
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbohydrates as enantioinduction components in stereoselective catalysis
Alexander S.
Henderson
,
John F.
Bower
* and
M. Carmen
Galan
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol. BS8 1TS, UK. E-mail: John.Bower@bristol.ac.uk; M.C.Galan@bristol.ac.uk
First published on 31st March 2016
Abstract
Carbohydrate derivatives are readily available chiral molecules, yet they are infrequently employed as enantioinduction components in stereoselective catalysis. In this review, synthetic approaches to carbohydrate-based ligands and catalysts are outlined, along with example applications in enantioselective catalysis. A wide range of carbohydrate-based functionality is covered, and key trends and future opportunities are identified.
 Alexander S. Henderson | Alexander S. Henderson graduated from the University of Bristol in 2011 with an MSci degree in Chemistry. Shortly after, he began a PhD degree, supervised by John F. Bower and M. Carmen Galan, researching carbohydrate-based NHCs for asymmetric catalysis. Alexander has won several awards during his studies, and, in 2013 he was selected by the Japanese Society for the Promotion of Science to undertake a summer research project at Kyoto University. He is predominantly interested in organic synthesis, carbohydrate chemistry, catalysis, and the interplay between these fields. |
 John F. Bower | John F. Bower obtained his MSci degree in Chemistry in 2003 from the University of Bristol, where he remained to study for his PhD degree (2007). His first postdoctoral appointment (2007–2008) was with Professor Michael Krische at the University of Texas at Austin, and his second was with Professor Timothy Donohoe at the University of Oxford. In 2010, he was awarded a Royal Society University Research Fellowship and commenced his independent career at the University of Bristol. Bower's research has been recognized by a number of awards, including the 2013 Royal Society of Chemistry Harrison-Meldola Memorial Prize and the 2015 Royal Society of Chemistry Hickinbottom Award. |
 M. Carmen Galan | M. Carmen Galan is currently a Reader in Organic and Biological Chemistry and an EPSRC Career Acceleration Fellow in the School of Chemistry at the University of Bristol. Prior to that, she held a four-year Royal Society Dorothy Hodgkin fellowship. Carmen received her PhD in Organic Chemistry from the Complex Carbohydrate Research Center at the University of Georgia, USA, under the supervision of Prof. Geert-Jan Boons. She then moved to California to pursue post-doctoral research with Prof. Chi-Huey Wong at the Scripps Research Institute. After that, she continued her post-doctoral training at M.I.T. with Prof. Sarah O'Connor. Carmen moved to the UK in October 2006 to start her independent career in Bristol. |
1. Introduction
Enantioselective catalysis has become the dominant approach to the asymmetric synthesis of chiral molecules. Relaying “chiral information” from a sub-stoichiometric source, by way of a useful chemical transformation, will underpin future advances in asymmetric chemistry.1 The development of new chiral metal–ligand complexes and organocatalysts, which surpass established enantioinduction benchmarks or provide new enantioselective transformations, is of huge importance because of the vital role of homochiral molecules in drug design. A large proportion of chiral catalysts are prepared directly from biologically derived sources (e.g. amino acids). Carbohydrates are a class of abundant and readily modifiable “chiral pool” building blocks. However, these stereochemically rich biomolecules continue to be underexploited in catalyst design.2 This represents something of a chemical paradox, which has not arisen through lack of effort from synthetic chemists. Many reports describe the use of carbohydrates in stereoselective transformations, and this area has been reviewed previously.3 A preconception may exist that carbohydrates are challenging to work with, and therefore largely limited to glycobiological applications.4 However, carbohydrates are innately chiral and possess a valuable array of stereochemistry, functionality and scaffold diversity, which can all be exploited in catalyst design.This review aims to break the stigma associated with carbohydrate chemistry, by highlighting expedient, modular and diversifiable synthetic routes to carbohydrate-based catalysts for application in asymmetric organo- and transition-metal (TM) catalysis (Fig. 1). It is our hope that the successful strategies outlined here will stimulate the design and evaluation of novel carbohydrate-based systems in other areas of asymmetric catalysis. Although by no means exhaustive, the examples discussed herein contain key steps towards the ligand or catalyst, along with benchmark applications in enantioselective catalysis.5
 | ||
| Fig. 1 Carbohydrates as enantioinduction components. | ||
2. Selected examples
2.1 Phosphine ligands
Chiral phosphine ligands have underpinned progress in asymmetric TM catalysed reactions.6 Early homochiral bidentate phosphines, such as 2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane (DIOP),7 used chiral pool building blocks to construct the ligand backbone and this strategy has been extensively explored. Two main approaches have been developed to access carbohydrate-based systems embodying a P–Ccarbohydrate bond: (1) SN2 displacement of activated alcohols by P-based nucleophiles8 (Scheme 1) and (2) nucleophilic opening of anhydro-sugars9 (see Scheme 5). A complication often observed with the former approach is competing E2 elimination.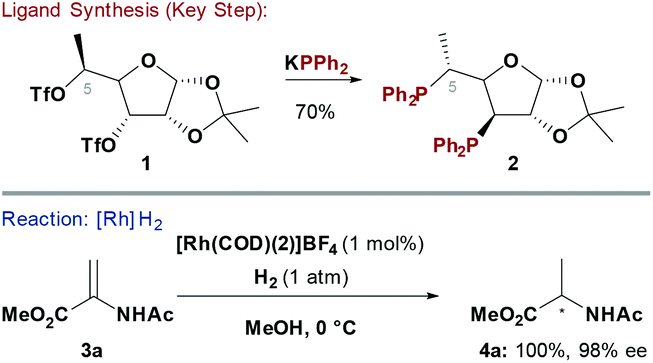 | ||
| Scheme 1 Synthesis and evaluation of a PR3 ligand. | ||
Diéguez and co-workers showed that furanoside-derived ligand 2 could be synthesised by double SN2 displacement of ditriflate 1 with KPPh2 (Scheme 1).10 Chiral phosphine 2 was effective for Rh-catalysed enantioselective hydrogenations of “classical” chelating alkene substrates. For example, reduction of methyl 2-acetamidoacrylate (3a) proceeded with excellent levels of enantioselectivity (98% ee) at 0 °C to deliver 4a in quantitative yield. Notably, the C-5 methyl group of 2 was critical to the efficiency of the process. In the absence of this substituent low conversions were observed, whereas the corresponding C-5 epimer of 2 provided significantly lower levels of enantioselectivity.10,11 The synthetic route to 2 is modular, such that further variations of ligand structure can be easily envisaged by varying the P-based nucleophile or the furanoside substituents.
Ligand systems where the phosphine is not directly attached to the carbohydrate unit have also been reported. For example, Ruffo and co-workers accessed 7 by amide coupling of carboxylic acid containing phosphine 6 with protected 2,3-glucodiamine 5 (Scheme 2).12 Given that 7 closely resembles the highly successful Trost Ligand,13 it was unsurprising that it performed efficiently in Pd-catalysed asymmetric allylic alkylation (AAA) reactions. For example, desymmetrisation of 8 proceeded smoothly to generate carbamate 9 in excellent yield and 97% ee. Carbohydrate diamine 5 could be considered “greener” than trans-diaminocyclohexanes commonly used for Trost ligand synthesis because the latter require resolution to access enantiopure material. A D-mannose derived pseudo-enantiomeric variant of 7 was also reported.12 It is pertinent to note that diamine 5 has also been used in the synthesis of salen ligands for Mn-catalysed epoxidation of styrenes.14
 | ||
| Scheme 2 Synthesis and evaluation of a PR3 ligand. | ||
2.2 Phosphinite ligands
P–O bonds are generally easier to form than P–C variants and this enables the direct attachment of carbohydrate scaffolds to phosphorous centres (cf.Scheme 1).3b In early work, Selke synthesised a range of hexapyranoside-derived phosphinites and examined how their configuration affected enantioselectivity in Rh-catalysed asymmetric hydrogenations of functionalised alkenes. It was found that systems with all equatorial substituents (e.g. β-glucose derivatives) were most effective.15 Later, RajanBabu and co-workers exploited the potential of modular P–O bond formation to synthesise a library of phosphinite ligands by reacting β-glucoside-based backbone 10 with electronically distinct chlorodiarylphosphines (Scheme 3).16 For the reduction of dehydroamino ester precursors 3, electron rich P(aryl)2 groups induced the highest levels of asymmetry. For example, modification of a cationic Rh pre-catalyst with ligand 11 provided a system that was effective for a wide range of β-aryl substituted amino esters (e.g.4c, 97% ee). For alkyl substituted systems, enantioselectivity exhibited a greater substrate dependency, but, nevertheless, 4a (97% ee) and 4b (91% ee) were both accessed in an efficient manner. It is pertinent to note that a 3,4-O-diphosphinite glucopyranoside functioned as a pseudo-enantiomeric version of 11.16 Similar ligand systems have been evaluated in asymmetric Ni-catalysed hydrocyanations of vinyl-arenes.17 | ||
| Scheme 3 Synthesis and evaluation of a R2POR ligand. | ||
2.3 Phosphite ligands
The facile synthesis of P–O bonds has also enabled the modular construction of phosphite ligands from carbohydrate alcohols and diols. An early example by Reetz and co-workers relied on reaction of isomannide (12), which has a rigid concave structure, with various diaryl phosphorochloridates to furnish bidentate ligands such as 13 (Scheme 4).18 Phosphite 13 was evaluated in Rh-catalysed asymmetric hydrogenation of dimethyl itaconate (14) and afforded 15 in high ee (96%). The alternate (S)-BINOL derivative of 13 functioned as an efficient pseudo-enantiomeric ligand (15: 87% ee). Interestingly, ligands derived from achiral diaryl phosphorochloridates and 12 also induced high levels of asymmetry (see below). The option of modifying 12 with stereodefined diaryl phosphorochloridates is a useful strategy to fine-tune asymmetric induction and control product stereochemistry. Reetz and co-workers also showed that mono-phosphite ligands based on a similar scaffold to 13 are useful ligands for enantioselective metal catalysed transformations.19 | ||
| Scheme 4 Synthesis and evaluation of a P(OR)3 ligand. | ||
In a complementary approach by the groups of Diéguez and Claver, glucose derived furanoside diols, featuring a range of stereochemistries, were reacted with axially chiral diaryl phosphorochloridates to give a range of carbohydrate-based diphosphites in a modular manner.20 These ligands were evaluated in Rh-catalysed hydroformylation of styrenes giving branched aldehydes in up to 98% regioselectivity and 91% ee.
2.4 Mixed P ligands
Given the wide range of methods discussed above, it is not surprising that several mixed P-bidentate carbohydrate ligand systems have been developed. The sheer number of possible variations is high, hence only one example is illustrated below. Ruiz and co-workers reported phosphine–phosphite ligands in an attempt to merge favourable characteristics (e.g. high ee and catalyst turnover frequency (TOF)) found in the individual catalytic systems (Scheme 5).21 There has been growing demand for chiral electron deficient P-based ligand systems,22 and carbohydrate building blocks can provide facile access to a wide range of variants.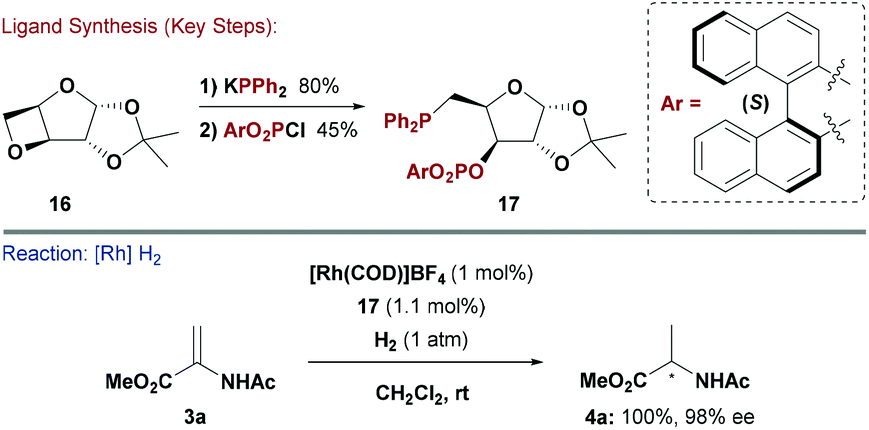 | ||
| Scheme 5 Synthesis and evaluation of a P(OR)3-PR3 ligand. | ||
The Ruiz approach hinged on derivatising modified D-furanoside xylose core 16. The PR3 unit was installed by oxetane ring opening of 16 with KPPh2. This step released a hydroxyl group, which could then be exploited for P–O bond formation by reaction with a range of diaryl phosphorochloridates.21 A Rh-system modified with 17 gave high enantioselectivities in the hydrogenation of various substituted acetamidoacrylates. Similarly to studies by Reetz and co-workers,18 it was found that the stereochemistry of product (4a) was controlled by the axial chirality of the (S)- or (R)-BINOL motif in 17. However, again, systems derived from achiral biaryls (in place of BINOL) were also efficient, presumably because the chirality of the carbohydrate backbone controlled the conformation of this unit.21 Studies by the groups of Claver and Diéguez utilised similar synthetic steps to access bidentate phosphite–phosphoramidite and diphosphoramidate ligands. These showed good results in asymmetric Rh-catalysed hydrogenations of chelating alkene substrates.23
2.5 P,alkene ligands
Due to their affinity for metal centres through cooperative binding modes, alkenes are widely employed as substrates in late-TM catalysis. This property has also stimulated the development of chiral diene and P,alkene ligands.24 Unsaturated carbohydrates are readily available (e.g. glycals and 2,3-unsaturated glycosides) and Boysen and co-workers have endeavoured to exploit these feedstocks for the easy synthesis of P,alkene ligands. These systems are of use for a variety of asymmetric reactions, including Rh-catalysed 1,4-additions of arylboronic acids to enones.25–28Ferrier rearrangement of commercially available acetylated galactal 18, followed by global deprotection and selective tritylation (Tr) of the primary alcohol gave 19 in good overall yield (Scheme 6). Reaction of the allylic alcohol of 19 with ClPPh2 effected P–O bond formation to generate phosphinite-alkene ligand 20.26 This was evaluated in challenging Rh-catalysed asymmetric additions of heteroaryl MIDA and pinacol boronates to cyclohexenone (21).28 Excellent enantioselectivities were observed using a wide range of heteroaryl nucleophiles, albeit often in modest yield. For example, thiophene derivative 22 was delivered in 56% yield and 96% ee. The modularity of the ligand synthesis and the availability of different carbohydrate precursors allowed Boysen and co-workers to conduct extensive structure-activity studies, which also led to pseudo-enantiomeric ligand systems.26,27
 | ||
| Scheme 6 Synthesis and evaluation of a R2POR-Alkene ligand. | ||
2.6 P,N ligands
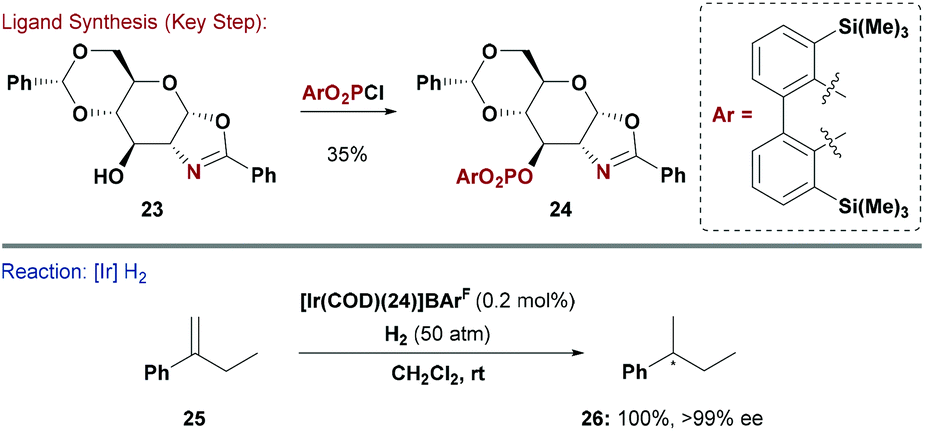
Chiral mimics of Crabtree's catalyst have been highly successful for enantioselective hydrogenation of unfunctionalised olefins, and numerous other asymmetric transformations now use P,N ligands.29 The possible permutations of P and N donors in these kind of systems is large. For example the P-based unit can be a phosphine, phosphinite or phosphite, whereas the N-donor is often part of an oxazoline or a pyridine.30 The trans-effect that such systems impart when ligated to a metal is crucial to their efficacy in enantioselective processes.31 Ohe, Uemura and co-workers reported carbohydrate P,N ligands based on a glucosamine derived scaffold. Their system offers high structural flexibility as both the oxazoline heterocycle and P-based substituents can be easily modified.32The groups of Diéguez and Andersson capitalised on this approach and synthesised a series of phosphite-oxazoline ligands, which were evaluated in Ir-catalysed hydrogenations of unfunctionalised olefins (Scheme 7).33 A range of substituted gluco-oxazolines (e.g.23) were reacted with diaryl phosphorochloridates to give the corresponding P,N ligands (e.g.24). By combining these with a cationic Ir-source, excellent enantioselectivities could be obtained for the hydrogenation of a broad range of tri- and 1,1-di-substituted olefins. Critical to the success of this approach was the ability to rapidly modify both the P and N donors. Ligand 24 and related derivatives are also effective in asymmetric Heck reactions.34
 | ||
| Scheme 7 Synthesis and evaluation of a P,N ligand. | ||
2.7 S-based ligands
In a similar vein to P,N ligands, P,S ligands have been evaluated in a wide range of asymmetric processes.3b The incorporation of S functional groups into carbohydrates is well established due to the synthetic importance of thioglycosides as glycosyl donors for oligosaccharide synthesis.35 Glycosylation with thiols is very general and provides a modular approach to diversifying any potential ligand library. In this context, Khiar and co-workers synthesised a range of mono-hydroxylated thio-galactopyranosides (e.g.27) and then used the free-OH to give a series of chiral thio-phosphinite ligands (e.g.28) (Scheme 8).36 Interestingly, ligation of 28 to a Pd-centre afforded selectively a single diastereomer, resulting from preferential coordination of one of the two diastereotopic sulfur lone pairs.36,37 This system was evaluated in Pd-catalysed AAA of dimethylmalonate with (E)-1,3-diphenylallyl acetate (29) and gave 30 in high yield and 92% ee. The synthesis of a pseudo-enantiomeric version of 28, where a D-arabinose core was used to invert the stereo-relationship between the S and P groups, was also described.36 Both systems were also evaluated in asymmetric Rh-catalysed hydrogenations of enamides. | ||
| Scheme 8 Synthesis and evaluation of a P,S ligand. | ||
In a related study, Khiar and co-workers glycosylated dithiols with peracylated glycosyl donors to give chelating C2-symmetric carbohydrate-bis(thioether) ligands, which were evaluated in Pd AAA.38 Additionally, Pregosin and co-workers have reported carbohydrate derived S-oxazoline ligands. These were accessed by alkylating anomeric thiols with oxazoline-based alkyl chlorides. These systems afforded high enantioselectivities in Pd-catalysed AAA reactions.39
An alternative strategy by Pàmies, Diéguez and co-workers exploited the facile displacement of the primary alkyl triflate of D-xylose derivative 31 with thiolate nucleophiles to create a library of furanoside-based thioether ligands (Scheme 9).40 The secondary alcohol of the product was then utilised for P–O bond formation to give various thio-phosphite ligands (e.g.32). This highly modular approach led to the discovery that ligand 32 is useful for asymmetric Pd-catalysed C–C, C–N and C–O bond formations between allylic acetates and various nucleophiles. Notably, 32 could induce high asymmetry for processes involving both cyclic (e.g.33) and acyclic allylic acetates.
 | ||
| Scheme 9 Synthesis and evaluation of a P,S ligand. | ||
2.8 Oxazoline ligands
C 2-Symmetric bis(oxazoline) ligands have revolutionalised asymmetric Lewis acid and TM catalysed reactions. This ligand class is especially popular due to its modularity and synthetic accessibility.41 Most C2-symmetric chiral oxazoline ligands use amino acid derived 1,2-amino-alcohols as the source of chirality. Surprisingly, amino-sugars, such as glucosamine, have not been widely exploited in the synthesis of this ligand class.42Boysen and co-workers have published extensive studies on a series of bis(oxazoline) ligands such as 37 (Scheme 10).42,43 The nature of the C3–OH appendage impacted asymmetric induction during catalysis and inversion or removal of this group gave decreased selectivities.43b One synthetic route facilitated late-stage modification of derivative 35 at the crucial C3–OH with a variety of groups (Ac, Piv, Me, Bn etc.), such as formyl (Scheme 10). Treatment of 36 with NIS and TfOH resulted in bis(oxazoline) 37, via double cyclisation of the amides.43b A complex formed in situ between 37 and CuOTf was efficient in catalysing the asymmetric cyclopropanation (Cp) of styrenes with diazoacetates.42,43 One of the more challenging processes, involving aliphatic alkene 38 and diazoacetate 39, proceeded smoothly to deliver 40 in 75% yield and 90% ee for the trans-diastereomer (73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 trans:cis). This intermediate could be further elaborated to (+)-grenadamide.44 Related carbohydrate pybox and thiazoline ligands have also been explored, with the former delivering high enantioselectivities in Cu-catalysed alkynylations of imines.45
:27 trans:cis). This intermediate could be further elaborated to (+)-grenadamide.44 Related carbohydrate pybox and thiazoline ligands have also been explored, with the former delivering high enantioselectivities in Cu-catalysed alkynylations of imines.45
 | ||
| Scheme 10 Synthesis and evaluation of a bis(oxazoline) ligand. | ||
2.9 N-heterocyclic carbene (NHC) ligands
NHCs have rapidly found wide-ranging applications in organo- and TM-catalysed transformations.46 In particular, chiral variants of this ligand class have been extensively researched for asymmetric processes.47 These studies have included systems where carbohydrates serve as the source of homochirality.Early approaches to carbohydrate-based NHC ligands exploited glycosylation of imidazoles with pyranoside-based anomeric bromides to give un-symmetrical C-1 linked NHCs. These were evaluated in alkene metathesis reactions and organocatalysis.48 However, this approach was limited as there were few options for modification of the carbohydrate hydroxyls. An alternative approach developed by Henderson, Bower and Galan, used glucosamine derivative 41, which could be O-alkylated with a variety of different groups, thereby providing a short and diversifiable approach to carbohydrates such as 42 (Scheme 11).49 Conversion of 42 to C2-symmetric imidazolium 43 was readily achieved, and ligation to Rh proceeded smoothly. The resulting complex showed promising results in asymmetric hydrosilylations of ketones (e.g.44). For example, alcohol 45 was obtained in 89% yield and 60% ee.49 The effects of carbohydrate stereochemistry were also examined using related mannosamine derived NHC ligands, but these provided inferior results.
 | ||
| Scheme 11 Synthesis and evaluation of an NHC ligand. | ||
An alternate and very elegant strategy was developed by Sollogoub and co-workers who selectively synthesised β-cyclodextrin 47·HCl by SN2 displacement of parent bis-mesylate 46 with imidazole (Scheme 12).50 A similar bis-alkylation with benzimidazole provided access to a core modified variant. The Ag-complex of 47 was used to transmetallate the NHC onto AuCl, and the resulting system was applied to an asymmetric Au-catalysed enyne cyclisation/cyclopropanation reaction, which gave 49 in 77% yield and 59% ee.50
 | ||
| Scheme 12 Synthesis and evaluation of a cyclodextrin-based NHC ligand. | ||
2.10 O-based ligands
Alkoxides generate hard anions and so their deployment as ligands for late-TMs is somewhat limited. However, when used in conjunction with oxophilic metals, the resulting Lewis acidic (LA) or basic (LB) complexes can be versatile catalysts for chiral transformations.51Shibasaki and co-workers synthesised a range of deoxy-glucose-based ligands (e.g.52) which possess free hydroxyl groups.52 Initially, the catechol moiety in 52 was installed by SNAr substitution of a Cr-complexed fluoro-arene.52a,b However, this approach did not allow modular access to a ligand library so an alternate sequence was employed. Reduction of D-glucal, inversion of the C3–OH and formation of the C6–OTs gave deoxy-allose scaffold 50 ready for functionalisation (Scheme 13). This was converted to cyclic sulfate 51, which could be opened stereospecifically with different catechol derivatives.52c The ligand class was designed for TM- or lanthanide-catalysed asymmetric cyanosilylation of ketones (Scheme 13).52 Indeed, catalytic quantities of 52 and Ti(Oi-Pr)4 effected 1,2-addition of TMSCN to acetophenone to deliver product (R)-53 in 85% yield and 92% ee. An analogous reaction, using a 2:1 ratio of 52 and Gd(Oi-Pr)3 as the catalyst, gave (S)-53 in 92% yield and 92% ee.53 The combination of a bimetallic Gd complex with 52 was far more reactive than the corresponding Ti system, and has since been employed in enantioselective Strecker reactions of ketoimines,54 1,4-additions of cyanide to α,β-unsaturated N-acylpyrroles,55 and desymmetrisations of N-acyl aziridines with TMSCN.56
 | ||
| Scheme 13 Synthesis and evaluation of an O ligand. | ||
2.11 N-based ligands
Homochiral 1,2-amino-alcohols can be used to construct other ligand classes, such as oxazolines, or one can directly exploit the ligation ability of the NH and OH groups in TM catalysis. This approach has seen widest application in asymmetric transfer hydrogenation (ATH).57Woggon and co-workers reported several β-cyclodextrin derived 1,2-amino-alcohol ligands for Ru-catalysed ATH of challenging alkyl–alkyl ketones (Scheme 14).58 Ligand 55 was accessed by regioselective opening of epoxide 54 with methylamine (other amines could also be used).58b1H NMR indicated that the resulting anti-relationship between the C-2 and C-3 substituents caused carbohydrate 55 to ring flip to the indicated conformer. The C-3NH and C-2OH of 55 ligated to [Ru(C6H6)Cl2]2, and the resulting homochiral complex effected high asymmetric induction in ketone reduction; for example, reduction of 56 generated 57 in 93% ee. The efficacy of the system was attributed to encapsulation of the ketone substrate within the cyclodextrin pore.58b
 | ||
| Scheme 14 Synthesis and evaluation of an N,O cyclodextrin-based ligand. | ||
In an alternate strategy, the groups of Diéguez and Adolfsson pursued a modular approach to a new family of N,heteroatom donors. Here, carbohydrate derived amines were N-acylated with protected amino acids to give hydroxy-amide type ligands, such as 60 (Scheme 15).59 The C-6 amine of 58 was installed by reduction of the corresponding azide, itself accessed by SN2 displacement of a primary tosylate. The modularity of the approach allowed the introduction of different amino-acid derivatives (e.g.59) with varying steric demands and stereochemistry. Ligand 60 was used in Ru-catalysed ATH of aryl–alkyl ketones (e.g.61), and delivered the product alcohols in very high enantioselectivity (generally >99% ee), including sterically hindered variants, such as 62. Interestingly, the carbohydrate unit of 60 is primarily responsible for asymmetric induction. A system in which the valine unit was replaced with a glycine residue still gave high enantioselectivities, albeit with lower catalytic activity.59
 | ||
| Scheme 15 Synthesis and evaluation of an N,N,O ligand. | ||
Another popular ligand class for asymmetric ATH are thioamides (e.g.64, Scheme 16), which can be accessed by treatment of parent amide 63 with Lawesson's reagent. Diéguez and co-workers explored furanoside and pyranoside thioamides, using their previous strategy59 of coupling amino acids with carbohydrates (Scheme 15).60 This modular approach led to the identification of mannosamine-(S)-valine 64 as an efficient ligand for Rh-catalysed ATH of heteroaromatic ketones (e.g.65).60c Using this system, alcohol 66 was generated in high yield (90%) and enantioselectivity (99%). Interestingly, the (R)-valine derived diastereomer of 64 functioned efficiently as a pseudo-enantiomeric ligand.
 | ||
| Scheme 16 Synthesis and evaluation of an N,S ligand. | ||
Carbohydrate-modified pyridine ligands are sparsely documented, even though scaffolds such as bypridine (bpy) are ubiquitous in many TM catalysed protocols.61 An interesting example from Billard, Queneau and co-workers describes the synthesis of bipyridine-diesters (e.g.68) from the parent carboxylic acid, 67, and various carbohydrate alcohols, such as diacetone glucose (Scheme 17).62 When 68 was combined with Cu(OTf)2, β-keto ester 69 underwent electrophilic fluorination with NFSI to give products, such as 70, in moderate yields and low enantioselectivity (27%). These results suggest that this approach might have further potential for optimisation.
 | ||
| Scheme 17 Synthesis and evaluation of a bipyridine ligand. | ||
2.12 Organocatalysts
Asymmetric organocatalysis has emerged as a powerful strategy to access enantioenriched molecules.46c,63 The well-established Shi epoxidation system has been the focus of several in-depth reviews and will not be discussed further.5b,c H-bond donors, such as (thio)ureas, have proved immensely popular since their inception.64 A wide variety of chiral pool building blocks have been evaluated as the source of chirality for these systems. A seminal report by Kunz and co-workers described the use of carbohydrate-based urea organocatalysts in asymmetric Strecker and Mannich reactions.65 Later, carbohydrate based primary-amine thiourea organocatalysts were also developed. For example, Ma and co-workers disclosed systems for enantioselective addition of aromatic ketones to nitro-olefins.66 Independently, Zhou and co-workers developed similar tertiary amine-thiourea catalysts for asymmetric aza-Henry reactions.67 The discovery of these carbohydrate-based amine thiourea organocatalysts was facilitated by their modular construction.66,67To highlight an example, the addition of functionalised chiral amine 72, serving as the variational motif, to anomeric thioisocyanate–carbohydrate 71 delivered thiourea 73 (Scheme 18).66 Ma and co-workers applied organocatalyst 73 to a decarboxylative Mannich reaction (DMR) between β-keto acids (e.g.75) and ketimines (e.g.74). This afforded products such as 76 in high yields (98%) and enantioselectivities (93%).68 Elaboration of 76 into anti-HIV drug DPC 083 was also reported. However, organocatalyst 73, derived from common D-glucose, gave the incorrect enantiomer and so the antipode of thiourea 73 was used. In a related Mannich reaction, the structural effects of the carbohydrate unit were probed and found to be crucial. In the absence of this unit, the levels of asymmetric induction were inferior.69
 | ||
| Scheme 18 Synthesis and evaluation of a thiourea organocatalyst. | ||
It is pertinent to note that several carbohydrate-based amines have been developed for enamine catalysis. Benchmark applications have focussed on aldol reactions between cyclohexanone or acetone and electron deficient aryl aldehydes.70
More recently Morken and co-workers have reported enantioselective diborations of alkenes catalysed by de-oxy carbohydrates (Scheme 19).71 Key diol 79 was synthesised in 4-steps in 65% yield on a gram scale from commercially available D-glucal 77. Hydrogenation of the double bond followed by enzyme-mediated O-6 selective deacetylation afforded 78. Silylation of the free hydroxyl group and then global deacetylation gave 79 concisely. Given the wide range of silyl protecting groups available, it is easy to envisage modular access to derivatives of the diol catalyst. By exploiting the increased reactivity of the homochiral diboron reagent formed from the reversible displacement of the neopentyl (neo) ligands in 81 with 79 ((79)2B2), the enantioselective diboration of alkenes (e.g.80) was achieved in high ee's and good yields. The intermediate boronic esters (e.g.82) can undergo stereospecific oxidation to give the corresponding diol (e.g.83), or, alternatively, can be modified by site selective Pd-catalysed cross-coupling reactions (not shown). It is pertinent to note that an L-rhamnal-derived diol functioned as an efficient pseudo-enantiomeric catalyst.72
 | ||
| Scheme 19 Synthesis and evaluation of a diol organocatalyst. | ||
3. Conclusions and outlook
The aim of this review is to exemplify synthetic routes to carbohydrate derivatives that have been used as enantioinducing components in catalysis. This survey reveals the frequent use of glucosyl scaffolds, in both the furan- and pyranoside forms, alongside a plethora of C–X (C–S, C–N and C–P), O–P and N–P bond formations. The resulting “toolbox” of methods provides flexible entries to a wide range of normally bidentate ligand systems. The carbohydrate unit can also be combined with other readily available homochiral sources, such as amino acids or conformationally restricted biaryls. This approach provides a high degree of modularity and often easy access to pseudo-enantiomeric systems.Because carbohydrate chemistry has evolved as a methodology for oligosaccharide synthesis, it is not surprising that common protecting groups (esters, acetals and alkyl ethers) are routinely featured. However, for chiral catalyst systems, the C–OH functionality provides an opportunity for modification or tuning, rather than simply requiring protection. For example, “clicking” in aryl groups by SNAr would deliver carbohydrate–aryl ethers, where sterics and electronics could be varied to produce ligand/catalyst libraries in a rapid manner.2c
Although access to the carbohydrate-based systems in the examples described here is generally expedient, the asymmetric reactions chosen for evaluation are largely benchmarks and not new transformations. Applications to asymmetric hydrogenation of chelating olefins or AAA reactions addresses a mature field. The deployment of carbohydrate-based catalysts in asymmetric methodologies that embody novel or underutilised reactivity modes would greatly elevate their importance in asymmetric catalysis.
Acknowledgements
ASH thanks EPSRC Bristol Chemical Synthesis CDT EP/G036764/1, JFB thanks Royal Society for a University Research Fellowship and MCG thanks the ERC and EPSRC CAF EP/L001926/1 and EPSRC EP/J002542/1 for funding.Notes and references
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (b) R. Gawley and J. Aubé, Principles of Asymmetric Synthesis, Elsevier, Oxford, 2nd edn, 2012 Search PubMed.
- (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed; (b) Privileged Chiral Ligands and Catalysts, ed. Q. L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) A. S. Henderson, S. Medina, J. F. Bower and M. C. Galan, Org. Lett., 2015, 17, 4846 CrossRef CAS PubMed.
- (a) M. Diéguez, O. Pàmies, A. Ruiz, Y. Díaz, S. Castillón and C. Claver, Coord. Chem. Rev., 2004, 248, 2165 CrossRef; (b) M. Diéguez, O. Pàmies and C. Claver, Chem. Rev., 2004, 104, 3189 CrossRef PubMed; (c) M. M. K. Boysen, Chem. – Eur. J., 2007, 13, 8648 CrossRef CAS PubMed; (d) M. Diéguez, C. Claver and O. Pàmies, Eur. J. Org. Chem., 2007, 4621 CrossRef; (e) V. Benessere, R. Del Litto, A. De Roma and F. Ruffo, Coord. Chem. Rev., 2010, 254, 390 CrossRef CAS; (f) S. Woodward, M. Diéguez and O. Pàmies, Coord. Chem. Rev., 2010, 254, 2007 CrossRef CAS; (g) T. Lehnert, G. Özüduru, H. Grugel, F. Albrecht, S. M. Telligmann and M. M. K. Boysen, Synthesis, 2011, 2685 CAS; (h) V. Benessere, M. Lega and F. Ruffo, J. Organomet. Chem., 2014, 771, 105 CrossRef CAS; (i) Carbohydrates: Tools for Stereoselective Synthesis, ed. M. M. K. Boysen, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) M. C. Galan, D. Benito-Alifonso and G. M. Watt, Org. Biomol. Chem., 2011, 9, 3598 RSC; (b) M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599 RSC; (c) S. Oscarson and L. Somsák, Carbohydr. Res., 2011, 346, 1357 CrossRef CAS PubMed; (d) P. Stallforth, B. Lepenies, A. Adibekian and P. H. Seeberger, J. Med. Chem., 2009, 52, 5561 CrossRef CAS PubMed.
- Heavily researched systems, such as the Shi organocatalyst or tartaric acid derived ligands, will not be discussed given the wealth of literature already in existence. See: (a) H. Pellissier, Tetrahedron, 2008, 64, 10279 CrossRef CAS; (b) M. Frohn and Y. Shi, Synthesis, 2000, 1979 CrossRef CAS; (c) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed.
- B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5348 CrossRef CAS PubMed.
- T. P. Dang and H. B. Kagan, J. Chem. Soc. D, 1971, 481 RSC.
- V. Šunjić, I. Habuš and G. Snatzke, J. Organomet. Chem., 1989, 370, 295 CrossRef.
- C. Li, B. Bernet, A. Vasella, E. A. Broger and A. Meili, Carbohydr. Res., 1992, 216, 149 CrossRef.
- M. Diéguez, O. Pàmies, A. Ruiz, S. Castillón and C. Claver, Tetrahedron: Asymmetry, 2000, 11, 4701 CrossRef.
- O. Pàmies, G. Net, A. Ruiz and C. Claver, Eur. J. Inorg. Chem., 2000, 2000, 2011 CrossRef.
- F. Ruffo, R. Del Litto, A. De Roma, A. D'Errico and S. Magnolia, Tetrahedron: Asymmetry, 2006, 17, 2265 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed.
- (a) C. Borriello, R. Del Litto, A. Panunzi and F. Ruffo, Tetrahedron: Asymmetry, 2004, 15, 681 CrossRef CAS; (b) F. Ruffo, A. Bismuto, A. Carpentieri, M. E. Cucciolito, M. Lega and A. Tuzi, Inorg. Chim. Acta, 2013, 405, 288 CrossRef CAS.
- (a) R. Selke, J. Prakt. Chem., 1987, 329, 717 CrossRef CAS; (b) A. Kumar, G. Oehme, J. P. Roque, M. Schwarze and R. Selke, Angew. Chem., Int. Ed. Engl., 1994, 33, 2197 CrossRef.
- T. V. RajanBabu, T. A. Ayers, G. A. Halliday, K. K. You and J. C. Calabrese, J. Org. Chem., 1997, 62, 6012 CrossRef CAS.
- (a) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1992, 114, 6265 CrossRef CAS; (b) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1996, 118, 6325 CrossRef CAS.
- M. T. Reetz and T. Neugebauer, Angew. Chem., Int. Ed., 1999, 38, 179 CrossRef CAS.
- M. T. Reetz and G. Mehler, Angew. Chem., Int. Ed., 2000, 39, 3889 CrossRef CAS.
- M. Diéguez, O. Pàmies, A. Ruiz, S. Castillón and C. Claver, Chem. – Eur. J., 2001, 7, 3086 CrossRef.
- (a) O. Pàmies, M. Diéguez, G. Net, A. Ruiz and C. Claver, Chem. Commun., 2000, 2383 RSC; (b) O. Pàmies, M. Diéguez, G. Net, A. Ruiz and C. Claver, J. Org. Chem., 2001, 66, 8364 CrossRef.
- J. Ansell and M. Wills, Chem. Soc. Rev., 2002, 31, 259 RSC.
- (a) M. Diéguez, A. Ruiz and C. Claver, Chem. Commun., 2001, 2703 Search PubMed; (b) M. Coll, O. Pàmies and M. Diéguez, Dalton Trans., 2012, 41, 3038 RSC.
- C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482 CrossRef CAS PubMed.
- T. Minuth and M. M. K. Boysen, Org. Lett., 2009, 11, 4212 CrossRef CAS PubMed.
- H. Grugel, F. Albrecht, T. Minuth and M. M. K. Boysen, Org. Lett., 2012, 14, 3780 CrossRef CAS PubMed.
- H. Grugel, F. Albrecht and M. M. K. Boysen, Adv. Synth. Catal., 2014, 356, 3289 CrossRef CAS.
- F. Albrecht, O. Sowada, M. Fistikci and M. M. K. Boysen, Org. Lett., 2014, 16, 5212 CrossRef CAS PubMed.
- (a) P. J. Guiry and C. P. Saunders, Adv. Synth. Catal., 2004, 346, 497 CrossRef CAS; (b) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819 RSC.
- (a) B. Gläser and H. Kunz, Synlett, 1998, 53 CrossRef; (b) H. Steinhagen, M. Reggelin and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1997, 36, 2108 CrossRef CAS.
- K. Yonehara, T. Hashizume, K. Mori, K. Ohe and S. Uemura, Chem. Commun., 1999, 415 RSC.
- (a) M. Diéguez, J. Mazuela, O. Pàmies, J. J. Verendel and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 7208 CrossRef PubMed; (b) J. Mazuela, P.-O. Norrby, P. G. Andersson, O. Pàmies and M. Diéguez, J. Am. Chem. Soc., 2011, 133, 13634 CrossRef CAS PubMed.
- (a) Y. Mata, M. Diéguez, O. Pàmies and C. Claver, Org. Lett., 2005, 7, 5597 CrossRef CAS PubMed; (b) Y. Mata, O. Pàmies and M. Diéguez, Chem. – Eur. J., 2007, 13, 3296 CrossRef CAS PubMed.
- (a) P. J. Garegg, in Adv. Carbohydr. Chem. Biochem., ed. D. Horton, Academic Press, Cambridge, 1997, vol. 52, p. 179 Search PubMed; (b) M. C. Galan, C. Brunet and M. Fuensanta, Tetrahedron Lett., 2009, 50, 442 CrossRef CAS; (c) M. C. Galan, A. T. Tran and S. Whitaker, Chem. Commun., 2010, 46, 2106 RSC.
- (a) N. Khiar, B. Suárez, M. Stiller, V. Valdivia and I. Fernández, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1253 CrossRef CAS; (b) N. Khiar, B. Suárez, V. Valdivia and I. Fernández, Synlett, 2005, 2963 CrossRef CAS.
- A. Albinati, P. S. Pregosin and K. Wick, Organometallics, 1996, 15, 2419 CrossRef CAS.
- N. Khiar, C. Serrano Araújo, B. Suárez and I. Fernández, Eur. J. Org. Chem., 2006, 1685 CrossRef CAS.
- K. Boog-Wick, P. S. Pregosin and G. Trabesinger, Organometallics, 1998, 17, 3254 CrossRef CAS.
- M. Coll, O. Pàmies and M. Diéguez, Org. Lett., 2014, 16, 1892 CrossRef CAS PubMed.
- (a) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS PubMed; (b) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284 CrossRef CAS PubMed.
- M. Irmak, A. Groschner and M. M. K. Boysen, Chem. Commun., 2007, 177 RSC.
- (a) T. Minuth, M. Irmak, A. Groschner, T. Lehnert and M. M. K. Boysen, Eur. J. Org. Chem., 2009, 997 CrossRef CAS; (b) T. Minuth and M. M. K. Boysen, Beilstein J. Org. Chem., 2010, 6, 23 Search PubMed.
- T. Minuth and M. M. K. Boysen, Synthesis, 2010, 2799 CAS.
- (a) M. Irmak and M. M. K. Boysen, Adv. Synth. Catal., 2008, 350, 403 CrossRef CAS; (b) M. Irmak, T. Lehnert and M. M. K. Boysen, Tetrahedron Lett., 2007, 48, 7890 CrossRef CAS.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS; (b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (c) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS PubMed.
- (a) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (b) F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS.
- (a) F. Tewes, A. Schlecker, K. Harms and F. Glorius, J. Organomet. Chem., 2007, 692, 4593 CrossRef CAS; (b) B. K. Keitz and R. H. Grubbs, Organometallics, 2009, 29, 403 CrossRef.
- A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2014, 12, 9180 CAS.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213 CrossRef CAS PubMed.
- (a) M. Shibasaki and M. Kanai, Org. Biomol. Chem., 2007, 5, 2027 RSC; (b) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117 CrossRef CAS PubMed.
- (a) Y. Hamashima, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 7412 CrossRef CAS; (b) Y. Hamashima, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2001, 42, 691 CrossRef CAS; (c) S. Masumoto, K. Yabu, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2002, 43, 2919 CrossRef CAS.
- K. Yabu, S. Masumoto, S. Yamasaki, Y. Hamashima, M. Kanai, W. Du, D. P. Curran and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 9908 CrossRef CAS PubMed.
- (a) S. Masumoto, H. Usuda, M. Suzuki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 5634 CrossRef CAS PubMed; (b) N. Kato, M. Suzuki, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2004, 45, 3147 CrossRef CAS.
- T. Mita, K. Sasaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 127, 514 CrossRef PubMed.
- T. Mita, I. Fujimori, R. Wada, J. Wen, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 11252 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- (a) A. Schlatter, M. K. Kundu and W.-D. Woggon, Angew. Chem., Int. Ed., 2004, 43, 6731 CrossRef CAS PubMed; (b) A. Schlatter and W.-D. Woggon, Adv. Synth. Catal., 2008, 350, 995 CrossRef CAS.
- M. Coll, O. Pàmies, H. Adolfsson and M. Diéguez, Chem. Commun., 2011, 47, 12188 RSC.
- (a) M. Coll, K. Ahlford, O. Pàmies, H. Adolfsson and M. Diéguez, Adv. Synth. Catal., 2012, 354, 415 CrossRef CAS; (b) M. Coll, O. Pàmies, H. Adolfsson and M. Diéguez, ChemCatChem, 2013, 5, 3821 CrossRef CAS; (c) M. Coll, O. Pàmies and M. Diéguez, Adv. Synth. Catal., 2014, 356, 2293 CrossRef CAS.
- (a) C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553 CrossRef CAS PubMed; (b) H.-L. Kwong, H.-L. Yeung, C.-T. Yeung, W.-S. Lee, C.-S. Lee and W.-L. Wong, Coord. Chem. Rev., 2007, 251, 2188 CrossRef CAS.
- A. Assalit, T. Billard, S. Chambert, B. R. Langlois, Y. Queneau and D. Coe, Tetrahedron: Asymmetry, 2009, 20, 593 CrossRef CAS.
- E. I. Balmond, M. C. Galan and E. M. McGarrigle, Synlett, 2013, 2335 CAS.
- (a) M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef CAS; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- C. Becker, C. Hoben and H. Kunz, Adv. Synth. Catal., 2007, 349, 417 CrossRef CAS.
- K. Liu, H.-F. Cui, J. Nie, K.-Y. Dong, X.-J. Li and J.-A. Ma, Org. Lett., 2007, 9, 923 CrossRef CAS PubMed.
- C. Wang, Z. Zhou and C. Tang, Org. Lett., 2008, 10, 1707 CrossRef CAS PubMed.
- H.-N. Yuan, S. Wang, J. Nie, W. Meng, Q. Yao and J.-A. Ma, Angew. Chem., Int. Ed., 2013, 52, 3869 CrossRef CAS PubMed.
- B. Qiao, Y. J. Huang, J. Nie and J. A. Ma, Org. Lett., 2015, 17, 4608 CrossRef CAS PubMed.
- Selected examples: (a) A. Tsutsui, H. Takeda, M. Kimura, T. Fujimoto and T. Machinami, Tetrahedron Lett., 2007, 48, 5213 CrossRef CAS; (b) S. Pedatella, M. De Nisco, D. Mastroianni, D. Naviglio, A. Nucci and R. Caputo, Adv. Synth. Catal., 2011, 353, 1443 CrossRef CAS; (c) L. Wynands, S. Delacroix, A. Nguyen Van Nhien, E. Soriano, J. Marco-Contelles and D. Postel, Tetrahedron, 2013, 69, 4899 CrossRef CAS.
- L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508 CrossRef CAS PubMed.
- S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |