling, the most widely used one is the 4-amino-1-oxyl-2,2,6,6,-tetramethyl-piperidine-4-carboxylic acid (TOAC 1, Fig. 3B) spin label.71,72 This label is exceptionally rigid and allows flexibility of the paramagnetic center only by flipping of the piperidine moiety. However, this rigidity also bears the potential of perturbing peptide secondary structures.
As a possible solution to this problem, the 4-(3,3,5,5-tetramethyl-2,6-dioxo-4-oxylpiperazin-1-yl)-L-phenylglycine spin label 2 (TOPP, Fig. 3B)73 has been developed that lowers this risk, but with an expectedly higher flexibility. Both labels however can only be introduced into peptides by solid phase synthesis and have low67 or unknown stabilities in biological environments, respectively.
The by far most popular nitroxide for SDSL of proteins by conjugation to canonical amino acids is the sulfhydryl-reactive methanethiosulfonate spin label 3 (MTSSL, Fig. 3C).74 MTSSL readily reacts with accessible cysteine residues in proteins under formation of the side chain R1 (Fig. 3C). The conformational properties of R1 are well characterized and the label has been used in a vast variety of proteins.75,76 Moreover, the 1-oxyl-2,2,5,5-tetramethyl-pyrroline moiety of MTSSL is comparably stable in reducing/biological environments.64 However, since R1 bears a disulfide linkage that is by itself redox-sensitive, alternative labels with stable linkers for cysteine labeling have been introduced, among them the maleimides and iodoacetamides 4 and 5 (Fig. 3C). Compared to TOAC and TOPP, all cysteine-reactive nitroxides result in side chains with increased flexibility. However, an interesting concept to improve the rigidity of such labels are double sulfhydryl-reactive labels that can be used for conjugation to two cysteines in spatial vicinity.77 Here, the side chain RX formed by reaction with 6 (Fig. 3C) has been introduced into different secondary structural elements of T4 lysozyme and exhibited reduced flexibility compared to R1. To overcome inherent limitations of cysteine labeling, ncAA with reactive side chains for bioorthogonal conjugation reactions can be incorporated into proteins in vivo by nonsense codon suppression using orthogonal tRNA/aminoacyl-tRNA-synthetase (aaRS) pairs.55,56 The first study in this direction employed an evolved tyrosyl-tRNA-synthetase from Methanocaldococcus jannaschii and the genetically encoded ketone-bearing ncAA p-acetyl-L-phenylalanine (p-AcF). This ncAA was reacted with the aminooxy-functionalized nitroxide 7via oxime formation in the context of T4 lysozyme.57 This resulted in side chain K1 (Fig. 3D) that exhibited useful spectroscopic properties, though compared to R1 with increased flexibility. The requirement for low pH or for the presence of high concentration of catalyst for oxime formation and the moderate kinetics of the reaction make it however desirable to employ faster and more bioorthogonal chemistries such as copper-free click reactions as available for genetically encoded ncAA.56,78 Indeed, a proof of principle has been demonstrated for the conjugation of the cyclooctyne-bearing nitroxide 8 that was reacted with the aryl azide ncAA p-azido-L-phenylalanine (p-AzF) in T4 lysozyme, resulting in side chain T1.60 However, the very large size of T1 is suggestive of a high flexibility, and the spectroscopic properties of T1 have not yet been thoroughly studied. Finally, the nitroxide ncAA 9 (SLK-1, Fig. 4) has been genetically encoded using an evolved Methanosarcina mazei pyrrolysyl-tRNA-synthetase mutant,79 allowing for the co-translational incorporation of nitroxides directly in E. coli cells.59,61 SLK-1 was introduced into multiple sites of GFP and thioredoxin and exhibited similar flexibility as MTSSL in simulations and in DEER distance measurements.60
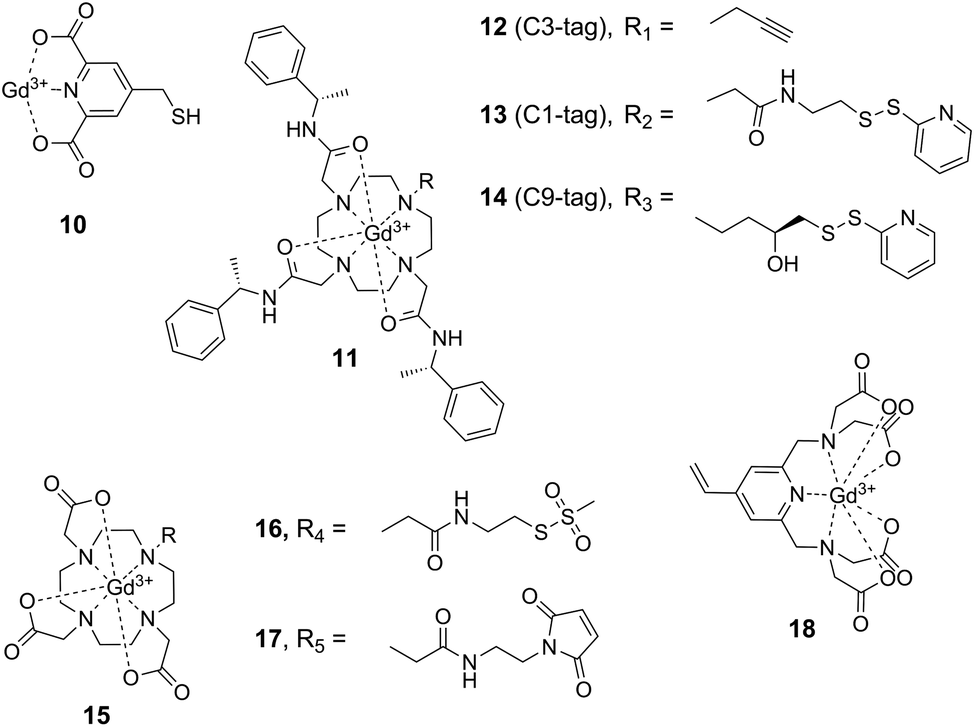 | ||
| Fig. 4 Commonly applied Gd3+-labels in SDSL of proteins. | ||
Spin labels based on metal cations
A major drawback of nitroxide-based spin labels is their limited lifetime in intracellular environments60,67 which limits their use for in-cell EPR structural studies on proteins (see below). A promising approach to circumvent this limitation involves the application of reduction-stable, paramagnetic metal cations and their attachment to proteins using conjugation or complexation strategies. Lanthanide tags based on the complexation of Gd3+ ions have proven highly useful for EPR distance measurements on proteins in vitro,49 but also in intracellular environments.50,51,80 Prerequisite for the application of paramagnetic cations in SDSL is a suitable complexation approach. Since free Gd3+ ions decrease data quality in DEER experiments,81 high affinity chelating agents are necessary to stably complex the Gd3+ ion. Affinity is largely controlled by the denticity of the complex, imposing the necessity of rather bulky chelating ligands. This increases the overall size of the label and thus the potential to perturb the structure of the protein under study. A variety of Gd3+ spin labels have been developed and have been shown to be applicable for protein SDSL when introduced via sulfhydryl-directed conjugation chemistries (Fig. 4).The first report of Gd3+-labeled proteins in DEER measurements was based on pre-activating cysteine residues with Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid)) and subsequently conjugating 4-mercaptomethyldipicolinic acid 10 (4MMDPA) followed by Gd3+ complexation (Fig. 4).82 To avoid titration with Gd3+ ions after labeling and thus to reduce free Gd3+ species, phenylethyl-derived Gd3+–DOTA 11 (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) pre-complexes were also applied in SDSL approaches (Fig. 4).83–85 The bulkiness of this complex is sterically reducing the mobility of the paramagnetic Gd3+ center; assuming the structure to remain unperturbed by the size of the complex, the steric restriction may provide a narrower distance distribution.85 The activated disulfide-bond reagent 13 (C1-tag, Fig. 4) was applied for labeling unique cysteine residues, and DEER measurements were performed to investigate chaperon ERp29 in W-band.83 Further improving the spectroscopic properties of these labels, a more rigid linker was then established and employed in the cysteine-reactive label 14 (C9-tag, Fig. 4) to resolve rather narrow distance distributions in DEER experiments with high resolution.85 Gd3+–DOTA complexes 15 have also been introduced into proteins via methanethiosulfonate84,86 and maleimide51 moieties 16 and 17 for cysteine conjugation, the latter strategy offering redox-stable linkages for in cell EPR measurements.51 Moreover, the sulfhydryl-reactive vinyl-derived Gd3+-complex 18 has been shown to be applicable for in-cell distance measurements.50
Similar to nitroxide spin labeling, the range of conjugation chemistries has also been extended for metal cation-based labeling, from targeting cysteine to targeting ncAA with alternative bioorthogonal chemistries. By employing an evolved orthogonal tyrosyl-tRNA synthetase from M. jannaschii for nonsense codon suppression in vivo, the ncAA p-AzF was site-specifically incorporated into various sites of the E. coli aspartate/glutamate binding protein.87 Labeling with the alkyne-modified label 12 (C3-tag, Fig. 4) by copper click chemistry resulted in a conformational fixation of the complex via the participation of the formed triazole ring in Gd3+-complexation, facilitating theoretical predictions due to the rigidity of the complex.88 Though being potentially interesting for in cell applications, this approach suffers from chemical conversion of the azide moiety inside cells and could thus only be applied using a cell-free protein synthesis approach.89 Until today, the labeling of proteins using Gd3+-tags could not be demonstrated inside cells, making invasive microinjection or transfection protocols necessary for intracellular studies.
Besides Gd3+, the transition metals Cu2+ and Mn2+ have been shown to be interesting paramagnetic centers in protein SDSL EPR experiments. In a first study, Mn2+–EDTA methanethiosulfonate tags 19 were covalently attached to unique cysteine residues on the death domain of a neurotrophin receptor, and subjected to Mn2+–Mn2+ distance measurements (Fig. 5A).90 To overcome limitations concerning the stability of the formed disulfide-bonds, the Mn2+ binding ligands 20–22 based on a PEDTA chelator (N-(pyrid-2-yl-methyl)ethylene-diamine-N,N′,N′-triacetic acid) were developed to introduce the Mn2+ label via C–S bond formation into proteins (L1, L2 and L3, Fig. 5A).91 Finally, Mn2+ ions have been introduced site-specifically into proteins via the genetically encoded ncAA 8-hydroxyquinoline alanine that was co-translationally incorporated into proteins in vivo by nonsense suppression (Fig. 5B).92,93 Labeling proteins with Cu2+ ions has been achieved by EDTA and TETAC complexation (Fig. 5A and C)94 or recently, by exploiting a rigid, double-histidine binding motif in combination with iminodiacetate to resolve distances with remarkable resolution (Fig. 5D).54 These transition metal labeling strategies can also be applied in combination with nitroxide spin labels to measure distance distributions.23
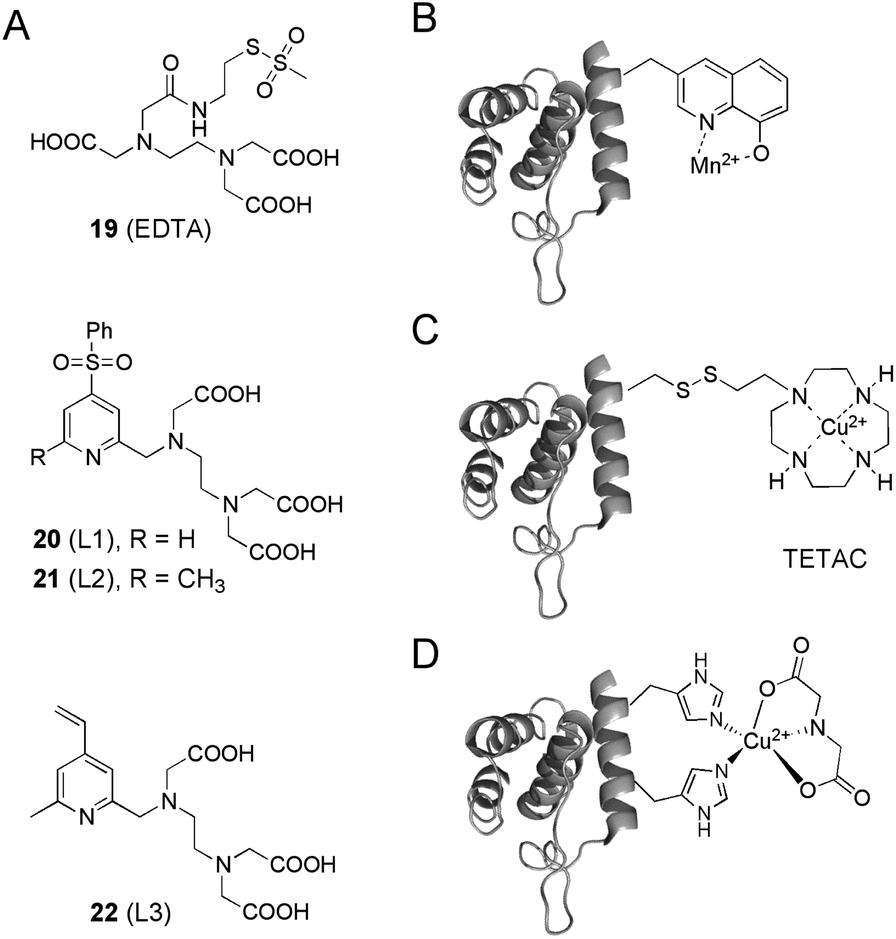 | ||
| Fig. 5 Labels and labeling strategies to introduce Mn2+ or Cu2+ cations into proteins. | ||
Trityl-based spin-labels
Since EPR distance measurements usually have to be performed at cryogenic temperatures, trityl radical spin-labels were investigated in SDSL and distance measurements at physiologically relevant temperatures to overcome this limitation. Measurements at ambient temperature can potentially resolve conformational sub-states, which might not be accessible following traditional measurements in frozen solution. Especially tetrathiatriarylmethyl-based spin-labels feature beneficial spectroscopic properties, showing one narrow signal with a line width of 90 mG (ref. 95) and a prolonged transverse relaxation time in the microsecond range, making them suitable candidates for measurements at ambient temperature.96Interestingly, increased stabilities in human blood were reported, featuring half-lives of up to 24 h.97 Besides their applicability as oxygen sensors,97–99 trityl-based spin labels could be applied in distance measurements of polymers100 and nucleic acids (at 37 °C).101 Recently, the first report of distance measurements using a doubly TAM-labeled protein could demonstrate that protein distance measurements in liquid solution can be feasible. A cysteine-reactive TAM-reagent (Fig. 6) was applied to label the rigid C helix of T4-lysozyme, which was immobilized and subjected to double quantum coherence (DQC) distances measurements at 4 °C.26 Although a sharp distance distribution of 2 nm could be resolved, further improvements to increase the Tm of the TAM-label have to be conducted to expand the range of accessible distances beyond 2 nm.
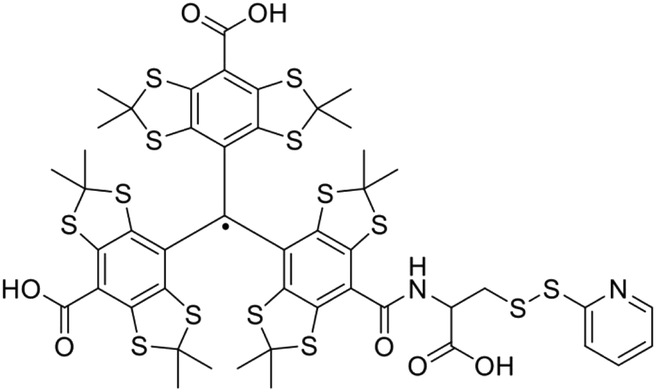 | ||
| Fig. 6 Cysteine-reactive TAM-reagent, which was applied in SDSL of T4-lysozyme. | ||
In cell SDSL EPR studies of proteins
In cell EPR and in particular in cell DEER distance measurements in combination with SDSL can provide insights into the structural dynamics of proteins in living cells as basis of their physiological function. This field has so far been approached using both nitroxide and metal cation-based spin labels, and using different labeling strategies. First studies used a complex transfection of previous in vitro chemically spin-labeled organic macromolecules in model cells (e.g. Xenopus oocytes).102–104 Apart from the considerable experimental effort of such experiments, Xenopus oocytes represent an artificial environment for all non-Xenopus proteins, i.e. without natural processing of the target protein by folding, transport, post-translational modifications and degradation.A bacterial outer membrane protein was spin-labeled via traditional MTSSL labeling of unique cysteine residues in live E. coli cells, and the proteins conformational flexibility upon ligand binding was studied.105,106 Here, the difficulties concerning the reduction of nitroxide radicals (and disulfide bridges) in cells were circumvented by studying proteins on the cell surface rather than in the cytoplasm. In contrast, the nitroxide ncAA 9 (SLK-1) was used for the intracellular biosynthesis of E. coli thioredoxin in the cytoplasm of live E. coli cells by translation with an expanded genetic code. However, the experiment was limited to the selective detection of the protein in live E. coli cells and no in cell DEER distance measurements were demonstrated. The limited stability of nitroxides in cells has led to strategies using redox-stable Gd3+ labels for in cell DEER distance measurements. This approach has led to successful in cell distance measurements in proteins in several cases, highlighting the superior stability of the employed Gd3+ complexes in the tested cell types. However, these labels can so far only be introduced chemically into proteins and thus require microinjection or transfection procedures to deliver the labeled protein into living cells.50,51 This does not allow for studying proteins in their truly natural state, that is, after natural translation, folding, transport, modification and degradation.
Conclusions and outlook
Though a large variety of spin labels and strategies for their introduction into proteins are now available for SDSL EPR studies, there is still no ideal approach for all applications. Instead, available approaches have unique profiles with specific strengths and weaknesses in respect to label size, flexibility, stability, and to the potential to study proteins in their natural biological environments. One of the most exciting perspectives of SDSL EPR is the emerging field of in cell EPR. Here, the use of stable Gd3+ labels has recently enabled important breakthroughs,50,51,107 and is expected to greatly advance the significance of EPR for cell biology. However, the current requirement to deliver chemically labeled proteins into cells does not fully meet the ultimate goal of in cell EPR, that is, studying endogenous, naturally translated and processed proteins directly in their natural host cells. The perspective of studying membrane proteins by chemical spin labeling, though limited to the cell surface, does offer this potential, and could deliver insights into biological processes based on proteins that are otherwise difficult to study due to size and crystallization properties. Moreover, the use of genetically encoded ncAA offers advancements for bioorthogonal cell surface (and even cytoplasmic) labelling by rapid click chemistries, using both nitroxide or Gd3+ labels. Alternatively, the direct genetic encoding of stable spin labeled ncAA offers the potential for further simplifications and may allow the investigation of proteins directly in their natural cellular environment with minimal perturbation. These combined developments have the potential to redefine the role of EPR structural studies for cell biology by providing precise insights into the physiological function of proteins that are otherwise difficult to access.Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DR 743/7-1 and SU 726/4-2 in SPP1601, in SFB 969, and SU 726/2-2 in SPP1623).References
- C. Altenbach, S. L. Flitsch, H. G. Khorana and W. L. Hubbell, Biochemistry, 1989, 28, 7806–7812 CrossRef CAS PubMed.
- C. Altenbach, T. Marti, H. G. Khorana and W. L. Hubbell, Science, 1990, 248, 1088–1092 CAS.
- L. J. Berliner, Spin labeling: theory and applications, Academic Press, New York, 1976 Search PubMed.
- G. Jeschke, M. Sajid, M. Schulte, N. Ramezanian, A. Volkov, H. Zimmermann and A. Godt, J. Am. Chem. Soc., 2010, 132, 10107–10117 CrossRef CAS PubMed.
- C. Hintze, F. Schütze, M. Drescher and S. Mecking, Phys. Chem. Chem. Phys., 2015, 17, 32289–32296 RSC.
- G. W. Reginsson and O. Schiemann, in Encyclopedia of Biophysics, Springer, 2013, pp. 2429–2431 Search PubMed.
- J.-J. Yin, M. Pasenkiewicz-Gierula and J. S. Hyde, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 964–968 CrossRef CAS.
- J. Wertz, Electron spin resonance: elementary theory and practical applications, Springer Science & Business Media, 2012 Search PubMed.
- O. H. Griffith and A. Waggoner, Acc. Chem. Res., 1969, 2, 17–24 CrossRef CAS.
- J. H. Freed, Spin Labeling: theory and applications, 1976, vol. 1, pp. 53–132 Search PubMed.
- L. Berliner, Spin labeling II: Theory and applications, Academic, New York, 1979 Search PubMed.
- M. Brustolon, Electron Paramagnetic Resonance: A Practitioner's Toolkit, 2009, pp. 83–108 Search PubMed.
- W. L. Hubbell, H. S. McHaourab, C. Altenbach and M. A. Lietzow, Structure, 1996, 4, 779–783 CrossRef CAS PubMed.
- J. M. Isas, R. Langen, H. T. Haigler and W. L. Hubbell, Biochemistry, 2002, 41, 1464–1473 CrossRef CAS PubMed.
- Y. N. Molin, K. M. Salikhov and K. I. Zamaraev, Spin exchange, Springer-Verlag, 1980 Search PubMed.
- C. Altenbach, W. Froncisz, R. Hemker, H. Mchaourab and W. L. Hubbell, Biophys. J., 2005, 89, 2103–2112 CrossRef CAS PubMed.
- T. I. Smirnova and A. I. Smirnov, in ESR spectroscopy in Membrane Biophysics, Springer, 2007, pp. 165–251 Search PubMed.
- K. A. Earle, J. K. Moscicki, M. Ge, D. E. Budil and J. H. Freed, Biophys. J., 1994, 66, 1213 CrossRef CAS PubMed.
- O. Griffith, P. Dehlinger and S. Van, J. Membr. Biol., 1974, 15, 159–192 CrossRef CAS PubMed.
- H.-J. Steinhoff, A. Savitsky, C. Wegener, M. Pfeiffer, M. Plato and K. Möbius, Biochim. Biophys. Acta, 2000, 1457, 253–262 CrossRef CAS.
- G. Jeschke and H. W. Spiess, in Novel NMR and EPR techniques, Springer, 2006, pp. 21–63 Search PubMed.
- H. S. McHaourab, M. A. Lietzow, K. Hideg and W. L. Hubbell, Biochemistry, 1996, 35, 7692–7704 CrossRef CAS PubMed.
- Z. Yang, G. Jiménez-Osés, C. J. López, M. D. Bridges, K. N. Houk and W. L. Hubbell, J. Am. Chem. Soc., 2014, 136, 15356–15365 CrossRef CAS PubMed.
- A. Rajca, V. Kathirvelu, S. K. Roy, M. Pink, S. Rajca, S. Sarkar, S. S. Eaton and G. R. Eaton, Chem. – Eur. J., 2010, 16, 5778–5782 CrossRef CAS PubMed.
- V. Meyer, M. A. Swanson, L. J. Clouston, P. J. Boratynski, R. A. Stein, H. S. McHaourab, A. Rajca, S. S. Eaton and G. R. Eaton, Biophys. J., 2015, 108, 1213–1219 CrossRef CAS PubMed.
- Z. Yang, Y. Liu, P. Borbat, J. L. Zweier, J. H. Freed and W. L. Hubbell, J. Am. Chem. Soc., 2012, 134, 9950–9952 CrossRef CAS PubMed.
- J. E. Banham, C. M. Baker, S. Ceola, I. J. Day, G. H. Grant, E. J. Groenen, C. T. Rodgers, G. Jeschke and C. R. Timmel, J. Magn. Reson., 2008, 191, 202–218 CrossRef CAS PubMed.
- P. P. Borbat and J. H. Freed, Chem. Phys. Lett., 1999, 313, 145–154 CrossRef CAS.
- P. P. Borbat and J. H. Freed, in Distance measurements in biological systems by EPR, Springer, 2002, pp. 383–459 Search PubMed.
- S. S. Eaton and G. R. Eaton, in Distance Measurements in Biological Systems by EPR, ed. L. J. Berliner, G. R. Eaton and S. S. Eaton, Springer US, Boston, MA, 2002, pp. 347–381, DOI:10.1007/0-306-47109-4_8.
- K. V. Lakshmi and G. W. Brudvig, in Distance Measurements in Biological Systems by EPR, ed. L. J. Berliner, G. R. Eaton and S. S. Eaton, Springer US, Boston, MA, 2002, pp. 513–567, DOI:10.1007/0-306-47109-4_12.
- S. Razzaghi, E. K. Brooks, E. Bordignon, W. L. Hubbell, M. Yulikov and G. Jeschke, ChemBioChem, 2013, 14, 1883–1890 CrossRef CAS PubMed.
- A. Milov, K. Salikhov and M. Shirov, Fiz. Tverd. Tela, 1981, 23, 975–982 CAS.
- A. Milov, A. Ponomarev and Y. D. Tsvetkov, Chem. Phys. Lett., 1984, 110, 67–72 CrossRef CAS.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- G. Jeschke, A. Bender, H. Paulsen, H. Zimmermann and A. Godt, J. Magn. Reson., 2004, 169, 1–12 CrossRef CAS PubMed.
- A. M. Bowen, C. E. Tait, C. R. Timmel and J. R. Harmer, in Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences, Springer, 2013, pp. 283–327 Search PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419 CrossRef CAS PubMed.
- S. S. Eaton and G. R. Eaton, in Distance Measurements in Biological Systems by EPR, Springer, 2002, pp. 1–27 Search PubMed.
- A. Schweiger and G. Jeschke, Principles of pulse electron paramagnetic resonance, Oxford University Press, 2001 Search PubMed.
- R. Ward, A. Bowman, E. Sozudogru, H. El-Mkami, T. Owen-Hughes and D. G. Norman, J. Magn. Reson., 2010, 207, 164–167 CrossRef CAS PubMed.
- A. Bowman, C. M. Hammond, A. Stirling, R. Ward, W. Shang, H. El-Mkami, D. A. Robinson, D. I. Svergun, D. G. Norman and T. Owen-Hughes, Nucleic Acids Res., 2014, 42, 6038–6051 CrossRef CAS PubMed.
- P. Zou and H. S. Mchaourab, Biophys. J., 2010, 98, L18–L20 CrossRef CAS PubMed.
- C. W. Kay, R. Feicht, K. Schulz, P. Sadewater, A. Sancar, A. Bacher, K. Möbius, G. Richter and S. Weber, Biochemistry, 1999, 38, 16740–16748 CrossRef CAS PubMed.
- R. Bittl and S. G. Zech, J. Phys. Chem. B, 1997, 101, 1429–1436 CrossRef CAS.
- J. P. Klare and H. J. Steinhoff, Photosynth. Res., 2009, 102, 377 CrossRef CAS PubMed.
- J. P. Klare, Biol. Chem., 2013, 394, 1281–1300 CrossRef CAS PubMed.
- W. L. Hubbell, C. J. Lopez, C. Altenbach and Z. Yang, Curr. Opin. Struct. Biol., 2013, 23, 725–733 CrossRef CAS PubMed.
- A. Potapov, H. Yagi, T. Huber, S. Jergic, N. E. Dixon, G. Otting and D. Goldfarb, J. Am. Chem. Soc., 2010, 132, 9040–9048 CrossRef CAS PubMed.
- M. Qi, A. Gross, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS PubMed.
- A. Martorana, G. Bellapadrona, A. Feintuch, E. Di Gregorio, S. Aime and D. Goldfarb, J. Am. Chem. Soc., 2014, 136, 13458–13465 CrossRef CAS PubMed.
- C. F. W. Becker, K. Lausecker, M. Balog, T. Kalai, K. Hideg, H. J. Steinhoff and M. Engelhard, Magn. Reson. Chem., 2005, 43, S34–S39 CrossRef CAS PubMed.
- C. Baldauf, K. Schulze, P. Lueders, E. Bordignon and R. Tampe, Chemistry, 2013, 19, 13714–13719 CrossRef CAS PubMed.
- T. F. Cunningham, M. R. Putterman, A. Desai, W. S. Horne and S. Saxena, Angew. Chem., Int. Ed., 2015, 54, 6330–6334 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- M. R. Fleissner, E. M. Brustad, T. Kalai, C. Altenbach, D. Cascio, F. B. Peters, K. Hideg, S. Peuker, P. G. Schultz and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21637 CrossRef CAS PubMed.
- T. Kalai, M. R. Fleissner, J. Jeko, W. L. Hubbell and K. Hideg, Tetrahedron Lett., 2011, 52, 2747–2749 CrossRef CAS.
- M. J. Schmidt, A. Fedoseev, D. Summerer and M. Drescher, Methods Enzymol., 2015, 563, 483–502 CAS.
- M. J. Schmidt, A. Fedoseev, D. Bucker, J. Borbas, C. Peter, M. Drescher and D. Summerer, ACS Chem. Biol., 2015, 10, 2764–2771 CrossRef CAS PubMed.
- M. J. Schmidt, J. Borbas, M. Drescher and D. Summerer, J. Am. Chem. Soc., 2014, 136, 1238–1241 CrossRef CAS PubMed.
- V. W. Cornish, D. R. Benson, C. A. Altenbach, K. Hideg, W. L. Hubbell and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2910 CrossRef CAS.
- A. M. Shafer, T. Kalai, S. Q. B. Liu, K. Hideg and J. C. Voss, Biochemistry, 2004, 43, 8470 CrossRef CAS PubMed.
- J. T. Paletta, M. Pink, B. Foley, S. Rajca and A. Rajca, Org. Lett., 2012, 14, 5322–5325 CrossRef CAS PubMed.
- F. Hyodo, K. Matsumoto, A. Matsumoto, J. B. Mitchell and M. C. Krishna, Cancer Res., 2006, 66, 9921–9928 CrossRef CAS PubMed.
- L. Marx, R. Chiarelli, T. Guiberteau and A. Rassat, J. Chem. Soc., Perkin Trans. 1, 2000, 1181–1182, 10.1039/b001891k.
- M. Azarkh, O. Okle, P. Eyring, D. R. Dietrich and M. Drescher, J. Magn. Reson., 2011, 212, 450–454 CrossRef CAS PubMed.
- A. A. Bobko, I. A. Kirilyuk, I. A. Grigor'ev, J. L. Zweier and V. V. Khramtsov, Free Radicals Biol. Med., 2007, 42, 404–412 CrossRef CAS PubMed.
- M. Emoto, F. Mito, T. Yamasaki, K. I. Yamada, H. Sato-Akaba, H. Hirata and H. Fujii, Free Radical Res., 2011, 45, 1325–1332 CrossRef CAS PubMed.
- A. P. Jagtap, I. Krstic, N. C. Kunjir, R. Hänsel, T. F. Prisner and S. T. Sigurdsson, Free Radical Res., 2015, 49, 78–85 CrossRef CAS PubMed.
- A. Rassat and P. Rey, Bull. Soc. Chim. Fr., 1967, 3, 815–818 CAS.
- C. R. Nakaie, G. Goissis, S. Schreier and A. C. Paiva, Braz. J. Med. Biol. Res., 1981, 14, 173–180 CAS.
- S. Stoller, G. Sicoli, T. Y. Baranova, M. Bennati and U. Diederichsen, Angew. Chem., Int. Ed., 2011, 50, 9743–9746 CrossRef CAS PubMed.
- C. Altenbach, T. Marti, H. G. Khorana and W. L. Hubbell, Science, 1990, 248, 1088 CAS.
- M. R. Fleissner, D. Cascio and W. L. Hubbell, Protein Sci., 2009, 18, 893–908 CrossRef CAS PubMed.
- L. Columbus, T. Kalai, J. Jeko, K. Hideg and W. L. Hubbell, Biochemistry, 2001, 40, 3828–3846 CrossRef CAS PubMed.
- M. R. Fleissner, M. D. Bridges, E. K. Brooks, D. Cascio, T. Kalai, K. Hideg and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16241–16246 CrossRef CAS PubMed.
- M. J. Schmidt and D. Summerer, ChemBioChem, 2012, 13, 1553–1557 CrossRef CAS PubMed.
- M. J. Schmidt, A. Weber, M. Pott, W. Welte and D. Summerer, ChemBioChem, 2014, 15, 1755–1760 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- D. Goldfarb, Phys. Chem. Chem. Phys., 2014, 16, 9685–9699 RSC.
- X. C. Su, T. Huber, N. E. Dixon and G. Otting, ChemBioChem, 2006, 7, 1599–1604 CrossRef CAS PubMed.
- H. Yagi, D. Banerjee, B. Graham, T. Huber, D. Goldfarb and G. Otting, J. Am. Chem. Soc., 2011, 133, 10418–10421 CrossRef CAS PubMed.
- E. Matalon, T. Huber, G. Hagelueken, B. Graham, V. Frydman, A. Feintuch, G. Otting and D. Goldfarb, Angew. Chem., Int. Ed., 2013, 52, 11831–11834 CrossRef CAS PubMed.
- E. H. Abdelkader, M. D. Lee, A. Feintuch, M. R. Cohen, J. D. Swarbrick, G. Otting, B. Graham and D. Goldfarb, J. Phys. Chem. Lett., 2015, 6, 5016–5021 CrossRef CAS PubMed.
- D. Thonon, V. Jacques and J. F. Desreux, Contrast Media Mol. Imaging, 2007, 2, 24–34 CrossRef CAS PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- C. T. Loh, K. Ozawa, K. L. Tuck, N. Barlow, T. Huber, G. Otting and B. Graham, Bioconjugate Chem., 2013, 24, 260–268 CrossRef CAS PubMed.
- E. H. Abdelkader, A. Feintuch, X. Yao, L. A. Adams, L. Aurelio, B. Graham, D. Goldfarb and G. Otting, Chem. Commun., 2015, 51, 15898–15901 RSC.
- D. Banerjee, H. Yagi, T. Huber, G. Otting and D. Goldfarb, J. Phys. Chem. Lett., 2012, 3, 157–160 CrossRef CAS.
- A. Martorana, Y. Yang, Y. Zhao, Q. F. Li, X. C. Su and D. Goldfarb, Dalton Trans., 2015, 44, 20812–20816 RSC.
- S. H. Park, V. S. Wang, J. Radoicic, A. A. De Angelis, S. Berkamp and S. J. Opella, J. Biomol. NMR, 2015, 61, 185–196 CrossRef CAS PubMed.
- H. S. Lee, G. Spraggon, P. G. Schultz and F. Wang, J. Am. Chem. Soc., 2009, 131, 2481–2483 CrossRef CAS PubMed.
- T. F. Cunningham, M. D. Shannon, M. R. Putterman, R. J. Arachchige, I. Sengupta, M. Gao, C. P. Jaroniec and S. Saxena, J. Phys. Chem. B, 2015, 119, 2839–2843 CrossRef CAS PubMed.
- T. J. Reddy, T. Iwama, H. J. Halpern and V. H. Rawal, J. Org. Chem., 2002, 67, 4635–4639 CrossRef CAS PubMed.
- R. Owenius, G. R. Eaton and S. S. Eaton, J. Magn. Reson., 2005, 172, 168–175 CrossRef CAS PubMed.
- J. H. Ardenkjaer-Larsen, I. Laursen, I. Leunbach, G. Ehnholm, L. G. Wistrand, J. S. Petersson and K. Golman, J. Magn. Reson., 1998, 133, 1–12 CrossRef CAS PubMed.
- Y. Liu, F. A. Villamena, J. Sun, Y. Xu, I. Dhimitruka and J. L. Zweier, J. Org. Chem., 2008, 73, 1490–1497 CrossRef CAS PubMed.
- S. Xia, F. A. Villamena, C. M. Hadad, P. Kuppusamy, Y. Li, H. Zhu and J. L. Zweier, J. Org. Chem., 2006, 71, 7268–7279 CrossRef CAS PubMed.
- G. W. Reginsson, N. C. Kunjir, S. T. Sigurdsson and O. Schiemann, Chemistry, 2012, 18, 13580–13584 CrossRef CAS PubMed.
- G. Y. Shevelev, O. A. Krumkacheva, A. A. Lomzov, A. A. Kuzhelev, O. Y. Rogozhnikova, D. V. Trukhin, T. I. Troitskaya, V. M. Tormyshev, M. V. Fedin, D. V. Pyshnyi and E. G. Bagryanskaya, J. Am. Chem. Soc., 2014, 136, 9874–9877 CrossRef CAS PubMed.
- I. Krstic, R. Hansel, O. Romainczyk, J. W. Engels, V. Dotsch and T. F. Prisner, Angew. Chem., Int. Ed., 2011, 50, 5070 CrossRef CAS PubMed.
- M. Azarkh, O. Okle, V. Singh, I. T. Seemann, J. S. Hartig, D. R. Dietrich and M. Drescher, ChemBioChem, 2011, 12, 1992 CrossRef CAS PubMed.
- R. Igarashi, T. Sakai, H. Hara, T. Tenno, T. Tanaka, H. Tochio and M. Shirakawa, J. Am. Chem. Soc., 2010, 132, 8228–8229 CrossRef CAS PubMed.
- B. Joseph, A. Sikora, E. Bordignon, G. Jeschke, D. S. Cafiso and T. F. Prisner, Angew. Chem., Int. Ed., 2015, 54, 6196–6199 CrossRef CAS PubMed.
- B. Joseph, A. Sikora and D. S. Cafiso, J. Am. Chem. Soc., 2016, 138(6), 1844–1847 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum and D. Goldfarb, Nature, 2016, 530(7588), 45–50 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |