
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDelivering aminopyridine ligands into cancer cells through conjugation to the cell-penetrating peptide BP16†
M.
Soler
ab,
M.
González-Bártulos
ac,
E.
Figueras
b,
A.
Massaguer
*c,
L.
Feliu
*b,
M.
Planas
*b,
X.
Ribas
*a and
M.
Costas
*a
aQBIS-CAT Research Group, Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus Montilivi, E-17071 Girona, Catalonia, Spain. E-mail: xavi.ribas@udg.edu; miquel.costas@udg.edu
bLIPPSO, Departament de Química, Universitat de Girona, Campus Montilivi, E-17071 Girona, Catalonia, Spain. E-mail: lidia.feliu@udg.edu; marta.planas@udg.edu
cDepartament de Biologia, Universitat de Girona, Campus Montilivi, E-17071 Girona, Catalonia, Spain. E-mail: anna.massaguer@udg.edu
First published on 4th April 2016
Abstract
Peptide conjugates incorporating the N-based ligands Me2PyTACN or (S,S′)-BPBP at the N- or the C-terminus of the cell-penetrating peptide BP16 were synthesized (PyTACN–BP16 (BP341), BP16-PyTACN (BP342), BPBP–BP16 (BP343), and BP16-BPBP (BP344)). Metal binding peptides bearing at the N-terminus the ligand, an additional Lys and a β-Ala were also prepared (PyTACN-βAK–BP16 (BP345) and BPBP-βAK–BP16 (BP346)). Moreover, taking into account the clathrin-dependent endocytic mechanism of BP16, the enzymatic cleavable tetrapeptide Gly-Phe-Leu-Gly was incorporated between the ligand and the N- or C-terminus of BP16 (BPBP-GFLG-BP16 (BP347) and BP16-GLFG-BPBP (BP348)). Analysis of the cytotoxicity of all the peptide conjugates showed that: (i) the position of the ligand influenced the IC50 values, (ii) the incorporation of the βAla-Lys dipeptide rendered non active sequences, (iii) peptide conjugates derived from the (S,S′)-BPBP ligand were more active than those bearing Me2PyTACN, and (iv) the introduction of the cleavable tetrapeptide significantly enhanced the activity of the BPBP conjugates (IC50 of 4.3 to 11.7 μM (BP347 and BP348) compared to 26.0 to >50 μM (BP343, BP344 and BP346)). The most active peptide was BPBP-GFLG-BP16 (BP347) (IC50 of 4.3 to 5.0 μM). This high activity was attributed to its high internalization in MCF-7 cells, as shown by flow cytometry, and to the subsequent release of the ligand by the intracellular cleavage of the enzyme-labile spacer, as observed in cathepsin B enzymatic assays. Therefore, these results pave the way for the design of novel peptide conjugates to be used in pro-oxidant anticancer therapies.
Introduction
Great strides have been made in recent years in finding effective cancer treatments. Despite the significant efforts invested in this field, current anticancer therapies are hindered by the poor solubility of drugs as well as their low cellular uptake and lack of selectivity. In this sense, targeted drug delivery has emerged as a convenient strategy to overcome these limitations.1–6 Peptide-mediated drug delivery constitutes a powerful and versatile tool to enhance the uptake and trigger the release of therapeutic agents into cancer cells.7–9 In particular, one of the most common approaches to facilitate the cellular uptake of anticancer drugs is based on their conjugation to a cell-penetrating peptide (CPP).10–23 On the other hand, the efficacy of the drug can be further increased by its conjugation to a CPP bearing an enzyme-cleavable moiety which could allow its intracellular release.8 Despite the exact mode of action of CPPs is not well understood, it is generally accepted that CPPs are mainly internalized by endocytosis.24 Following this pathway, they will end up inside lysosomes where lysosomal proteases are expected to hydrolytically cleave drugs from CPPs incorporating specific cleavage sites recognized by these enzymes.7,8,13 For example, cathepsin B is able to promote the pH-sensitive release of therapeutic agents from CPPs holding the tetrapeptide Gly-Phe-Leu-Gly.25 The resulting peptide conjugates have been successfully used in drug delivery strategies.26–30Recently, we have identified KKLFKKILKKL-NH2 (BP16) from a library of antimicrobial undecapeptides as a novel CPP. BP16 efficiently internalizes into cancer cells mainly through a clathrin-dependent endocytic mechanism and is an excellent vector for the intracellular delivery of the DNA-alkylating drug chlorambucil (CLB).31 Besides, the incorporation of the cleavable spacer Gly-Phe-Leu-Gly in CLB–BP16 conjugates allows the selective release of CLB, leading to a further increase of the efficacy of this drug obtaining IC50 values in the very low micromolar range.30
Transition metal complexes can interfere in cellular redox chemistry through metal or ligand based redox processes. Targeting the redox balance in cancer cells might promote irreversible damage through reactive oxygen species (ROS)-mediated mechanisms.32–35 In this context, nitrogen rich tetradentate aminoyridine ligands 1,4-dimethyl-7-(2-pyridylmethyl)-1,4,7-triazacyclononane (Me2PyTACN) and (2S,2S′)-1,1′-bis(pyrid-2-ylmethyl)-2,2′-bipyrrolidine ((S,S′)-BPBP) and their transition metal complexes have been described to form powerful oxidation species upon reaction with peroxides. These species resemble those operating in nonheme iron dependent enzymes, and have found use in the catalytic oxidation of organic substrates.36–44 Interestingly, it has been recently demonstrated that Me2PyTACN and (S,S′)-BPBP ligands are able to chelate intracellular iron in cancer cells and that the resulting redox-active moieties promote apoptosis via iron-dependent pro-oxidant mechanisms.45 Therefore, we envisioned that the intracellular delivery of Me2PyTACN and (S,S′)-BPBP ligands into cancer cells could constitute an effective approach to obtain promising redox-directed anticancer agents.
On the other hand, complexes of these ligands and metal ions, such as Cu(II) and Zn(II), have been successfully conjugated to a cationic tetrapeptide using a versatile stepwise approach by means of solid-phase peptide synthesis (SPPS).46 Moreover, it has been found that Cu metallopeptides show an enhanced nuclease activity compared to the parent complexes [Cu(PyTACN)]2+ and [Cu(BPBP)]2+, leading to ∼4-fold and ∼23 rate accelerations, respectively. Furthermore, additional mechanistic studies propose that an oxidative cleavage mechanism is operative, based on the generation of ROS species, causing the DNA damage.
Based on the above considerations, we envisaged that the conjugation of a Me2PyTACN or a (S,S′)-BPBP ligand to a CPP, such as BP16, could enhance their cellular uptake improving their biological activity. In this study, we report the solid-phase synthesis of BP16 conjugates incorporating a Me2PyTACN or a (S,S′)-BPBP ligand at either the N- or the C-terminus. Considering the clathrin-dependent endocytic mechanism of BP16, metal binding peptides bearing the ligand and the enzymatic cleavable tetrapeptide Gly-Phe-Leu-Gly at both ends of BP16 were also prepared. The cytotoxicity of all these sequences was evaluated in cancer and healthy cell lines. In addition, the internalization of the corresponding 5(6)-carboxyfluorescein-labeled metal binding peptides was examined by flow cytometry. Finally, ligand release from the metal binding peptides incorporating the tetrapeptide Gly-Phe-Leu-Gly was demonstrated by cathepsin B enzymatic digestion.
Results and discussion
Design of metal binding peptides
We previously reported families of tetradentate ligands derived from PyTACN and BPBP that formed iron and manganese complexes with high catalytic activities in oxidation reactions.36–44 Mechanistic studies in these reactions showed evidences in favor of the implications of oxygenase-like metal based oxidants, which in turn confer interesting selectivity properties. Moreover, biological analysis of Me2PyTACN and (S,S′)-BPBP ligands have shown their ability to chelate intracellular labile Fe(II) in cancer cells and that the resulting redox-active complexes induce apoptosis through pro-oxidant mechanisms.45 In this work we envisioned that the conjugation of these ligands to a CPP could facilitate their uptake into cancer cells increasing their activity. Recently, we described the undecapeptide BP16 as a CPP able to favor the translocation of biologically active compounds across the cell membrane.30,31 Based on this, herein we designed peptide conjugates resulting from the combination of the Me2PyTACN or the (S,S′)-BPBP ligands with BP16.Previous studies revealed the importance of evaluating the influence of the position of a drug in a peptide sequence on its activity.30,47–49 Thus, we first designed conjugates PyTACN–BP16 (BP341), BP16-PyTACN (BP342), BPBP–BP16 (BP343) and BP16-BPBP (BP344), incorporating the Me2PyTACN or the (S,S′)-BPBP ligand at either the N- or the C-terminal end of BP16 (Fig. 1). Besides, we also included in this study metal binding peptides PyTACN-βAK–BP16 (BP345) and BPBP-βAK–BP16 (BP346), bearing at the N-terminus an additional Lys, a β-Ala and the corresponding ligand. The Lys serves as a site for labeling and the β-Ala residue was introduced as a spacer between the ligand and the peptide sequence. With the aim of promoting the intracellular release of the ligand, the cathepsin B cleavable sequence Gly-Phe-Leu-Gly was incorporated on the sequences BPBP–BP16 (BP343) and BP16-BPBP (BP344), rendering peptide conjugates BPBP-GFLG-BP16 (BP347) and BP16-GLFG-BPBP (BP348), respectively. In addition, to study the cellular uptake properties of the N-terminal metal binding peptides BP346 and BP347, they were labeled with 5(6)-carboxyfluorescein (CF) leading to compounds BPBP-βAK(CF)-BP16 (BP349) and BPBP-GFLG-BP16-CF (BP350), respectively. In derivatives BP342, BP344, BP348 and BP350 containing at the C-terminus the ligands Me2PyTACN or (S,S′)-BPBP, the GLFG-BPBP or the CF moieties, respectively, these moieties were attached to the Nε-amino group of an additional Lys residue, which was introduced to maintain the cationic charges of the parent sequence.
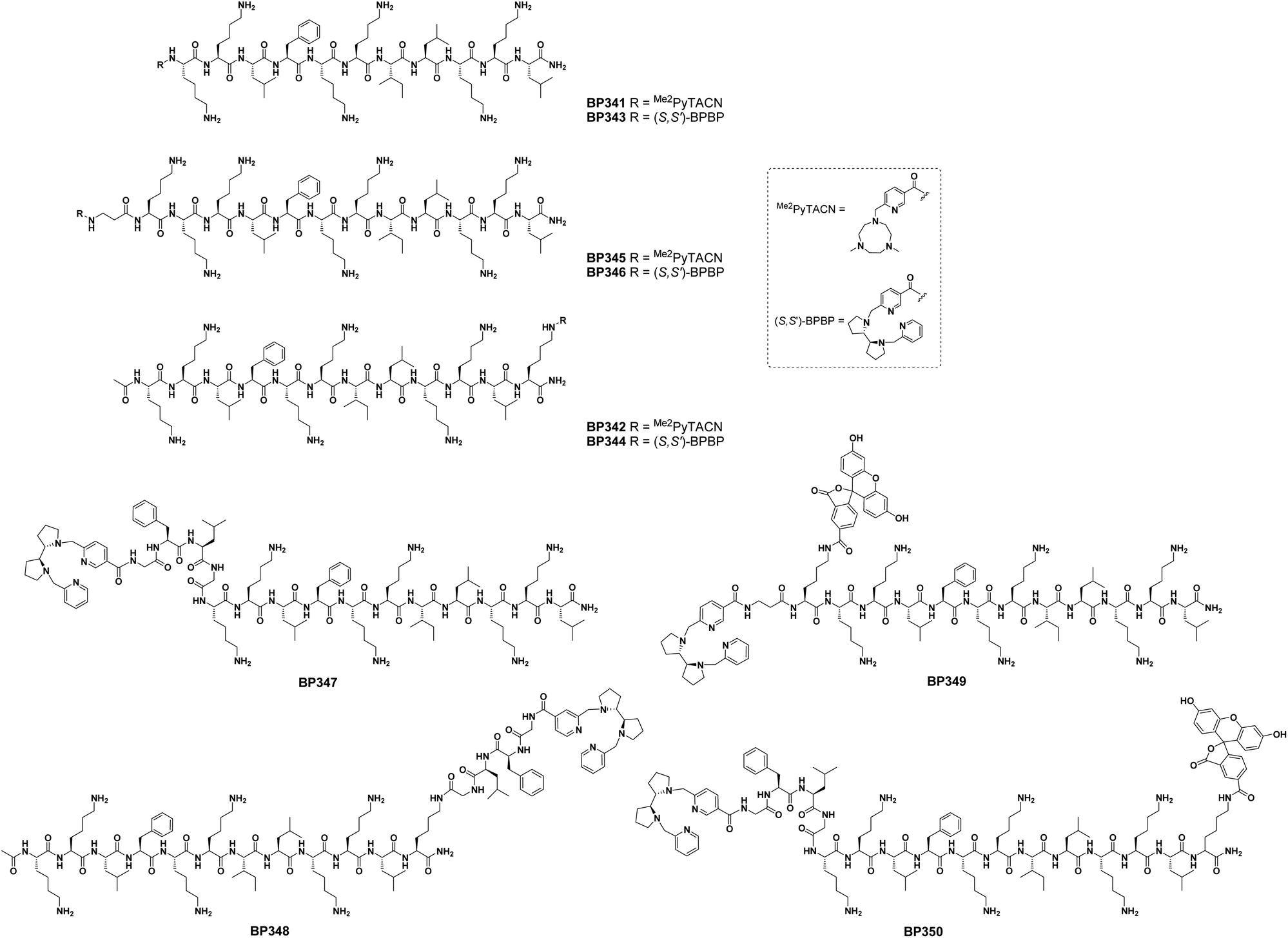 | ||
| Fig. 1 Structure of the metal binding peptides described in this work. | ||
Synthesis of metal binding peptides
We first studied the synthesis of the metal binding peptides BP341 and BP343 bearing the Me2PyTACN or the (S,S′)-BPBP ligand at the N-terminus, respectively (Scheme 1). For this purpose, the peptidyl resin H-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Rink-MBHA was prepared from a Fmoc-Rink-MBHA resin following a standard Fmoc/tBu strategy. The Fmoc group removal step was performed by exposing the peptidyl resin to a piperidine/DMF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) solution. Couplings of the conveniently protected Fmoc amino acids were mediated by ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma) and N,N′-diisopropylcarbodiimide (DIPCDI) in N,N-dimethylformamide (DMF). Peptide elongation proceeded through sequential Fmoc removal and coupling of the corresponding amino acid. Incorporation of the Me2PyTACN or the (S,S′)-BPBP ligand was assayed following the protocol previously described for the preparation of metal binding tetrapeptides.46 Thus, the peptidyl resin H-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Rink-MBHA was treated with 6-[(tert-butyldimethylsilyloxy)methyl]nicotinic acid (1)46 in the presence of COMU, Oxyma and N,N′-diisopropylethylamine (DIPEA) in N-methyl-2-pyrrolidinone (NMP) at 80 °C for 36 h. The TBS group was removed by exposing the resulting resin to a solution of tetrabutylammonium fluoride (TBAF) and acetic acid (AcOH) in THF at room temperature for 6 h. Chlorination of the hydroxymethyl group was achieved by subjecting the resin to three treatments of 6 h with LiCl, p-toluenesulfonyl chloride (TsCl) and DIPEA in THF. Finally, alkylation with the secondary amine 1,4-dimethyl-1,4,7-triazacyclononane (Me2TACN) (2) or (2S,2S′)-1-(pyrid-2-ylmethyl)-2,2′-bipyrrolidine (3) was performed in presence of NaI (0.04 equiv.) and DIPEA (24 equiv.) in NMP under microwave irradiation at 125 °C for 1 h. After acidolytic cleavage with trifluoroacetic acid (TFA)/H2O/triisopropylsilane (TIS) (95:2.5:2.5), the crude reaction mixtures were dissolved in water and washed with CH2Cl2 to remove TsCl impurities. The aqueous phases were lyophilized and conjugates BP341 and BP343 were obtained in excellent purities (>99 and 94%, respectively). Metal binding peptides BP345 and BP346, bearing an additional Lys, a β-Ala and the Me2PyTACN or the (S,S′)-BPBP ligand at the N-terminus of BP16, were prepared following the same strategy and were obtained in >99% purity. Their identities were confirmed by ESI-MS and HRMS.
:7) solution. Couplings of the conveniently protected Fmoc amino acids were mediated by ethyl 2-cyano-2-(hydroxyimino)acetate (Oxyma) and N,N′-diisopropylcarbodiimide (DIPCDI) in N,N-dimethylformamide (DMF). Peptide elongation proceeded through sequential Fmoc removal and coupling of the corresponding amino acid. Incorporation of the Me2PyTACN or the (S,S′)-BPBP ligand was assayed following the protocol previously described for the preparation of metal binding tetrapeptides.46 Thus, the peptidyl resin H-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Rink-MBHA was treated with 6-[(tert-butyldimethylsilyloxy)methyl]nicotinic acid (1)46 in the presence of COMU, Oxyma and N,N′-diisopropylethylamine (DIPEA) in N-methyl-2-pyrrolidinone (NMP) at 80 °C for 36 h. The TBS group was removed by exposing the resulting resin to a solution of tetrabutylammonium fluoride (TBAF) and acetic acid (AcOH) in THF at room temperature for 6 h. Chlorination of the hydroxymethyl group was achieved by subjecting the resin to three treatments of 6 h with LiCl, p-toluenesulfonyl chloride (TsCl) and DIPEA in THF. Finally, alkylation with the secondary amine 1,4-dimethyl-1,4,7-triazacyclononane (Me2TACN) (2) or (2S,2S′)-1-(pyrid-2-ylmethyl)-2,2′-bipyrrolidine (3) was performed in presence of NaI (0.04 equiv.) and DIPEA (24 equiv.) in NMP under microwave irradiation at 125 °C for 1 h. After acidolytic cleavage with trifluoroacetic acid (TFA)/H2O/triisopropylsilane (TIS) (95:2.5:2.5), the crude reaction mixtures were dissolved in water and washed with CH2Cl2 to remove TsCl impurities. The aqueous phases were lyophilized and conjugates BP341 and BP343 were obtained in excellent purities (>99 and 94%, respectively). Metal binding peptides BP345 and BP346, bearing an additional Lys, a β-Ala and the Me2PyTACN or the (S,S′)-BPBP ligand at the N-terminus of BP16, were prepared following the same strategy and were obtained in >99% purity. Their identities were confirmed by ESI-MS and HRMS.
 | ||
| Scheme 1 Synthesis of metal binding peptides BP341 and BP343. | ||
The preparation of metal binding peptide BP347, incorporating the tetrapeptide Gly-Phe-Leu-Gly and the (S,S′)-BPBP ligand was accomplished by coupling the (S,S′)-BPBP derivative 4 to the N-terminus of the corresponding peptidyl resin (Scheme 2). Synthesis of 4 was achieved through N-alkylation of the secondary amine 3 with methyl 6-(chloromethyl)nicotinate hydrochloride, which was obtained following previously described protocols (Scheme S1†).46 Hydrolysis of the resulting methyl ester provided the (S,S′)-BPBP derivative 4 in 51% overall yield. Coupling of 4 to the peptidyl resin H-Gly-Phe-Leu-Gly-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Rink-MBHA mediated by COMU, Oxyma, DIPEA in NMP at 80 °C for 36 h afforded metal binding peptide BP347 in 95% purity, which was characterized by HRMS.
 | ||
| Scheme 2 Synthesis of metal binding peptide BP347. | ||
Metal binding peptides BP342 and BP344, containing the Me2PyTACN and (S,S′)-BPBP ligands, respectively, at the side-chain of a C-terminal Lys residue were then prepared (Scheme S2†). For its selective derivatization, this Lys residue was incorporated as Fmoc-Lys(Dde)-OH (Dde = N-[1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl]). Peptidyl resin H-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Lys(Dde)-Rink-MBHA was synthesized and then acetylated at the N-terminus. After Dde group removal by treatment with hydrazine, the nicotinic acid derivative 1 was coupled with COMU, Oxyma and DIPEA in NMP. Subsequent TBS group removal, chlorination and alkylation with secondary amines 2 and 3 were performed as described above for metal binding peptides BP341 and BP343. Acidolytic cleavage of the resulting resins afforded conjugates BP342 and BP344 in 98 and 90% purity, respectively, and were characterized by HRMS.
Synthesis of metal binding peptide BP348, incorporating the GLFG-(S,S′)-BPBP moiety at the side-chain of a C-terminal Lys, also started from peptidyl resin H-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Lys(Dde)-Rink-MBHA (Scheme S3†). After N-terminal acetylation, selective Dde group removal and subsequent coupling of the amino acids of the GLFG moiety, the (S,S′)-BPBP derivative 4 was incorporated using COMU, Oxyma and DIPEA in NMP at 80 °C for 36 h. Conjugate BP348 was obtained in 90% purity and was characterized by HRMS.
For the preparation of the fluorescein-labeled BPBP conjugates BP349 (BPBP-βAK(CF)-BP16) and BP350 (BPBP-GFLG-BP16-CF), the Lys residue to be labeled was incorporated as Fmoc-Lys(Dde)-OH. Thus, BP349 and BP350 were prepared from peptidyl resins H-β-Ala-Lys(Dde)-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Rink-MBHA and H-Gly-Phe-Leu-Gly-Lys(Boc)-Lys(Boc)-Leu-Phe-Lys(Boc)-Lys(Boc)-Ile-Leu-Lys(Boc)-Lys(Boc)-Leu-Lys(Dde)Rink-MBHA, respectively (Scheme S4†). These peptidyl resins were treated with the (S,S′)-BPBP derivative 4 and then subjected to Dde group removal with hydrazine followed by 5(6)-carboxyfluorescein coupling in presence of DIPCDI, Oxyma in CH2Cl2/NMP. The resulting resins were washed with piperidine/NMP to remove overincorporated carboxyfluorescein moieties and then treated with TFA/H2O/TIS (95:2.5:2.5) to afford BP349 and BP350 in >99 and 94% purity, respectively, which were characterized by HRMS.
Cell cytotoxicity of BP16 conjugates incorporating a Me2PyTACN or (S,S′)-BPBP ligand
The cytotoxicity of the metal binding peptides was examined in MCF-7 and CAPAN-1 tumour cells, and in 1BR3G non-malignant cells. Our aim was to determine the influence on the activity of incorporating: (i) the ligand at the N- or at the C-terminus of BP16; (ii) the dipeptide β-Ala-Lys between the ligand and the N-terminus of BP16; and (iii) the Gly-Phe-Leu-Gly enzymatic cleavable sequence between the ligand and the N- or the C-terminal end of BP16. The IC50 values were determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after 48 h of peptide exposure at concentrations ranging from 0 to 50 μM (Table 1).| Compound | Sequencea | Notation | IC50b (μM) |
||
|---|---|---|---|---|---|
| 1BR3G | MCF-7 | CAPAN-1 | |||
| a All sequences are C-terminal amides. b The IC50 values were determined by the MTT assay after 48 h of peptide exposure. Data represents the mean ± SD of at least three independent experiments made in triplicates. | |||||
| BP341 | Me2PyTACN-KKLFKKILKKL | PyTACN–BP16 | >50 | >50 | >50 |
| BP342 | Ac-KKLFKKILKKLK(Me2PyTACN) | BP16-PyTACN | 35.5 ± 1.9 | >50 | 41.5 ± 0.7 |
| BP343 | (S,S)-BPBP-KKLFKKILKKL | BPBP–BP16 | 36.8 ± 7.2 | >50 | 45.8 ± 1.1 |
| BP344 | Ac-KKLFKKILKKLK((S,S)-BPBP) | BP16-BPBP | 26.0 ± 3.4 | 34.5 ± 5.0 | >50 |
| BP345 | Me2PyTACN-βAK-KKLFKKILKKL | PyTACN-βAK–BP16 | >50 | >50 | >50 |
| BP346 | (S,S)-BPBP-βAK-KKLFKKILKKL | BPBP-βAK–BP16 | >50 | >50 | >50 |
| BP347 | (S,S)-BPBP-GFLG-KKLFKKILKKL | BPBP-GFLG-BP16 | 4.7 ± 1.4 | 5.0 ± 0.2 | 4.3 ± 0.8 |
| BP348 | Ac-KKLFKKILKKLK(GLFG-(S,S)-BPBP) | BP16-GLFG-BPBP | 9.5 ± 0.8 | 11.7 ± 0.5 | 6.9 ± 0.7 |
| (S,S′)-BPBP | (S,S′)-BPBP | 12.7 ± 1.6 | 7.4 ± 0.2 | 9.9 ± 1.0 | |
| Me2 PyTACN | Me2PyTACN | 78.0 ± 8.5 | 88.5 ± 0.7 | 32.0 ± 10.4 | |
The metal binding peptide BP341, incorporating the Me2PyTACN ligand at the N-terminus of BP16, was not active against the cell lines tested, whereas the C-terminal derivative BP342 (BP16-PyTACN) showed IC50 values of 35.5 and 41.5 μM against 1BR3G and CAPAN-1 cells, respectively. The introduction of the dipeptide βAla-Lys between the ligand and the N-terminus of BP16 did not improve the results, the metal binding peptide BP345 exhibited IC50 values >50 μM against the three cell lines.
Regarding the BPBP-metal binding peptides, the conjugate BP343 (BPBP–BP16), bearing the ligand at the N-terminus, displayed IC50 of 36.8 and 45.8 μM against 1BR3G and CAPAN-1 cells, respectively, and the C-terminal BPBP derivative (BP344) was active against 1BR3G and MCF-7 cells with IC50 of 26.0 and 34.5 μM, respectively (Table 1). Similarly to the Me2PyTACN derivatives, the incorporation of the dipeptide βAla-Lys also rendered a non-active peptide (BP346).
The analysis of these results revealed that the activity of the metal binding peptides was consistent with that observed for the ligands alone.45 Thus, BP16 conjugates incorporating the (S,S′)-BPBP ligand were more active than the sequences bearing the Me2PyTACN metal binding moiety. However, the conjugation of these ligands to BP16 decreased their activity. Moreover, contrary to our expectations, the incorporation of a dipeptide spacer between the ligand and BP16 resulted in non-active conjugates. These results suggest that the peptide sequence adopts a conformation that could interfere with the metal binding ability of the ligand. In addition and in agreement with previous results, a different behaviour of the N- and C-terminal derivatives was observed. This fact points out that the position of a cargo in a peptide sequence influences the biological activity of the resulting conjugate, constituting a crucial factor in the design of biologically active peptide conjugates.30,47–49
The higher activity of the BPBP-metal binding peptides compared to the Me2PyTACN conjugates prompted us to focus next studies on sequences bearing the former ligand. Remarkably, the incorporation of the cathepsin B cleavable sequence Gly-Phe-Leu-Gly in conjugates BP343 and BP344 resulted in metal binding peptides BP347 and BP348, respectively, with an excellent activity (IC50 = 5.0–11.7 μM). The best derivative was BP347, bearing the BPBP-GFLG moiety at the N-terminus of BP16. This derivative displayed IC50 values ranging from 4.3 to 5.0 μM against the three cell lines, comparable to those of widely used anticancer drugs such as cisplatin or doxorubicin.50,51 Moreover, this metal binding conjugate showed an enhanced cytotoxic activity compared to that determined for the (S,S′)-BPBP ligand alone against the same cell lines (Table 1). These results can be reasoned taken into account the uptake mechanism described for BP16, which internalizes into cancer cells by clathrin-dependent endocytosis being lysosomes the final stage of this pathway.31 Therefore, the high activity observed for BP347 and BP348 could be attributed to the selective release of the ligand in the lysosomal compartment promoted by cathepsin B. A similar effect was previously reported for conjugates incorporating the anticancer drug chlorambucil (CLB) and the tetrapeptide Gly-Phe-Leu-Gly linked to BP16.30 Moreover, the behaviour of BP347 and BP348 compared to conjugates BP343 and BP344 reinforces our previous hypothesis that the peptide sequence could hamper the activity of the ligand. Concerning the possible mechanism of action, on the basis of a previous study,45 the selective release of the (S,S′)-BPBP ligand would facilitate the chelation of intracellular labile Fe(II) inside the cell. Then, chemically reminiscent to activated bleomycin, the cytotoxic activity of this moiety would rely on the generation of ROS, DNA fragmentation through oxidative mechanisms and further induction of cell cycle arrest and apoptosis. Unfortunately, the conjugation of the (S,S′)-BPBP ligand to the cell-penetrating peptide BP16 did not increase the selectivity of the ligand towards tumour cells. However, BP347 and BP348 highlight the benefit of designing conjugates that include peptide systems able to deliver and release their cargo into the cells.
Cellular uptake of BPBP peptide conjugates
To analyze if the cytotoxic activity observed for these metal binding peptides correlated with their cell internalization, we studied the cellular uptake properties of the most active conjugate BP347 and of the non-active sequence BP346. For this purpose, the internalization ability of the corresponding 5(6)-carboxyfluorescein (CF)-labeled conjugates BP350 (BPBP-GFLG-BP16-CF) and BP349 (BPBP-βAK(CF)-BP16) was examined by flow cytometry (Fig. 2). These experiments were performed by incubating MCF-7 cells with the corresponding CF-labeled peptide conjugate at 25 μM for 6 h at 37 °C. | ||
| Fig. 2 Uptake of 5(6)-carboxyfluorescein (CF) labeled conjugates BP349 (BPBP-βAK(CF)-BP16) and BP350 (BPBP-GFLG-BP16-CF) in MCF-7 cells. Cells were exposed to the peptide conjugate at 25 μM for 6 h at 37 °C. Each column in the graph represents the mean fluorescence intensity of the cells determined in three independent experiments ± SD. | ||
As represented in Fig. 2, important differences between the cellular uptake of labeled conjugates BP349 and BP350 were observed. Remarkably, the internalization ability of conjugate BP350 was about 5-fold higher than that of BP349 (1427.20 ± 72 vs. 310.31 ± 0.1). This result correlated with the cytotoxic activity exhibited by the corresponding non-labeled sequences BP346 and BP347, respectively. Therefore, the high activity of the metal binding peptide BP347 could be attributed to its high cellular uptake and to the subsequent release of the (S,S′)-BPBP ligand by the intracellular cleavage of the enzyme-labile spacer Gly-Phe-Leu-Gly.
Cathepsin B enzymatic assays
To determine the intracellular release of the ligand, enzymatic digestions with the lysosomal enzyme cathepsin B were carried out with conjugates BP347 (BPBP-GFLG-BP16) and BP348 (BP16-GLFG-BPBP) (Fig. 3). Conditions for digestion were chosen to mimic the lysosomal medium (pH 4.5–5.5) and followed a previously reported method.47 Experiments were performed by exposure of BP347 and BP348 to a solution of cathepsin B in phosphate buffer (pH 5) containing 25 mM L-Cys and 1 mM EDTA. Enzymatic digestions were monitored by HPLC-MS after 10, 30, 60, 90 and 120 min. In the case of the N-terminal binding peptide BP347 (BPBP-GFLG-BP16), the main fragments split by cathepsin B were (S,S′)-BPBP-Gly-OH ([M + H]+ = 424.2) and (S,S′)-BPBP-Gly-Phe-OH ([M + H]+ = 571.3) (Fig. 3A). These fragments eluted at a retention time of 2.6 and 3.4 min, respectively, and increased over time, remaining stable after 90 min of digestion. Our results suggested that the main cleavage sites were between the Gly and the Phe residues, and between the Phe and the Leu amino acids in the tetrapeptide sequence. | ||
| Fig. 3 Enzymatic digestion of BP347 (BPBP-GFLG-BP16) and BP348 (BP16-GLFG-BPBP) by cathepsin B. (A) Extracted ion chromatogram resulting from the enzymatic digestion of BP347 after 0, 10, 30, 60, 90 and 120 min of exposure to the enzyme; ESI-MS spectrum of the peaks at tR = 2.6 and 3.4 min after 90 min. (B) Extracted ion chromatogram resulting from the enzymatic digestion of BP348 after 0, 10, 30, 60, 90 and 120 min of exposure to the enzyme; ESI-MS spectrum of the peaks at tR = 2.6 and 3.4 min after 90 min. | ||
The digestion of the C-terminal binding peptide BP348 also revealed the release of the fragments (S,S′)-BPBP-Gly-OH ([M + H]+ = 424.2) and (S,S′)-BPBP-Gly-Phe-OH ([M + H]+ = 571.3) in a time-dependent manner (Fig. 3B). The intensity of the peaks corresponding to these fragments pointed out that the hydrolysis of the amide bond between the Gly and the Phe residues occurred faster than that of the Phe–Leu bond.
These findings confirm that the release of the ligand could occur in the lysosomes under enzymatic conditions and correlate with the high cytotoxicity observed for these conjugates. These data are in agreement with previous studies based on conjugates bearing chlorambucil attached to BP16 through the cathepsin B-cleavable tetrapeptide Gly-Phe-Leu-Gly.30
Conclusions
In summary, we have shown that conjugation of the (S,S′)-BPBP ligand to the enzymatic cleavable tetrapeptide Gly-Phe-Leu-Gly at the N- or the C-terminus of the cell-penetrating peptide BP16 renders peptide conjugates with high cytotoxic activity against cancer cells. The role of BP16 in these sequences is to deliver the ligand into the cancer cells while the presence of the tetrapeptide allows a very efficient enzyme-triggered release of the ligand inside the cells. These findings confirm the usefulness of BP16 as cell-penetrating peptide and, at the same time, suggest that (S,S′)-BPBP represents a promising pro-oxidant therapeutic strategy in the search for novel redox-directed anticancer treatments.Experimental section
Synthesis of methyl (2S,2S′)-6-[1′-(pyrid-2-ylmethyl)-[2,2′-bipyrrolidin]-1-ylmethyl] nicotinate
(2S,2S′)-1-(Pyrid-2-ylmethyl)-2,2′-bipyrrolidine (3)46 (0.79 g, 3.41 mmol) was dissolved in H2O (5.80 mL) and CH2Cl2 (5.80 mL). NaOH pellets (0.31 g, 7.84 mmol) and methyl 6-(chloromethyl) nicotinate hydrochloride (0.63 g, 3.41 mmol)46 were added. The mixture was stirred during 24 h at room temperature and the reaction was monitored by TLC. After consumption of the starting material, 1 M NaOH (35 mL) was added and the aqueous layer was extracted with CH2Cl2 (2 × 25 mL). The combined organic layers were dried with anhydrous MgSO4 and the solvent was evaporated under reduced pressure yielding the expected nicotinate derivative as an orange oil (0.70 g, 54% yield). 1H NMR (400 MHz, CDCl3): δ = 1.69–1.82 (m, 8 H, 2 × H3′, 2 × H3, 2 × H4′, 2 × H4), 2.18–2.26 (m, 2 H, H2, H2′), 2.75–2.83 (m, 2 H, 2 × H5′), 2.94–3.02 (m, 2 H, 2 × H5), 3.52 (d, J = 14.3 Hz, 1 H, NCH2Pyr), 3.57 (d, J = 15.2 Hz, 1 H, NCH2Pyr), 3.92 (s, 3 H, OCH3), 4.18 (d, J = 14.3 Hz, 1 H, NCH2Pyr), 4.26 (d, J = 15.2 Hz, 1 H, NCH2Pyr), 7.07–7.09 (m, 1 H, pyr-5′), 7.36 (d, J = 8.2 Hz, 1 H, pyr-3′), 7.49 (d, J = 8.2 Hz, 1 H, pyr-5), 7.57 (td, J = 2.0 Hz and 8.2 Hz, 1 H, pyr-4′), 8.17 (dd, J = 1.6 Hz and 8.2 Hz, 1 H, pyr-4), 8.46–8.47 (m, 1 H, pyr-6′), 9.07 (d, J = 1.6 Hz, 1 H, pyr-2). 13C NMR (100 MHz, CDCl3) δ = 23.61, 23.68 (C4, C4′), 26.25, 26.31 (C3, C3′), 52.25 (CH3), 55.28, 55.32 (C5, C5′), 61.15, 61.20 (C2, C2′), 65.82 (NCH2pyr), 121.72 (pyr-5′), 122.07, 122.82, 124.10 (pyr-3, pyr-5, pyr-3′), 136.34, 137.39 (pyr-4, pyr-4′), 148.82 (pyr-6′), 150.10 (pyr-2), 160.03 (pyr-2′), 165.27 (pyr-6), 165.95 (CO2CH3) ppm. MS (ESI): m/z = 381.2 [M + H]+.Synthesis of (2S,2S′)-6-[1′-(pyrid-2-ylmethyl)-[2,2′-bipyrrolidin]-1-ylmethyl] nicotinic acid (4)
An aqueous solution of LiOH (1.6 M, 5.55 mmol, 3.55 mL) was added dropwise to a solution of methyl (2S,2S′)-6-[1′-(pyrid-2-ylmethyl)-[2,2′-bipyrrolidin]-1-ylmethyl] nicotinate (1 g, 1.85 mmol) in THF/CH3OH (1:1, 7.2 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and at room temperature for 4 h. After this time, the solvent was evaporated under reduced pressure to provide compound 4 as a white solid (0.65 g, 95% yield). 1H NMR (400 MHz, CD3OD): δ = 1.68–1.74 (m, 8 H, 2 × H3′, 2 × H3, 2 × H4′, 2 × H4), 2.20–2.30 (m, 2 H, H2, H2′), 2.76–2.82 (m, 2 H, 2 × H5′), 2.94–2.98 (m, 2 H, 2 × H5), 3.53 (d, J = 19.4 Hz, 2 H, NCH2Pyr), 4.21 (d, J = 19.4 Hz, 1 H, NCH2Pyr), 4.25 (d, J = 19.4 Hz, 1 H, NCH2Pyr), 7.22–7.30 (m, 1 H, pyr-5′), 7.48 (d, J = 10.6 Hz, 2 H, pyr-5, pyr-3′), 7.75 (td, J = 2.4 and 7.2 Hz, 1 H, pyr-4′), 8.21 (dd, J = 2.7 and 10.6 Hz, 1 H, pyr-4), 8.39–8.40 (m, 1 H, pyr-6′), 8.94 (d, J = 2.7 Hz, 1 H, pyr-2). MS (ESI): m/z = 367.2 [M + H]+. HRMS (ESI): calcd for C21H27N4O2 [M + H]+ 367.2129; found 367.2119; calcd for C21H26N4O2Na [M + Na]+ 389.1948; found 389.1934.
Synthesis of the metal binding peptides
:7, 2 + 10 min). For longer sequences, Fmoc group removal was carried out with piperidine/NMP (3:7, 1 × 3 min + 3 × 10 min). After each coupling and deprotection step, the resin was washed with DMF (6 × 1 min) and CH2Cl2 (6 × 1 min), and air dried. After the coupling of the eleventh residue, NMP was used instead of DMF. Peptide elongation was performed by repeated cycles of Fmoc group removal, coupling and washings.
:2.5:2.5) for 3 h at room temperature. Following TFA evaporation and cold Et2O extraction, the crude was dissolved in H2O (5 mL) and extracted with CH2Cl2 (6 × 3 mL). The aqueous phase was lyophilized and the residue was dissolved in H2O/CH3CN (1:1) affording the conjugate peptide BP341. To remove trifluoroacetate counterions, a StratoSpheres™ SPE device containing a PL-HCO3 MP resin was used. This resin was initially pre-conditioned with CH3OH (1 mL). Then, an aqueous solution of BP341 was treated with the SPE device, which was washed with H2O. The aqueous solution was lyophilized affording the desired TFA-free amine conjugate, which was tested for purity by HPLC. Detection was performed at 220 nm. Solvent A was 0.1% aq. TFA and solvent B was 0.1% TFA in CH3CN. Analysis was carried out with a Kromasil 100 C18 (4.6 mm × 40 mm, 3.5 μm) column with a 2–100% B linear gradient over 7 min at a flow rate of 1.0 mL min−1. ESI-MS and HRMS (ESI) were used to confirm peptide identity. BP341 was obtained in >99% purity. tR = 6.85 min. MS (ESI): m/z = 554.4 [M + 3H]3+, 831.1 [M + 2H]2+, 1661.1 [M + H]+. HRMS (ESI): m/z calcd for C84H154N22O12 [M + 4H]4+ 415.8024; found 415.8037; calcd for C84H153N22O12 [M + 3H]3+ 554.0674; found 554.0696; calcd for C84H152N22O12 [M + 2H]2+ 830.5975; found 830.5994.
:1:1 v/v, 2 × 30 min) under stirring. The resin was then washed with NMP (6 × 1 min) and CH2Cl2 (6 × 1 min), and the Kaiser test was used to test the completion of the reaction.52 The resulting peptidyl resin was treated with hydrazine/NMP (2:98, 5 × 20 min). After these treatments the resin was washed with NMP (6 × 1 min) and CH2Cl2 (1 × 1 min). Conjugation of the Me2PyTACN ligand at the free amino group, acidolytic cleavage, removal of trifluoroacetate counterions and characterization were carried out following the procedure described for BP341. The metal binding peptide BP342 was obtained in 98% purity. tR = 7.23 min. MS (ESI): m/z = 1831.2 [M + H]+. HRMS (ESI): m/z calcd for C92H169N24O14 [M + 5H]5+ 366.8645; found 366.8653; calcd for C92H168N24O14 [M + 4H]4+ 458.3287; found 458.3293; calcd for C92H167N24O14 [M + 3H]3+ 610.7692; found 610.7719.
:2.5:2.5) for 3 h at room temperature. Following TFA evaporation and cold Et2O extraction, the crude was dissolved in H2O (5 mL) and lyophilized. Removal of trifluoroacetate counterions and characterization was carried out following the procedure described for BP341, leading to the metal binding peptide BP347 in >99% purity. tR = 7.24 min. HRMS (ESI): m/z calcd for C109H183N26O16 [M + 5H]5+ 422.4856; found 422.4859; calcd for C109H182N26O16 [M + 4H]4+ 527.8551; found 527.8563; calcd for C109H181N26O16 [M + 3H]3+ 703.4711; found 703.4738.
:1:1 v/v, 2 × 30 min) under stirring. The resin was then washed with NMP (6 × 1 min) and CH2Cl2 (6 × 1 min), and the Kaiser test was used to test the completion of the reaction.52 The resulting peptidyl resin was treated with hydrazine/NMP (2:98, 5 × 20 min). After these treatments the resin was washed with NMP (6 × 1 min) and CH2Cl2 (1 × 1 min). The amino acids Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Phe-OH and Fmoc-Gly-OH were sequentially coupled following the protocol described above for the synthesis of peptidyl resins. After Fmoc group removal, coupling of the nicotinic acid derivative 4 was performed as described for BP347. The resulting peptidyl resin was cleaved with TFA/H2O/TIS (95:2.5:2.5) for 3 h at room temperature. Following TFA evaporation and cold Et2O extraction, the crude was dissolved in H2O (5 mL) and lyophilized. Removal of trifluoroacetate counterions and characterization were carried out following the procedure described for BP341, leading to the metal binding peptide BP348 in 97% purity. tR = 7.89 min. HRMS (ESI): m/z calcd for C117H198N28O18 [M + 6H]6+ 380.5901; found 380.5931; C117H197N28O18 [M + 5H]5+ 456.5067; found 456.5078; calcd for C117H196N28O18 [M + 4H]4+ 570.3815; found 570.3816; calcd for C117H195N28O18 [M + 3H]3+ 760.1729; found 760.1737.
Cytotoxicity assays
Cytotoxicity of peptides conjugates BP341–BP348 in CAPAN-1, and MCF-7 tumour cells, and in 1BR3G non-malignant cells was determined by the MTT assay. Appropriate aliquots of these solutions were diluted in the cell culture medium to obtain the final working concentrations. Aliquots of 10 000 CAPAN-1 cells, 6000 MCF-7 cells and 5000 1BR3G cells were seeded on 96-well plates 24 h prior to the treatments. Then, cells were treated for 48 h with the corresponding compound at concentrations ranging from 0 to 50 μM. After removal of the treatment, cells were washed with PBS and incubated for additional 2 h in the darkness with fresh culture medium (100 μL) with MTT (10 μL). The medium was discarded and DMSO (100 μL) was added to each well to dissolve the purple formazan crystals. Plates were agitated at room temperature for 10 min and the absorbance of each well was determined with an absorbance microplate reader (ELx800, BioTek, Winooski, USA) at a wavelength of 570 nm. Three replicates for each compound were used, and all treatments were tested at least in three independent experiments. For each treatment, the cell viability was determined as a percentage of the control untreated cells, by dividing the mean absorbance of each treatment by the mean absorbance of the untreated cells. The IC50 was established for each compound by standard non-linear regression and curve fitting using GraphPad Prism (Graph Pad software Inc., La Jolla, CA).Flow cytometry
The uptake efficiency of BP349 (BPBP-βAK(CF)-BP16) and BP350 (BPBP-GFLG-BP16-CF) by MCF-7 cells was quantified by flow cytometry. Aliquots of 50000 cells were seeded in 24 well-plates and allowed to attach for 24 h. Next, cells were treated with BP349 and BP350 at 25 μM for 6 h at 37 °C. The cells were harvested by trypsinization and gently washed with 2% FBS in cold PBS. The fluorescence of the cells, corresponding to the cellular uptake of the carboxyfluorescein labeled peptides, was analyzed using FACSCalibur (Becton Dickinson Immunocytometry Systems, San Jose, CA) equipped with the CellQuest™ software (Becton Dickinson). The mean fluorescence intensity was represented on a four orders of magnitude log scale (1–10000). Ten thousand cells were analyzed in each experiment.
Cathepsin B enzymatic assays
The enzymatic digestion of metal binding peptides BP347 (BPBP-GFLG-BP16) and BP348 (BP16-GLFG-BPBP) was evaluated using a previously reported method with minor modifications.47 A cathepsin B stock solution (1 × 104 U L−1, 10 μL) was added to phosphate buffer (pH 5.0, 25 mM L-Cys, 1 mM EDTA; 940 μL) and preactivated for 10 min at 37 °C. Next, BP347 and BP348 (1.8 mM, 50 μL) were added to the mixture. Aliquots of this mixture (110 μL) were sampled at 0, 10, 30, 60, 90 and 120 min, flash frozen in liquid nitrogen and analyzed by HPLC-MS. The analytical HPLC conditions used for these analyses were the same as described above for BP341.Acknowledgements
This work was supported by Consolider Ingenio CSD/CSD2010-00065 from MICINN of Spain. We also thank the Catalan DIUE of the Generalitat de Catalunya (2014 SGR 862). X. R. thanks financial support from INNPLANTA project INP-2011-0059-PCT-420000-ACT1. M. C. and X. R. thank ICREA Academia Awards. We also acknowledge the Serveis Tècnics de Recerca of the University of Girona for technical support.Notes and references
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC.
- L. W. Kleiner, J. C. Wright and Y. Wang, J. Controlled Release, 2014, 181, 1–10 CrossRef CAS PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- Y. Singh, M. Palombo and P. J. Sinko, Curr. Med. Chem., 2008, 15, 1802–1826 CrossRef CAS PubMed.
- R. Karaman, Chem. Biol. Drug Des., 2013, 82, 643–668 CAS.
- S. Aluri, S. M. Janib and J. A. Mackay, Adv. Drug Delivery Rev., 2009, 61, 940–952 CrossRef CAS PubMed.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS PubMed.
- E. Vivès, J. Schmidt and A. Pèlegrin, Biochim. Biophys. Acta, 2008, 1786, 126–138 Search PubMed.
- S. B. Fonseca, M. P. Pereira and S. O. Kelley, Adv. Drug Delivery Rev., 2009, 61, 953–964 CrossRef CAS PubMed.
- Y. Huang, Y. Jiang, H. Wang, J. Wang, M. C. Shin, Y. Byun, H. He, Y. Liang and V. C. Yang, Adv. Drug Delivery Rev., 2013, 65, 1299–1315 CrossRef CAS PubMed.
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS PubMed.
- I. Nakase, Y. Kawaguchi, N. Nomizu and S. Futaki, Trends Glycosci. Glycotechnol., 2015, 27, 81–88 CrossRef.
- K. Kurrikoff, M. Gestin and U. Langel, Expert Opin. Drug Delivery, 2016, 13, 373–387 CrossRef CAS PubMed.
- R. Weinstain, E. N. Savariar, C. N. Felsen and R. Y. Tsien, J. Am. Chem. Soc., 2014, 136, 874–877 CrossRef CAS PubMed.
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed.
- B. M. deRonde, A. Birke and G. N. Tew, Chem. – Eur. J., 2015, 21, 3013–3019 CrossRef CAS PubMed.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- I. Nakase, H. Akita, K. Kogure, A. Gräslund, U. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelly, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972–1994 CrossRef CAS PubMed.
- M. Fotin-Mleczek, R. Fischer and R. Brock, Curr. Pharm. Des., 2005, 11, 3613–3628 CrossRef CAS PubMed.
- M. M. Mohamed and B. F. Sloane, Nat. Rev. Cancer, 2006, 6, 764–775 CrossRef CAS PubMed.
- K. B. Bai, O. Láng, E. Orbán, R. Szabó, L. Köhidai, F. Hudecz and G. Mezö, Bioconjugate Chem., 2008, 19, 2260–2269 CrossRef CAS PubMed.
- K. Splith, W. Hu, U. Schatzschneider, R. Gust, I. Ott, L. A. Onambele, A. Prokop and I. Neundorf, Bioconjugate Chem., 2010, 21, 1288–1296 CrossRef CAS PubMed.
- R. Shankar, A. Samykutty, C. Riggin, S. Kannan, U. Wenzel and R. Kolhatkar, Mol. Pharm., 2013, 10, 3776–3788 CrossRef CAS PubMed.
- M. Moreno, E. Zurita and E. Giralt, J. Controlled Release, 2014, 182, 13–21 CrossRef CAS PubMed.
- M. Soler, M. González-Bártulos, E. Figueras, X. Ribas, M. Costas, A. Massaguer, M. Planas and L. Feliu, Org. Biomol. Chem., 2015, 13, 1470–1480 CAS.
- M. Soler, M. González-Bártulos, D. Soriano-Castell, X. Ribas, M. Costas, F. Tebar, A. Massaguer, L. Feliu and M. Planas, Org. Biomol. Chem., 2014, 12, 1652–1663 CAS.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97–110 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- I. Romero-Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed.
- A. Company, L. Gómez, X. Fontrodona, X. Ribas and M. Costas, Chem. – Eur. J., 2008, 14, 5727–5731 CrossRef CAS PubMed.
- A. Company, Y. Feng, M. Güell, X. Ribas, J. M. Luis, L. Que, Jr. and M. Costas, Chem. – Eur. J., 2009, 15, 3359–3362 CrossRef CAS PubMed.
- L. Gómez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720–5723 CrossRef PubMed.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- I. Garcia-Bosch, L. Gómez, A. Polo and X. Ribas, Adv. Synth. Catal., 2012, 354, 65–70 CrossRef CAS.
- I. Prat, L. Gómez, M. Canta, X. Ribas and M. Costas, Chem. – Eur. J., 2013, 19, 1908–1913 CrossRef CAS PubMed.
- Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042–8047 CrossRef PubMed.
- L. Gómez, M. Canta, D. Font, I. Prat, X. Ribas and M. Costas, J. Org. Chem., 2013, 78, 1421–1433 CrossRef PubMed.
- O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871–14878 CrossRef PubMed.
- M. González-Bártulos, C. Aceves-Luquero, J. Qualai, O. Cussó, M. A. Martínez, S. Fernández de Mattos, J. A. Menéndez, P. Villalonga, M. Costas, X. Ribas and A. Massaguer, PLoS One, 2015, 10, e0137800 Search PubMed.
- M. Soler, E. Figueras, J. Serrano-Plana, M. González-Bártulos, A. Massaguer, A. Company, M. Á. Martínez, J. Malina, V. Brabec, L. Feliu, M. Planas, X. Ribas and M. Costas, Inorg. Chem., 2015, 54, 10542–10558 CrossRef CAS PubMed.
- P. Zhang, A. G. Cheetham, L. L. Lock and H. Cui, Bioconjugate Chem., 2013, 24, 604–613 CrossRef CAS PubMed.
- A. Brieger, G. Plotz, I. Hinrichsen, S. Passmann, R. Adam and S. Zeuzem, PLoS One, 2012, 7, e31863 CAS.
- W. Zhang, J. Song, L. Mu, B. Zhang, L. Liu, Y. Xing, K. Wang, Z. Li and R. Wang, Bioorg. Med. Chem. Lett., 2011, 21, 1452–1455 CrossRef CAS PubMed.
- A. Grau-Campistany, A. Massaguer, D. Carrion-Salip, F. Barragán, G. Artigas, P. López-Senín, V. Moreno and V. Marchán, Mol. Pharm., 2013, 10, 1964–1976 CrossRef CAS PubMed.
- V. Kuete, T. Eichhorn, B. Wiench, B. Krusche and T. Efferth, Cell Div., 2012, 7, 16 CrossRef CAS PubMed.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; cell lines; synthesis of the metal binding peptides BP343, BP344, BP345, BP346, BP349 and BP350; schemes of the synthesis of the (S,S′)-BPBP derivative 4 and of the metal binding peptides BP342, BP344, BP348 and BP350; NMR and mass spectra of compounds for the synthesis of 4; HPLC, ESI-MS and HRMS of peptide conjugates; HPLC and HRMS of 5(6)-carboxyfluorescein-labeled peptides. See DOI: 10.1039/c6ob00470a |
| This journal is © The Royal Society of Chemistry 2016 |