
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-assisted addition of a nitrate anion to bis(oxy)enamines. A general approach to the synthesis of α-nitroxy-oxime derivatives from nitronates†
Yana A.
Naumovich
a,
Victoria Emily
Buckland
b,
Dmitry A.
Sen'ko
c,
Yulia V.
Nelyubina
d,
Yulia A.
Khoroshutina
a,
Alexey Yu.
Sukhorukov
*a and
Sema L.
Ioffe
a
aN.D. Zelinsky Institute of Organic Chemistry, Leninsky prospect 47, 119991, Moscow, Russia. E-mail: sukhorukov@ioc.ac.ru; Fax: +7 499 1355328; Tel: +7 499 1355329
bNational Junior College, Hillcrest Road 37, 288913, Singapore
cMoscow Chemical Lyceum, Tamozhenniy proezd 4, 111033, Moscow, Russia
dA.N. Nesmeyanov Institute of Organoelement Compounds, Vavilov str. 28, 119991, Moscow, Russia
First published on 29th March 2016
Abstract
The synthesis of α-nitroxy-substituted oxime derivatives has been achieved by an unprecedented metal-assisted addition of a nitrate anion to bis(oxy)enamines, which are readily available from nitronates or nitroalkanes. The method has a broad scope and provides access to α-nitroxy-oximes and their cyclic ethers including nitroxy-substituted isoxazolines and dihydro-1,2-oxazines, which are of interest as potential NO-donors and intermediates in the synthesis of bioactive molecules.
Introduction
Nitrate esters are widely used in pharmaceuticals as efficient NO donors1 with vasodilative,1a vasorelaxant,1b and anti-tumor activities.1c Since NO is associated with the cytotoxic and antimicrobial functions of the immune system, the introduction of a nitroxy group in natural molecules and pharmaceuticals significantly enhances their efficacy.1d This strategy is widely applied in the design of hybrid drugs.1d–hIn organic synthesis, nitrates are considered convenient substrates for nucleophilic substitution reactions, due to the good leaving group ability of the ONO2 fragment.2 Nitrates are employed as intermediates in the synthesis of alcohols3 and aldehydes4 from the corresponding halides. Furthermore, nitrates of small alcohols are also well-known high-energy materials and components of explosive compositions.5
Traditional methods for the introduction of the ONO2 fragment in organic molecules are based either on the direct nitration of alcohols6 or on the nucleophilic SN1-type substitution of halides in haloalkanes, upon the action of silver or mercury nitrate.7 Another method substantially less explored is nucleophilic addition of a nitrate anion to double carbon–carbon bonds.8,9 Such processes are usually realized under oxidative conditions (Scheme 1, eqn (1)),8 or initiated by the addition of an electrophile to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, followed by the nucleophilic attack of a nitrate anion on the carbocation or onium ion (Scheme 1, eqn (2)).9 However, examples of direct addition of a nitrate anion to an activated carbon–carbon double bond as well as its participation in SN′ type substitution reactions are, to the best of our knowledge, unprecedented to date.
C bond, followed by the nucleophilic attack of a nitrate anion on the carbocation or onium ion (Scheme 1, eqn (2)).9 However, examples of direct addition of a nitrate anion to an activated carbon–carbon double bond as well as its participation in SN′ type substitution reactions are, to the best of our knowledge, unprecedented to date.
 | ||
| Scheme 1 Addition of a nitrate anion to a C,C-double bond. | ||
The carbon–carbon double bond in bis(oxy)enamines 1, readily available by silylation of nitronates,10 is easily attacked by some nucleophiles, thus forming α-substituted oximes and their ethers as a result of a formal SN′ substitution of the silyloxy group.11 We were interested in employing nitrate anions as nucleophiles in reactions with bis(oxy)enamines, in order to obtain α-nitroxy-oximes and their cyclic ethers (Scheme 1, eqn (3)). These products are virtually unknown in the literature,12 though first attempts to synthesize simple α-nitroxy-oximes (so-called “nitrosates”) by nitration of some olefins with N2O4 date back to the 19th century in the studies of Guthrie,13a Wallach13b and Ipatiew.13c However, in those times the structure of “nitrosates” could not be determined unambiguously.
Results and discussion
Recently, we have demonstrated that the reactions of bis(oxy)enamines with halide anions are promoted by Lewis acids, in particular transition metal and magnesium salts.14 Therefore, to achieve the desired transformation we studied the interactions of model cyclic bis(oxy)enamine 1a with a series of main and transition group metal nitrates (Table 1). In our experiments, crystal hydrates were usually used as they were the most readily available form of metal nitrates. The latter were dissolved in tetrahydrofuran (in the event of insolubility, dimethylformamide was used as a solvent instead) and treated with a solution of 1a in dichloromethane.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Metal nitrate M(NO3)n | Solvent | Time (h) | Yield of 2aa (%) | Yields of by-productsa (%) | |||
| 3a | 4a | 5a | 6a | |||||
| a Yields of all products were determined by 1H NMR with an internal standard. b Conversion – 27%. c By-products were not identified. d Conversion – 82%. | ||||||||
| 1 | LiNO3 | THF/CH2Cl2 | 2.5 | 17b | — | 8 | — | — |
| 2 | Mg(NO3)2·6H2O | THF/CH2Cl2 | 2.5 | 42 | 17 | 7 | — | — |
| 3 | Al(NO3)3·9H2O | DMF/CH2Cl2 | 2.5 | 53 | 43 | — | — | — |
| 4 | Ga(NO3)3·9H2O | THF/CH2Cl2 | 2.5 | 59 | 19 | — | — | — |
| 5 | Pb(NO3)2 | DMF/CH2Cl2 | 2.5 | 40 | 23 | 18 | 4 | — |
| 6 | Bi(NO3)3·5H2O | DMF/CH2Cl2 | 2.5 | 22 | — | — | 35 | — |
| 7 | Mn(NO3)2·H2O | THF/CH2Cl2 | 2.5 | 42 | 19 | 18 | — | 4 |
| 8 | Fe(NO3)3·9H2O | THF/CH2Cl2 | 2.5 | 43c | — | — | — | — |
| 9 | Co(NO3)2·6H2O | THF/CH2Cl2 | 2.5 | 59 | 32 | — | — | 6 |
| 10 | Co(NO3)2 | THF/CH2Cl2 | 3 | 55 | — | — | — | 6 |
| 11 | Ni(NO3)2·6H2O | THF/CH2Cl2 | 3 | 60 | 29 | — | — | 6 |
| 12 | Cu(NO3)2·3H2O | THF/CH2Cl2 | 2.5 | 54 | 11 | 5 | — | — |
| 13 | Zn(NO3)2·6H2O | THF/CH2Cl2 | 3 | 53 | 32 | 7 | — | — |
| 14 | Cr(NO 3 ) 3 ·9H 2 O | THF/CH 2 Cl 2 | 2.5 | 71 | 21 | — | — | 6 |
| 15 | Cr(NO3)3·9H2O | DMF/CH2Cl2 | 2.5 | 41 | 25 | 5 | — | — |
| 16 | AgNO3 | DMF/CH2Cl2 | 2.5 | 10 | 6 | — | — | — |
| 17 | Y(NO3)3·6H2O | THF/CH2Cl2 | 3 | 24 | 56 | — | — | 7 |
| 18 | La(NO3)3·6H2O | THF/CH2Cl2 | 2.5 | 16 | 60 | 13 | — | — |
| 19 | Eu(NO3)3·6H2O | THF/CH2Cl2 | 2.5 | 21 | 56 | 4 | — | 6 |
| 20 | NH4NO3 | DMF/CH2Cl2 | 1 | 35 | 24 | 14 | — | — |
| 21 | HNO3 | THF/CH2Cl2 | 2.5 | 58 | 13 | 5 | — | — |
| 22 | 3 equiv. KNO3 + 1 equiv. BF3·Et2O | DMF/CH2Cl2 | 2.5 | 26 | 37 | — | — | — |
| 23 | LiNO3 + 10 mol% Cr(NO3)3·9H2O | THF/CH2Cl2 | 3 | 56d | 4 | 18 | — | 5 |
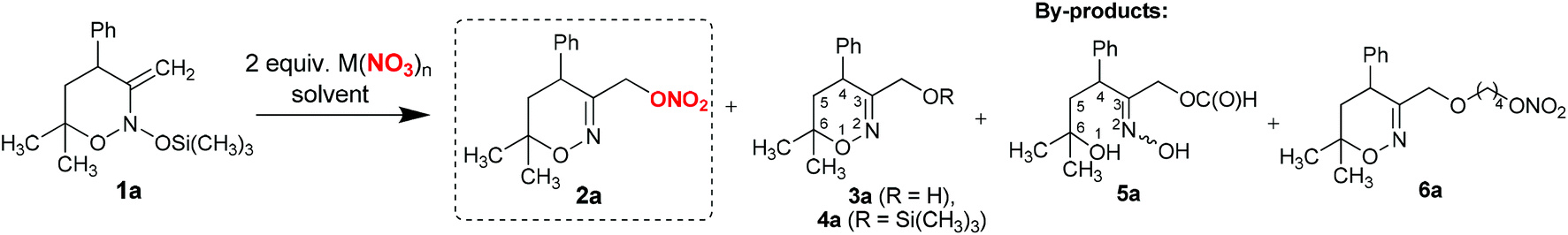
As can be seen from the data in Table 1, almost all studied metal nitrates produced the target product 2a with full conversion of enamine 1a. An exception was lithium nitrate which gave low conversion of the starting material (Table 1, entry 1). The major by-products identified in experiments were 3-hydroxymethyl-substituted 1,2-oxazine 3a, its trimethylsilyl ether 4a, as well as formate 5a and 1,4-butanediol ether nitrate 6a. Products 5a and 6a arise from competitive addition of solvent to bis(oxy)enamine 1a (vide infra). In reactions with magnesium, aluminium and gallium nitrates, the desired nitro-ester 2a was obtained in 42–59% yields (Table 1, entries 2–4). The reaction of bis(oxy)enamine 1a with lead and bismuth nitrates (Table 1, entries 5 and 6) produced formiate 5a in addition to dihydrooxazines 2a, 3a and 4a. In the case of bismuth nitrate, formiate 5a was obtained as the major product (35% yield). Nitrates of early transition metals afforded nitrate ester 2a in moderate to good yields, while by-product 3a was usually formed in 10–30% yield (Table 1, entries 7–16). It should be noted that employing anhydrous nitrates allows for the suppression of the formation of 3a, but does not lead to the increase of the yield of nitrate 2a due to the formation of unidentified by-products (cf. entries 9 and 10 in Table 1).
The best result was achieved with chromium(III) nitrate nonahydrate, which gave product 2a in 71% yield (Table 1, entry 14). Changing of solvent to dimethylformamide led to a decrease of nitrate 2a yield (Table 1, entry 15).
Yttrium, lanthanum and europium nitrates produced 3-hydroxymethyl-substituted 1,2-oxazine 3a as the major product, while the yield of target 2a did not exceed 24% (Table 1, entries 17–19). The formation of nitrate ester 2a was also observed in reactions of enamine 1a with ammonium nitrate and nitric acid (yield of 35% and 58% respectively, entries 20 and 21 in Table 1). Our attempts to use other non-metal based Lewis acids such as boron trifluoride to promote the addition of nitrate to enamine 1a were not successful and led to 3a as the major product (entry 22 in Table 1).
Reaction with chromium nitrate was subsequently studied in more detail. UV-Vis titration of chromium nitrate solution in THF with bis(oxy)enamine 1a revealed that the majority of chromium is present in the form of the aqueous complex [Cr(H2O)6]3+ (see the ESI†). This implies that a catalytic process could be designed using a catalytic amount of Cr(NO3)3, in combination with a cheaper nitrate source. Indeed, addition of 10 mol% of chromium nitrate in the reaction of 1a with LiNO3 led to a high conversion of the starting material (cf. entries 1 and 23 in Table 1). However, the yield of target 2a was only 56% due to the formation of significant amounts of by-products.
Using the procedure with chromium nitrate in THF (procedure i, Table 2), a series of 3-nitroxymethyl-substituted 1,2-oxazines and isoxazolines 2a–j from the corresponding bis(oxy)enamines 1a–j were prepared. As can be seen from Table 2, the reaction is well-tolerated with various substituents in bis(oxy)enamines 1 and usually provides products in good yields. The structure of compound 2g was confirmed by single-crystal X-ray diffraction analysis.
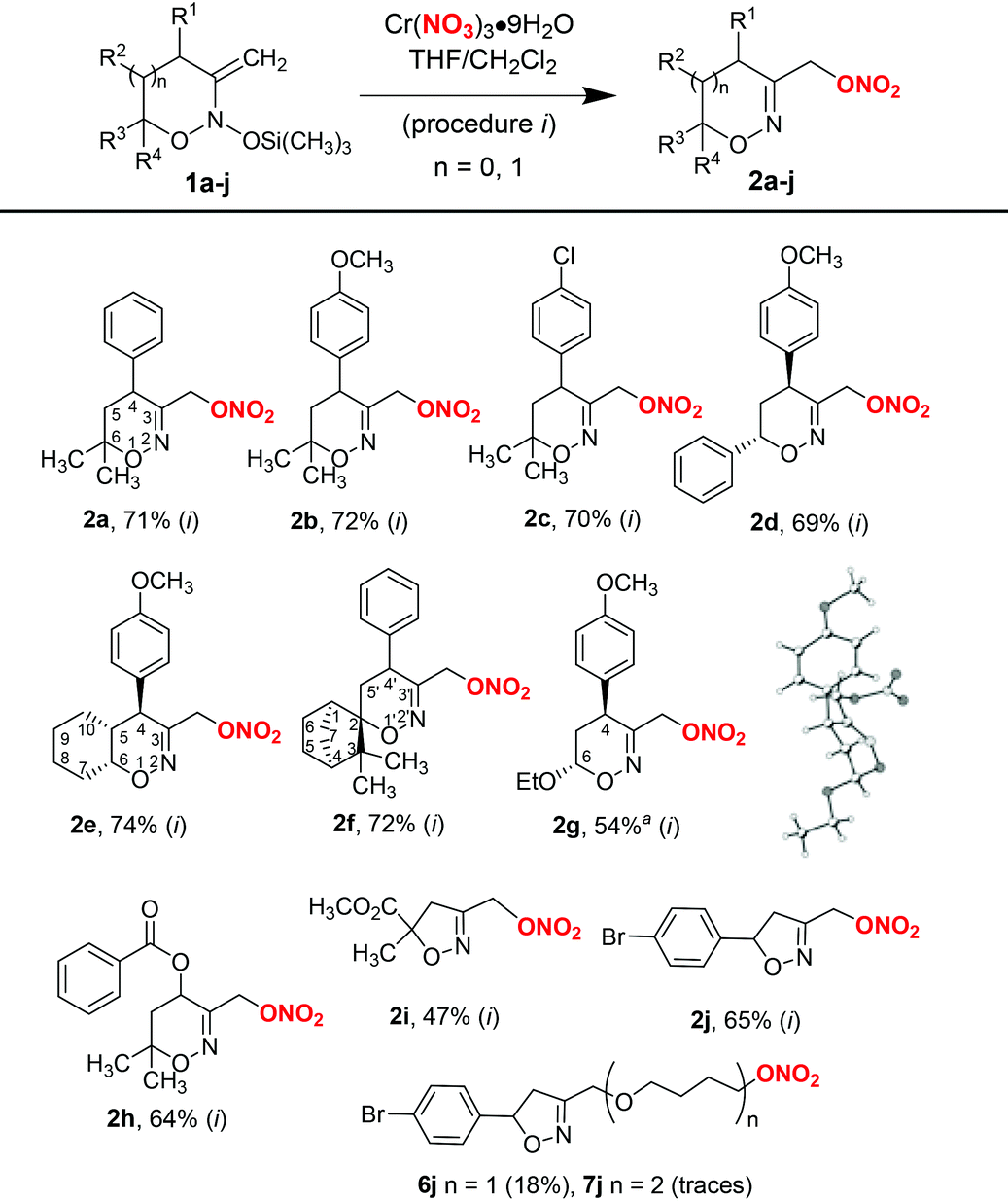
Similar to 1a, the major by-products in reactions with bis(oxy)enamines 1b–i were 3-hydroxymethyl-substituted 1,2-oxazines 3 (typically, not more than 15%) and products of tetrahydrofuran insertion 6 (5–10%), which in some cases were isolated and characterized. In the case of bis(oxy)enamine 1j, ether 6j was isolated in 18% yield. Furthermore, diether 7j resulting from formal insertion of two tetrahydrofuran molecules was detected in trace amounts by high-resolution mass-spectrometry (Table 2).
The synthesis of nitrates 2 can be accomplished directly from the corresponding cyclic nitronates 8, which are synthetic precursors of bis(oxy)enamines 1 (Scheme 2, procedure ii). In this procedure, bis(oxy)enamines are generated in situ by silylation of nitronates 8 with trimethylsilyl triflate and triethylamine, followed by treatment of the reaction mixture with chromium nitrate solution in tetrahydrofuran. Such a one-pot process is operationally simpler, however the yield of product 2a is somewhat lower compared to a two-step procedure with isolation of bis(oxy)enamine 1a (cf. Scheme 2, eqn (1) and entry 14 in Table 1). Still, the application of a one-pot procedure is beneficial in cases when bis(oxy)enamine 1 is labile upon isolation (for example bis(oxy)enamine generated from nitronate 8k, see Scheme 2, eqn (2)).
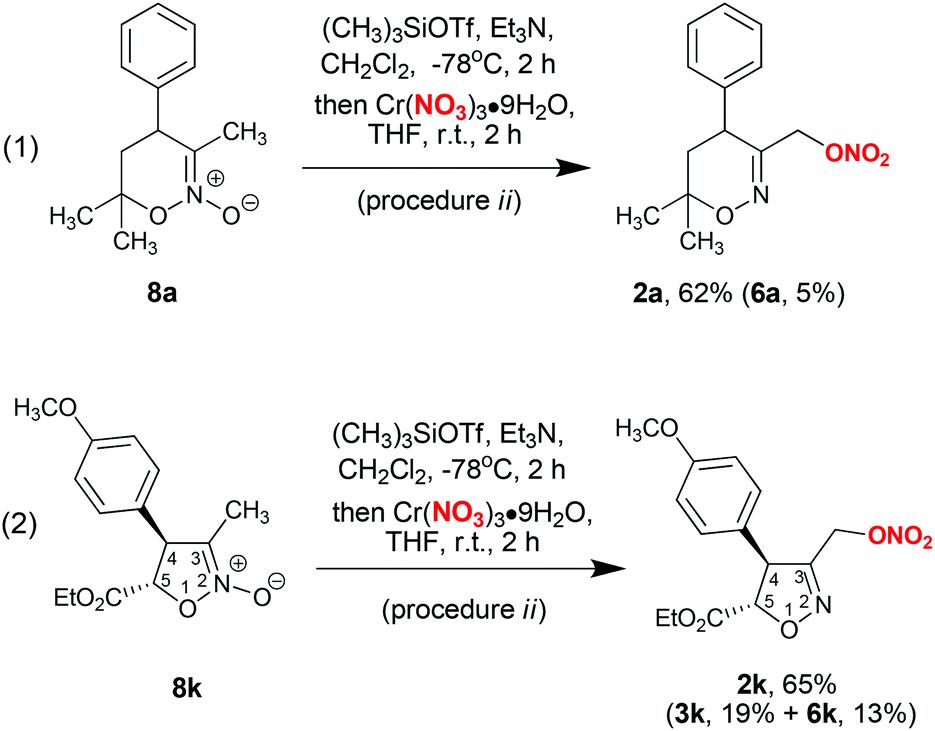 | ||
| Scheme 2 One-pot synthesis of nitrates 2 from nitronates 8. | ||
Procedure i did not prove to be applicable to acyclic bis(silyloxy)enamines 1l–o (Schemes 3 and 4). In the corresponding experiments, only the formation of indecipherable product mixtures was observed. Interestingly, switching to anhydrous cobalt nitrate (procedure iii) in reaction with bis(oxy)enamine 1l produced the desired labile trimethylsilyl ether of α-nitroxy-oxime 2l in 65% yield (Scheme 3). However, in reactions with acyclic bis(silyloxy)enamines 1m,o the corresponding nitrates 2m,o were identified in reaction mixtures by 1H NMR and GC-MS in trace amounts (see the ESI†).
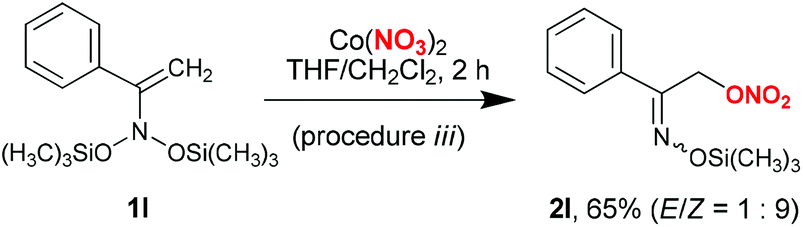 | ||
| Scheme 3 Synthesis of silyl ether 2l from bis(oxy)enamine 1l. | ||
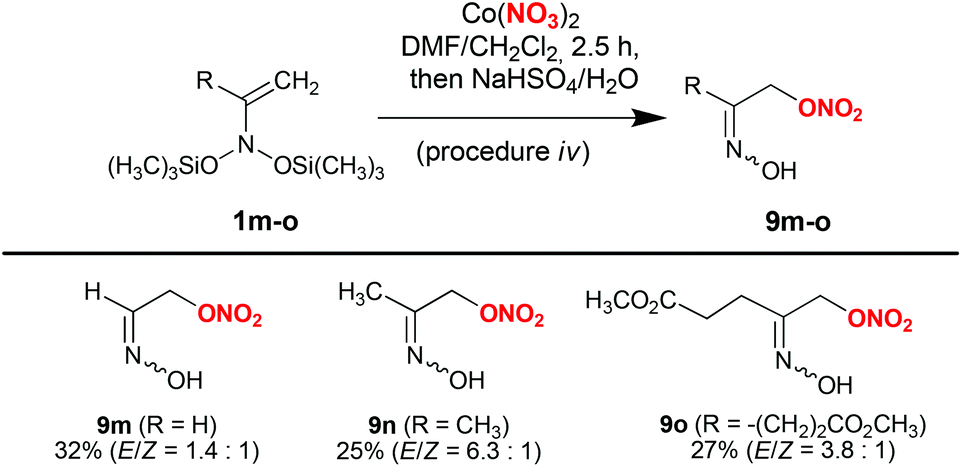 | ||
| Scheme 4 Synthesis of α-nitroxy-oximes 9m–o from enamines 1m–o. | ||
By conducting reactions with Co(NO3)2 in DMF, followed by desilylation with aqueous NaHSO4 solution, labile α-nitroxy-oximes 9 were obtained in reasonable yields (Scheme 4). In particular, by using this procedure, the simplest α-nitroxy-oxime 9m (2-(hydroxyimino)ethyl nitrate) was obtained from bis(oxy)enamine 1m available from nitroethane. This product can be considered as a prospective intermediate in the synthesis of high-energy compounds. α-Nitroxy-oximes 9m–o are viscous liquids, which are unstable upon heating and slowly decompose at room temperature.
The mechanism of α-nitroxy-oxime 2 formation deserves special discussion. In fact, depending on the nature of metal nitrate, the mechanism can be different. It is likely that the process involves initial Lewis acid-mediated cleavage of weak N–O bonds in initial bis(oxy)enamine 1, and the subsequent addition of the nitrate anion to the resulting N-vinyl-N-alkoxy-nitrenium cation A15 (Scheme 5). This is confirmed by isolation of products 5 and 6, which result from competitive addition of solvent to cation A. Thus, nitrates 2 are probably formed as a result of SN1′ substitution of the trimethylsilyloxy group in bis(oxy)enamines 1 for the nitrate anion.
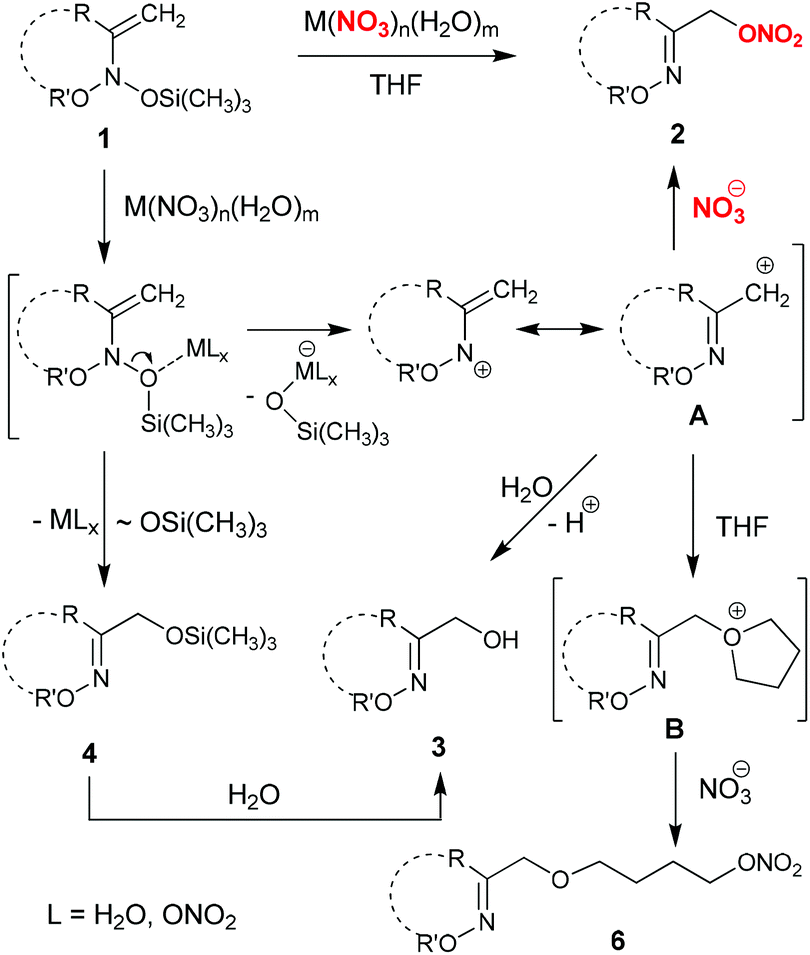 | ||
| Scheme 5 Plausible mechanism for the formation of α-nitroxy-oxime ethers 2 from bis(oxy)enamines 1. | ||
The observed formation of α-hydroxy-oxime silyl ethers 4 can be explained by LA-mediated 1,3-migration of the trimethylsilyloxy group in initial bis(oxy)enamines 1.15d On the other hand, α-hydroxy-oxime cyclic ethers 3 may originate from either the addition of water to cation A or the hydrolysis of the corresponding trimethylsilyl ethers 4 (Scheme 5). The formation of an open-chain oxime 5a in the reaction of 1a with bismuth and lead nitrates (Table 1, entries 5 and 6) demonstrates the possibility of competitive coordination of these metals by endo- and exo-cyclic oxygen atoms in cyclic bis(oxy)enamines 1.
Nitrate esters are frequently employed as intermediates in the total synthesis of pharmacologically relevant compounds.3,4b,c,7b,c,8a,b,16 Recently, we developed the synthesis of oxazolidinone CMPO (a highly potent phosphodiesterase 4 inhibitor) via the nitrate intermediate 2p (Scheme 6).17a The synthesis of nitrate 2p was achieved by the nucleophilic substitution of bromine in 3-bromomethyl-substituted 1,2-oxazine 10p. The latter was synthesized from cyclic nitronate 8p with poor yield and conversion, as well as a high degree of epimerization of the acetal center C-6 (Scheme 6).17a Employing the process developed here, we were able to optimize the synthesis of nitrate 2p from nitronate 8p by the silylation of the latter and treatment of the resulting bis(oxy)enamine 1p with chromium nitrate. In this sequence, the yield of product 2p is considerably higher and the degree of epimerization of C-6 is lower (cf. both approaches in Scheme 6). Unfortunately, one-pot synthesis of nitrate 2p from nitronate 8p using procedure ii was less efficient than the two-step procedure via isolation of enamine 1p (Scheme 6).
 | ||
| Scheme 6 Two approaches to the synthesis of nitrate 2p – key precursor of the PDE 4b inhibitor CMPO. | ||
Conclusions
In conclusion, we have demonstrated that the reaction of readily available N,N-bis(oxy)enamines with metal nitrates results in the addition of a nitrate anion to a double carbon–carbon bond, with the elimination of the trimethylsilyloxy-group from the nitrogen atom. Using reactions of bis(oxy)enamines with chromium and cobalt nitrates, a general method for the synthesis of poorly accessible α-nitroxy-oximes of cyclic and acyclic structures was developed. These oximes are prospective NO donors and intermediates in the synthesis of bioactive molecules. The simplest α-nitroxy-oxime, 2-(hydroxyimino)ethyl nitrate, a potential intermediate in the synthesis of high energy materials, has been synthesized in two steps from nitroethane. The efficiency of the suggested method for the total synthesis of bioactive compounds was demonstrated by the optimization of the synthesis of the highly potent PDE 4 inhibitor.Experimental
All reactions were performed in oven-dried (150 °C) glassware. Tetrahydrofuran was distilled first from LiAlH4, stored under sodium benzophenone ketyl and distilled using the vacuum trap-to-trap technique prior to use. CH2Cl2, MeCN, Et3N, and (CH3)3SiBr were distilled from CaH2. Hexane and EtOAc were distilled without drying agents. Pentane was commercial grade and used as received.Inorganic reagents were commercial grade and were used as received. Anhydrous cobalt(II) nitrate was prepared by thermal decomposition of the corresponding hexahydrate as described in ref. 18. Initial cyclic nitronates 8a10c and 8p17b were synthesized according to literature methods. Bis(oxy)enamines 1a,10cb,10hc,10gd,10fe,10cf,14g,10ch,10fi,10ij,10i1l,10e and 1m–o10d were obtained by silylation of the corresponding nitronates according to previously published procedures.
Column chromatography was performed using Kieselgel 40–60μm 60A. Analytical thin-layer chromatography was performed on silica gel plates with QF-254. Visualization was accomplished with UV light and a solution of anisaldehyde/H2SO4 in ethanol.
1D and 2D NMR spectra were recorded at room temperature in CDCl3 on a Bruker AM 300 spectrometer. The chemical shifts (1H, 13C) are given in ppm (δ) relative to the solvent signal, and liquid ammonia was used as a reference compound in the 14N NMR spectra. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). Trichloroethylene was used as an internal standard in quantitative NMR measurements.
FTIR spectra were recorded on a Bruker Alpha-T spectrometer. Peaks in IR-spectra data are reported in cm−1 with the following relative intensities: s (strong), m (medium), w (weak), br (broad), and sh (shoulder). UV-VIS spectra were recorded on a Shimadzu UVmini-1240 spectrometer (data are reported in nm). Elemental analysis (average of two combustions) was performed by the Analytical Laboratory of the Institute of Organic Chemistry. HRMS were measured on an electrospray ionization (ESI) instrument with a time-of-flight (TOF) detector. GC-MS was performed on a Chromatec 5000 with the Agilent DB-1MS column 122-0132. EI mass spectra were recorded on a Finnigan MAT Incos 50 spectrometer (70 eV). Optical rotation angles were measured on a JASCO P2000 polarimeter. Concentrations c are given in g per 100 mL. [α]D values are given in 10−1 deg cm2 g−1.
rel-(4S,6S)-4-(3-(Cyclopentyloxy)-4-methoxyphenyl)-3-methylene-6-((1S,2R)-2-phenylcyclohexyloxy)-2-(trimethylsilyloxy)morpholine (1p)
To a stirred solution of nitronate rac-8p17b (0.20 g, 0.416 mmol) and Et3N (0.14 mL, 1.0 mmol) in 4.8 mL of CH2Cl2 was added Me3SiBr (0.12 mL, 0.91 mmol) at −78 °C under an argon atmosphere. The mixture was kept at −78 °C for 60 h, then diluted with hexane (5 mL) and poured into a mixture of hexane (50 mL) and 0.25 M aqueous NaHSO4 solution (50 mL). The aqueous layer was back-extracted with hexane (20 mL). Combined organic layers were washed with water (30 mL), and brine (30 mL), dried (Na2SO4), and evaporated in a vacuum. The residue was dried in a vacuum until constant weight to give 0.202 g (88%) of bis(oxy)enamine 1p as colorless oil (unstable at r.t.). 1H NMR (CDCl3, 300.13 MHz, COSY, HSQC): 0.31 (s, 9 H, (CH3)3Si), 1.25–1.47, 1.50–1.63 and 1.72–2.00 (3 m, 16 H, CH-5, CH2-9, CH2-10, CH2-11, CH-12, CH2-22, CH2-23), 2.10 (ddd, J = 12.0, 8.9, 4.3 Hz, 1 H, HC-5), 2.40 (m, 1 H, HC-12), 2.67 (ddd, J = 11.5, 10.3, 2.9 Hz, 1 H, HaxC-8), 3.53 (br s, 1 H, HC-4), 3.84 (s, 3 H, OCH3), 3.94 (br s, 1 H, HC-13), 4.09 (ddd, J = 10.3, 10.1, 3.5 Hz, 1 H, HaxC-7), 4.75 (br m, 2 H, HC-13 and HC-21), 5.42 (dd, J = 4.6, 4.3 Hz, 1 H, HeqC-6), 6.68 (d, J = 8.0 Hz, 1 H, HC-19), 6.69 (s, 1 H, HC-15), 6.81 (d, J = 8.0 Hz, 1 H, HC-18), 7.19–7.37 (m, 5 H, o-, m-, p-C6H5). 29Si NMR (CDCl3, 59.63 MHz): 25.9. 13C NMR (CDCl3, 75.47 MHz, HSQC): −0.51 (CH3)3Si), 24.0, 24.8 and 26.2 (CH2-9, CH2-10 and CH2-23), 30.6 (CH2-12), 32.8 (CH2-22), 34.5 and 36.9 (CH2-5 and CH2-11), 40.0 (br, CH-4), 50.2 (CH-8), 56.1 (OCH3), 76.2 (CH-7), 80.4 (CH-21), 93.9 (CH-6), 96.9 (br, CH2-3), 111.9 (CH-18), 116.0 (CH-15), 120.7 (CH-19), 125.8, 127.9 and 128.0 (o-, m-, p-C6H5), 132.3 (br, C-14), 144.2 (i-C6H5), 147.4 and 149.0 (C-16 and C-17), 159.3 (br, C-13). 29Si NMR (CDCl3, 59.63 MHz): 26.0. HRMS: m/z Calcd for [C32H46NO5Si+] 552.3140 ([M + H]+). Found: 552.3136.rel-(4S,5S)-5-(Ethoxycarbonyl)-4-(4-methoxyphenyl)-3-methyl-4,5-dihydroisoxazole 2-oxide (8k)
To a stirred solution of sulfonium ylide19 (5 mL in CHCl3, c = 0.14 g mL−1, 4.8 mmol) derived from carbethoxymethyl-dimethylsulfonium bromide and K2CO3 was added (E)-1-methoxy-4-(2-nitroprop-1-enyl)benzene (0.776 g, 4 mmol). After 2.5 h at r.t., an additional 2.5 ml of sulfonium ylide solution in CHCl3 was added (c = 0.14 g mL−1, 2.4 mmol). After 2 h the resulting solution was poured into a mixture of EtOAc (100 mL) and water (100 mL). The aqueous layer was back-extracted with EtOAc (50 mL). Combined organic layers were washed with water (50 mL), and brine (50 mL), dried (Na2SO4), and evaporated in a vacuum. The residue was triturated with an Et2O/pentane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) mixture and dried in a vacuum until constant weight to give 0.992 g (89%) of nitronate 8k as a white solid. Mp = 93–95 °C. 1H NMR (CDCl3, 300.13 MHz, HSQC, NOESY): 1.36 (t, J = 7.1 Hz, 3 H, CH3CH2), 1.90 (s, 3 H, CH3), 3.85 (s, 3 H, OCH3), 4.34 (q, J = 7.1 Hz, 2 H, CH3CH2), 4.51 (br d, J = 4.0 Hz, 1 H, CH-4), 4.81 (d, J = 4.0 Hz, 1 H, CH-5), 6.96 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.21 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, HSQC): 10.6 (CH3), 14.0 (CH3CH2), 55.3 (OCH3 and CH-4), 62.2 (CH3CH2), 78.5 (CH-5), 113.3 (CN), 114.9 (o-C6H4OCH3), 128.6 (m-C6H4OCH3), 128.9 (p-C6H4OCH3), 159.9 (C–O), 168.7 (CO). Characteristic 2D NOESY correlations: CH-5/m-C6H4OCH3. Anal. Calcd for C14H17NO5: C, 60.21; H, 6.14; N, 5.02. Found: C, 60.43; H, 6.07; N, 5.05.
:10) mixture and dried in a vacuum until constant weight to give 0.992 g (89%) of nitronate 8k as a white solid. Mp = 93–95 °C. 1H NMR (CDCl3, 300.13 MHz, HSQC, NOESY): 1.36 (t, J = 7.1 Hz, 3 H, CH3CH2), 1.90 (s, 3 H, CH3), 3.85 (s, 3 H, OCH3), 4.34 (q, J = 7.1 Hz, 2 H, CH3CH2), 4.51 (br d, J = 4.0 Hz, 1 H, CH-4), 4.81 (d, J = 4.0 Hz, 1 H, CH-5), 6.96 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.21 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, HSQC): 10.6 (CH3), 14.0 (CH3CH2), 55.3 (OCH3 and CH-4), 62.2 (CH3CH2), 78.5 (CH-5), 113.3 (CN), 114.9 (o-C6H4OCH3), 128.6 (m-C6H4OCH3), 128.9 (p-C6H4OCH3), 159.9 (C–O), 168.7 (CO). Characteristic 2D NOESY correlations: CH-5/m-C6H4OCH3. Anal. Calcd for C14H17NO5: C, 60.21; H, 6.14; N, 5.02. Found: C, 60.43; H, 6.07; N, 5.05.
General procedure for the synthesis of nitrates 2a–j,p (procedure i)
To a stirred solution of Cr(NO3)3·9H2O (0.8 g, 2.0 mmol) in THF (4 mL) was added 2 mL of 0.5 M solution of bis(oxy)enamines 1a–j,p. The resulting solution was stirred for 2 h at room temperature and poured into a mixture of EtOAc (50 mL) and 0.25 M NaHSO4 solution (50 mL). The aqueous layer was back-extracted with EtOAc (50 mL). Combined organic layers were washed with 0.25 M NaHSO4 solution (50 mL), water (50 mL), and brine (50 mL), dried (Na2SO4), and evaporated in a vacuum. The residue was subjected to column chromatography on silica gel to give nitrates 2a–j,p. For determining analytical properties, products were recrystallized from pentane/Et2O mixtures.General procedure for the synthesis of nitrates 2a,k from cyclic nitronates 8 (procedure ii)
To a stirred solution of nitronates 8a,k (1.0 mmol) and triethylamine (0.17 mL, 1.2 mmol) in CH2Cl2 (3 mL) was added trimethylsilyl triflate (0.2 mL, 1.1 mmol) at −78 °C under an argon atmosphere. After stirring the reaction mixture for 2 h at −78 °C, a solution of Cr(NO3)3·9H2O (0.8 g, 2.0 mmol) in THF (4 mL) was added via a syringe and the cooling bath was removed. After 2 h the resulting dark-colored solution was poured into a mixture of EtOAc (50 mL) and 0.25 M NaHSO4 solution (50 mL). The aqueous layer was back-extracted with EtOAc (50 mL). Combined organic layers were washed with 0.25 M NaHSO4 solution (50 mL), water (50 mL), and brine (50 mL), dried (Na2SO4), and evaporated in a vacuum. The residue was subjected to column chromatography on silica gel to give nitrates 2a,k as well as by-products 6a and 3k, 6k, respectively.:1). 1H NMR (CDCl3, 300.13 MHz): 1.31 and 1.41 (2 s, 6 H, 2 CH3), 1.95 (dd, J = 12.0, 13.5 Hz, 1 H, HaxC-5), 2.13 (dd, J = 7.8, 13.5 Hz, 1 H, HeqC-5), 3.51 (dd, J = 7.8, 12.0 Hz, 1 H, HaxC-4), 4.75 (d, J = 12.4 Hz, 1 H, HC–ONO2), 4.84 (d, J = 12.4 Hz, 1 H, HC–ONO2), 7.22 (d, J = 6.7 Hz, 2 H, o-C6H5), 7.32–7.38 (m, 3 H, m- and p-C6H5). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.6 and 28.3 (2 CH3), 37.8 (CH-4), 39.7 (CH2-5), 72.0 (CH2–ONO2), 75.7 (C-6), 127.9, 128.1 and 129.4 (o-, m- and p-C6H5), 138.6 (i-C6H5), 151.3 (CN). 14N NMR (CDCl3, 21.69 MHz): −45.0. FTIR (KBr): 1643 (s, νas ONO2). UV-VIS (CH2Cl2): λmax, nm 203, 237. Anal. Calcd for C13H16N2O4: C, 59.08; H, 6.10; N, 10.60. Found: C, 59.12; H, 6.22; N, 10.53.
:1). 1H NMR (CDCl3, 300.13 MHz): 1.31 and 1.35 (2 s, 6 H, 2 CH3), 1.58 and 1.74 (2 m, 4 H, CH2–CH2), 1.91 (dd, J = 12.3, 13.3 Hz, 1 H, HaxC-5), 2.09 (dd, J = 7.9, 13.3 Hz, 1 H, HeqC-5), 3.18 and 3.40 (2 m, 2 H, CH2–CH2–O), 3.56 (dd, J = 7.9, 12.3 Hz, 1 H, CH-4), 3.81 (s, 2 H, CH2–O), 4.43 (t, J = 6.5 Hz, 2 H, CH2–CH2–ONO2), 7.19 (d, J = 6.7 Hz, 2 H, o-C6H5), 7.27–7.38 (m, 3 H, m- and p-C6H5). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.8 and 28.5 (2 CH3), 23.8 and 25.7 (CH2–CH2), 37.4 (CH-4), 40.2 (CH2-5), 69.6, 70.6 and 73.1 (3 CH2–O), 74.6 (C-6), 127.2, 128.4 and 128.9 (o-, m- and p-C6H5), 140.1 (i-C6H5), 156.1 (CN). 14N NMR (CDCl3, 21.69 MHz): −41.1. FTIR (thin layer): 1636 (s, νas ONO2). HRMS: m/z Calcd for [C17H24N2O5Na+] 359.1577 ([M + Na]+). Found: 359.1577.
:1). Rf = 0.7 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.28 and 1.38 (2 s, 6 H, 2 CH3), 1.91 (dd, J = 12.1, 13.5 Hz, 1 H, HaxC-5), 2.09 (dd, J = 7.8, 13.5 Hz, 1 H, HeqC-5), 3.47 (dd, J = 7.8, 12.1 Hz, 1 H, HaxC-4), 3.80 (s, 3 H, OCH3), 4.74 (d, J = 12.2 Hz, 1 H, HC–ONO2), 4.80 (d, J = 12.2 Hz, 1 H, HC–ONO2), 6.94 (d, J = 8.7 Hz, 2 H, o-C6H4OCH3), 7.11 (d, J = 8.7 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.6 and 28.3 (2 CH3), 37.0 (CH-4), 39.6 (CH2-5), 55.3 (OCH3), 71.9 (CH2–ONO2), 75.8 (C-6), 114.7 (o-C6H4OCH3), 129.2 (m-C6H4OCH3), 130.3 (p-C6H4OCH3), 151.6 (C–O), 159.2 (CN). 14N NMR (CDCl3, 21.69 MHz): −45.0. FTIR (KBr): 1644 (s, νas ONO2). Anal. Calcd for C14H18N2O5: C, 57.13; H, 6.16; N, 9.52. Found: C, 57.02; H, 6.05; N, 9.44.
:1). Rf = 0.7 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.29 and 1.39 (2 s, 6 H, 2 CH3), 1.89 (dd, J = 12.0, 13.3 Hz, 1 H, HaxC-5), 2.14 (dd, J = 7.8, 13.3 Hz, 1 H, HeqC-5), 3.52 (dd, J = 7.8, 12.0 Hz, 1 H, HaxC-4), 4.72 (d, J = 12.5 Hz, 1 H, HC–ONO2), 4.84 (d, J = 12.5 Hz, 1 H, HC–ONO2), 7.16 (d, J = 8.3 Hz, 2 H, C6H4Cl), 7.35 (d, J = 8.3 Hz, 2 H, C6H4Cl). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.6 and 28.2 (2 CH3), 37.2 (CH-4), 39.6 (CH2-5), 71.8 (CH2-7), 75.7 (C-6), 129.5 and 129.6 (o- and m-C6H4Cl), 133.8 and 137.1 (p-C6H4Cl and C–Cl), 150.7 (CN). 14N NMR (CDCl3, 21.69 MHz): −45.6. FTIR (thin layer): 1629 (s, νas ONO2). Anal. Calcd for C13H15ClN2O4: C, 52.27; H, 5.06; N, 9.38. Found: C, 52.35; H, 5.06; N, 9.30.
:1). 1H NMR (CDCl3, 300.13 MHz): 1.29 and 1.37 (2 s, 6 H, 2 CH3), 1.59 and 1.75 (2 m, 4 H, CH2–CH2), 1.86 (dd, J = 12.1, 13.3 Hz, 1 H, HaxC-5), 2.08 (dd, J = 7.8, 13.3 Hz, 1 H, HeqC-5), 3.20 and 3.38 (2 m, 2 H, CH2–CH2–O), 3.55 (dd, J = 7.8, 12.1 Hz, 1 H, CH-4), 3.80 (s, 2 H, CH2–O), 4.44 (t, J = 6.4 Hz, 2 H, CH2–CH2–ONO2), 7.14 (d, J = 8.3 Hz, 2 H, C6H4Cl), 7.32 (d, J = 8.3 Hz, 2 H, C6H4Cl). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.7 and 28.4 (2 CH3), 23.8 and 25.7 (CH2–CH2), 36.8 (CH-4), 40.0 (CH2-5), 69.6, 70.5 and 73.0 (3 CH2–O), 74.6 (C-6), 129.1 and 129.7 (o- and m-C6H4Cl), 133.1 and 138.6 (p-C6H4Cl and C–Cl), 155.5 (CN). 14N NMR (CDCl3, 21.69 MHz): −40.9. FTIR (KBr): 1641 (s, νas ONO2). HRMS: m/z Calcd for [C17H23ClN2O5Na+] 393.1188 ([M + Na]+). Found: 393.1182.
:1). Rf = 0.7 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 2.13 (ddd, J = 13.6, 2.1, 1.5 Hz, 1 H, HeqC-5), 2.38 (ddd, J = 13.6, 11.3, 6.3 Hz, 1 H, HaxC-5), 3.66 (dd, J = 6.3, 1.5 Hz, 1 H, HeqC-4), 3.84 (s, 3 H, OCH3), 4.91 (dd, J = 11.1, 2.1 Hz, 1 H, HaxC-6), 4.96 (d, J = 12.8 Hz, 1 H, HC–ONO2), 5.09 (d, J = 12.8 Hz, 1 H, HC–ONO2), 6.96 (d, J = 8.3 Hz, 2 H, o-C6H4OCH3), 7.17 (d, J = 8.3 Hz, 2 H, m-C6H4OCH3), 7.31–7.42 (m, 5 H, o-, m-, p-C6H5). 13C NMR (CDCl3, 75.47 MHz, DEPT): 34.2 (CH2-5), 36.7 (CH-4), 55.4 (OCH3), 71.8 (CH2–ONO2), 74.0 (CH-6), 114.8 (o-C6H4OCH3), 126.6, 128.5 and 128.6 (o-, m-, p-C6H5), 129.3 (m-C6H4OCH3), 132.0 (p-C6H4OCH3), 138.7 (i-C6H5), 150.6 (C−O), 159.2 (CN). 14N NMR (CDCl3, 21.69 MHz): −45.8. FTIR: 1643 (s, νas ONO2). Anal. Calcd for C18H18N2O5: C, 63.15; H, 5.30; N, 8.18. Found: C, 62.63; H, 5.26; N, 7.97.
:1). Rf = 0.8 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz, COSY, HSQC): 1.22–1.53, 1.54–1.69 and 1.70–1.82 (3 m, 4 H, 2 H and 2 H, HC-5, HC-7, H2C-8, H2C-9, H2C-10), 2.03–2.12 (m, 1 H, HC-7), 3.12 (br s, 1 H, HeqC-4), 3.80 (s, 3 H, H3CO), 4.06 (br m, 1 H, HC-6), 4.86 (d, J = 12.7 Hz, 1 H, HC–ONO2), 5.02 (d, J = 12.7 Hz, 1 H, HC–ONO2), 6.89 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.07 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, COSY, HSQC, DEPT): 19.9 and 24.8 (CH2-9 and CH2-10), 27.5 (CH2-8), 29.0 (CH2-7), 38.8 (CH-5), 43.5 (CH-4), 55.3 (OCH3), 69.7 (CH-6), 72.1 (CH2-ONO2), 114.5 (o-C6H4OCH3), 129.1 (m-C6H4OCH3), 132.1 (p-C6H4OCH3), 148.9 (C−O), 159.0 (C-3). 14N NMR (CDCl3, 21.69 MHz): −45.1. FTIR (KBr): 1639 (s, νas ONO2). Anal. Calcd for C16H20NO5: C, 59.99; H, 6.29; N, 8.74. Found: C, 60.03; H, 6.36; N, 8.73.
:1). [α]D = +48.1 (EtOAc, c = 1.0, 24 °C). 1H NMR (CDCl3, 300.13 MHz, COSY, HSQC): 0.91 (s, 3 H, CH3), 1.16 (s, 3 H, CH3), 1.19 (d, J = 10.1 Hz, 1 H, HC-7), 1.24–1.46 (m, 2 H, CH2-6), 1.48–1.64 (m, 2 H, CH2-5), 1.83 (dd, J = 13.6, 12.7 Hz, 1 H, H′axC-5′), 1.85 (br s, 1 H, HC-4), 2.18 (d, J = 10.1 Hz, 1 H, HC-7), 2.33 (dd, J = 13.6, 7.0 Hz, 1 H, H′′eqC-5′), 2.38 (d, J = 4.4 Hz, 1 H, HC-1), 3.42 (dd, J = 12.7, 7.0 Hz, 1 H, HaxC-4′), 4.74 (d, J = 12.4 Hz, 1 H, HC–ONO2), 4.80 (d, J = 12.4 Hz, 1 H, HC–ONO2), 7.22 (d, J = 7.7 Hz, 2 H, o-C6H5), 7.30–7.42 (m, 3 H, m- and p-C6H5). 13C NMR (CDCl3, 75.47 MHz, HSQC, DEPT): 22.3 and 24.2 (2 CH3), 22.3 and 23.8 (CH2-5 and CH2-6), 30.8 (CH2-5′), 34.6 (CH2-7), 39.9 (CH-4′), 42.7 (CH-1), 44.7 (C-3), 49.3 (CH-4), 72.0 (CH2-8), 87.8 (C-2), 127.9, 128.1 and 129.4 (o-, m-, p-C6H5), 138.8 (i-C6H5), 151.9 (C-3′). 14N NMR (CDCl3, 21.69 MHz): −45.0. FTIR (KBr): 1647 (s, νas ONO2). Anal. Calcd for C19H24N2O4: C, 66.26; H, 7.02; N, 8.13. Found: C, 66.28; H, 7.00; N, 8.03.
:1). Rf = 0.7 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.25 (t, J = 7.1 Hz, 3 H, CH3), 2.12 (ddd, J = 2.6, 12.0, 13.5 Hz, 1 H, HaxC-5), 2.28 (ddd, J = 2.4, 7.6, 13.5 Hz, 1 H, HeqC-5), 3.68 (dd, J = 7.6, 12.0 Hz, 1 H, HaxC-4), 3.63 and 3.90 (2 m, 2 H, OCH2CH3), 3.82 (s, 3 H, OCH3), 4.73 (d, J = 13.2 Hz, 1 H, HC–ONO2), 4.81 (d, J = 13.2 Hz, 1 H, HC–ONO2), 5.20 (dd, J = 2.4 and 2.6 Hz, 1 H, HeqC-6), 6.91 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.13 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, DEPT): 15.0 (CH3CH2–O), 32.4 (CH2-5), 34.0 (CH-4), 55.3 (OCH3), 64.0 (CH3CH2–O), 71.4 (CH2–ONO2), 96.1 (CH-6), 114.8 (o-C6H4OCH3), 129.3 (m-C6H4OCH3), 130.0 (p-C6H4OCH3), 154.3 (C–O), 159.2 (CN). 14N NMR (CDCl3, 21.69 MHz): −45.8. FTIR (KBr): 1643 (s, νas ONO2). Anal. Calcd for C14H18N2O6: C, 54.19; H, 5.85; N, 9.03. Found: C, 54.22; H, 5.89; N, 8.98. Single crystals for X-ray analysis (CCDC 1419082) were obtained by crystallization from pentane/Et2O = 5:1 at −30 °C. Minor 4,6-cis-isomer: 1H NMR (CDCl3, 300.13 MHz, characteristic signals): 2.45 (ddd, J = 3.1, 7.8, 13.8 Hz, 1 H, HaxC-5), 4.80 (d, J = 12.6 Hz, 1 H, HC–ONO2), 4.90 (d, J = 12.6 Hz, 1 H, HC–ONO2), 5.10 (dd, J = 2.9, 5.7 Hz, 1 H, HeqC-6).
:1). 1H NMR (CDCl3, 300.13 MHz): 1.43 and 1.44 (2 s, 6 H, 2 CH3), 2.08 (dd, J = 5.8, 14.3 Hz, 1 H, HC-5), 2.33 (dd, J = 6.6, 14.3 Hz, 1 H, HC-5), 5.16 (d, J = 12.7 Hz, 1 H, HC–ONO2), 5.30 (d, J = 12.7 Hz, 1 H, HC–ONO2), 5.62 (dd, J = 5.8, 6.6 Hz, 1 H, HC-4), 7.51 (dd, J = 7.5, 7.6 Hz, 2 H, m-C6H5), 7.66 (t, J = 7.5 Hz, 1 H, p-C6H5), 8.06 (d, J = 7.6 Hz, 2 H, m-C6H5). 13C NMR (CDCl3, 75.47 MHz, DEPT): 25.2 and 25.9 (2 CH3), 35.9 (CH2-5), 60.1 (CH-4), 70.7 (CH2–ONO2), 75.8 (C-6), 128.7, 129.7 and 133.8 (o-, m-, p-C6H5 and i-C6H5), 147.6 (CN), 165.3 (CO). 14N NMR (CDCl3, 21.69 MHz): −46.6. FTIR (thin layer): 1724 (s, CO), 1643 (s, νas ONO2). Anal. Calcd for C14H16N2O6: C, 54.54; H, 5.23; N, 9.09. Found: C, 54.40; H, 4.88; N, 8.90.
:3). 1H NMR (CDCl3, 300.13 MHz): 1.66 (s, 3 H, CH3), 2.92 (d, J = 17.6 Hz, 1 H, CHH), 3.57 (d, J = 17.6 Hz, 1 H, CHH), 3.81 (s, 3 H, OCH3), 5.22 (s, 2 H, CH2ONO2). 13C NMR (CDCl3, 75.47 MHz, DEPT): 23.2 (CH3), 44.4 (CH2), 53.1 (OCH3), 66.4 (CH2ONO2), 86.8 (C−O), 151.7 (CN), 171.6 (CO). 14N NMR (CDCl3, 21.69 MHz): −47.1. FTIR (thin layer): 1647 (s, νas ONO2). HRMS: Calcd for [C7H10N2O6K+]: m/z 257.0170 ([M + K]+). Found: 257.0171.
:1). Rf = 0.7 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 2.99 (dd, J = 17.3, 8.5 Hz, 1 H, HC), 3.49 (dd, J = 17.3, 11.0 Hz, 1 H, HC), 5.27 (s, 2 H, CH2–ONO2), 5.66 (dd, J = 11.0, 8.5 Hz, 1 H, HC–Ar), 7.20 (d, J = 8.2 Hz, 2 H, C6H4Br), 7.52 (d, J = 8.2 Hz, 2 H, C6H4Br). 13C NMR (CDCl3, 75.47 MHz, DEPT): 42.7 (CH2), 66.6 (CH2–ONO2), 82.4 (CH), 122.5 (p-C6H4Br), 127.4 and 132.0 (o-C6H4Br and m-C6H4Br), 139.0 (C–Br), 151.4 (CN). 14N NMR (CDCl3, 21.69 MHz): −47.0. FTIR (KBr): 1640 (s, νas ONO2), 525 (m, C–Br). Anal. Calcd for C10H9BrN2O4: C, 39.89; H, 3.01; N, 9.30. Found: C, 39.71; H, 3.01; N, 9.11.
:1). Oil. Rf = 0.6 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.60–2.99 (m, 4 H, CH2–CH2), 2.96 (dd, J = 17.3, 8.0 Hz, 1 H, HC), 3.46 (dd, J = 17.3, 10.5 Hz, 1 H, HC), 3.47 (dd, J = 6.2, 5.1 Hz, 2 H, CH2–CH2–O), 4.26 (s, 2 H, CH2–O), 4.46 (dd, J = 6.2, 6.2 Hz, 2 H, CH2–ONO2), 5.58 (dd, J = 10.5, 8.0 Hz, 1 H, HC–Ar), 7.22 (d, J = 8.2 Hz, 2 H, C6H4Br), 7.50 (d, J = 8.2 Hz, 2 H, C6H4Br). 13C NMR (CDCl3, 75.47 MHz, DEPT): 23.8 and 25.8 (CH2–CH2), 43.4 (CH2), 65.2, 70.0 and 73.0 (CH2–O, CH2–CH2–O and CH2–ONO2), 81.3 (CH), 122.1 (p-C6H4Br), 127.4 and 131.9 (o-C6H4Br and m-C6H4Br), 139.9 (C–Br), 156.0 (CN). 14N NMR (CDCl3, 21.69 MHz): −41.4. IR: 1627 (s, νas ONO2). HRMS: m/z Calcd for [C14H17BrN2O5Na+]: 395.0213 and 397.0193 ([M + Na]+). Found: 395.0216 and 397.0194. Contains diether 7j in trace amounts: Calcd for [C18H25BrN2O6Na+]: 467.0794 and 469.0776 ([M + Na]+). Found: 467.0789 and 469.0775.
:1). 1H NMR (CDCl3, 300.13 MHz): 1.32 (t, J = 7.1 Hz, 3 H, OCH2CH3), 3.81 (s, 3 H, OCH3), 4.29 (q, J = 7.1 Hz, 2 H, OCH2CH3), 4.63 (d, J = 5.3 Hz, 1 H, CH-4), 4.95 (d, J = 13.9 Hz, 1 H, HC–ONO2), 4.97 (d, J = 5.3 Hz, 1 H, CH-5), 5.21 (d, J = 13.9 Hz, 1 H, HC–ONO2), 6.91 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.12 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, DEPT): 14.0 (OCH2CH3), 55.4 (OCH3), 57.1 (CH-4), 62.2 and 64.8 (2 CH2), 86.4 (CH-5), 115.0 (o-C6H4OCH3), 127.5 (p-C6H4OCH3), 128.7 (m-C6H4OCH3), 154.1 (C–O), 159.9 (CN), 169.9 (CO). 14N NMR (CDCl3, 21.69 MHz): −47.6. FTIR (thin layer): 1741 (s, CO), 1650 (s, νas ONO2). HRMS: m/z Calcd for [C14H17N2O7+] 325.1030 ([M + H]+). Found: 325.1021. Anal. Calcd for C14H16N2O7: C, 51.85; H, 4.97; N, 8.64. Found: C, 51.86; H, 4.90; N, 8.60.
:1.0). Oil. Rf = 0.5 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.30 (t, J = 7.1 Hz, 3 H, OCH2CH3), 2.76 (br, 1 H, OH), 3.79 (s, 3 H, OCH3), 4.11 (d, J = 14.3 Hz, 1 H, CH–OH), 4.25 (q, J = 7.1 Hz, 2 H, OCH2CH3), 4.37 (d, J = 14.3 Hz, 1 H, CH–OH), 4.68 (d, J = 5.2 Hz, 1 H, CH-4), 4.87 (d, J = 5.2 Hz, 1 H, CH-5), 6.89 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.13 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, DEPT): 14.0 (OCH2CH3), 55.3 (OCH3), 57.5 (CH-4), 56.3 and 62.0 (2 CH2), 85.5 (CH-5), 114.8 (o-C6H4OCH3), 128.7 (m-C6H4OCH3 and p-C6H4OCH3), 159.6 and 160.5 (C–O and CN), 169.8 (CO). HRMS: m/z Calcd for [C14H18NO5+] 280.1179 ([M + H]+). Found: 280.1179.
:1). Oil. Rf = 0.55 (EtOAc/hexane = 1:1). 1H NMR (CDCl3, 300.13 MHz): 1.35 (t, J = 7.1 Hz, 3 H, OCH2CH3), 1.65 and 1.77 (2 m, 4 H, CH2–CH2), 3.37 and 3.48 (2 m, 2 H, CH2–CH2–O), 3.84 (s, 3 H, OCH3), 4.04 (d, J = 13.0 Hz, 1 H, CH2–O), 4.20 (d, J = 13.0 Hz, 1 H, CH2–O), 4.31 (q, J = 7.1 Hz, 2 H, OCH2CH3), 4.46 (t, J = Hz, 2 H, CH2–CH2–ONO2), 4.64 (d, J = 5.0 Hz, 1 H, CH-4), 4.92 (d, J = 5.0 Hz, 1 H, CH-5), 6.93 (d, J = 8.6 Hz, 2 H, o-C6H4OCH3), 7.16 (d, J = 8.6 Hz, 2 H, m-C6H4OCH3). 13C NMR (CDCl3, 75.47 MHz, DEPT): 14.1 (OCH2CH3), 23.7 and 25.6 (CH2–CH2), 55.3 (OCH3), 57.6 (CH-4), 62.0, 63.3, 69.7 and 73.0 (4 CH2–O), 85.4 (CH-5), 114.7 (o-C6H4OCH3), 128.7 (m-C6H4OCH3 and p-C6H4OCH3), 158.4 (C–O), 159.6 (CN), 169.7 (CO). 14N NMR (CDCl3, 21.69 MHz): −41.4. FTIR (thin layer): 1740 (s, sh, CO), 1649 (s, sh, νas ONO2). HRMS: m/z Calcd for [C18H25N2O8+] 397.1605 ([M + H]+). Found: 397.1606.
:1 (procedure i), 5.8:1 (procedure ii), HPLC). Oil. Pure 2p can be obtained by crystallization from pentane/Et2O mixtures (Mp = 110–114 °C). 1H NMR (CDCl3, 300 MHz, COSY, HSQC): 1.22–1.45, 1.49–1.75 and 1.77–2.10 (3 m, 17 H, H2C(5), H2C(9), H2C(10), H2C(11), HC(12), H2C(22) and H2C(23)), 2.40 (m, 1 H, HC(12)), 2.61 (dd, J = 11.2, 10.7 Hz, 1 H, HaxC(8)), 2.90 (dd, J = 10.9, 7.9 Hz, 1 H, HaxC(4)), 3.84 (s, 3 H, H3C(20)), 3.99 (dd, J = 10.7, 9.9 Hz, 1 H, HaxC(7)), 4.22 (d, J = 13.3 Hz, 1 H, HC(13)), 4.39 (d, J = 13.3 Hz, 1 H, HC(13)), 4.73 (br m, 1 H, HC(21)), 5.41 (br s, 1 H, HeqC(6)), 6.54 (s, 1 H, HC(15)), 6.58 (d, J = 7.1 Hz, 1 H, HC(19)), 6.79 (d, J = 7.1 Hz, 1 H, HC(18)), 7.16–7.39 (m, 5 H, C6H5). 13C NMR (CDCl3, 75.47 MHz, HSQC): 24.0 and 24.6 (CH2-9 and CH2-23), 26.1 (CH2-10), 30.5 (CH2-5), 31.8 (CH2-12), 32.7 and 32.8 (CH2-22), 33.6 (CH-4), 34.0 (CH2-11), 50.9 (CH-8), 56.1 (CH3-20), 70.6 (CH2-13), 76.3 (CH-7), 80.6 (CH-21), 91.1 (CH-6), 112.4 (CH-18), 114.8 (CH-15), 120.6 (CH-19), 125.9 (p-C6H5), 127.9 and 128.1 (o- and m-C6H5), 130.1 (C-14), 144.5 (i-C6H5), 148.2 and 149.7 (C-16 and C-17), 153.1 (C-3). 14N NMR (CDCl3, 21.69 MHz, rac-2p): −44.2. 1H NMR spectra of rac-2p are in accordance with previously published data. 1H NMR (CDCl3, 300.13 MHz, rac-2p′, characteristic signals): 3.20 (dd, J = 8.3, 8.1 Hz, 1 H, HeqC-4), 3.84 (s, 3 H, OCH3), 4.28 (dd, J = 6.5, 2.8 Hz, 1 H, HeqC-6), 4.68 (d, J = 12.9 Hz, 1 H, HC–ONO2), 4.80 (d, J = 12.9 Hz, 1 H, HC–ONO2).
Reactions of model bis(oxy)enamine 1a with metal nitrates
To a stirred solution of metal nitrate (0.5 mmol) in 1 mL of solvent indicated in Table 1 was added 0.5 ml of 0.5 M solution of bis(oxy)enamine (0.25 mmol) in CH2Cl2. The mixture was stirred at r.t. for the time indicated in Table 1, and then poured into a mixture of EtOAc (25 mL) and 0.25 M NaHSO4 solution (25 mL). The aqueous layer was back-extracted with EtOAc (25 mL). Combined organic layers were washed with 0.25 M NaHSO4 solution (25 mL), water (25 mL), and brine (25 mL), dried (Na2SO4), and evaporated in a vacuum. The residue was analyzed by 1H NMR with an internal standard. 1H NMR of by-product 3a is in accordance with published data.10h A sample of pure 4a was prepared by standard silylation (r.t., 18 h) of 3a (0.3 mmol) with (CH3)3SiCl (0.6 mmol)/Et3N (0.75 mmol) in CH2Cl2 (1 mL) followed by evaporation and extraction of the product with pentane (yield: 99%). M.p. = 51–55 °C. 1H NMR (CDCl3, 300.13 MHz): −0.04 (s, 9 H, (CH3)3Si), 1.33 and 1.37 (2 s, 6 H, 2 CH3), 1.90 (dd, J = 12.8, 12.2 Hz, 1 H, HC-5), 2.11 (dd, J = 12.8, 8.1 Hz, 1 H, HC-5), 3.66 (dd, J = 12.2, 8.1 Hz, 1 H, HC-4), 3.95 (d, J = 11.7 Hz, 1 H, CHO), 4.03 (d, J = 11.7 Hz, 1 H, CHO), 7.10–7.38 (m, 5 H, o-, m-, p-C6H5). 13C NMR (CDCl3, 75.47 MHz, HSQC): −0.37 ((CH3)3Si), 22.9 and 28.6 (2 CH3), 36.9 (CH-4), 40.6 (CH2-5), 63.0 (CH2–O), 74.5 (C-6), 127.1, 128.6 and 128.8 (o-, m-, p-C6H5), 140.5 (i-C6H5), 158.0 (CN). 29Si NMR (CDCl3, 59.63 MHz): 19.9. HRMS: m/z Calcd for [C16H26NO2Si+] 292.1727 ([M + H]+). Found: 292.1725.
:1 → 5:1 → 1:1) to give 0.045 g (34%) of 5a. Colorless oil. A mixture of E,Z-isomers, ratio 12:1.0. Rf = 0.38, 0.32 (2 isomers, EtOAc/hexane = 1:1). E-Isomer: 1H NMR (CDCl3, 300.13 MHz,): 1.26 and 1.30 (2 s, 6 H, 2 CH3), 1.84 (dd, J = 15.0, 3.8 Hz, 1 H, HC-5), 2.1 (br, 1 H, OH), 2.48 (dd, J = 15.0, 10.9 Hz, 1 H, HC-5), 3.98 (dd, J = 10.9, 3.8 Hz, 1 H, HC-4), 4.58 (d, J = 14.6 Hz, 1 H, HC–O), 5.17 (d, J = 14.6 Hz, 1 H, HC–O), 7.12–7.38 (m, 5 H, o-, m-, p-C6H5), 7.91 (s, 1 H, HCO), 10.7 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 27.9 and 31.5 (2 CH3), 45.1 (CH-4), 47.3 (CH2-5), 57.4 (CH2O), 70.6 (C-6), 127.2, 127.9 and 129.2 (o-, m-, p-C6H5), 141.6 (i-C6H5), 156.9 (CN), 160.0 (HCO). Z-Isomer: 1H NMR (CDCl3, 300.13 MHz): 1.39 and 1.47 (2 s, 6 H, 2 CH3), 2.1 (br, 1 H, OH), 2.14 (dd, J = 12.8, 3.8 Hz, 1 H, HC-5), 2.58 (dd, J = 12.8, 12.6 Hz, 1 H, HC-5), 3.53 (m, 1 H, HC-4), 4.76 (d, J = 14.7 Hz, 1 H, HC–O), 5.53 (d, J = 14.7 Hz, 1 H, HC–O), 7.12–7.38 (m, 5 H, o-, m-, p-C6H5), 7.91 (s, 1 H, HCO), 10.7 (br, 1 H, OH). 13C NMR (CDCl3, 75.47 MHz, DEPT, characteristic signals): 40.3 and 43.7 (CH-4 and CH2-5), 63.3 (CH2O). Both isomers: HRMS: m/z Calcd for [C14H20NO4+] 266.1387 ([M + H]+). Found: 266.1377.
:1. Z-Isomer: 1H NMR (CDCl3, 300.13 MHz): 0.35 (s, 9 H, (CH3)3Si), 5.69 (s, 2 H, CH2–ONO2), 7.39–7.46 and 7.63–7.67 (2 m, 4 H and 1 H, o-, m-, p-C6H5). 13C NMR (CDCl3, 75.47 MHz, DEPT): −0.76 ((CH3)3Si), 64.3 (CH2–ONO2), 126.8, 128.6 and 129.9 (o-, m-, p-C6H5), 132.9 (i-C6H5), 154.8 (CN). 29Si NMR (CDCl3, 59.63 MHz): 28.4. 14N NMR (CDCl3, 21.69 MHz): −44.7. E-Isomer: 1H NMR (CDCl3, 300.13 MHz, characteristic signals): 0.34 (s, 9 H, (CH3)3Si), 5.37 (s, 2 H, CH2–ONO2). 13C NMR (CDCl3, 75.47 MHz, DEPT, characteristic signals): −0.64 ((CH3)3Si), 62.2 (CH2–ONO2). Both isomers: FTIR (thin layer): 1644 (s, νas ONO2). MS (EI): m/z (%) = 268 (8) [M]+˙, 207 (95) [M − CH3 − NO2]+, 192 (73) [M − CH2ONO2]+, 103 (20) [M − Ph − CH2ONO2]+, 77 (26) [Ph]+, 73 (100) [(CH3)3Si]+.
Synthesis of acyclic α-nitroxyoximes 9m–o (procedure iv)
To a stirred solution of anhydrous Co(NO3)2 (0.732 g, 4.0 mmol) in a mixture of DMF (4 mL) and CH2Cl2 (4 mL) was added 4 mL of 1 M solution of bis(trimethysilyloxy)enamines 1m–o (4.0 mmol) in CH2Cl2 at r.t. under an argon atmosphere. The mixture was stirred for 2.5 h at r.t., then poured into a mixture of AcOEt (100 mL) and 0.25 M aqueous solution of NaHSO4 (100 mL) and intensively shaken for ca. 5 min. The aqueous layer was back-extracted with EtOAc (50 mL). The combined organic layers were washed with a 0.25 M aqueous solution of NaHSO4 (70 mL), water (70 mL), and brine (70 mL), dried (Na2SO4), and concentrated in a vacuum. The residue was dissolved in 5 mL of Et2O. Water (1 mL) was added and the mixture was intensively shaken for 1 min. The organic layer was separated and the water phase was washed with 5 mL of Et2O. Combined organic layers were dried over Na2SO4 and the volatiles were evaporated. The residue was dried in a vacuum (10 Torr) to give α-nitroxy-oximes 9m–o. Caution! Low molecular weight organic nitrates are explosive. No attempts to prepare more than 0.2 g were made.:1. E-Isomer: 1H NMR (CDCl3, 300.13 MHz): 5.05 (d, J = 5.5 Hz, 2 H, CH2ONO2), 7.54 (t, J = 5.5 Hz, 1 H, CH), 8.39 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 68.6 (CH2ONO2), 143.7 (CN). Z-Isomer: 1H NMR (CDCl3, 300.13 MHz): 5.29 (d, J = 3.7 Hz, 2 H, CH2ONO2), 6.91 (t, J = 3.7 Hz, 1 H, CH), 8.74 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 65.8 (CH2ONO2), 145.1 (CN). Both isomers: 14N NMR (CDCl3, 21.69 MHz): −45.8. FTIR (thin layer): 3306 (s, br, OH), 2917 (m, CH), 1645 (s, νas ONO2), 1426 (m), 1277 (s), 935 (m, sh), 852 (s). MS (EI): m/z = 76 [CH2ONO2]+, 58 [M − NO3]+, 57 [M − HNO3]+˙, 46 [NO2]+, 44 [M − CH2ONO2]+.
:1. E-Isomer: 1H NMR (CDCl3, 300.13 MHz): 1.98 (s, CH3), 4.98 (s, CH2ONO2), 9.12 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 12.0 (CH3), 73.1 (CH2ONO2), 151.2 (CN). Z-Isomer: 1H NMR (CDCl3, 300.13 MHz): 1.96 (s, CH3), 5.33 (s, CH2ONO2), 9.12 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT, characteristic signals): 15.7 (CH3), 64.5 (CH2ONO2). Both isomers: 14N NMR (CDCl3, 21.69 MHz): −45.2. FTIR (thin layer): 3272 (s, br, OH), 2923 (m, CH), 1730 (m), 1644 (s, νas ONO2), 1438 (m), 1288 (s), 945 (m, sh), 921 (m), 851 (s). MS (EI): m/z = 135 [M + H]+, 76 [CH2ONO2]+, 58 [M − NO3]+, 46 [NO2]+, 44 [M − CH2ONO2]+.
:1) on silica gel at −15 °C provided a sample of individual 12c, which was used for further analysis. A mixture of E/Z-isomers, ratio 3.8:1. Rf = 0.55 (EtOAc/hexane = 1:1). E-Isomer: 1H NMR (CDCl3, 300.13 MHz): 2.50–2.70 (m, 4 H, CH2–CH2), 3.70 (s, 3 H, OCH3), 5.07 (s, 2 H, CH2ONO2), 9.18 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 22.0 and 29.7 (CH2–CH2), 52.0 (OCH3), 72.5 (CH2ONO2), 153.0 (CN), 173.2 (CO). Z-Isomer: 1H NMR (CDCl3, 300.13 MHz): 2.50–2.70 (m, 4 H, CH2–CH2), 3.80 (s, 3 H, OCH3), 5.33 (d, J = 3.7 Hz, 2 H, CH2ONO2), 9.03 (br, 1 H, NOH). 13C NMR (CDCl3, 75.47 MHz, DEPT): 25.7 and 29.9 (CH2–CH2), 52.0 (OCH3), 66.8 (CH2ONO2), 155.1 (CN), 173.9 (CO). Both isomers: 14N NMR (CDCl3, 21.69 MHz): −45.2. FTIR (thin layer): 3382 (s, br, OH), 2957 (m, sh, CH), 1733 (s, CO), 1644 (s, νas ONO2), 1440 (m), 1368 (m), 1283 (s, sh), 1203 (m), 1176 (m), 983 (m, sh), 851 (s), 640 (w). HRMS: m/z Calcd for [C6H10N2O6Na+] 229.0431 ([M + Na]+). Found: 229.0434. MS (EI): m/z = 206 [M]+˙.
Acknowledgements
This work was supported by the Russian Science Foundation (Grant 14-50-00126).Notes and references
- (a) A. Koenig, K. Lange, J. Konter, A. Daiber, D. Stalleicken, E. Glusa and J. Lehmann, J. Cardiovasc. Pharmacol., 2007, 50, 68–74 CrossRef CAS PubMed; (b) K. Lange, A. Koenig, C. Roegler, A. Seeling and J. Lehmann, Bioorg. Med. Chem. Lett., 2009, 19, 3141–3144 CrossRef CAS PubMed; (c) M. W. Dewhirst, J. S. Stamler, T. J. Mcmahon and P. Sonveaux, Patent WO2006/113540A2, 2006 (Duke University) Search PubMed; (d) I. V. Serkov and V. V. Bezuglov, Russ. Chem. Rev., 2009, 78, 407–429 CrossRef CAS; (e) P. L. Bosquesi, T. R. F. Melo, E. O. Vizioli, J. Leandro dos Santos and M. C. Chung, Pharmaceuticals, 2011, 4, 1450–1474 CrossRef CAS; (f) S. G. Zlotin, A. M. Churakov, O. A. Luk'yanov, N. N. Makhova, A. Yu. Sukhorukov and V. A. Tartakovsky, Mendeleev Commun., 2015, 25, 399–409 CrossRef CAS; (g) L. L. Fershtat, M. A. Epishina, A. S. Kulikov, I. V. Ovchinnikov, I. V. Ananyev and N. N. Makhova, Tetrahedron, 2015, 71, 6764–6775 CrossRef CAS; (h) G. A. Smirnov, P. B. Gordeev, S. V. Nikitin, G. V. Pokhvisneva, T. V. Ternikova and O. A. Luk'yanov, Russ. Chem. Bull., Int. Ed., 2015, 64, 1057–1061 CrossRef CAS.
- (a) A. K. Shaikh, A. J. A. Cobb and G. Varvounis, Org. Lett., 2012, 14, 584–587 CrossRef CAS PubMed; (b) A. K. Shaikh and G. Varvounis, Org. Lett., 2014, 16, 1478–1481 CrossRef CAS PubMed; (c) T. H. Graham, C. M. Jones, N. T. Jui and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 16494–16495 CrossRef CAS PubMed.
- (a) R. Caderas, R. Lett, L. Overman, M. H. Rabinowitz, L. A. Robinson, M. Z. Sharp and J. Zablocki, J. Am. Chem. Soc., 1996, 118, 9073–9082 CrossRef; (b) Z. Qin, J. Luo, L. Vandevrede, E. Tavassoli, M. Fa, A. F. Teich, O. Arancio and G. R. J. Thatcher, J. Med. Chem., 2012, 55, 6784–6801 CrossRef CAS PubMed.
- (a) N. Kornblum and H. W. Frazier, J. Am. Chem. Soc., 1966, 88, 865 CrossRef CAS; (b) D. L. Lindner, J. B. Doherty, G. Shoham and R. B. Woodward, Tetrahedron Lett., 1982, 23, 5111–5114 CrossRef CAS; (c) M. J. Melnick and S. M. Weinreb, J. Org. Chem., 1988, 53, 850–854 CrossRef CAS.
- (a) J. Prakash Agrawal, High Energy Materials: Propellants, Explosives and Pyrotechnics, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) D. E. Chavez, M. A. Hiskey, D. L. Naud and D. Parrish, Angew. Chem., Int. Ed., 2008, 47, 8307–8309 CrossRef CAS PubMed.
- G. A. Olah, R. Malhotra and S. C. Narang, Nitration. Methods and Mechanisms (Organic Nitro Chemistry Series), VCH Verlagsgesellschaft, Weinheim, VCH Publishers, New York, 1989 Search PubMed.
- (a) A. F. Ferris, K. W. McLean, I. G. Marks and W. D. Emmons, J. Am. Chem. Soc., 1953, 75, 4078–4078 CrossRef CAS; (b) D. Garvey, L. Letts, R. Earl, M. Ezawa, X. Fang, R. Gaston, S. Khanapure, C.-E. Lin, R. Ranatunge, C. Stevenson and S.-J. Wey, Patent US20060189603A1, 2006(Nitromed, Inc.) Search PubMed; (c) L. Lazzarato, M. Donnola, B. Rolando, K. Chegaev, E. Marini, C. Cena, A. Di Stilo, R. Fruttero, S. Biondi, E. Ongini and A. Gasco, J. Med. Chem., 2009, 52, 5058–5068 CrossRef CAS PubMed; (d) A. McKillop and M. E. Ford, Tetrahedron, 1974, 30, 2467–2475 CrossRef CAS.
- (a) X.-T. Chen, D. Sames and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 7760–7769 CrossRef CAS; (b) S. Lee and J. N. P. Rosazza, Org. Lett., 2004, 6, 365–368 CrossRef CAS PubMed; (c) E. Baciocchi, C. Rol, G. V. Sebastiani and A. Zampini, J. Chem. Soc., Chem. Commun., 1982, 1045–1047 RSC; (d) M.-Y. Chang, C.-Y. Lin and T.-C. Wu, Tetrahedron Lett., 2006, 47, 5445–5449 CrossRef CAS; (e) V. Nair, S. B. Panicker, A. Augustine, T. G. George, S. Thomas and M. Vairamani, Tetrahedron, 2001, 57, 7417–7422 CrossRef CAS.
- (a) A. Michael and G. H. Carlson, J. Am. Chem. Soc., 1935, 57, 1268–1276 CrossRef CAS; (b) I. Dunstan, J. V. Griffiths and S. A. Harvey, J. Chem. Soc., 1965, 1319–1324 RSC; (c) J. Barluenga, J.-M. Martinez-Gallo, C. Najera and M. Yus, J. Chem. Soc., Chem. Commun., 1985, 20, 1422–1423 RSC; (d) R. J. Lewis and R. B. Moodie, J. Chem. Soc., Perkin Trans. 2, 1996, 1315–1320 RSC; (e) H. Wieland and F. Rahn, Berichte, 1921, 54, 1770–1776 Search PubMed; (f) A. A. Stotskii, V. V. Kirichenko and L. I. Bagal, J. Org. Chem. USSR, 1973, 9, 2486–2491 Search PubMed.
- (a) A. A. Tishkov, A. D. Dilman, V. I. Faustov, A. A. Birukov, K. S. Lysenko, P. A. Belyakov, S. L. Ioffe, Y. A. Strelenko and M. Yu. Antipin, J. Am. Chem. Soc., 2002, 124, 11358–11367 CrossRef CAS PubMed; (b) H. Feger and G. Simchen, Liebigs Ann. Chem., 1986, 1456–1465 CrossRef CAS; (c) A. A. Tishkov, A. V. Lesiv, Yu. A. Khomutova, Yu. A. Strelenko, I. D. Nesterov, M. Yu. Antipin, S. L. Ioffe and S. E. Denmark, J. Org. Chem., 2003, 68, 9477–9480 CrossRef CAS PubMed; (d) A. D. Dilman, A. A. Tishkov, I. M. Lyapkalo, S. L. Ioffe, Yu. A. Strelenko and V. A. Tartakovsky, Synthesis, 1998, 181–184 CrossRef CAS; (e) A. D. Dilman, S. L. Ioffe and H. Mayr, J. Org. Chem., 2001, 66, 3196–3200 CrossRef CAS PubMed; (f) M. S. Klenov, A. V. Lesiv, Yu. A. Khomutova, I. D. Nesterov and S. L. Ioffe, Synthesis, 2004, 1159–1170 CAS; (g) A. A. Tabolin, A. V. Lesiv, Yu. A. Khomutova, Yu. V. Nelyubina and S. L. Ioffe, Tetrahedron, 2009, 65, 4578–4592 CrossRef CAS; (h) A. A. Tabolin, A. V. Lesiv and S. L. Ioffe, Synthesis, 2009, 3099–3105 CAS; (i) A. A. Mikhaylov, A. D. Dilman, M. I. Struchkova, Yu. A. Khomutova, A. A. Korlyukov, S. L. Ioffe and V. A. Tartakovsky, Tetrahedron, 2011, 67, 4584–4594 CrossRef CAS; (j) A. A. Mikhaylov, P. A. Zhmurov, A. S. Naumova, Y. A. Khoroshutina, A. Yu. Sukhorukov and S. L. Ioffe, Mendeleev Commun., 2015, 25, 449–451 CrossRef CAS.
- (a) S. L. Ioffe, in Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis: Novel Strategies in Synthesis, ed. H. Feuer, Wiley, Hoboken, 2nd edn, 2008, p. 658 Search PubMed; (b) A. Yu. Sukhorukov and S. L. Ioffe, Chem. Rev., 2011, 111, 5004–5041 CrossRef CAS PubMed; (c) A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed. For a related process see: (d) T. Miyoshi, T. Miyakawa, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2011, 123, 958–961 CrossRef.
- Only one example of acyclic α-nitroxy-oxime has been authentically reported: O. A. Luk'yanov and G. V. Pokhvisneva, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1991, 40, 2439–2444 CrossRef CAS . Only one example of 5-membered cyclic α-nitroxy-oxime ether has been obtained as a by-product: J. F. Hansen and P. J. Georgiou, J. Heterocycl. Chem., 1994, 31, 1487–1492 CrossRef.
- (a) F. Guthrie, Ann. Chem. Pharm., 1860, 116, 234–249 CrossRef; (b) O. Wallach, Ann. Chem., 1887, 241, 288–315 CrossRef; (c) V. N. Ipatiew and A. A. Solonina, Zh. Russ. Fiz.-Khim. O-va., 1901, 33, 496–501 Search PubMed.
- A. Yu. Sukhorukov, M. A. Kapatsyna, T. Lim Ting Yi, H. Park, Ya. A. Naumovich, P. A. Zhmurov, Yu. A. Khomutova, S. L. Ioffe and V. A. Tartakovsky, Eur. J. Org. Chem., 2014, 8148–8159 CrossRef CAS.
- For processes involving N-alkoxynitrenium cations as intermediates see: (a) V. F. Rudchenko, Chem. Rev., 1993, 93, 725–739 CrossRef CAS; (b) J. J. Campbell, S. A. Glover, G. P. Hammond and C. A. Rowbottom, J. Chem. Soc., Perkin Trans. 2, 1991, 2067–2079 RSC; (c) V. O. Smirnov, Yu. A. Khomutova, V. A. Tartakovsky and S. L. Ioffe, Eur. J. Org. Chem., 2012, 3377–3384 CrossRef CAS; (d) A. A. Tabolin, A. V. Lesiv, Yu. A. Khomutova, P. A. Belyakov, Yu. A. Strelenko and S. L. Ioffe, Synthesis, 2005, 1656–1662 CAS.
- For some other applications of the ONO2 group in total synthesis see: (a) P. W. Glunz, S. Hintermann, L. J. Williams, J. B. Schwarz, S. D. Kuduk, V. Kudryashov, K. O. Lloyd and S. J. Danishefsky, J. Am. Chem. Soc., 2000, 122, 7273–7279 CrossRef CAS; (b) T. K. Jones, H. Mrozik and M. H. Fisher, J. Org. Chem., 1992, 57, 3248–3250 CrossRef CAS; (c) A. L. Parry, N. A. Clemson, J. Ellis, S. S. R. Bernhard, B. G. Davis and N. R. Cameron, J. Am. Chem. Soc., 2013, 135, 9362–9365 CrossRef CAS PubMed.
- (a) P. A. Zhmurov, A. Yu. Sukhorukov, V. I. Chupakhin, Yu. A. Khomutova, S. L. Ioffe and V. A. Tartakovsky, Org. Biomol. Chem., 2013, 11, 8082–8091 RSC; (b) A. Yu. Sukhorukov, Y. D. Boyko, Yu. A. Khomutova, Yu. V. Nelyubina, S. L. Ioffe and V. A. Tartakovsky, J. Org. Chem., 2011, 76, 7893–7900 CrossRef CAS PubMed.
- C. Ehrhardt, M. Gjikaj and W. Brockner, Thermochim. Acta, 2005, 432, 36–40 CrossRef CAS.
- H. Wong, P. Hanselmann and W. Wenger, Patent WO2010/45764A1, 2010 (Lonza Ltd) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR and UV-Vis spectra for new compounds, X-ray data for 2g. CCDC 1419082. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob00388e |
| This journal is © The Royal Society of Chemistry 2016 |