
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric nucleophilic dearomatization of diazarenes by anion-binding catalysis†
Theresa
Fischer
a,
Julia
Bamberger
ab and
Olga García
Mancheño
*ab
aInstitute for Organic Chemistry, University of Regensburg, 93053 Regensburg, Germany
bStraubing Center of Science for Renewable Resources, 94315 Straubing, Germany. E-mail: olga.garcia-mancheno@chemie.uni-regensburg.de
First published on 31st March 2016
Abstract
The first anion-binding organocatalyzed enantioselective Reissert-type dearomatization of diazarenes has been developed. This reaction represents a synthetic challenge since diazarenes have various reactive sites. The use of a chiral tetrakistriazole as a C–H-based hydrogen-donor catalyst allowed the straightforward highly regio- and enantioselective synthesis of a variety of chiral diazaheterocycles.
Introduction
Chiral diazaheterocycles and their partial unsaturated derivatives are important naturally occurring substances and building blocks for the synthesis of bioactive compounds with a broad activity spectrum.1 A few examples of relevant natural and synthetic bioactive di-nitrogen-containing chiral heterocycles are shown in Fig. 1. | ||
| Fig. 1 Selected bioactive chiral diazaheterocycles. | ||
Among some interesting quinazoline derivatives, letermovir2 is one of the top-selling antiviral drugs developed for the treatment of Cytomegalovirus infections and the alkaloid vasicine3 is a cardiac-depressant. Moreover, based on a pyrazine moiety, matlystain B shows collagenase inhibitor properties.4 Other di- or tetrahydro-structures based on diazarenes such as quinoxaline, naphthyridine or phthalazine present relevant biological activities such as CETP inhibition against atherosclerosis,5 anti-dyslipidemia6 or dihydrofolate reductase inhibition towards antibiotic-resistant Gram-positive bacteria.7
Despite the great diversity of applications of chiral diazaheterocycles, there is still a demand of simple, mild and direct synthesis methods. Most of the common routes to chiral diazaheterocycles require long and tedious synthesis from chiral starting materials and normally involve the generation of at least one of the N-heterocyclic rings.1 A more appealing and straightforward approach consists of the enantioselective dearomatization of readily available diazarenes (Scheme 1).8
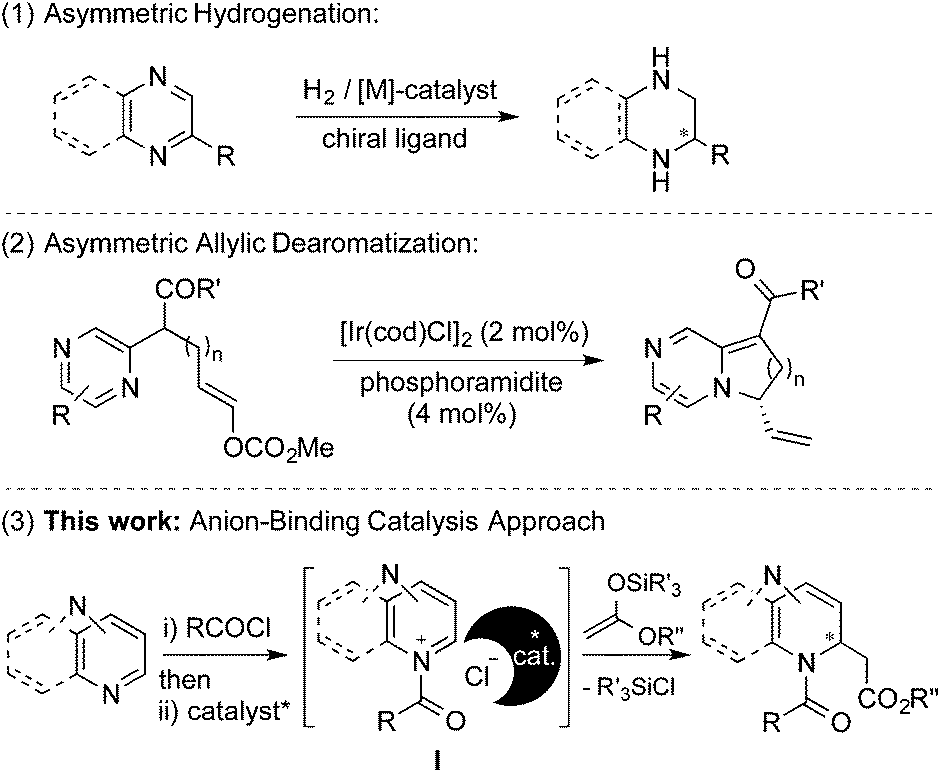 | ||
| Scheme 1 Asymmetric dearomatization of diazarenes. | ||
In this regard, the main method for inducing chirality relies on catalyzed asymmetric hydrogenation reactions of substituted azarenes (Scheme 1, (1)).9 Several methods based on enantioselective nucleophilic additions have been developed for mono N-heteroarenes.10 However, to the best of our knowledge only one example for diazarenes, the intramolecular allylic amination of pyrazines, has been described to date (Scheme 1, (2)).11 This fact could be attributed to the more challenging dearomatization of diazarenes due to the presence of a larger number of reactive sites and the possible generation of a complex mixture of products.
Recently, we have described the use of a family of triazole-based H-bond donors12 as efficient anion-binding catalysts13 for the asymmetric nucleophilic dearomatization of N-heteroarenes such as isoquinolines, quinolines and pyridines.14 Aiming at the development of a new entry for the synthesis of chiral diazaheterocycles, we decided to explore these H-donor catalysts for the related dearomatization of various types of 6-membered ring-containing diazarenes (Scheme 1, (3)). Accordingly, we anticipated successful chiral transfer from a contact ion-pair I formed between an ionic intermediate and the catalyst-counter anion complex. In this article, we present a highly enantioselective dearomatization of in situ generated N-acyldiazarene chloride salts (Reissert-type reaction)15 with silyl ketene acetals catalyzed by a chiral tetrakistriazole.
Results and discussion
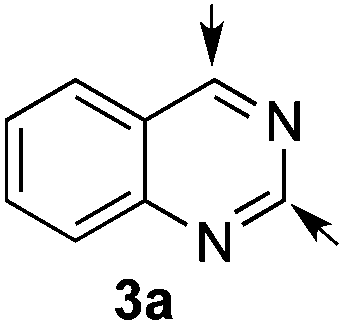
Our studies started with quinazoline (3a) as the model substrate (Table 1). Various chiral H-donor catalysts such as tetrakistriazole 1a,14 Jacobsen's thiourea 2a,16 squaramide 2b17 and bifunctional thiourea-cinchona alkaloid 2c18 were initially explored (Fig. 2). | ||
| Fig. 2 H-donor catalysts tested in this study. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Yieldb (%) |
5a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6a:7ac :6a:7ac |
5a, erd |
| a Conditions: (i) 3a (1 equiv.) and TrocCl (1 equiv.) were stirred in an appropriate dry solvent at 0 °C for 30 min; then (ii) catalyst 1 or 2 (10 mol%) and 4a (2 equiv.) were added at −78 °C and stirred for 18 h while allowing to reach slowly rt. b Isolated yield. c Isomeric ratios determined by 1H-NMR of the crude reaction. d Enantiomeric ratios determined by chiral HPLC. e Isomer 7a was not detected by NMR. f Reaction using 5 mol% of catalyst 1a. | ||||||
| 1 | — | MTBE | −78–rt | 56 | 91:9:—e |
— |
| 2 | 1a | MTBE | −78–rt | 65 | 92:8:—e |
96:4 |
| 3 | 2a | MTBE | −78–rt | 34 | 92:8:—e |
61:39 |
| 4 | 2b | MTBE | −78–rt | 21 | 92:8:—e |
46:54 |
| 5 | 2c | MTBE | −78–rt | 13 | 92:8:—e |
45:55 |
| 6 | 1a | Et2O | −78–rt | 61 | 91:9:—e |
84:16 |
| 7 | 1a | MTBE | −78 | 88 | 92:8:—e |
96:4 |
| 8 | 1a | MTBE | −40 | 54 | 92:8:—e |
86:14 |
| 9 | 1a | MTBE | −78–rt | 66 | 92:8:—e |
91:9f |

Following previously reported procedures,14,16 2,2,2-trichloroethyl chloroformate (TrocCl) was employed to generate in situ the required quinazolinium chloride salt in MTBE at 0 °C. Subsequent addition of the silyl ketene acetal 4 and the H-donor catalyst 1 or 2 at −78 °C (allowing the reaction mixture to warm up to room temperature overnight) delivered the dearomatized product. It is worth mentioning that there was an appreciable background reaction in the absence of a catalyst (entry 1, 56%). Fortunately the heterocycle 5a was formed regioselectively, not observing the formation of other possible isomers 6a and 7a. The catalytic reactions also showed complete regioselectivity towards 5a. From the catalysts tested in this study (entries 2–5), the triazole-based H-donor 1a proved to be the most efficient in terms of both reactivity and enantioselectivity. Thus, 5a was obtained in 65% yield and 96:4 er (entry 2),19 whereas the other catalysts delivered the dearomatized product in significantly low yields (13–34%) and low to moderate enantiomeric inductions (45:55–61:39 er vs. 96:4 er). The change to other ethereal solvents such as Et2O was not beneficial, hampering the enantioselectivity (84:16 er, entry 6). When the reaction was carried out at a continuous temperature of −78 °C, the same enantiomeric result (96:4 er, entry 7) was obtained. A similar procedure at −40 °C led to 5a in a lower 86:14 er (entry 8). Lastly, the use of 5 mol% of catalyst 1a provided an inferior chiral induction (91:9 er, entry 9). Therefore, 10 mol% of catalyst loading and a slow temperature-gradient (from −78 °C to r.t.) were employed as optimal conditions for further studies.
Next, screening of the acylation reagents and silyl ketene acetals 4 was carried out (Table 2). CbzCl and methoxycarboxylic chloride could also be employed as acylating reagents (entries 2 and 3). However, a significantly lower enantioselectivity and a deficient conversion accompanied by a poor regioselectivity were respectively observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1C(O)Cl | XR2/R3 | Product 5 | Yieldb (%) | 5, erc |
|
a Conditions: (i) 3a (1 equiv.) and R1COCl (1 equiv.) were stirred in dry MTBE at 0 °C for 30 min; then (ii) catalyst 1a (10 mol%) and 4 (2 equiv.) were added at −78 °C and stirred for 18 h while allowing to reach slowly rt.
b Isolated yield.
c Enantiomeric ratios determined by chiral HPLC.
d Isomeric ratios 5:6 determined by 1H–NMR of the crude reaction in brackets.
e An inseparable mixture of 5ac, 6ac and staring material 3a.
f Reaction with a 1:0.8 isomeric mixture of silyl ketene acetal 4d. The diastereomeric ratio of 5af was determined by 1H-NMR of the crude reaction. n.d. = not determined.
|
|||||
| 1 | TrocCl | OiPr/H |
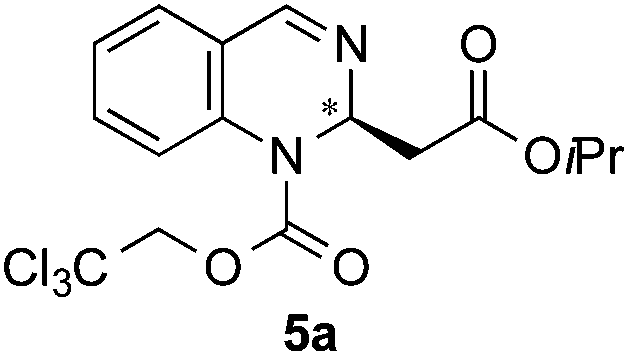
|
65 | 96:4 (98:2)d |
| 2 | CbzCl | OiPr/H |

|
71 | 60:40 (n.d.)d |
| 3 | MeOCOCl | OiPr/H |
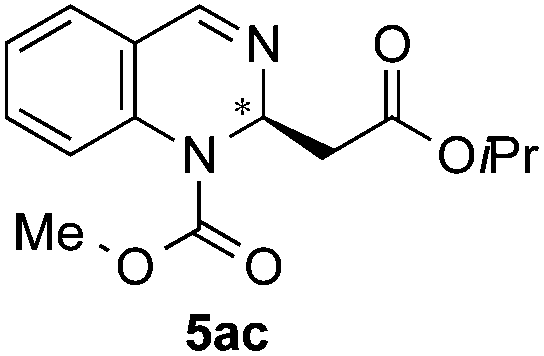
|
n.d.e | n.d.e (84:16)d |
| 4 | TrocCl | OMe/H |

|
72 | 72:28 (94:6)d |
| 5 | TrocCl | OtBu/H |
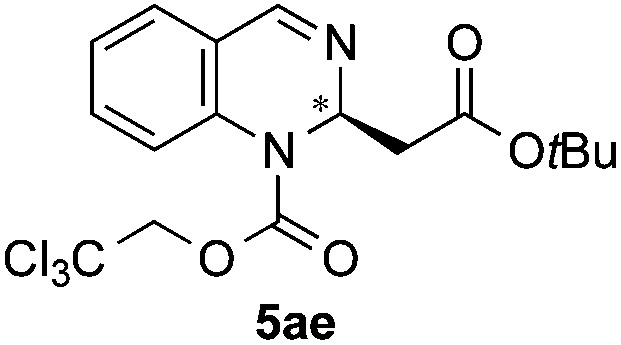
|
62 | 83:17 (95:5)d |
| 6 | TrocCl | OEt/Me |
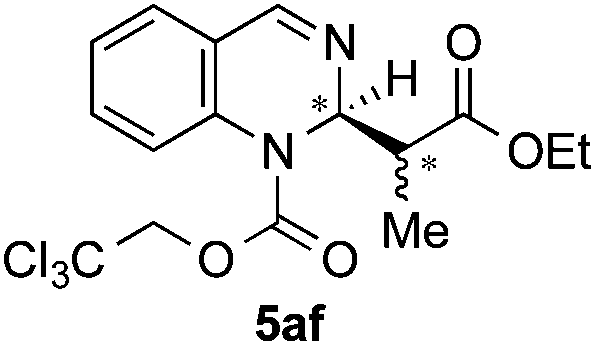
|
91 | 88:12 (major) |
| 73:27 (minor) |
|||||
| [5:1 dr]f |
|||||
Silyl ketene acetals 4 derived from acetic acid presenting different substitution such as less hindered MeO (4b, entry 4) or bulkier tBuO (4c, entry 5) groups, as well as a propionic acid derivative (4d, entry 6) were then explored. Moderate to good enantioselectivities were achieved (72:28 to 88:12 er), where the initial iPrO-substituted ketene acetal 4a remained the most efficient nucleophile.
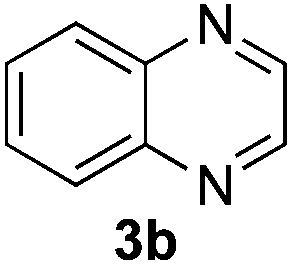
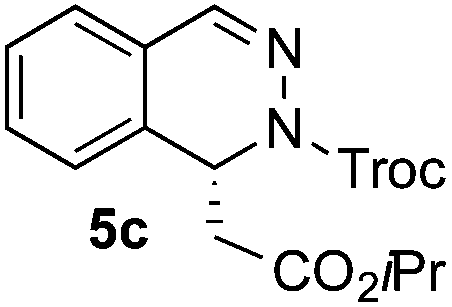
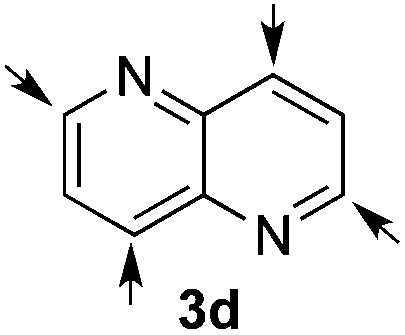
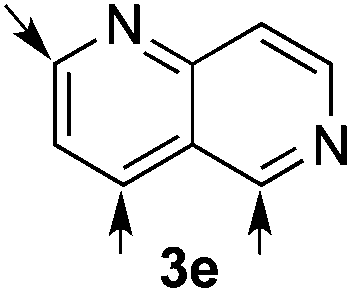
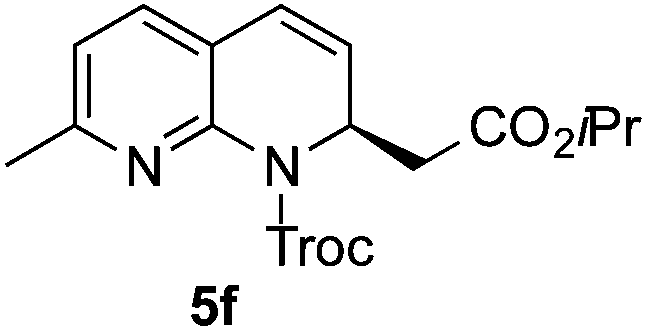
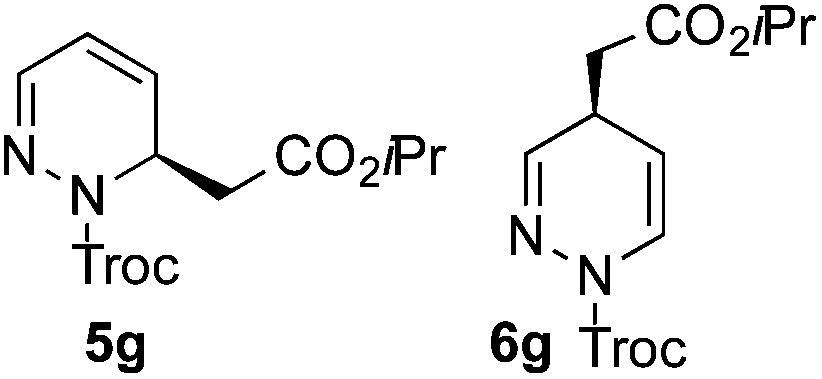
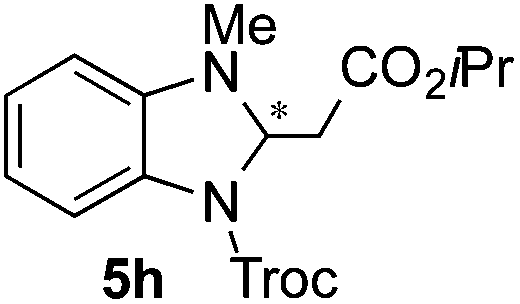
Based on these results, the screening of the substrate scope was next carried out with catalyst 1a, TrocCl, silyl ketene acetal 4a and a number of representative, readily available mono- and bicyclic diazarenes in MTBE (Table 3).19 It is important to mention that the reaction could be scaled-up to approximately 40 times (0.5 g scale) without any significant detriment to the enantioselectivity of the reaction (92:8 er, entry 1). Moreover, the catalyst could be re-isolated in a good 74% yield and reused, delivering the same reactivity and stereochemical results. The study continued with the dearomatization of the analogous diazarene quinoxaline (3b), which also presents both nitrogen atoms in the same aromatic unit (entry 2). Although this substrate reminds of the structure of quinoline, only a complex mixture was obtained, in which the double addition of the TrocCl to both nitrogen atoms could also be observed. The dearomatization of the highly symmetric phthalazine (3c), exhibiting only one equivalent reactive α-position, yielded compound 5c in a good 93% yield and 76:24 enantiomeric ratio (entry 3). In the case of 1,5-naphthyridine (3d), which has one nitrogen atom in each ring, the challenge was again the control of the regioselectivity since both the C4 and C2 positions of each heteroaromatic ring are prone to nucleophilic addition (entry 4). A good regioselectivity of 95:5 was obtained in favour of the desired C2-addition product 5d. After the separation from the minor 4-addition product 6d, compound 5d was obtained in a 86% yield and a good 83:17 enantiomeric ratio. Next, 1,6-naphthyridine (3e) was explored as a substrate (entry 5). Since this compound contains both the quinoline and the isoquinoline unit, it was interesting to get a deeper understanding about the reactivity, regioselectivity and enantioselectivity of this type of mixed structure. Due to the higher intrinsic reactivity of the benzylic position within the isoquinoline core, a high regioselectivity could be expected. Consequently, 5e was obtained as a single isomer and with high enantioselectivity (80:20 er). The reaction with methyl-substituted 1,8-naphthyridine (3f) proceeded smoothly, providing exclusively compound 5f in a good 74% yield and a significantly lower enantioselectivity (63:37 er, entry 6). This unexpected result compared to other naphthyridines cannot be easily rationalized, since in the previous work the related monoazarene quinolines provided very high enantioselectivities for this type of reaction (typically >95:5 er).14a As the dearomatization of the bicyclic diazarenes showed a good performance and a moderate to excellent enantioselectivity, a more challenging six-membered monocyclic heteroarene was next explored. Pyridazine (3g) was again nicely enrolled in the catalytic dearomatization reaction, providing a good 93% overall yield and a 94:6 mixture of the 2- (5g) and 4-addition (6g) products (entry 7). Remarkably, an acceptable 73:27 enantiomeric ratio was obtained for the more interesting 2-addition product 5g, whereas for the minor regioisomer 6g an almost racemic compound was formed. This can be explained by the greater distance of the newly introduced stereocenter at the C4 with respect to the C2 position to the positive nitrogen present in the key ionic intermediate. Consequently, the catalyst-chloride anion complex should stay in close proximity to the nitrogen atom and therefore, the chirality transfer might be more efficient in the adjacent C2-position. Lastly, the reaction with five membered diazarenes was carried out. While N-methyl benzimidazole provided the desired dearomatized heterocycle 5h in a good yield and moderate enantioselectivity (72%, 66:34 er; entry 8), N-methyl pyrazole led to a complex mixture of decomposition products.20
|
|
|||||
|---|---|---|---|---|---|
| Entry | Diazarene 3 | Products 5/6 | Yieldb (%) [5:6]c |
5, erd | 6, erd |
| a Conditions: (i) 3 (1 equiv.) and TrocCl (1 equiv.) were stirred in dry MTBE at 0 °C for 30 min; (ii) catalyst 1a (10 mol%) and 4 (2 equiv.) were added at −78 °C, and stirred for 18 h while allowing to reach slowly rt. b Isolated yield. c Isomeric ratios determined by 1H–NMR. d Enantiomeric ratios determined by chiral HPLC. e Scale-up reaction in brackets: 3a (500 mg, 3.85 mmol). f Other possible isomers were not detected by NMR. g Reaction at −78 °C in brackets. | |||||
| 1 |
|

|
65% (42%),e5a [92:8] |
96:4 (92:8)e |
— |
| 2 |
|

|
5b, decomp. | — | — |
| 3 |

|
|
93% (45%),g5c | 76:24 (79:21)g |
— |
| 4 |
|

|
86%, 5d | 83:17 |
62:38 |
| 5%, 6d [95:5] |
|||||
| 5 |
|

|
56%, 5ef | 80:20 |
— |
| 6 |

|
|
74% (38%),g5f | 63:37 (61:39)g |
— |
| 7 |

|
|
87%, 5g | 73:27 |
52:48 |
| 6%, 6g [94:6] |
|||||
| 8 |

|
|
72%, 5h | 66:34 |
— |
| 9 |

|

|
5i, decomp. | — | — |

Finally, the synthetic utility of this method was demonstrated by the derivatization of 5a. Thus, the corresponding tetrahydro derivative 8 was synthesized by reduction with NaBH4 in the presence of B(OH)321,22 and the dimethyl derivative 9 by trans-esterification with in situ generated KOMe with K2CO3 in MeOH (Scheme 2). Moreover, the Troc protecting group could easily be removed from 5a using Zn and NH4OAc at room temperature, providing the corresponding N-deprotected product 10 in 97% yield.
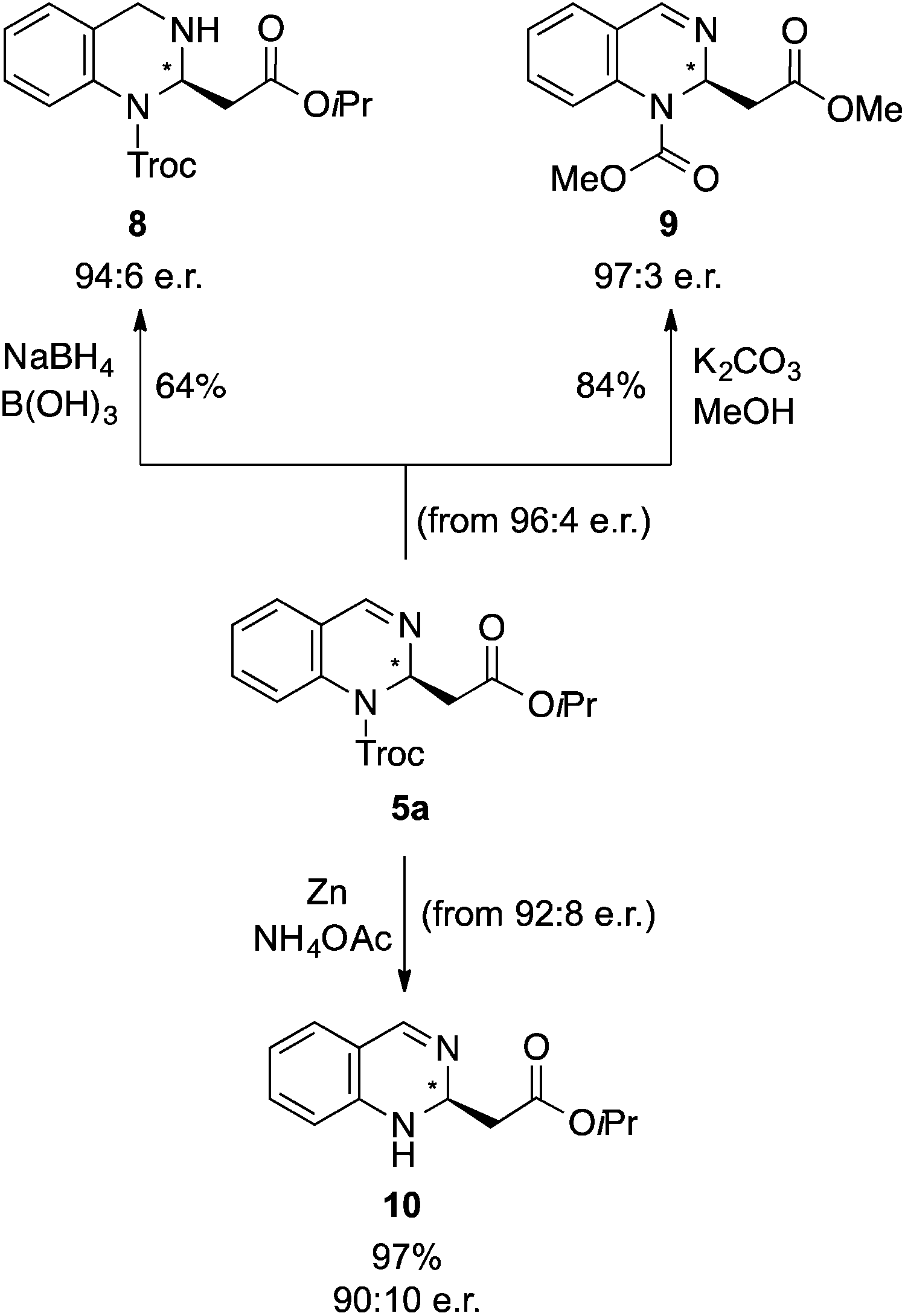 | ||
| Scheme 2 Derivatization of the quinazoline derivative 5a. | ||
Conclusions
In conclusion, the first enantioselective nucleophilic dearomatization of diazarenes using an anion-binding organocatalysis approach has been developed. Tetrakistriazole-based H-bond donor catalysts were superior to other known hydrogen-bond donors, providing the corresponding products in high regioselectivities and up to 96:4 er. This method allows rapid access to substituted chiral di- or tetrahydro diazaheterocycles.
Experimental section
General methods
1H- and 13C-NMR spectra were recorded in CDCl3 (reference signals: 1H = 7.26 ppm, 13C = 77.16 ppm) on a Bruker ARX-300 and a Varian AV-300, 400 or 600 MHz. Chemical shifts (δ) are given in ppm and spin–spin coupling constants (J) are given in Hz. Analytical thin layer chromatography was performed using silica gel 60 F254 and a solution of KMnO4 served as the staining agent. Column chromatography was performed on silica gel 60 (0.040–0.063 mm). Exact masses (HRMS) were recorded on an Agilent Q-TOF 6540 UHD spectrometer using electrospray (ES) or chemical (CI) ionization techniques. Chiral High Pressure Liquid Chromatography (HPLC) analyses were performed on an Agilent 1200 series instrument.MTBE and Et2O were distilled and dried over Na. The catalysts 1a14 and 2a–c,16–18 and the silyl ketene acetals 4,14a,16 were prepared following the known literature procedures. The starting materials and other commercially available reagents were used without further purification.
General organocatalytic procedure
The diazarene 3 (0.10 mmol, 1.0 equiv.) was dissolved in freshly distilled anhydrous MTBE (1 mL, 0.1 M) and cooled to 0 °C. After the addition of 2,2,2-trichloroethyl chloroformate (14 μL, 0.10 mmol, 1.0 equiv.) the reaction was stirred for 30 min at 0 °C and then cooled to −78 °C. Isopropyl TBS-ketene acetal 4a (51 μL, 0.20 mmol, 2.0 equiv.) and the catalyst 1a (11.2 mg, 0.01 mmol, 10 mol%) were added and stirred overnight. The solution was allowed to warm slowly to room temperature during that time. The crude product was purified by flash column chromatography (petrol ether/EtOAc 10:1).
:2 mixture of 5a and 6a. The main product 5a (26.5 mg, 0.065 mmol, 65%) was isolated by column chromatography. The enantiomeric ratio was determined as 96:4 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 14.0 min, tr (major): 22.2 min]. [α]20589: −91.5 (c 0.1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.08 (s, 1H), 7.36–7.31 (m, 2H), 7.25–7.15 (m, 2H), 5.77–5.60 (m, 1H), 5.14–4.75 (m, 3H), 2.81–2.70 (m, 1H), 2.70–2.58 (m, 1H), 1.16 (d, J = 6.3 Hz, 3H), 1.13 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 168.9, 150.8, 140.7, 138.7, 129.2, 127.7, 126.1, 126.2, 124.8, 94.3, 75.7, 68.6, 50.7, 41.8, 21.7, 21.6; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0333.
:40 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 26.6 min, tr (major): 38.2 min]. [α]20589: −13.2 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.07 (s, 1H), 7.44–7.35 (m, 5H), 7.32–7.28 (m, 2H), 7.19 (m, 2H), 5.66 (t, J = 6.3 Hz, 1H), 5.32 (d, J = 3.5 Hz, 2H), 4.97–4.84 (sept, 1H), 2.63 (m, 2H), 1.14 (d, J = 5.9 Hz, 3H), 1.12 (d, J = 6.3 Hz, H); 13C NMR (100 MHz, CDCl3): 169.1, 145.6, 141.8, 134.8, 129.1, 128.8, 128.8, 128.4, 127.4, 126.1, 125.9, 125.0, 69.0, 68.5, 50.5, 42.0, 29.7, 21.6; HRMS (ESI): m/z calculated for [C21H23N2O4]+: 367.1652; found 367.1658.
:28 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 28.0 min, tr (major): 36.7 min]. [α]20589: −39.6 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.03 (s, 1H), 7.29 (m, 2H), 7.20–7.08 (m, 2H), 5.65 (t, J = 6.2 Hz, 1H), 4.87 (s, 2H), 3.58 (s, 3H), 2.68 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 169.8, 147.1, 138.7, 129.3, 127.8, 126.4, 126.1, 124.8, 94.3, 75.7, 52.1, 50.7, 29.7; HRMS (ESI): m/z calculated for [C14H14Cl3N2O4]+: 379.0014; found 379.0019.
:17 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 13.90 min, tr (major): 21.4 min]. [α]20589: −1.7 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.09 (s, 1H), 7.36–7.30 (m, 2H), 7.24–7.16 (m, 2H), 5.72–5.61 (t, J = 11.8, 1H), 4.91 (s, 2H), 2.80–2.53 (m, 2H), 1.35 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 168.56, 150.85, 138.68, 129.20, 127.67, 126.28, 124.89, 94.36, 81.48, 75.67, 50.80, 40.86, 27.89; HRMS (ESI): m/z calculated for [C17H20Cl3N2O4]+: 421.0483; found 421.0487.
:0.8 E/Z mixture) were added according to the general procedure, leading to the desired product 5af (74.5 mg, 0.18 mmol, 91%) as a 5:1 mixture of diastereomers. The enantiomeric ratio was determined as 88:12 er for the major diastereoisomer and 73:27 for the minor diastereoisomer by chiral HPLC [Chiralcel OD-H, hexane/iPrOH (95:5), 1.0 mL min−1, λ = 290 nm: major isomer: tr (minor): 9.23 min, tr (major): 16.54 min; minor: tr (minor): 7.78 min, tr (major): 11.76 min]. 1H NMR (300 MHz, CDCl3) (major): δ 8.13 (s, 1H), 7.33 (d, J = 3.9 Hz, 2H), 7.21 (m, 1H), 7.02 (d, J = 7.3 Hz, 1H), 5.70 (bs, 1H), 5.03–4.79 (m, 2H), 4.15 (q, J = 7.1 Hz, 2H), 2.89 (bs, 1H), 1.27 (t, J = 7.2 Hz, 3H), 0.99 (d, J = 7.1 Hz, 3H); 1H NMR (300 MHz, CDCl3) (minor): δ 8.11 (s, 1H), 7.33 (d, J = 3.9 Hz, 2H), 7.21 (m, 1H), 7.12 (d, J = 7.5 Hz, 1H), 5.54 (bs, 1H), 5.03–4.77 (m, 2H), 4.07–3.95 (m, 2H), 2.80–2.69 (m, 1H), 1.15 (t, J = 4.5 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 172.8, 172.2, 172.1, 151.4, 141.3, 139.4, 129.3, 129.2, 127.6, 126.6, 126.5, 126.2, 126.1, 94.3, 75.8, 75.7, 61.1, 55.3, 46.6, 24.0, 14.2, 14.0; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0329.
:24 er by chiral HPLC [Chiralcel OD-H, hexane/iPrOH (99:1), 1.0 mL min−1, λ = 290 nm: tr (minor): 44.4 min, tr (major): 46.8 min]. [α]20589: −145.0 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.81 (bs, 1H), 7.56–7.38 (m, 2H), 7.36–7.29 (m, 2H), 5.97 (t, J = 7.1 Hz, 1H), 5.12–5.00 (m, 1H), 4.94 (sept, J = 6.3 Hz, 1H), 4.90–4.81 (m, 1H), 2.83–2.50 (m, 2H), 1.20 (d, J = 6.2 Hz, 3H), 1.11 (d, J = 6.2 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 168.9, 145.1, 144.2, 132.4, 132.0, 128.9, 126.5, 126.3, 123.2, 95.0, 75.6, 68.5, 50.7, 39.5, 21.7, 21.7; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0333.
:5 mixture of 5d and 6d. The mixture of isomers were separated and isolated by flash column chromatography to yield the 2-addition product 5d (35.6 mg, 0.086 mmol, 86%) and the 4-addition 6d (2.0 mg, 0.005 mmol, 5%).
5d: The enantiomeric ratio was determined as 83:17 er by chiral HPLC [Chiralcel OD-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 280 nm: tr (minor): 13.8 min, tr (major): 29.7 min]. [α]20589: −147.2 (c 0.1, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.33 (d, J = 4.6 Hz, 1H), 8.01 (bs, 1H), 7.19 (dd, J = 8.3, 4.8 Hz, 1H), 6.75 (d, J = 9.9 Hz, 1H), 6.43 (dd, J = 9.8, 5.8 Hz, 1H), 5.53 (dd, J = 13.4, 6.9 Hz, 1H), 5.01 (bs, 1H), 4.97 (sept, J = 6.3 Hz, 1H), 4.72 (bs, 1H), 2.60–2.40 (m, 2H), 1.17 (d, J = 8.9 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 169.1, 152.0, 145.7, 132.3, 127.3, 122.5, 94.8, 75.6, 68.5, 50.2, 38.9, 21.8, 21.7; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0332.
6d: The enantiomeric ratio was determined as 62:38 er by chiral HPLC [Chiralcel OD-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 230 nm: tr (minor): 8.9 min, tr (major): 9.7 min]. [α]20589: +4.8 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.43 (d, J = 8.5 Hz, 1H), 8.37 (dd, J = 4.7, 1.4 Hz, 1H), 7.21 (dd, J = 8.4, 4.6 Hz, 1H), 7.07 (dd, J = 8.0, 0.9 Hz, 1H), 5.50 (dd, J = 8.0, 4.5 Hz, 1H), 5.03 (sept., J = 6.3 Hz, 1H), 4.94 (d, J = 11.9 Hz (AB system), 1H), 4.80 (d, J = 11.9 Hz (AB system), 1H), 4.04 (dt, J = 9.2, 4.6 Hz, 1H), 2.92 (dd, J = 15.7, 5.1 Hz, 1H), 2.64 (dd, J = 15.7, 8.9 Hz, 1H), 1.21 (d, J = 6.3 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 171.0, 150.6, 150.5, 148.6, 146.0, 128.5, 124.7, 121.8, 113.1, 94.7, 75.6, 67.9, 40.9, 36.7, 21.8; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0331.
:1.6 mixture of rotamers of the titled product 5e (23.0 mg, 0.056 mmol, 56%). The enantiomeric ratio was determined as 80:20 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 290 nm: tr (minor): 15.8 min, tr (major): 23.2 min]. [α]20589: −65.0 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.45 (dd, J = 4.9, 1.5 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.21–7.14 (m, 1H), 7.13–7.04(m, 1H), 6.21 (d, J = 8.1 Hz, 1H, minor rotamer), 6.16 (d, J = 8.0 Hz, 1H, major rotamer), 5.86 (dd, J = 7.5, 6.3 Hz, 1H), 5.07–4.86 (m, 2H), 4.78 (bd, J = 15.2, 1H), 2.94–2.58 (m, 2H), 1.22–1.07 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 169.2, 150.8, 149.3, 134.4, 129.3, 129.2, 128.2, 126.5, 126.4, 122.03, 121.8, 111.0, 94.9, 75.8, 68.6, 52.9, 40.4, 39.6, 21.8; HRMS (ESI): m/z calculated for [C16H18Cl3N2O4]+: 407.0327; found 407.0334.
:37 er by chiral HPLC [Chiralcel OD-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 290 nm: tr (minor): 13.4 min, tr (major): 22.1 min]. [α]20589: +4.0 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.31 (d, J = 7.6 Hz, 1H), 6.94 (dd, J = 7.7, 0.4 Hz, 1H), 6.47 (d, J = 9.5 Hz, 1H), 6.16 (dd, J = 9.5, 5.8 Hz, 1H), 5.51 (dt, J = 10.0, 5.5 Hz, 1H), 5.05–4.92 (m, 1H), 4.99 (d, J = 11.9 Hz, 1H), 4.72 (d, J = 11.9 Hz, 1H), 2.64 (dd, J = 15.3, 5.3 Hz, 1H), 2.51 (s, 3H), 2.48 (dd, J = 15.3, 9.7 Hz, 1H), 1.22 (d, J = 6.7 Hz, 3H), 1.19 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 169.3, 156.7, 152.1, 146.6, 134.4, 128.3, 124.1, 120.4, 119.2, 94.9, 75.7, 68.3, 51.2, 39.3, 24.3, 21.8; HRMS (ESI): m/z calculated for [C17H20Cl3N2O4]+: 421.0483; found 421.0486.
:6 mixture of 5g and 6g. The mixture of isomers was separated and isolated by flash column chromatography to yield the 2-addition product 5g (31.0 mg, 0.087 mmol, 87%) and the 4-addition product 6g (2.2 mg, 0.006 mmol, 6%).
5g: The enantiomeric ratio of the main product was determined as 73:27 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 15.3 min, tr (major): 18.2 min]. [α]20589: −245.0 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.24 (bs, 1H), 6.39 (ddd, J = 9.6, 6.1, 1.7 Hz, 1H), 5.97 (dd, J = 9.7, 3.2 Hz, 1H), 5.43–5.23 (m, 1H), 5.16–4.61 (m, 3H), 2.83–2.30 (m, 2H), 1.22 (d, J = 6.3 Hz, 6H); 13C NMR (75 MHz, acetone-D6): δ 168.9, 152.7, 141.4, 132.0, 117.4, 95.0, 75.5, 68.5, 47.8, 38.1, 21.8; HRMS (ESI): m/z calculated for [C12H16Cl3N2O4]+: 357.0175; found 357.0175.
6g: The enantiomeric ratio was determined as 52:48 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 230 nm: tr (minor): 17.8 min, tr (major): 19.9 min]. [α]20589: −2.0 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.06 (d, J = 8.4 Hz, 1H), 7.02 (bs, 1H), 5.18–4.95 (m, 2H), 4.92 (s, 2H), 3.46–3.27 (m, 1H), 2.53 (dd, J = 16.1, 6.9 Hz, 1H), 2.43 (dd, J = 16.1, 7.3 Hz, 1H) 1.24 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 169.9, 154.0, 142.7, 123.2, 94.7, 75.5, 68.6, 39.7, 28.8, 21.8; HRMS (ESI): m/z calculated for [C12H16Cl3N2O4]+: 357.0175; found 357.0183.
:34 er by chiral HPLC [Chiralcel OJ-H, hexane/iPrOH (98:2), 1.0 mL min−1, λ = 300 nm: tr (minor): 10.5 min, tr (major): 12.0 min]. (Note: unstable compound. Partial decomposition occurred during the structural analysis.) [α]20589: −12.7 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.62–7.39 (m, 1H), 6.94 (dd, J = 7.7, 6.9 Hz, 1H), 6.71 (bd, J = 7.0 Hz, 1H), 6.48 (d, J = 7.7 Hz, 1H), 5.79 (bd, J = 9.8 Hz, 1H), 5.15–4.68 (m, 3H), 2.92 (s, 5H), 1.14 (d, J = 6.3 Hz, 3H), 1.08 (d, J = 6.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 169.3, 149.2, 142.3, 124.6, 118.9, 118.7, 114.1, 109.9, 107.3, 78.8, 78.3, 75.7, 75.0, 68.4, 40.3, 38.9, 34.7, 34.2, 21.8, 21.7; HRMS (ESI): m/z calculated for [C16H20Cl3N2O4]+: 409.0483; found 409.0480.
:4 er) in anhydrous MeOH (1 mL) at 0 °C, B(OH)3 (0.2 mmol, 12.2 mg, 2 equiv.) and NaBH4 (0.2 mmol, 7.2 mg, 2 equiv.) were added slowly and stirred for 1 h at room temperature. The reaction mixture was quenched with H2O (2 mL), filtered and washed with EtOAc (3 x3 mL). Purification by solid phase extraction (MeOH:Et3N 50:1) yielded the desired product 8 (26 mg, 0.064 mmol, 64%). The enantiomeric ratio was determined as 94:6 er by chiral HPLC [Chiralpack AD, hexane/iPrOH (90:10), 1.0 mL min−1, λ = 300 nm: tr (major): 18.1 min, tr (minor): 19.1 min]. [α]20589: −4.6 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.09 (t, J = 7.2 Hz, 2H), 6.82 (t, J = 7.1 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 5.64 (t, J = 6.9 Hz, 1H), 5.22 (d, J = 12.1 Hz, 1H), 5.00 (m, 1H), 4.77 (s, 1H), 4.43 (m, 1H), 2.84 (bs, 2H), 1.23 (d, J = 6.2 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 169.7, 152.8, 141.8, 128.1, 127.6, 122.9, 120.0, 117.4, 95.3, 75.3, 68.4, 51.4, 42.6, 29.7, 21.8; HRMS (ESI): m/z calculated for [C16H19Cl3N2NaO4]+: 431.0303; found 431.0300.
:4 er) in anhydrous MeOH (1 mL) was stirred for 1 h at room temperature. Afterwards, H2O (1 mL) was added, the mixture extracted with CHCl3 (3 × 3 mL), the organic phase washed with brine (3 × 3 mL) and the crude product was dried over Na2SO4. Purification by column chromatography (petrol ether/EtOAc 5:1) yielded the desired product 9 (11.0 mg, 0.04 mmol, 84%). The enantiomeric ratio was determined as 97:3 er by chiral HPLC [Chiralpack AD, hexane/iPrOH (90:10), 1.0 mL min−1, λ = 290 nm: tr (minor): 21.5 min, tr (major): 25.2 min]. [α]20589: −18.8 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.72 (bs, 1H), 7.45 (td, J = 7.5, 1.4 Hz, 1H), 7.41–7.36 (td, J = 7.5, 1.2 Hz, 1H), 7.32–7.25 (m, 2H), 6.01–5.88 (bm, 1H), 3.91 (s, 3H), 3.62 (s, 3H), 2.69 (dd, J = 14.5, 5.8 Hz, 1H), 2.63 (dd, J = 14.5, 8.2 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 170.1, 154.4, 143.1, 132.4, 131.9, 128.8, 126.3, 126.0, 123.5, 76.7, 54.0, 51.9, 39.1; HRMS (ESI): m/z calculated for [C13H15N2O4]+: 263.1026; found 263.1031.
:8 er) and Zn-powder (34.0 mg, 0.05 mmol, 10 equiv.) in NH4OAc (1.0 M)/THF (1/3; 1 mL) was stirred for 16 h at room temperature. After that time sat. aq. K2CO3 solution (1 mL) was added, extracted with CHCl3 (3 × 3 mL) and dried over Na2SO4 to yield the desired product 10 (11.2 mg, 0.048 mmol, 97%). The enantiomeric ratio was determined as 90:10 er by chiral HPLC [Chiralpack AD, hexane/iPrOH (90:10), 1.0 mL min−1, λ = 280 nm: tr (major): 8.6 min, tr (minor): 9.7 min]. [α]20589: +16.8 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3): δ 7.25–7.12 (m, 2H), 7.06–7.01 (m, 2H), 6.93 (dd, J = 8.3, 1.6 Hz, 1H), 5.11–4.99 (m, 1H), 5.05 (sept., J = 6.1 Hz, 1H), 4.63 (bs, 1H), 2.88 (dd, J = 16.8, 10.0 Hz, 1H), 2.59 (dd, J = 16.8, 3.2 Hz, 1H), 1.25 (d, J = 6.3 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 171.2, 146.7, 140.5, 128.6, 125.7, 125.0, 123.4, 123.2, 68.6, 49.0, 44.1, 21.8; HRMS (APCI): m/z calculated for [C13H16N2O2]+m/z: 233.1285, found 233.1290.
Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged for financial support.Notes and references
- (a) P. Mátyus and P. Tapolcsányi, in Asymmetric Synthesis of Nitrogen Heterocycles, ed. J. Royer, Wiley-VCH, Weinheim, 2009, p. 293 Search PubMed; (b) K. A. Rinderspacher, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elservier, Amsterdam, 2015, vol. 27, p. 393 Search PubMed.
- S. Chou, Curr. Opin. Infect. Dis., 2015, 28, 293 CrossRef CAS PubMed.
- K. Nepali, S. Sharma, R. Ojha and K. L. Dhar, Med. Chem. Res., 2012, 22, 1 CrossRef.
- K. Tamaki, K. Tanzawa, S. Kurihara, T. Oikawa, S. Monma, K. Shmada and Y. Sugimura, Chem. Pharm. Bull., 1995, 43, 1883 CrossRef CAS PubMed.
- See for examples: (a) M.-C. Fernandez, A. Escribano, A. I. Mateo, S. Parthasarathy, E. M. Martin de la Nava, X. Wang, S. L. Cockerham, T. P. Beyer, R. J. Schmidt, G. Cao, Y. Zhang, T. M. Jones, A. Borel, S. A. Sweetana, E. A. Cannady, G. Stephenson, S. Frank and N. B. Mantlo, Bioorg. Med. Chem. Lett., 2012, 22, 3056 CrossRef CAS PubMed; (b) H. Kubota, Y. Nakamura, T. Higashijima, Y. Yamamoto, K. Oka and S. Igarashi, U.S. Pat. Appl. Publ, US20070032485A120070208, 2007 Search PubMed.
- M. Bell, G. Cao, A. Escribano, M. Fernandez, P. Lander, N. Mantlo, E. Martin de la Nava, A. Mateo Herranz, D. Mayhugh and X. Wang, U.S. Pat. Appl. Publ, US2007013526A120070726, 2007 Search PubMed.
- C. R. Bourne, E. W. Barrow, R. A. Bunce, P. C. Bourne, K. D. Berlin and W. W. Barrow, Antimicrob. Agents Chemother., 2010, 54, 3825 CrossRef CAS PubMed.
- For some reviews on asymmetric dearomatizations, see: (a) M. Ahamed and M. H. Todd, Eur. J. Org. Chem., 2010, 5935 CrossRef CAS; (b) C.-X. Zhuo, W. Zhan and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed.
- A recent review on asymmetric hydrogenation of azarenes: D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef CAS PubMed.
- For some recent organocatalytic and dual catalytic examples, see: (a) L. Mengozzi, A. Guanlandi and P. G. Cozzi, Chem. Sci., 2014, 5, 3915 RSC; (b) F. Berti, F. Malossi, F. Marchetti and M. Pineschi, Chem. Commun., 2015, 51, 13694 RSC; (c) S. Sun, Y. Mao, H. Lou and L. Liu, Chem. Commun., 2015, 51, 10691 RSC; (d) C. M. R. Volla, E. Fava, I. Atoderesei and M. Rueping, Chem. Commun., 2015, 51, 15788 RSC.
- (a) Z.-P. Yang, Q.-F. Wu and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 6986 CrossRef CAS PubMed. For a review on allylic dearomatizations, see: (b) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed.
- For the initial non-asymmetric studies: (a) S. Beckendorf, S. Asmus, C. Mück-Lichtenfeld and O. García Mancheño, Chem. – Eur. J., 2013, 19, 1581 CrossRef CAS PubMed; (b) S. Asmus, S. Beckendorf, M. Zurro, C. Mück-Lichtenfeld and O. García Mancheño, Chem. – Asian J., 2014, 9, 2178 CrossRef CAS PubMed.
- See reviews: (a) S. Beckendorf, S. Asmus and O. García Mancheño, ChemCatChem, 2012, 4, 926 CrossRef CAS; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed; (c) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed; (d) T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 2633 CrossRef CAS.
- (a) M. Zurro, S. Asmus, S. Beckendorf, C. Mück-Lichtenfeld and O. García Mancheño, J. Am. Chem. Soc., 2014, 136, 13999 CrossRef CAS PubMed; (b) O. García Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823 CrossRef PubMed; (c) M. Zurro, S. Asmus, J. Bamberger, S. Beckendorf and O. García Mancheño, Chem. – Eur. J., 2016, 22, 3785–3793 CrossRef CAS PubMed.
- (a) A. Reissert, Ber. Dtsch. Chem. Ges., 1905, 38, 1603 CrossRef CAS; (b) W. E. McEwen and R. L. Cobb, Chem. Rev., 1955, 55, 511 CrossRef CAS.
- M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700 CrossRef CAS PubMed.
- V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203 RSC.
- V. Benedek, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967 CrossRef PubMed.
- Assuming the same behaviour, the absolute configuration (R) was assigned by comparison with the one obtained with (R,R)-1a as the catalyst for the related mono-azarene quinolines, pyridines and isoquinolines. See ref. 14.
. - The reaction with other diazarenes such as 6-membered ring pyrazine and 5-bromopyrimidine, or five-membered derivatives N-methyl imidazole and N-methyl pyrazole proceeded; however the products showed a high instability and they could not be isolated pure.
- The reduction of 5a with Et3SiH/TFA or the H2–Pd/C system under different conditions did not occur or lead to decomposition products.
- For the reduction of imines with the boric acid-activated NaBH4 system, see for example: B. T. Cho and S. K. Kang, Tetrahedron, 2005, 61, 5725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HPLC collections. See DOI: 10.1039/c6ob00248j |
| This journal is © The Royal Society of Chemistry 2016 |