
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic transhalogenation of dendritic RGD peptide constructs with the fluorinase†
Stephen
Thompson
 a,
Ian N.
Fleming
b and
David
O'Hagan
*a
a,
Ian N.
Fleming
b and
David
O'Hagan
*a
aSchool of Chemistry and Biomedical Sciences Research Centre, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: do1@st-andrews.ac.uk
bAberdeen Biomedical Imaging Centre, School of Medicine and Dentistry, University of Aberdeen, Foresterhill, Aberdeen, AB25 2ZD, UK
First published on 15th February 2016
Abstract
The substrate scope of fluorinase enzyme mediated transhalogenation reactions is extended. Substrate tolerance allows a peptide cargo to be tethered to a 5′-chloro-5′-deoxynucleoside substrate for transhalogenation by the enzyme to a 5′-fluoro-5′-deoxynucleoside. The reaction is successfully extended from that previously reported for a monomeric cyclic peptide (cRGD) to cargoes of dendritic scaffolds carrying two and four cyclic peptide motifs. The RGD peptide sequence is known to bind upregulated αVβ3 integrin motifs on the surface of cancer cells and it is demonstrated that the fluorinated products have a higher affinity to αVβ3 integrin than their monomeric counterparts. Extending the strategy to radiolabelling of the peptide cargoes by tagging the peptides with [18F]fluoride was only moderately successful due to the poor water solubility of these higher order peptide scaffolds although the strategy holds promise for peptide constructs with improved solubility.
1. Introduction
The fluorinase enzyme (E.C. 2.5.1.63) was originally isolated1 from the soil bacterium Streptomyces cattleya and has recently been identified in a number of other bacterial species.2 The enzyme shows promise as a biocatalyst for the incorporation of fluorine-18 into [18F]-labelled PET radiotracers, and has the singular attribute that the C–F bond forming reaction is carried out under neutral aqueous conditions.3–6 The fluorinase catalyses the combination of fluoride ion and S-adenosyl-L-methionine 1 (SAM) to generate 5′-fluoro-5′-deoxyadenosine 2 (FDA) and L-(S)-methionine 3 (L-Met) (Scheme 1A).1,7 We have recently demonstrated that the enzyme has a specificity tolerance at the 2-position of the adenine base.5 Replacement of hydrogen with an acetylene group at this position, as shown in Scheme 1B, gave the modified substrate ClDEA 4 which was an efficient substrate for transhalogenation in the presence of Se-adenosyl-L-selenomethionine (SeSAM).7 This transformation was utilised to prepare [18F]FDEA [18F]-6 under ambient aqueous conditions. The pendant acetylene moiety was then efficiently reacted with an azido-cRGD (cyclic arginyl-glycinyl-aspartyl) peptide using the CuAAC (Cu-catalysed alkyne–azide cycloaddition) click reaction, to generate a [18F]-radiolabelled cRGD construct.6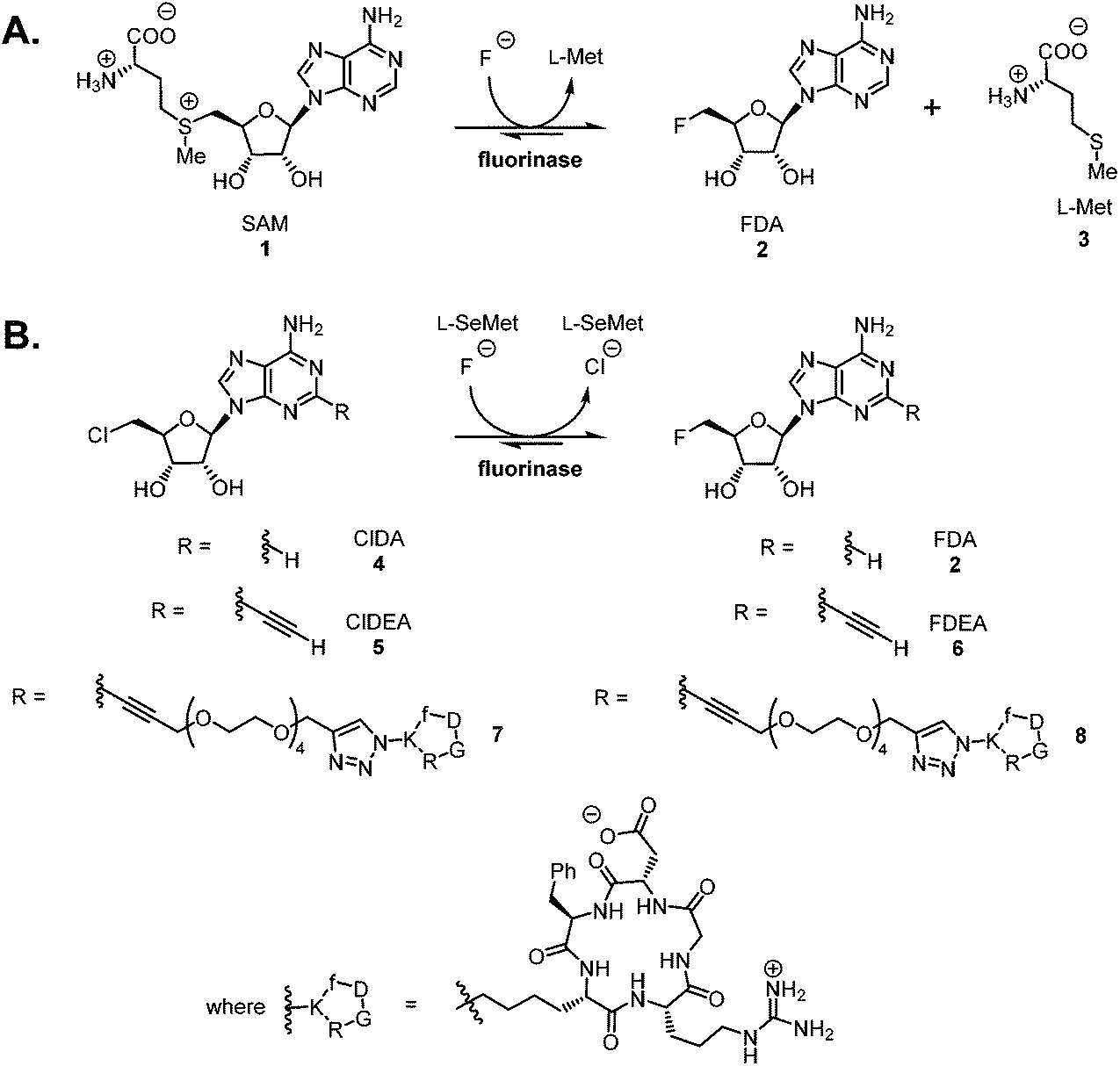 | ||
| Scheme 1 A. The native reaction catalysed by the fluorinase enzyme. B. Fluorinase catalysed transhalogenation reactions with C-2 modified substrates. | ||
While two step strategies involving prosthetic groups remain widely used for radiolabelling of peptides and proteins with fluorine-18,8 it is attractive to consider the development of straightforward, “last step” radiolabelling protocols. Such protocols need to be relatively rapid due to the short half-life of fluorine-18 (t1/2 = 109.7 min), and be conducted under conditions that preserve the structural and functional integrity of the biomolecule.9 The poor nucleophilicity of aqueous [18F]fluoride ion, the form most readily prepared on a cyclotron with an [18O]H2O target,10 often precludes direct labelling of biomolecules using a “last step” approach. To circumvent the limited reactivity of aqueous [18F]fluoride, fluoride sequestering strategies have been explored. For example boron,11 aluminium12 and silicon13 based methods have been developed in an effort to achieve “last step” radiolabelling of peptides by bioconjugation approaches which secure these elements to the peptide in advance. The strategies have proven successful and these technologies are being further developed,14,15 but there are challenges as the conditions required to achieve fluoride ion sequestration require various combinations of low pH, organic solvents and heating. The fluorinase has the capacity to catalyse C–F bond formation at ambient temperature and near neutral pH. Within this context, we have previously demonstrated5 a fluorinase enzyme-based system for “last step” radiolabelling of a small cRGD peptide 7. The peptide was tethered to a 5′-chloro-5′-deoxynucleoside substrate through a PEG linker prior to enzymatic radiolabelling with [18F]fluoride, as shown in Scheme 1B. This proved successful and the radiolabelled construct [18F]-8 was shown to be stable to defluorination in vivo (in a rat), and the cRGD moiety retained high affinity for αVβ3 integrins. In order to explore this approach further, we wished to examine whether the substrate scope at the C-2 position would extend to larger, more complex peptides.
The search for high affinity ligands for biological receptors has led to the development of imaging agents based on multivalent antibody fragments16 or multivalent small peptides.17 Multimeric interactions play a key role in recognition events between biological ligands and receptors.18 Weak non-covalent interactions between monovalent ligands and individual receptors can be enhanced by attaching multiple ligands to a single scaffold.19,20 Incorporating the RGD (arginyl–glycinyl–aspartyl) tripeptide motif into cyclic pentapeptides gives cRGDs, a class of high affinity ligands for αVβ3 integrin, a cell surface protein identified as a biomarker of angiogenesis and strongly associated with malignant tumours.21,22 Multimeric cRGD peptides are validated constructs employed for PET imaging using either fluorine-18 or heavy metal PET isotopes.19,23,24 Usually the cRGD peptide is conjugated to a metal chelating group for co-ordination to radiometals, often at the final step in the synthesis of such a tracer. Fewer fluorine-18-based multivalent radiotracers have been developed, as incorporation of fluorine-18 into such scaffolds often requires a multi-step, prosthetic group-based strategy.25–28 However, two multimeric cRGD-based radiotracers have been reported recently, synthesised using a “last step” radiofluorination protocol. These are the aluminium chelates, [18F]alfatide I14,29 and [18F]alfatide II15,30 which were prepared using a kit-like31 methodology and they have been evaluated in humans.14,15 In addition, the CuAAC reaction between an alkyne and an azide-modified cRGD peptide has also proved useful for the assembly of multimeric cRGD peptides, which were subsequently labelled with radiometals.32–35
Herein, we report our initial results on “last step” fluorination of structurally complex multimeric cRGD peptides with the fluorinase enzyme. A primary objective at this stage was to establish if the fluorinase could tolerate higher molecular weight cargoes attached to the C-2 position of the adenine base. Although successful biotransformations could be achieved with fluoride, it became apparent that reduced solubility of higher molecular weight substrates proved challenging for efficient radiolabelled experiments, where the fluoride-18 concentrations are necessarily low.
2. Results and discussion
2.1. Synthesis of di- and tetra-alkynes for a CuAAC reaction
A multimeric peptide substrate for the fluorinase requires an enzyme recognition/reaction site attached to a linker to extend the peptide cargo away from the surface of the enzyme. In order to assemble such a substrate, we envisaged a strategy similar to that used for the synthesis of 5 and 7 (Scheme 1B).5 A Sonogashira coupling was identified for coupling a 2-iodonucleoside to an alkynyl-PEG linker, which had been pre-functionalised with either a protected dialkyne or a protected tetraalkyne. Following deprotection to the free terminal alkynes, a multi-CuAAC reaction with an azide bearing cRGD peptide, similar to that reported by Liskamp et al.,34 should furnish multimeric cRGD substrates. The synthesis of the protected alkynes and halogenated 2-iodo-nucleosides is summarised in Schemes 2 and 3 respectively.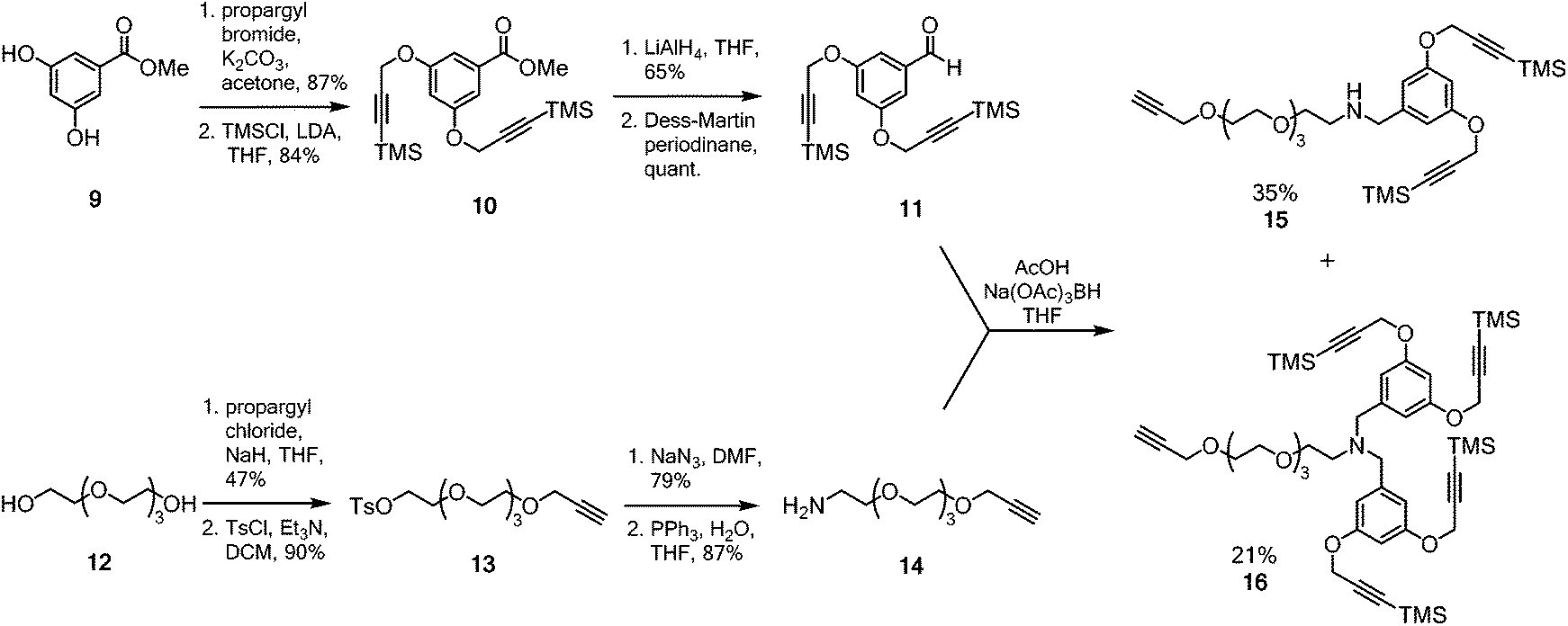 | ||
| Scheme 2 Synthesis of protected aldehyde 11 and amine 14 for reductive amination to generate the desired alkynyl frameworks 15 and 16. | ||
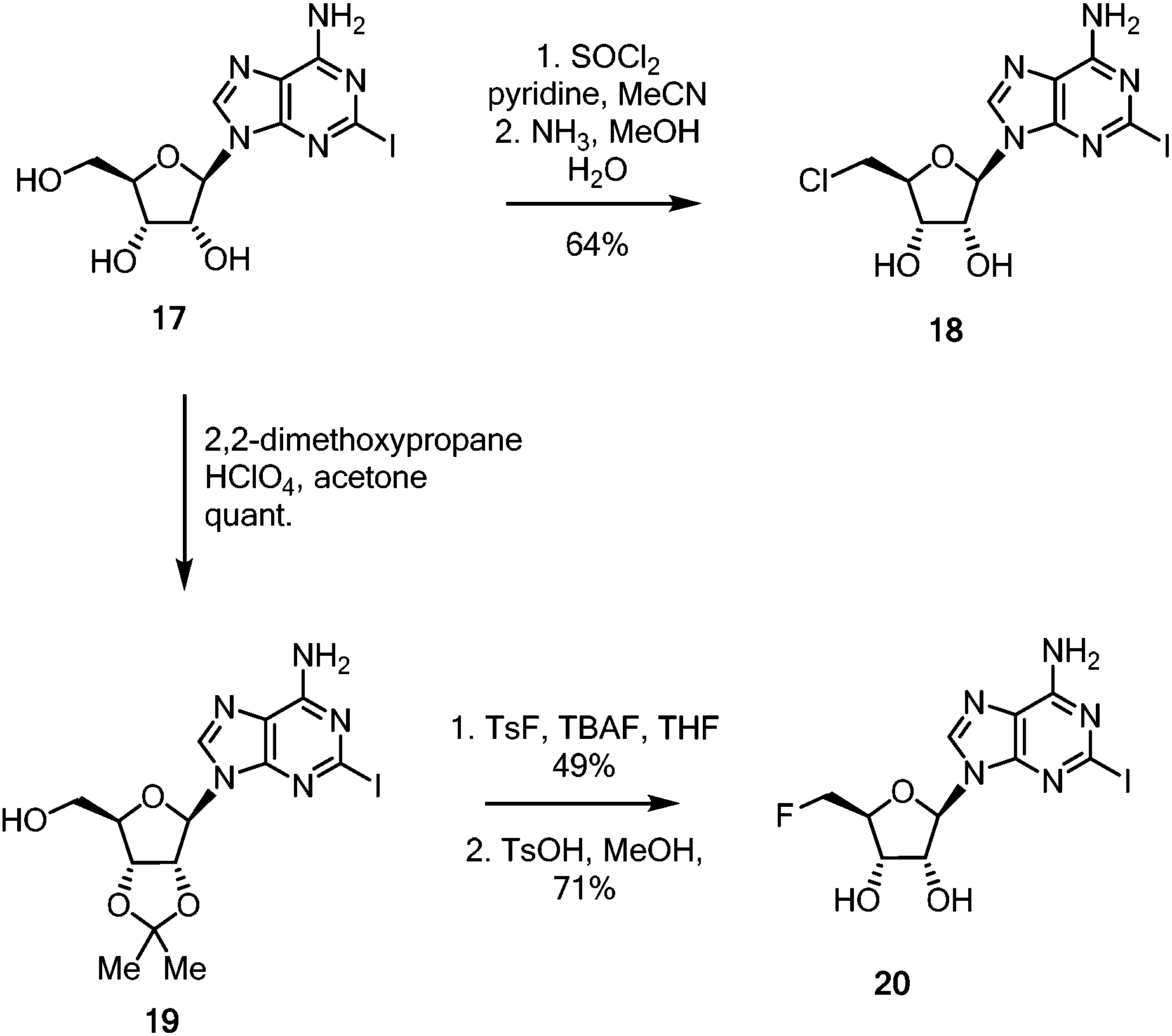 | ||
| Scheme 3 Synthesis of halogenated 2-iodo coupling partners 18 and 20 for Sonogashira coupling. | ||
The dendrimeric core was prepared by dialkylation of methyl 3,5-dihydroxybenzoate 9 with propargyl bromide (Scheme 2).34 The acetylenes of the newly installed propargylic ethers were then silylated to furnish ester 10. Ester 10 was then reduced to the corresponding benzylic alcohol and periodinane oxidation36 generated aldehyde 11 which was used directly in future reductive amination reactions without further purification. The linker was prepared by monoalkylation of tetraethylene glycol 12 with propargyl chloride. The free hydroxyl was converted to the corresponding tosyl ester and then nucleophilic displacement by azide gave the corresponding azido-PEG. Reduction under Staudinger conditions37 gave amine 14. With both aldehyde 11 and amine 14 in hand, attention turned to coupling the linker to the multimeric core.
The reductive amination after condensation of amine 14 with aldehyde 1138 generated both the mono- and di-alkylated products 15 and 16. Separation of the two products by column chromatography gave the di-alkyne linker-core assembly 15 in 35% yield (based on aldehyde 11), while the terta-alkyne linker-core assembly 16 was formed in 21% yield (based on aldehyde 11).
With these linker-core assemblies in hand, attention turned to the synthesis of the iodinated coupling partners. 2-Iodoadenosine 17,5 was treated with thionyl chloride in pyridine, followed by ammonia in aqueous methanol to furnish the 5′-chloro-5′-deoxy adenosine 18, as shown in Scheme 3.39 For the synthesis of the fluorinated nucleoside, 2-iodoadenosine 17 was protected as its 2′,3′-acetonide 19 before being fluorinated by the action of TsF and TBAF in refluxing THF. Deprotection to generate 20 was achieved in good yield with catalytic anhydrous TsOH in refluxing MeOH.40
Sonogashira couplings41–43 of the halogenated 2-iodonucleosides 18 and 20 with acetylenes 15 and 16 were now explored. 5′-Chloro-5′-deoxy-2-iodoadenosine 18 was added to an excess of either alkyne 15 or 16 in dry, degassed DMF, in the presence of Pd(PPh3)2Cl2 (10 mol%), CuI, and triethylamine. TLC and LC-MS was used to monitoring the formation of coupled products 21 and 25 (Scheme 4).
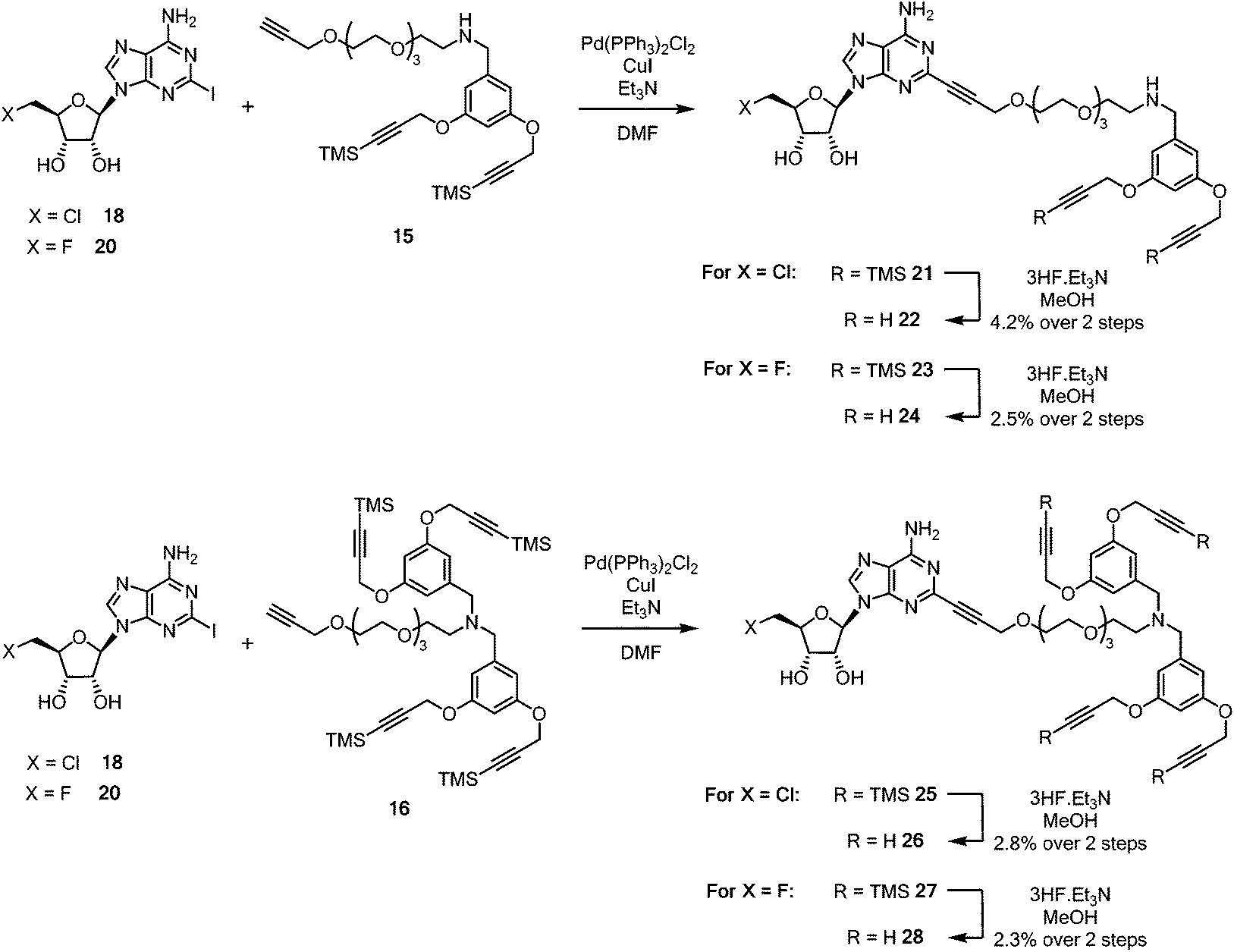 | ||
| Scheme 4 Synthesis of pendant di- and tetra-acetylene cores attached to 5′-halo-5′deoxyadenosine motifs. | ||
Coupled di-alkyne 21 and tetra-25 were subject to column chromatography before fluoride-based deprotection of the silyl groups using 3HF·Et3N in methanol. The products 22 and 26 were each passed through a plug of silica gel, then through a C18 reverse phase cartridge, before being purified by semi-preparative HPLC to give analytically pure samples of ClDEA-PEG-(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)222 and ClDEA-PEG-(CCH)426 for CuAAC reactions. Following a similar protocol, FDEA-PEG-(CCH)224 and FDEA-PEG-(CCH)428 were prepared and purified by semi-preparative HPLC.
CH)222 and ClDEA-PEG-(CCH)426 for CuAAC reactions. Following a similar protocol, FDEA-PEG-(CCH)224 and FDEA-PEG-(CCH)428 were prepared and purified by semi-preparative HPLC.
It became a focus to ‘click’ the multimeric alkynes to the cRGD peptides in aqueous CuAAC reactions with c(RGDfK[N3]) 29 as illustrated in Scheme 5A. The reactions were conducted with CuSO4·TBTA (TBTA = tris(benzyltriazolylmethyl)amine44) and sodium ascorbate as the catalyst, and were monitored by HPLC. The triazole products were trapped on a C18 cartridge, and eluted with MeCN/water mixture and purified by semi-preparative HPLC. This gave samples of ClDEA-PEG-(RGD)230 (1.8 mg, 78% yield) and FDEA-PEG-(RGD)231 (0.67 mg, 27% yield), the integrity of which was supported by MALDI-TOF MS analyses (see ESI†). The fluorinated analogue, FDEA-PEG-(RGD)231, was required as a standard for assessing affinity to targets.
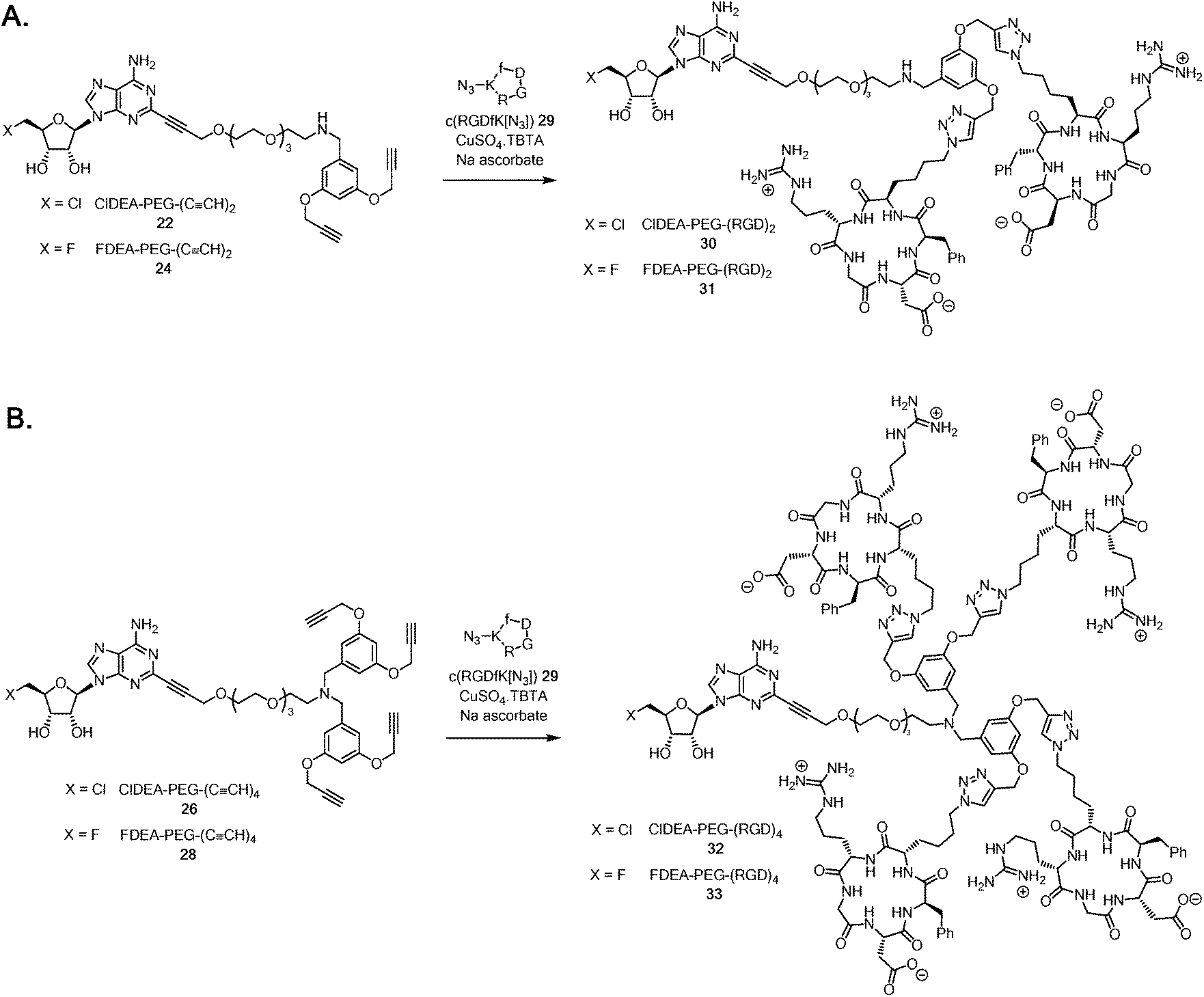 | ||
| Scheme 5 A. Synthesis of ClDEA-PEG-(RGD)230 and FDEA-PEG-(RGD)231. B. Synthesis of ClDEA-PEG-(RGD)432 and FDEA-PEG-(RGD)433. | ||
ClDEA-PEG-(CCH)426 was found to be poorly soluble in water and the CuAAC reaction with c(RGDfK[N3]) 29 (Scheme 5B) was sluggish, but the use of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 DMSO:water as a reaction solvent led to complete conversion within 70 minutes. The reaction product was trapped on a C18 cartridge, and was washed with water to remove residual DMSO, and then the product was eluted with MeCN:water mixtures. Purification by semi-preparative HPLC gave ClDEA-PEG-(RGD)432 (1.4 mg, 70% yield), the integrity of which was confirmed by MALDI–TOF MS (see ESI†). The tetra-acetylene, FDEA-PEG-(CCH)426 was similarly reacted in a mixture of DMSO:water, with c(RGDfK[N3]) 29. HPLC purification gave the desired reference compound FDEA-PEG-(RGD)433 (0.66 mg, 27% yield) with a m/z = 3412.6 ([M + H]+) as determined by MALDI-TOF MS (see ESI†).
:50 DMSO:water as a reaction solvent led to complete conversion within 70 minutes. The reaction product was trapped on a C18 cartridge, and was washed with water to remove residual DMSO, and then the product was eluted with MeCN:water mixtures. Purification by semi-preparative HPLC gave ClDEA-PEG-(RGD)432 (1.4 mg, 70% yield), the integrity of which was confirmed by MALDI–TOF MS (see ESI†). The tetra-acetylene, FDEA-PEG-(CCH)426 was similarly reacted in a mixture of DMSO:water, with c(RGDfK[N3]) 29. HPLC purification gave the desired reference compound FDEA-PEG-(RGD)433 (0.66 mg, 27% yield) with a m/z = 3412.6 ([M + H]+) as determined by MALDI-TOF MS (see ESI†).
2.2. Evaluation of multimeric RGDs as substrates for the fluorinase
With the multimeric cRGD fluorinase substrates in hand, enzyme catalysed transhalogenation reactions were explored. ClDEA-PEG-(RGD)230 (40 μM) was added to a mixture of L-SeMet (75 μM) and potassium fluoride (50 mM) in a 1 mL volume to explore the transformation illustrated in Fig. 1A. The reaction was initiated by addition of the fluorinase (1 mg mL−1).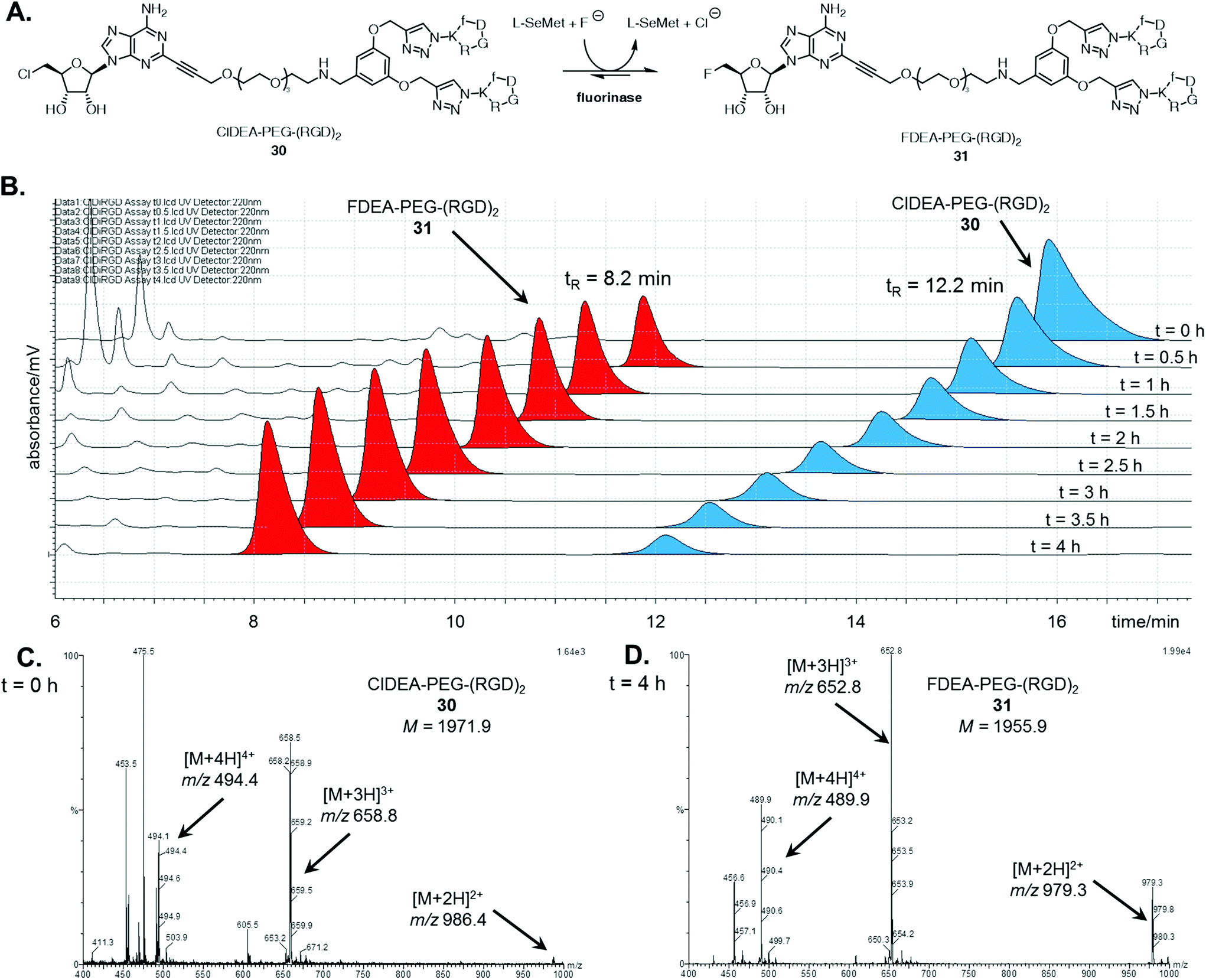 | ||
| Fig. 1 A. Fluorinase catalysed transhalogenation of ClDEA-PEG-(RGD)230 to FDEA-PEG-(RGD)231. B. HPLC time course (220 nm) of incubation of ClDEA-PEG-(RGD)230, blue, (tR = 12.2 min) with the fluorinase, showing samples taken every 0.5 h over 4 h. A new peak was evident at tR = 8.2 min, red, identified as FDEA-PEG-(RGD)231. Summed mass spectrum (m/z = 400–1000) of the LC-MS peaks corresponding to the tetrameric species from a sample taken at C. t = 0 h, showing peaks corresponding to multiply charged species of ClDEA-PEG-(RGD)230, and at D. t = 4 h, showing peaks corresponding to multiply charged species of FDEA-PEG-(RGD)231. | ||
Aliquots of the reaction were removed for HPLC assay over a 4 h period in order to assess conversion. The HPLC traces in Fig. 1B show the time-dependent appearance of the product at tR = 8.2 min, a peak which was identified as FDEA-PEG-(RGD)231 by LC-MS comparison with the synthetic reference previously prepared. The mass spectra of the HPLC peaks corresponding to the multimeric 5′-chloro substrates and 5′-fluoro products are shown in Fig. 1C and D respectively. The peaks for the product contain multiply charged ions of 31 ([M + 2H]2+, [M + 3H]3+, and [M + 4H]4+ respectively), confirming its identity as FDEA-PEG-(RGD)231.
A series of control experiments, where the reaction was conducted in the absence of either enzyme or L-SeMet, did not result in any conversion of 30 to 31, confirming an enzyme catalysed process. In the absence of fluoride, the consumption of 30 was observed, however, no fluorinated product was produced. This result is consistent with the generation of a SeSAM intermediate, which cannot be converted to product 31 in the absence of fluoride.
A similar analysis was conducted for the enzymatic reaction with ClDEA-PEG-(RGD)432, as illustrated in Fig. 2A. ClDEA-PEG-(RGD)432 (20 μM) was added to a mixture of L-SeMet (75 μM) and potassium fluoride (50 mM), in a total volume of 1 mL. The reaction was initiated by the addition of the fluorinase (1 mg mL−1) and monitored over 4 h. The resultant HPLC profile (Fig. 2B) indicated that 32 was consumed, while a new peak, at tR = 9.2 min, steadily increased throughout the assay. The retention time of this new peak was identical to that of synthetic FDEA-PEG-(RGD)433, and its identity was further confirmed by LC-MS.
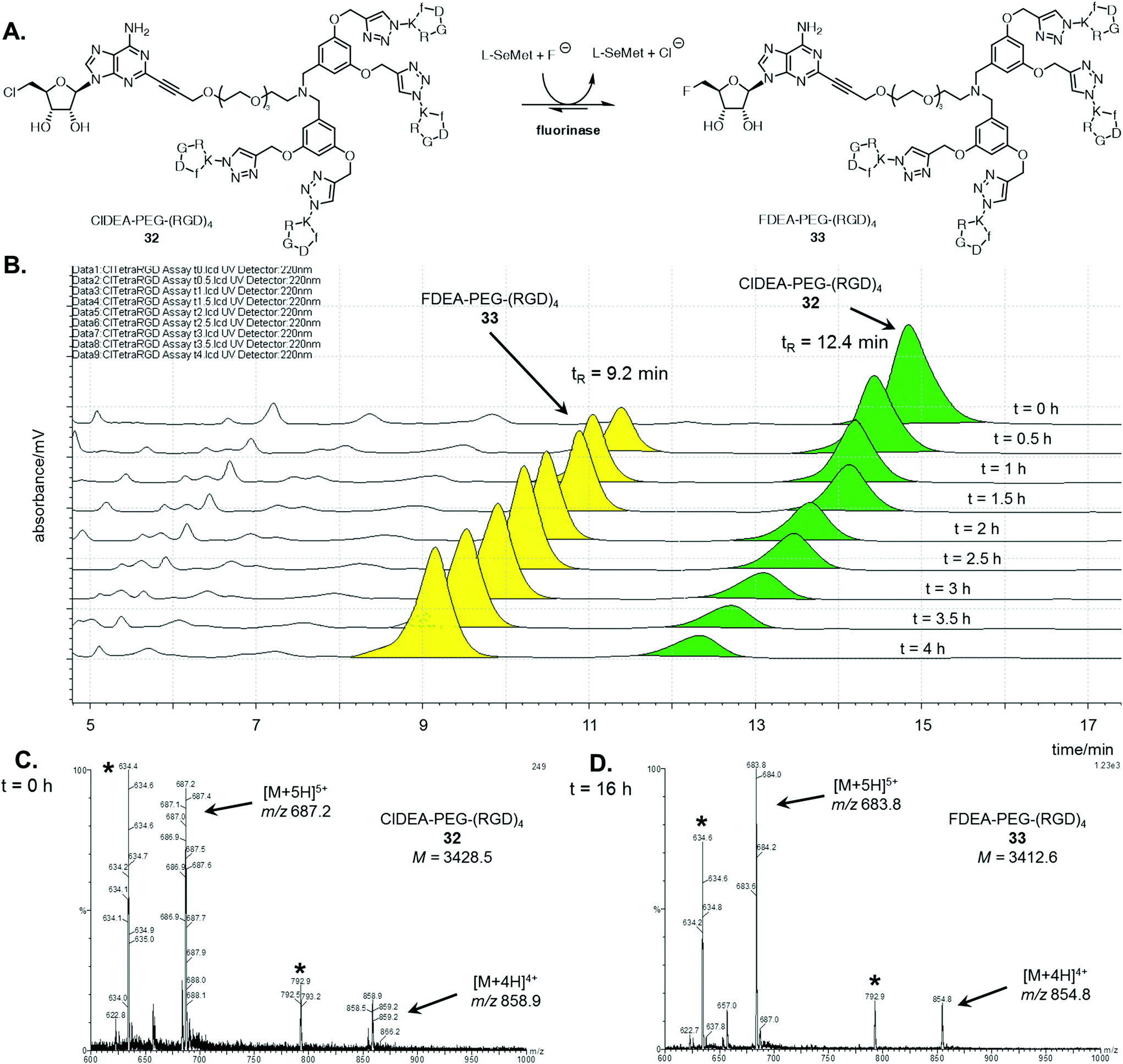 | ||
| Fig. 2 A. Fluorinase catalysed transhalogenation of ClDEA-PEG-(RGD)432 to FDEA-PEG-(RGD)433. B. HPLC time course (220 nm) of incubation of ClDEA-PEG-(RGD)432, green, (tR = 12.4 min) with the fluorinase, showing samples taken every 0.5 h over 4 h. A new peak was evident at tR = 9.2 min, yellow, identified as FDEA-PEG-(RGD)433. Summed mass spectrum (m/z = 600–1000) of the LC-MS peak corresponding to the tetrameric species of samples taken at B. t = 0 h, showing peaks corresponding to multiply charged species of ClDEA-PEG-(RGD)432 and at C. t = 16 h, showing peaks corresponding to multiply charged species of FDEA-PEG-(RGD)422. Identical fragmentation peaks (denoted with an asterisk (*)) appear in both spectra, thought to arise from fragmentation of the adenine (C-2)–(CCR) bond. | ||
Mass spectra the tetrameric substrate and product are shown in Fig. 2C and D respectively. In the sample analysed at t = 0 h (Fig. 2C), peaks corresponding to multiply charged species of ClDEA-PEG-(RGD)432 are observed at m/z = 858.9 ([M + 4H]4+) and 687.2 ([M + 5H]5+). In the sample analysed after an extended 16 h reaction (Fig. 2D), peaks at m/z = 854.8 ([M + 4H]4+) and 683.8 ([M + 5H]5+) are clearly observed. The presence of these ions is consistent with the formation of FDEA-PEG-(RGD)433 as the product of enzymatic transhalogenation.
Tolerance of the fluorinase to these large multimeric peptides suggests that the C-2 position of a chlorinated nucleoside represents a site for the attachment of a diverse range of peptide cargos for use in enzymatic fluorination. The constructs explored here consist of 10 and 20 amino acids, and they were efficiently fluorinated under neutral aqueous conditions. With the knowledge that the multimeric constructs were suitable substrates for fluorinase catalysed transhalogenation, it was of interest to evaluate the effect the multiple RGD motifs on binding affinity to αVβ3 integrin.
2.3. Binding affinities of RGD multimers to immobilised αVβ3 integrin
The binding affinities of the fluorinated multimers 31 and 33 to αVβ3 integrin were determined by their ability to displace a biotin-labelled cRGD peptide from immobilised αVβ3 integrin in an ELISA assay.45 The data are shown in Table 1 including the IC50 values of some reference peptides.5,6 The ELISA data indicate that for this series the multimers have a progressively higher integrin affinity (lower IC50) as the number of cRGD ligands increases e.g., monomeric 8 (IC50 = 74 nM), dimeric 31 (IC50 = 60 nM) and tetrameric 33 (IC50 = 24 nM). The IC50 values measured for the multimeric constructs are also lower than the parent azido-peptide 29 (IC50 = 90 nM) and the multimeric cRGD compounds have significantly lower IC50 values compared with reference compounds containing linear RGD-containing peptides.| Compound | IC50 ± s.e./nM | Q |
|---|---|---|
| a Affinities for these constructs were previously reported, and were measured using an identical assay. | ||
| RGDa | 8560 ± 2240 | 4.019 |
| GRGDSPKa | 2130 ± 410 | 1.000 |
| c(RGDfK[N3]) 29a | 90 ± 10 | 0.042 |
| FDEA-cRGDa | 330 ± 30 | 0.155 |
| FDEA-TEG-cRGD 8a | 74 ± 16 | 0.033 |
| FDEA-PEG-(cRGD)231 | 60 ± 13 | 0.027 |
| FDEA-PEG-(cRGD)433 | 24 ± 18 | 0.011 |
Going from one to two cRGD motifs resulted in a marginal increase in binding affinity however progression to a tetramer showed a significant decrease in IC50 and gave a compound with the highest affinity of those tested. The linker between the two or four cRGD units is not so extended and steric constraints will likely prevent simultaneous multivalent binding of cRGDs to more than one surface site. Instead the increased affinity is most likely due to an increased effective molarity at the surface.46 Some multimeric constructs reported in the literature have IC50 values in the 1–100 nM range,24 similar to this study although a direct comparison of different assays and dissimilar peptide motifs make accurate comparison difficult.
2.4. Enzymatic radiolabelling of multimers with fluorine-18
The ability of the fluorinase to employ these large peptide assemblies of 10 or 20 amino acids as substrates opened up prospects of labelling these peptides with [18F]fluoride for PET. Radiolabelling trials of ClDEA-PEG-(RGD)230 (0.04 mM) was initially investigated using similar reaction conditions to the assays described with [19F]fluoride, but with an aqueous [18F]fluoride solution generated on the cyclotron. The enzyme concentration was increased from 1 mg mL−1 (29 μM) to 10 mg mL−1 (290 μM), as it had previously been shown that high enzyme concentrations improves conversions in radiolabelling experiments.47 Notably [18F]fluoride ion concentrations are necessarily very low in the pico-molar range. Radiochemical incorporations (RCI) were found to be low under these conditions, and only traces of the product, [18F]FDEA-PEG-(RGD)2 [18F]-31 were observed by radio-HPLC. Optimisation trials were carried out with ClDA 4 to [18F]FDA [18F]-2 as a substrate, and the data suggest that an important factor for improving radiochemical incorporations is substrate concentration. A RCI of greater than 90% was observed when ClDA 4 was incubated at a concentration of 0.6 mM, compared with a 10% RCI at 0.04 mM (see ESI†).By contrast, ClDEA-PEG-(RGD)230 and ClDEA-PEG-(RGD)432 were found to be poorly soluble in water, and concentrations of 0.6 mM were not achievable in buffer. The upper concentration limit for the peptides was found to be ∼0.3 mM. ClDEA-PEG-(RGD)230 and ClDEA-PEG-(RGD)432 were incubated in separate experiments with L-SeMet (0.08 mM) and [18F]fluoride in water (138 μL–160 μL, 15.2–35.4 MBq). Some precipitation of the substrate was observed upon addition of aqueous [18F]fluoride. The reaction mixtures were incubated at 37 °C for 30 minutes, and then the enzyme was heat denatured and the mixture centrifuged. A sample of the clarified supernatant from each reaction was analysed by HPLC, and the resultant HPLC radio-chromatograms are shown in Fig. 3.
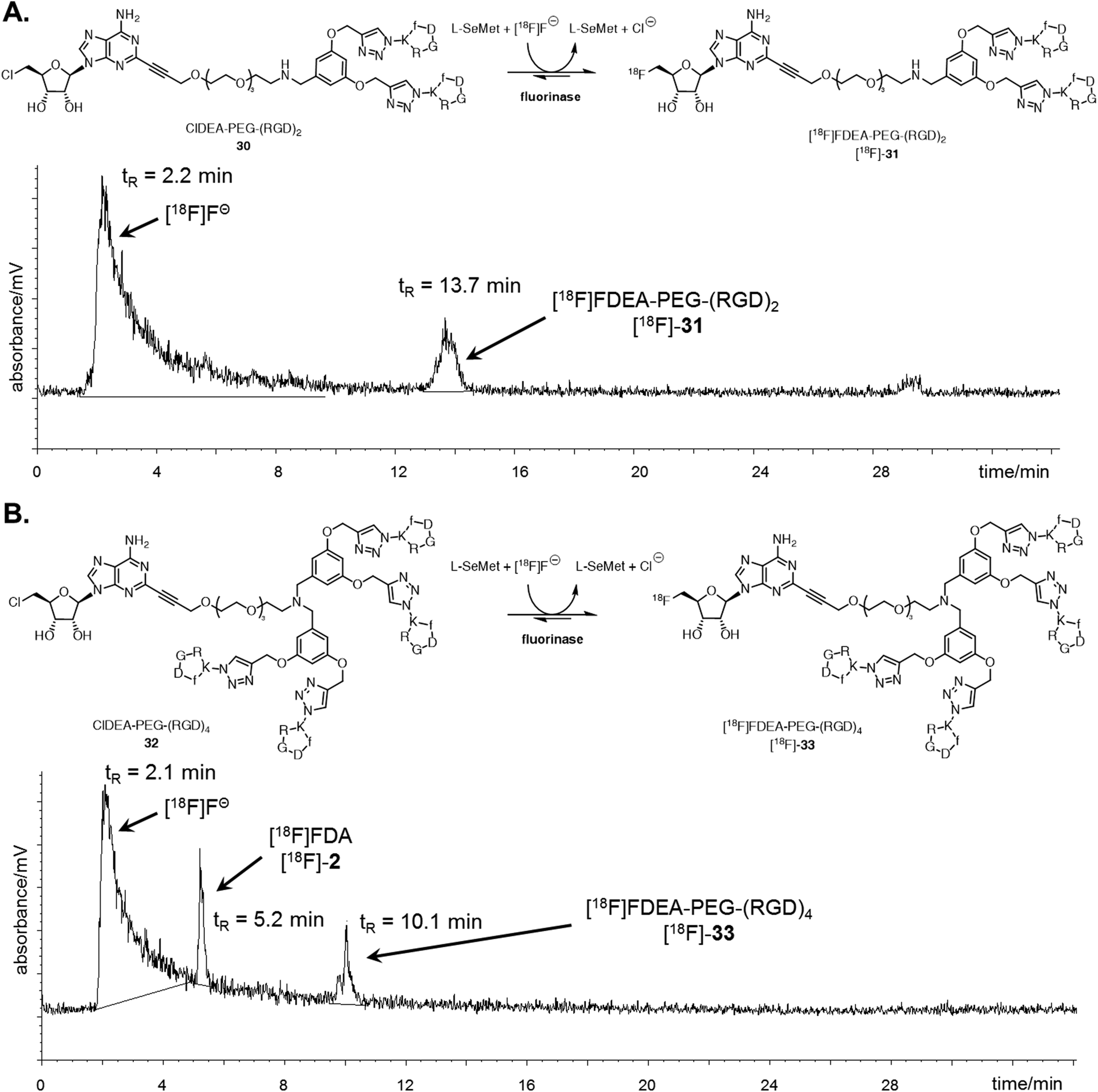 | ||
| Fig. 3 A. Reaction scheme and resultant isocratic HPLC radioactivity trace of an experiment where ClDEA-PEG-(RGD)230 (0.3 mM) was freeze dried with the fluorinase (20 mg mL−1) prior to reaction. [18F]FDEA-PEG-(RGD)2 [18F]-31 was observed at tR = 13.7 min, showing 10% RCI. B. Reaction scheme and resultant gradient HPLC radioactivity trace of an experiment where ClDEA-PEG-(RGD)432 (0.3 mM) was freeze dried with the fluorinase (20 mg mL−1) prior to reaction. [18F]FDEA-PEG-(RGD)4 [18F]-33 was observed at tR = 10.1 min, showing 5% RCI. An additional peak was also observed at tR = 5.2 min, identified as [18F]FDA [18F]-2, produced due to the presence of residual SAM 1 co-purified with the fluorinase.48 The peak is also present for ClDEA-PEG-(RGD)230 labelling experiments, but is not observed in A due to the HPLC conditions utilised (see ESI† for further discussion). | ||
Radio-HPLC of the two reaction mixtures (Fig. 3A and B) revealed the presence of the respective [18F]-products, along with a broad peak (tR = 2.2 min), identified as [18F]fluoride.49 The peak at tR = 13.7 min in Fig. 3A was identified as [18F]-31 by spiking and comparison of the retention time to that of a [19F]-reference sample. Similarly the peak at tR = 10.1 min in Fig. 3B was identified as [18F]-33 relative to a [19F]-reference. [18F]-FDA [18F]-2 was also observed in this experiment, which must arise as a consequence of low levels of L-AdoMet (SAM) bound to the enzyme and residual from the purification process. This outcome only arose in the reactions of low conversion. Reactions were also conducted with the addition of DMSO 1% (v/v) as a co-solvent, however this did not improve the radiochemical conversions and extending the reaction time or increasing the concentration of L-SeMet had no significant effect. Therefore despite the efficient conversions observed in the “cold” experiments at high [19F]fluoride ion concentrations, low [18F]fluoride concentrations require to be compensated for by high substrate concentrations, and this limited the efficiency of the radiolabelled experiments.
3. Conclusions
Enzymatic transhalogenations using the fluorinase was explored with relatively complex di- and tetra-cRGD constructs. This exploited the previously identified substrate tolerance at C-2 of the adenine base in the substrate motif. The multimeric assemblies, ClDEA-PEG-(RGD)230 and ClDEA-PEG-(RGD)432, were assessed as fluorinase substrates and both were found to undergo efficient enzyme-catalysed transhalogenations. The multimers also showed higher affinities to immobilised αVβ3 integrins, which opened up the prospect of using them as cancer imaging agents in [18F]-radiolabelled form. This proved only partially successful due to the relatively poor solubility of the multimeric constructs and low [18F]-fluoride concentrations. It follows that the development of this approach to larger peptides or proteins will depend on using more highly soluble peptide substrates in buffer. The peptides used here are ‘click’ derived with low solubility, however the approach should prove more useful with natural peptides and antibodies which have evolved high water solubility.Acknowledgements
We thank EPSRC and the Scottish Imaging Network (SINAPSE) for grants. DO'H thanks the Royal Society for a Wolfson Research Merit Award and ST is grateful to the John and Kathleen Watson Scholarship for financial support. We are grateful to Dr Catherine Botting and Dr Sally Shirran of the St Andrews Mass Spectrometry Service for MALDI-MS acquisitions. We also thank Dr Sally Pimlott of the University of Glasgow for the use of radiochemistry facilities.References
- D. O'Hagan, C. Schaffrath, S. L. Cobb and J. T. G. Hamilton, Nature, 2002, 416, 279 CrossRef PubMed.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, ChemBioChem, 2014, 15, 364–368 CrossRef CAS PubMed.
- M. Onega, J. Domarkas, H. Deng, L. F. Schweiger, T. A. D. Smith, A. E. Welch, C. Plisson, A. D. Gee and D. O'Hagan, Chem. Commun., 2010, 46, 139–141 RSC.
- S. Dall'Angelo, N. Bandaranayaka, A. D. Windhorst, D. J. Vugts, D. van der Born, M. Onega, L. F. Schweiger, M. Zanda and D. O'Hagan, Nucl. Med. Biol., 2013, 40, 464–470 CrossRef PubMed.
- S. Thompson, Q. Zhang, M. Onega, S. McMahon, I. Fleming, S. Ashworth, J. H. Naismith, J. Passchier and D. O'Hagan, Angew. Chem., Int. Ed., 2014, 53, 8913–8918 CrossRef CAS PubMed.
- S. Thompson, M. Onega, S. Ashworth, I. N. Fleming, J. Passchier and D. O'Hagan, Chem. Commun., 2015, 51, 13542–13545 RSC.
- H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O'Hagan, D. A. Robinson and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759–762 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- S. Richter and F. Wuest, Molecules, 2014, 19, 20536–20556 CrossRef PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094–13095 CrossRef CAS PubMed.
- C. A. D'Souza, W. J. McBride, R. M. Sharkey, L. J. Todaro and D. M. Goldenberg, Bioconjugate Chem., 2011, 22, 1793–1803 CrossRef PubMed.
- R. Schirrmacher, G. Bradtmöller, E. Schirrmacher, O. Thews, J. Tillmanns, T. Siessmeier, H. G. Buchholz, P. Bartenstein, B. Wängler, C. M. Niemeyer and K. Jurkschat, Angew. Chem., Int. Ed., 2006, 45, 6047–6050 CrossRef CAS PubMed.
- W. Wan, N. Guo, D. Pan, C. Yu, Y. Weng, S. Luo, H. Ding, Y. Xu, L. Wang, L. Lang, Q. Xie, M. Yang and X. Chen, J. Nucl. Med., 2013, 54, 691–698 CrossRef CAS PubMed.
- C. Yu, D. Pan, B. Mi, Y. Xu, L. Lang, G. Niu, M. Yang, W. Wan and X. Chen, Eur. J. Nucl. Med. Mol. Imaging, 2015, 42, 2021–2028 CrossRef CAS PubMed.
- A. Goel, J. Baranowska-Kortylewicz, S. H. Hinrichs, J. Wisecarver, G. Pavlinkova, S. Augustine, D. Colcher, B. J. Booth and S. K. Batra, J. Nucl. Med., 2001, 42, 1519–1527 CAS.
- S. Liu, D. S. Edwards, M. C. Ziegler, A. R. Harris, S. J. Hemingway and J. A. Barrett, Bioconjugate Chem., 2001, 12, 624–629 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- S. Liu, Mol. Pharm., 2006, 3, 472–487 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- C. J. Avraamides, B. Garmy-Susini and J. A. Varner, Nat. Rev. Cancer, 2008, 8, 604–617 CrossRef CAS PubMed.
- R. Haubner, R. Gratias, B. Diefenbach and S. Goodman, J. Am. Chem. Soc., 1996, 118, 7461–7472 CrossRef CAS.
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS PubMed.
- S. Liu, Bioconjugate Chem., 2015, 26, 1413–1438 CrossRef CAS PubMed.
- G. Thumshirn, U. Hersel, S. L. Goodman and H. Kessler, Chem. – Eur. J., 2003, 9, 2717–2725 CrossRef CAS PubMed.
- T. Poethko, M. Schottelius, G. Thumshirn, U. Hersel, M. Herz, G. Henriksen, H. Kessler, M. Schwaiger and H.-J. Wester, J. Nucl. Med., 2004, 45, 892–902 CAS.
- C. Hultsch, M. Berndt, R. Bergmann and F. Wuest, Appl. Radiat. Isot., 2007, 65, 818–826 CrossRef CAS PubMed.
- S. Liu, Z. Liu, K. Chen, Y. Yan, P. Watzlowik, H. J. Wester, F. T. Chin and X. Chen, Mol. Imaging Biol., 2010, 12, 530–538 CrossRef PubMed.
- W. Cheng, Z. Wu, S. Liang, H. Fu, S. Wu, Y. Tang, Z. Ye and H. Wang, PLoS One, 2014, 9, e100521 Search PubMed.
- J. Guo, N. Guo, L. Lang, D. O. Kiesewetter, Q. Xie, Q. Li, H. S. Eden, G. Niu and X. Chen, J. Nucl. Med., 2014, 55, 154–160 CrossRef CAS PubMed.
- W. J. McBride, C. A. D'Souza, H. Karacay, R. M. Sharkey and D. M. Goldenberg, Bioconjugate Chem., 2012, 23, 538–547 CrossRef CAS PubMed.
- J. A. F. Joosten, N. T. H. Tholen, F. Ait El Maate, A. J. Brouwer, G. W. van Esse, D. T. S. Rijkers, R. M. J. Liskamp and R. J. Pieters, Eur. J. Org. Chem., 2005, 3182–3185 CrossRef CAS.
- C. Wängler, S. Maschauer, O. Prante, M. Schäfer, R. Schirrmacher, P. Bartenstein, M. Eisenhut and B. Wängler, ChemBioChem, 2010, 11, 2168–2181 CrossRef PubMed.
- I. Dijkgraaf, A. Y. Rijnders, A. Soede, A. C. Dechesne, G. W. van Esse, A. J. Brouwer, F. H. M. Corstens, O. C. Boerman, D. T. S. Rijkers and R. M. J. Liskamp, Org. Biomol. Chem., 2007, 5, 935–944 CAS.
- H. Li, H. Zhou, S. Krieger, J. J. Parry, J. J. Whittenberg, A. V. Desai, B. E. Rogers, P. J. a. Kenis and D. E. Reichert, Bioconjugate Chem., 2014, 25, 761–772 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- M. J. Robins, F. Hansske, S. F. Wnuk and T. Kanai, Can. J. Chem., 1991, 69, 1468–1474 CrossRef CAS.
- V. Iaroshenko, D. Sevenard, A. Kotljarov, D. Volochnyuk, A. Tolmachev and V. Sosnovskikh, Synthesis, 2009, 731–740 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- M. Schilz and H. Plenio, J. Org. Chem., 2012, 77, 2798–2807 CrossRef CAS PubMed.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- M. Piras, I. Fleming, W. Harrison and M. Zanda, Synlett, 2012, 2899–2902 CAS.
- A. J. Beer, H. Kessler, H.-J. Wester and M. Schwaiger, Theranostics, 2011, 1, 48–57 CrossRef CAS PubMed.
- H. Deng, S. L. Cobb, A. D. Gee, A. Lockhart, L. Martarello, R. P. McGlinchey, D. O'Hagan and M. Onega, Chem. Commun., 2006, 652–654 RSC.
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- D. Ory, J. Van den Brande, T. de Groot, K. Serdons, M. Bex, L. Declercq, F. Cleeren, M. Ooms, K. Van Laere, A. Verbruggen and G. Bormans, J. Pharm. Biomed. Anal., 2015, 111, 209–214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00239k |
| This journal is © The Royal Society of Chemistry 2016 |