
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A one-pot, three-step process for the diastereoselective synthesis of aminobicyclo[4.3.0]nonanes using consecutive palladium(II)- and ruthenium(II)-catalysis†
Mohamed A. B.
Mostafa
,
Mark. W.
Grafton
,
Claire
Wilson
and
Andrew
Sutherland
*
WestCHEM, School of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
First published on 25th February 2016
Abstract
A diastereoselective synthesis of highly substituted aminobicyclo[4.3.0]nonanes has been attained using a one-pot multi-bond forming process. A four-step synthetic route was developed for the efficient synthesis of a series of C-7 substituted hept-2-en-6-yn-1-ols. These compounds were then investigated as substrates for a one-pot, three-step tandem process involving a palladium(II)-catalysed Overman rearrangement, a ruthenium(II)-catalysed ring closing enyne metathesis reaction followed by a hydrogen bond directed Diels–Alder reaction. The optimisation of the one-pot process has allowed the rapid preparation of a library of aminobicyclo[4.3.0]nonanes with significant molecular complexity and up to four stereogenic centres.
Introduction
A recent trend in identifying lead-hit compounds and small-molecule probes for medicinal chemistry and chemical biology has been the replacement of sp2-rich aromatic and heteroaromatic compounds with sp3-rich compounds.1 Partially saturated compounds with a higher degree of saturation have more suitable physicochemical properties such as solubility and allow a more efficient examination of three-dimensional chemical space.1,2 In this regard, saturated and partially saturated forms of amino substituted bicyclo[4.3.0]nonanes have exhibited wide-ranging biological and pharmacological properties.3 In particular, these compounds are found as components of natural products such as the guanidine alkaloid netamine A (1)4 and the antitumour antibiotic (+)-ptilocaulin (2) (Fig. 1).5 Amino-indanes are found as constituents of a range of medicinal agents including (+)-indatraline (3),6 a monoamine transporter inhibitor and rasagiline (Azilect), a drug used for the treatment of Parkinson's disease.7 Other amino-indane structural analogues can inhibit the proliferation of malignant cells8 and are used to treat HIV infections and AIDS.9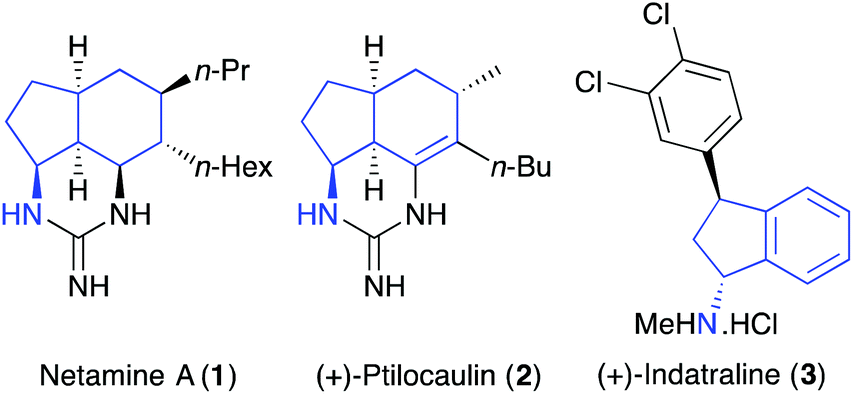 | ||
| Fig. 1 Biologically active amino bicyclo[4.3.0]nonanes and indanes. | ||
Due to these wide-ranging pharmacological activities, a number of synthetic approaches have been developed for the general preparation of these compounds.5,10–13 Diastereoselective syntheses have been achieved using an intramolecular 1,3-dipolar cycloaddition between an oxime and a cyclohexene5 and, using a Ru(I)-catalysed allenic cycloisomerisation of an alkynone, followed by a Diels–Alder reaction of the resulting 2-alkylidene-3-vinylcyclopentenone.10 Other diastereoselective syntheses include the C–H activation of a hexahydroindene that gave the corresponding secondary organoborane, which was then aminated to give the amino substituted bicyclo[4.3.0]nonane in good overall yield.11
As part of a research programme to develop new methods for rapid access to drug-like polycyclic scaffolds, we recently reported the diastereoselective synthesis of amino substituted bicyclo[4.3.0]nonanes using a one-pot multistep process involving a thermally-mediated Overman rearrangement of alkyne derived allylic alcohols, followed by a ring closing enyne metathesis (RCEYM) reaction of the resulting enyne and a hydrogen bond directed Diels–Alder reaction (Scheme 1).14 Using a range of dienophiles, this allowed the late-stage synthesis of a library of partially saturated indane ring systems. More recently the one-pot method has been extended to include a cross-metathesis step leading to the rapid preparation of C-4 substituted analogues with up to five stereogenic centres.15 While this approach permitted the facile preparation of a range of amino substituted bicyclo[4.3.0]nonanes, we found that some of the one-pot processes required particularly long reaction times and this was in part due to using thermal conditions to implement the Overman rearrangement (36 h).14,15 In previous studies, we found that alkene and alkyne derived allylic trichloroacetimidates would not undergo effective palladium(II)-catalysed rearrangements due to binding of the catalyst to the unsaturated side-chains.13,16 We were interested in exploring the structural requirements of alkyne derived allylic alcohols that could block catalyst side-chain binding and perform a Pd(II)-catalysed Overman rearrangement as part of a more rapid one-pot process leading to new C-5 substituted aminobicyclo[4.3.0]nonanes. We now report the synthesis of a series of C-7 substituted hept-2-en-6-yn-1-ols and the evaluation of these compounds to undergo a Pd(II)-catalysed Overman rearrangement. As well as using these allylic alcohols as substrates for a one-pot multistep process for the diastereoselective preparation of novel aminobicyclo[4.3.0]nonanes, we also report further functionalisation of these products to generate highly substituted sp3-rich, drug-like polycyclic scaffolds with up to six stereogenic centres.
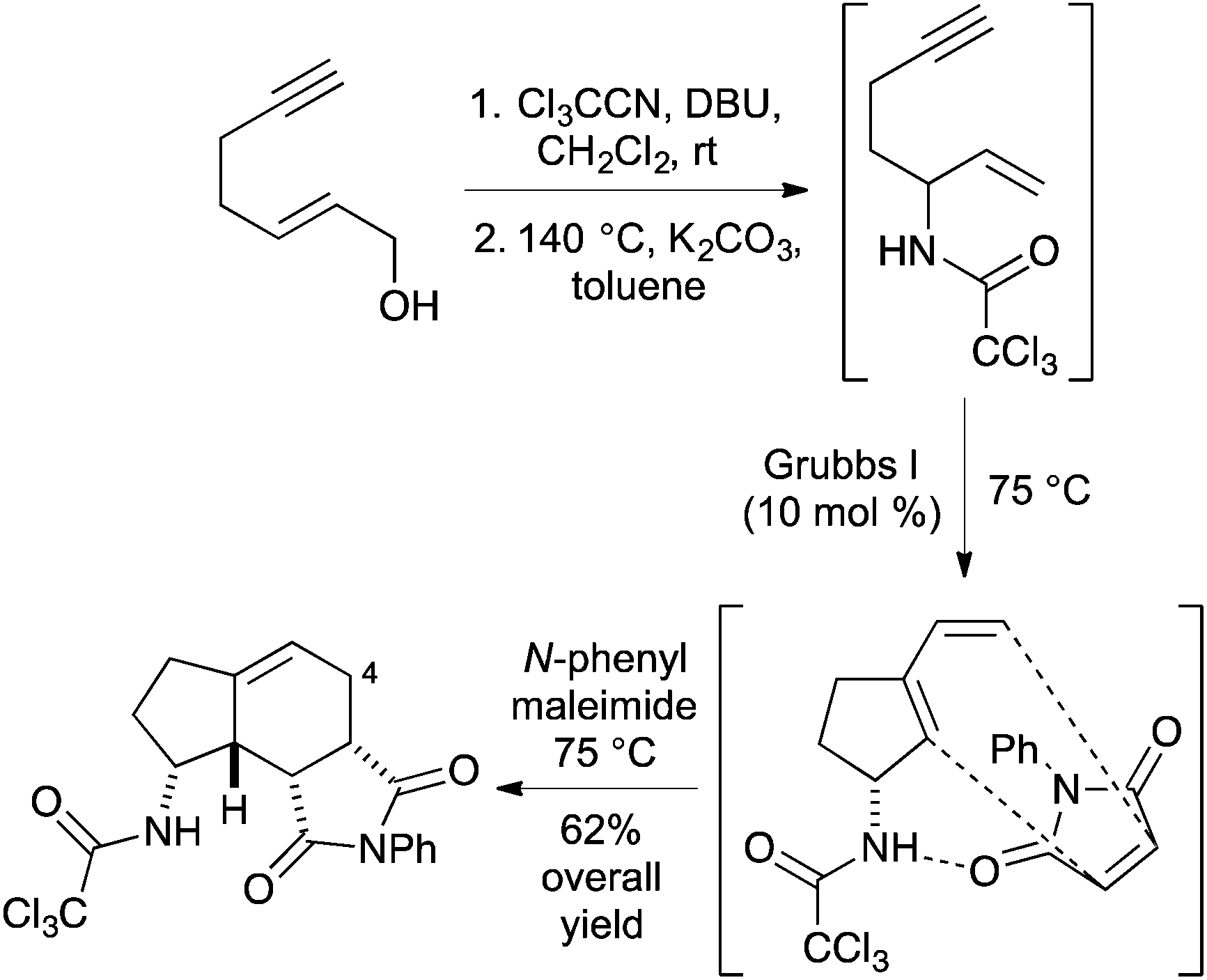 | ||
| Scheme 1 One-pot synthesis of amino substituted bicyclo[4.3.0]nonanes. | ||
Results and discussion
To investigate the requirements of alkyne substituents to block catalyst binding during a Pd(II)-catalysed Overman rearrangement, a series of C-7 substituted hept-2-en-6-yn-1-ols were prepared in four steps from pent-4-yn-1-ol (4) (Scheme 2). Sonogashira coupling with various aryl iodides in the presence of copper(I) iodide (2 mol%) and bis(triphenylphosphine)palladium(II) dichloride (1 mol%) gave disubstituted alkynes 5–7 in excellent yields.17 An alkyl analogue (9, R = Me) was also prepared by potassium tert-butoxide mediated isomerisation of hex-5-yn-1-ol (8).18 The various pent-4-yn-1-ols were then subjected to a one-pot Swern oxidation and Horner–Wadsworth–Emmons reaction under Masamune–Roush conditions which gave the corresponding (E)-α,β-unsaturated esters 10–14 in essentially quantitative yields.19,20 Reduction using DIBAL-H gave the desired hept-2-en-6-yn-1-ols 15–19 in excellent overall yield.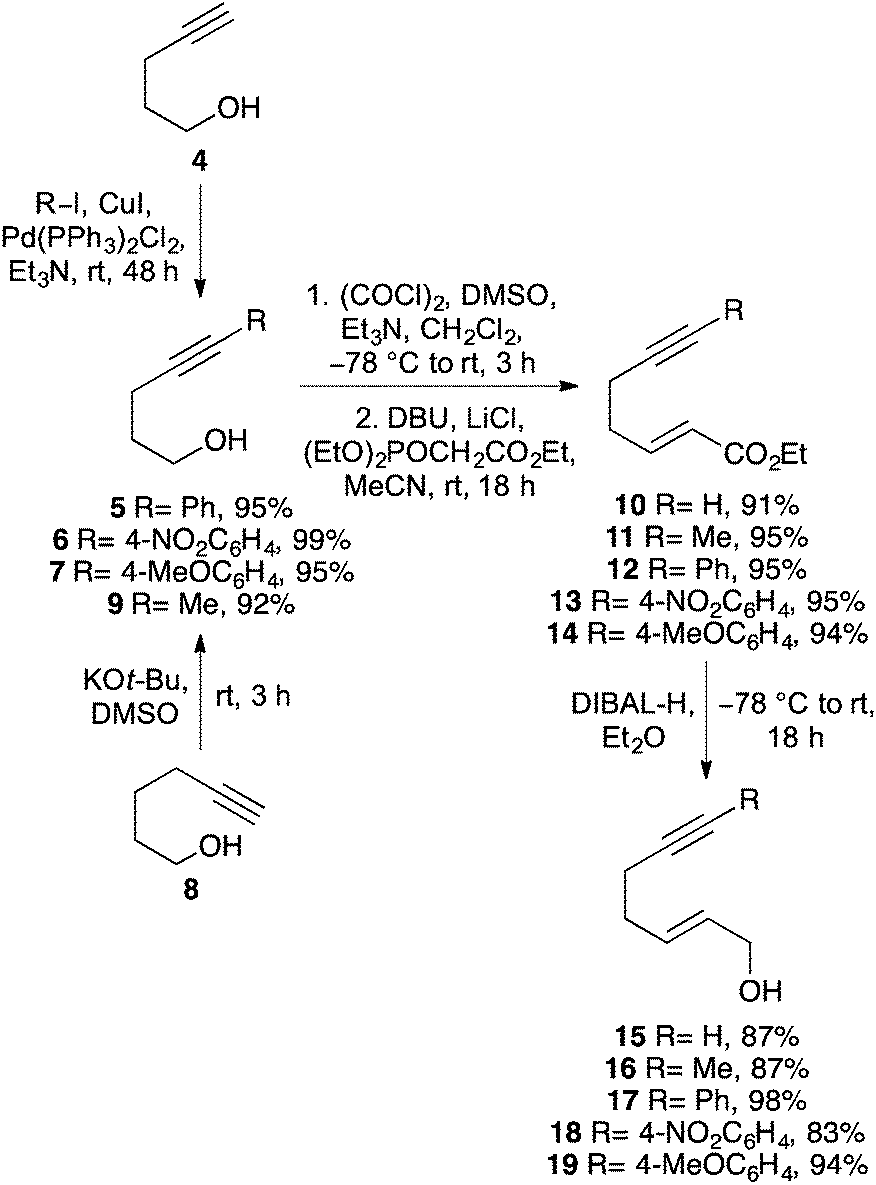 | ||
| Scheme 2 Synthesis of C-7 substituted hept-2-en-6-yn-1-ols. | ||
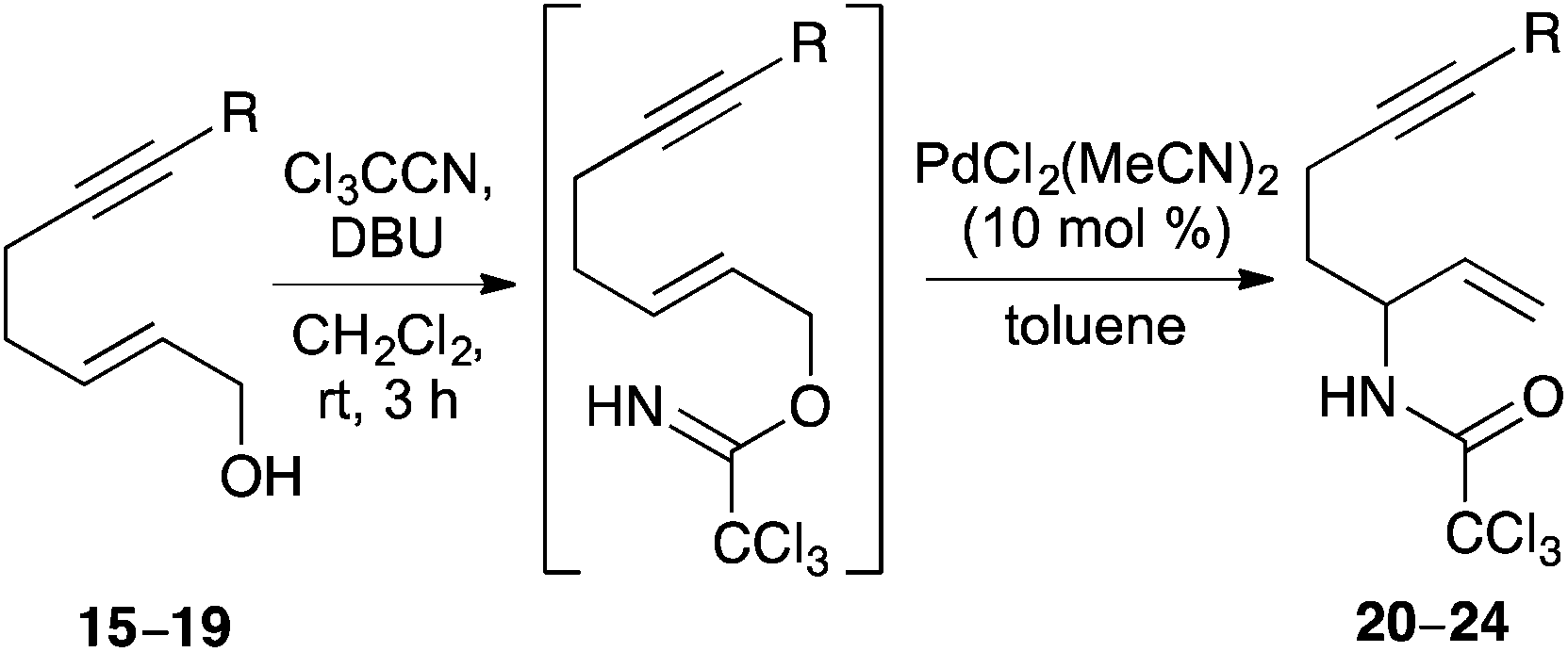
The ability of hept-2-en-6-yn-1-ols 15–19 to undergo a Pd(II)-catalysed Overman rearrangement was next investigated. The study began by exploring the rearrangement of mono-substituted alkyne, (2E)-hept-2-en-6-yn-1-ol (15) (Table 1, entry 1). Allylic alcohol 15 was converted to the corresponding allylic trichloroacetimidate using trichloroacetonitrile and a catalytic amount of DBU.21 Using standard conditions for a Pd(II)-catalysed Overman rearrangement (10 mol% catalyst loading at rt),22 only small amounts (<10%) of allylic trichloroacetamide 20 could be observed by NMR spectroscopy. A number of reactions were then performed to elucidate the optimal conditions for the preparation of 20. It was found that addition of a second batch of catalyst after 24 h and conducting the entire reaction at 40 °C gave allylic trichloroacetamide 20 in 34% yield after a reaction time of 48 h. The elevated temperature, high catalyst loading and long reaction time are exemplary of the conditions required for metal catalysed rearrangement of allylic trichloroacetamides bearing mono-substituted unsaturated side-chains. The rearrangement of disubstituted alkyne derived allylic trichloroacetamides was next investigated. While a methyl substituent is relatively small, the use of this group was sufficient to partially retard catalyst binding and allow rearrangement using only 10 mol% of catalyst at 20 °C (entry 2). This gave allylic trichloroacetamide 21 in 55% yield after 24 h. Using aryl groups with substantially more bulk proved effective and allowed the efficient synthesis of the corresponding allylic trichloroacetamides 22–24 in high yields after a 12 h reaction time (entries 3–5). Interestingly, the yields were independent of the electronic nature of the aryl groups indicating that the steric bulk of these substituents is primarily responsible for preventing binding of the catalyst to the alkyne moiety.
Having identified the structural requirements and optimal conditions for an efficient Overman rearrangement, these were incorporated into a one-pot multi-reaction process including a Ru(II)-catalysed RCEYM step23 and a Diels–Alder reaction for the preparation of novel aminobicyclo[4.3.0]nonanes (Scheme 3). Preliminary attempts at the one-pot preparation of 25 from phenyl substituted allylic alcohol 17 using Grubbs 2nd generation catalyst (7 mol%)24 for the RCEYM step and N-phenyl maleimide as a dienophile for the Diels–Alder step, under previously developed conditions for these reactions14,15 gave low yields of 25 (∼25%). Analysis of the 1H NMR spectrum of the reaction mixture showed the presence of the 1,6-enyne 22, indicating that the RCEYM step had not gone to completion. This was unsurprising as disubstituted, bulky alkynes often show suppressed reactivity during RCEYM reactions.23 Methods for improving this step were investigated. A combination of the use of 1,7-octadiene as an in situ source of ethylene25 and a higher reaction temperature (from 75 to 90 °C) resulted in an accelerated RCEYM reaction, allowing complete conversion of 1,6-enyne 22 to the corresponding cyclopentyl exo-diene. Using these modified conditions as part of the one-pot process gave 5-phenyl aminobicyclo[4.3.0]nonane 25 as a single diastereomer in 51% overall yield from allylic alcohol 17 (Scheme 3).26 As previously reported for the Diels–Alder reaction of trichloroacetamide derived cyclic exo-dienes, the reaction proceeds via a hydrogen bonding directed endo transition state, generating the syn-products with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).14 The relative stereochemistry of 25 was confirmed by difference NOE experiments, which showed the syn relationship of the hydrogen atoms at C-3a, C-8, C-8a and C-8b.27 For comparison, use of the optimised one-pot process was applied to methyl substituted allylic alcohol 16 which gave 26 in 25% overall yield. The significantly lower yield for 26 is a consequence of the less efficient Overman rearrangement for this analogue. Using phenyl derived allylic alcohol 17, the scope of the one-pot multistep process was explored using various dienophiles. In all cases, the compounds were formed as single diastereomers in good yields over the four steps (40–56%). It should be noted that the non-symmetrical dienophile, methyl acrylate gave indane 29 as a single regioisomer. This again is a direct consequence of the hydrogen bonding directed endo transition state.14
:1).14 The relative stereochemistry of 25 was confirmed by difference NOE experiments, which showed the syn relationship of the hydrogen atoms at C-3a, C-8, C-8a and C-8b.27 For comparison, use of the optimised one-pot process was applied to methyl substituted allylic alcohol 16 which gave 26 in 25% overall yield. The significantly lower yield for 26 is a consequence of the less efficient Overman rearrangement for this analogue. Using phenyl derived allylic alcohol 17, the scope of the one-pot multistep process was explored using various dienophiles. In all cases, the compounds were formed as single diastereomers in good yields over the four steps (40–56%). It should be noted that the non-symmetrical dienophile, methyl acrylate gave indane 29 as a single regioisomer. This again is a direct consequence of the hydrogen bonding directed endo transition state.14
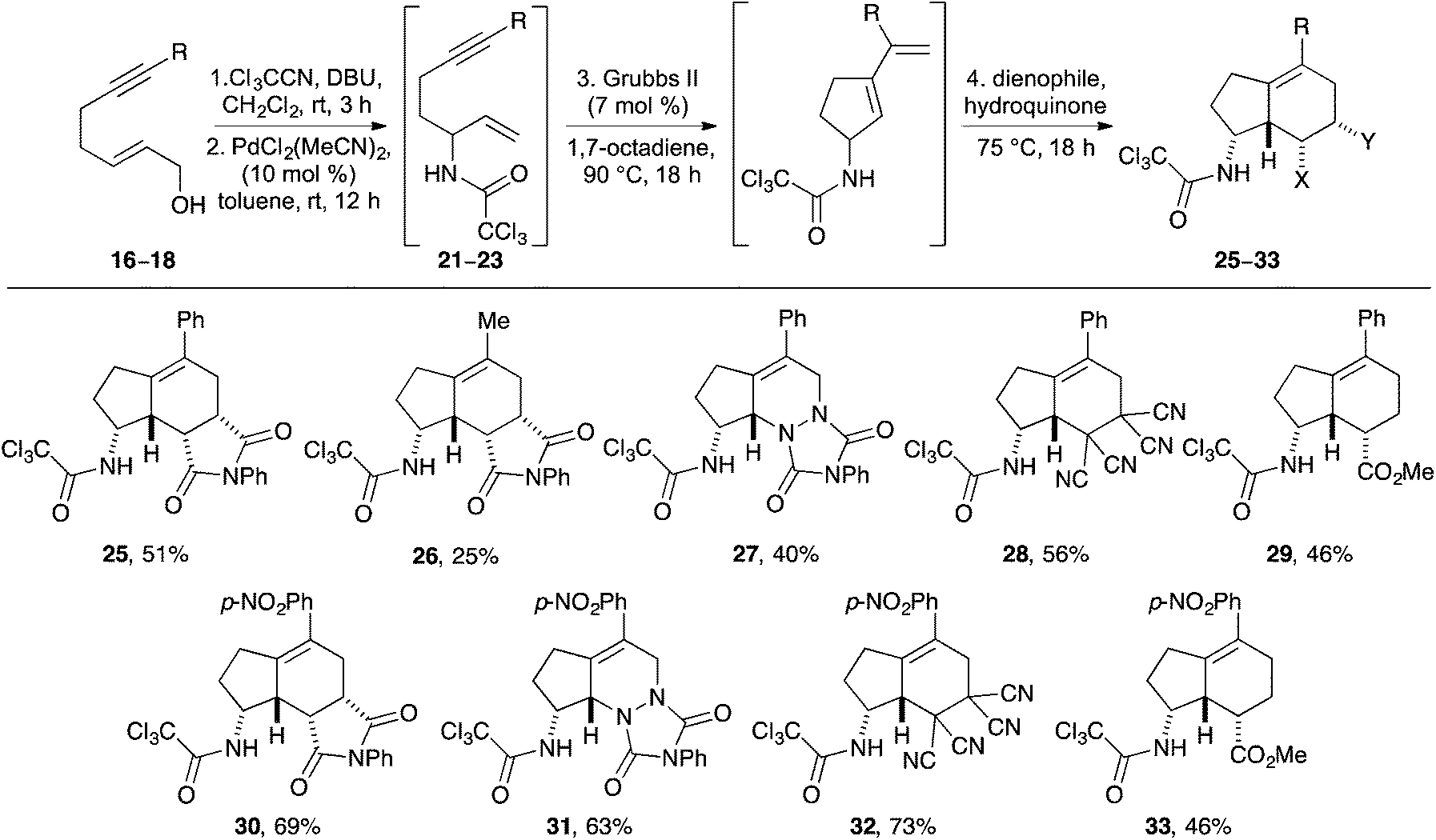 | ||
| Scheme 3 One-pot synthesis of aminobicyclo[4.3.0]nonanes 25–33. | ||
As well as developing a one-pot synthesis of aminobicyclo[4.3.0]nonanes using consecutive Pd(II)- and Ru(II)-catalysis, another major objective of this research programme was to probe the effect of electron-deficient and electron-rich aryl substituted alkynes on the outcome of the RCEYM step and the subsequent one-pot process. While 1,6-enynes bearing electron-deficient alkyne substituents have been shown to have a detrimental effect on RCEYM reactions,23a,28 examples with electron-poor aryl groups have given excellent yields under forcing conditions.28 Using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) as a substrate for the one-pot multistep process and N-phenyl maleimide as the dienophile, gave 4-nitrophenyl substituted aminobicyclo[4.3.0]nonane 30 as a single diastereomer in 69% yield. In a similar fashion, use of 4-phenyl-1,2,4-triazole-3,5-dione, tetracyanoethylene or methyl acrylate as dienophiles for the Diels–Alder step allowed the preparation of 4-nitrophenyl substituted aminobicyclo[4.3.0]nonanes 31–33 in good overall yields. Despite being sterically encumbered and electron-deficient, the 1,6-enyne produced during these one-pot processes seems able to undergo a highly effective RCEYM reaction under our optimal conditions. A more stable cyclopentyl exo-diene and a cleaner subsequent Diels–Alder reaction may account for the elevated yields of the 4-nitrophenyl series compared to the phenyl-substituted compounds.
Electron-rich allylic alcohol (2E)-7-(4′-methoxyphenyl)hept-2-en-6-yn-1-ol (19) was then converted to the allylic trichloroacetamidate and subjected to the one-pot, three-step process using N-phenyl maleimide as the dienophile. This gave the corresponding 4-methoxyphenyl substituted aminobicyclo[4.3.0]nonane 34 in only 34% yield. Analysis of the individual steps of the one-pot process revealed that while the electron-rich cyclopentyl exo-diene was readily formed during our optimised conditions for the RCEYM reaction, the temperature (90 °C) of this transformation was resulting in decomposition of the highly reactive diene. On screening various temperatures for the RCEYM reaction of electron-rich 1,6-enyne 24, it was found that the reaction proceeded to completion at a much lower temperature of 40 °C. Despite application of this optimised RCEYM step, one-pot reactions using allylic alcohol 19 still gave modest yields of the 4-methoxyphenyl substituted aminobicyclo[4.3.0]nonane 34. As such, the preparation of this final series of compounds was conducted as two separate processes. Following efficient large-scale preparation of allylic trichloroacetamide 24 (Table 1), this was subjected to a one-pot, two-step process involving the low temperature RCEYM step and a Diels–Alder reaction with various electron-deficient dienophiles (Scheme 4). This allowed the synthesis of 4-methoxyphenyl substituted aminobicyclo[4.3.0]nonanes 34–37 as single diastereomers in good yields over the two steps.
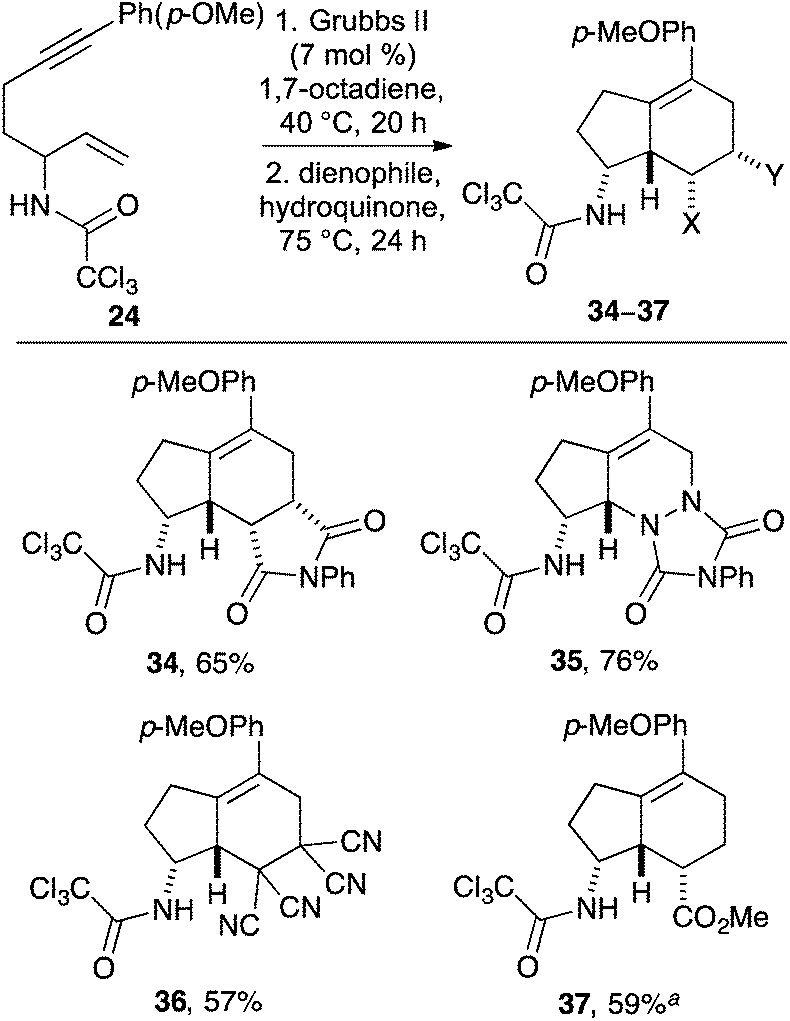 | ||
| Scheme 4 Synthesis of p-methoxyphenyl substituted aminobicyclo[4.3.0]nonanes 34–37. aDiels–Alder reaction was done at 111 °C for 5 days. | ||
Having synthesised a novel library of aminobicyclo[4.3.0]nonanes, a preliminary study was conducted to explore further functionalisation of these compounds and in particular, the reactivity of the tetra-substituted alkene moiety. Initially, hydrogenation of 25 was attempted under standard conditions (Scheme 5). However, after 48 h, partial reduction of the trichloromethyl group was the only change detected, giving the dichloroacetamide in 48% yield. Despite the resistance of the tetra-substituted alkene to undergo hydrogenation, oxidation of the moiety was readily observed. For example, reaction of 25 with osmium tetroxide in the presence of TMEDA under Donohoe conditions29 gave dihydroxy derivative 39 as a single diastereomer in quantitative yield after only 3 h. Based on the shape of aminobicyclo[4.3.0]nonane 25, it was expected that reactions of the alkene would take place from the more exposed convex face of the molecule. This was confirmed by X-ray crystallography. The (3aS*,5R*,5aR*,8R*,8aR*,8bR*)-stereoisomer 39 was found to crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the structure clearly shows the syn relationship of the hydrogen atoms at C-3a, C-8, C-8a and C-8b and the hydroxyl groups at C-5 and C-5a (Fig. 2).30,31 In a similar fashion, reaction of 25 with m-CPBA gave epoxide 40 as a single diastereomer in 65% yield.
and the structure clearly shows the syn relationship of the hydrogen atoms at C-3a, C-8, C-8a and C-8b and the hydroxyl groups at C-5 and C-5a (Fig. 2).30,31 In a similar fashion, reaction of 25 with m-CPBA gave epoxide 40 as a single diastereomer in 65% yield.
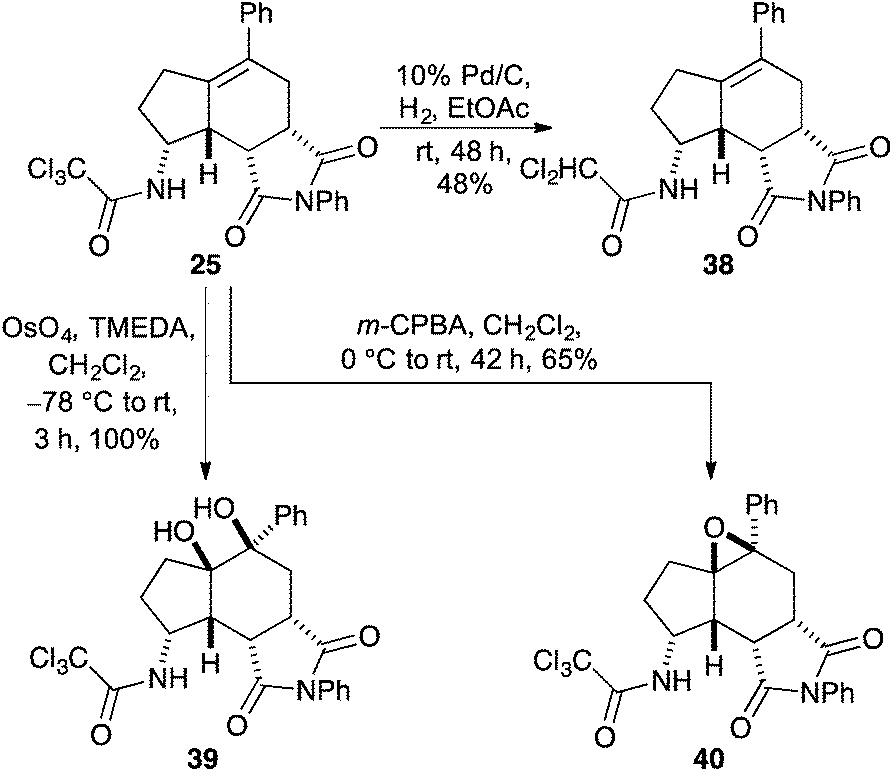 | ||
| Scheme 5 Functionalisation of aminobicyclo[4.3.0]nonane 25. | ||
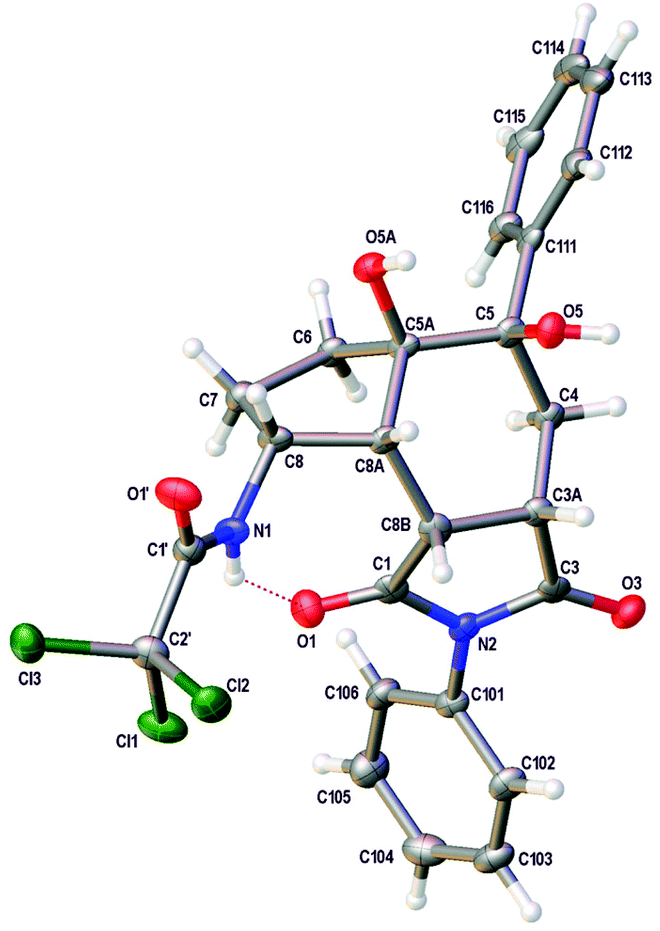 | ||
| Fig. 2 View showing the structure of one of the crystallographically independent molecules of 39. Atomic displacement ellipsoids are drawn at 50% probability level. | ||
Conclusions
In summary, a series of C-7 substituted hept-2-en-6-yn-1-ols have been examined as substrates for the one-pot diastereoselective synthesis of sp3-rich aminobicyclo[4.3.0]nonanes. The presence of the C-7 groups allowed an effective Pd(II)-catalysed Overman rearrangement to proceed. Incorporation of this transformation into a one-pot, three-step process involving a Ru(II)-catalysed RCEYM reaction and a hydrogen bonding directed Diels–Alder reaction gave a range of aminobicyclo[4.3.0]nonanes in good overall yields. The effect of the electronic nature of the aryl substituent on the RCEYM step was also studied and while an electron-poor analogue could be used as a substrate for the one-pot process using forcing conditions for the RCEYM step, the electron-rich cyclopentyl exo-diene was found to undergo decomposition during the multistep process. Nevertheless, a series of 4-methoxyphenyl substituted aminobicyclo[4.3.0]nonanes could be prepared efficiently using a one-pot, two-step process from the allylic trichloroacetamide. The reactivity of these novel compounds was also explored and aminobicyclo[4.3.0]nonane 25 was readily oxidised, generating dihydroxy and epoxide derivatives as single diastereomers in high yields. The combination of the one-pot, three-step multireaction process with the oxidations allowed the rapid preparation of these sp3-rich, drug-like polycyclic scaffolds with six stereogenic centres. With the development of an effective one-pot synthesis of these compounds using a Pd(II)-catalysed rearrangement, work is currently underway to incorporate chiral Pd(II)-catalysts for their asymmetric synthesis and preparation of natural product targets.Experimental
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a PureSolv 500 MD solvent purification system. All reactions were performed under an atmosphere of argon unless otherwise mentioned. Flash column chromatography was performed using Fisher matrix silica 60. Macherey-Nagel aluminium-backed plates pre-coated with silica gel 60 (UV254) were used for thin layer chromatography and were visualised by staining with KMnO4. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 spectrometer with chemical shift values in ppm relative to TMS (δH 0.00 and δC 0.0) or residual chloroform (δH 7.26 and δC 77.2) as standard. Proton and carbon assignments are based on two-dimensional COSY and DEPT experiments, respectively. Mass spectra were obtained using a JEOL JMS-700 spectrometer for EI and CI or a Bruker Microtof-q for ESI. Infrared spectra were obtained neat using a Shimadzu IRPrestige-21 spectrometer. Melting points were determined on a Reichert platform melting point apparatus.5-Phenylpent-4-yn-1-ol (5)32
Bis(triphenylphosphine)palladium(II) dichloride (0.022 g, 0.031 mmol) and copper iodide (0.012 g, 0.062 mmol) were dissolved in triethylamine (43 mL) and iodobenzene (0.42 mL, 3.71 mmol) was added and stirred at room temperature for 0.1 h. Pent-4-yn-1-ol (4) (0.26 g, 3.09 mmol) was added and the reaction mixture was stirred at room temperature for 48 h. The reaction mixture was concentrated in vacuo. Purification of the resulting residue by flash column chromatography (petroleum ether/ethyl acetate, 3:1) gave 5-phenylpent-4-yn-1-ol (5) (0.48 g, 95%) as a colourless oil. Spectroscopic data was consistent with the literature.32δH (400 MHz, CDCl3) 1.59 (1H, br s, OH), 1.86 (2H, quin, J 6.5 Hz, 2-H2), 2.54 (2H, t, J 6.5 Hz, 3-H2), 3.82 (2H, t, J 6.5 Hz, 1-H2), 7.24–7.30 (3H, m, 3 × ArH), 7.36–7.42 (2H, m, 2 × ArH); δC (101 MHz, CDCl3) 16.0 (CH2), 31.4 (CH2), 61.7 (CH2), 81.1 (C), 89.4 (C), 123.8 (C), 127.7 (CH), 128.2 (2 × CH), 131.6 (2 × CH); m/z (CI) 161 (MH+. 100%), 133 (20), 117 (28), 113 (13), 85 (28), 69 (39).
5-(4′-Nitrophenyl)pent-4-yn-1-ol (6)33
5-(4′-Nitrophenyl)pent-4-yn-1-ol (6) was synthesised as described for 5-phenylpent-4-yn-1-ol (5) using pent-4-yn-1-ol (4) (0.26 g, 3.09 mmol) and 4-iodo-1-nitrobenzene (0.92 g, 3.71 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 3:1) gave 5-(4′-nitrophenyl)pent-4-yn-1-ol (6) (0.63 g, 99%) as an orange solid. Mp 30–32 °C; spectroscopic data was consistent with the literature.33δH (500 MHz, CDCl3) 1.44 (1H, br t, J 6.8 Hz, OH), 1.89 (2H, quin, J 6.8 Hz, 2-H2), 2.59 (2H, t, J 6.8 Hz, 3-H2), 3.82 (2H, q, J 6.8 Hz, 1-H2), 7.49–7.54 (2H, m, 2′-H and 6′-H), 8.13–8.18 (2H, m, 3′-H and 5′-H); δC (126 MHz, CDCl3) 16.1 (CH2), 31.1 (CH2), 61.3 (CH2), 79.5 (C), 95.9 (C), 123.4 (2 × CH), 130.9 (C), 132.2 (2 × CH), 146.5 (C); m/z (ESI) 228 (MNa+. 100%), 199 (11), 176 (14), 166 (15), 152 (37), 144 (22), 138 (16), 102 (15).
5-(4′-Methoxyphenyl)pent-4-yn-1-ol (7)34
5-(4′-Methoxyphenyl)pent-4-yn-1-ol (7) was synthesised as described for 5-phenylpent-4-yn-1-ol (5) using pent-4-yn-1-ol (4) (0.26 g, 3.09 mmol) and 4-iodoanisole (0.87 g, 3.71 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 1:1) gave 5-(4′-methoxyphenyl)pent-4-yn-1-ol (7) (0.58 g, 95%) as a colourless oil. Spectroscopic data was consistent with the literature.34δH (400 MHz, CDCl3) 1.54 (1H, br t, J 6.8 Hz, OH), 1.85 (2H, quin, J 6.8 Hz, 2-H2), 2.52 (2H, t, J 6.8 Hz, 3-H2), 3.80 (3H, s, OCH3), 3.82 (2H, q, J 6.8 Hz, 1-H2), 6.78–6.84 (2H, m, 3′-H and 5′-H), 7.30–7.35 (2H, m, 2′-H and 6′-H); δC (126 MHz, CDCl3) 15.9 (CH2), 31.5 (CH2), 55.2 (CH3), 61.4 (CH2), 80.8 (C), 87.9 (C), 113.9 (2 × CH), 116.0 (C), 132.9 (2 × CH), 159.1 (C); m/z (EI) 190 (M+. 100%), 159 (24), 145 (75), 134 (55), 115 (38), 83 (59), 75 (23), 47 (16).
Hex-4-yne-1-ol (9)18
To a solution of hex-5-yn-1-ol (8) (0.20 g, 2.04 mmol) in dimethyl sulfoxide (7 mL) was added potassium tert-butoxide (0.46 g, 4.08 mmol). The reaction mixture was stirred at room temperature for 3 h. The reaction was quenched by the addition of 2 M hydrochloric acid and then extracted with diethyl ether (4 × 25 mL). The organic layers were combined, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (petroleum ether/ethyl acetate, 8:2) gave hex-4-yne-1-ol (9) (0.19 g, 92%) as a yellow oil. Spectroscopic data was consistent with the literature.18δH (400 MHz, CDCl3) 1.56 (1H, br s, OH), 1.73 (2H, quin, J 6.2 Hz, 2-H2), 1.78 (3H, t, J 2.6 Hz, 6-H3), 2.22–2.29 (2H, m, 3-H2), 3.75 (2H, t, J 6.2 Hz, 1-H2); δC (126 MHz, CDCl3) 3.4 (CH3), 15.3 (CH2), 31.5 (CH2), 61.7 (CH2), 76.1 (C), 78.5 (C); m/z (CI) 99 (MH+. 100%), 81 (10), 73 (15), 71 (8), 69 (7).
Ethyl (2E)-hept-2-en-6-ynoate (10)35
Dimethyl sulfoxide (3.16 mL, 44.5 mmol) was added to a stirred solution of oxalyl chloride (2.11 mL, 25.0 mmol) in dichloromethane (90 mL) at −78 °C. The mixture was stirred for 0.3 h before pent-4-yn-1-ol (4) (1.50 g, 17.8 mmol) in dichloromethane (20 mL) was slowly added. The mixture was stirred for a further 0.3 h before triethylamine (12.5 mL, 89.0 mmol) was added. This reaction mixture was stirred for 0.5 h at −78 °C and then allowed to warm to room temperature and stirred for a further 3 h. A solution of lithium chloride (1.36 g, 32.0 mmol), triethyl phosphonoacetate (6.35 mL, 32.0 mmol) and 1,8-diazabicyclo[5,4,0]undec-7-ene (4.79 mL, 32.0 mmol) in acetonitrile (60 mL) was then prepared and stirred for 1 h. The Swern solution was concentrated in vacuo, then the Horner Wadsworth Emmons solution was added and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with a saturated solution of ammonium chloride (45 mL) and concentrated to give an orange residue, which was then extracted with diethyl ether (4 × 60 mL). The organic layers were combined, dried (MgSO4), filtered and concentrated to give an orange oil. Purification by flash column chromatography (petroleum ether/diethyl ether, 7:3) gave ethyl (2E)-hept-2-en-6-ynoate (10) (2.45 g, 91%) as a yellow oil. Spectroscopic data was consistent with the literature.35δH (400 MHz, CDCl3) 1.30 (3H, t, J 7.1 Hz, OCH2CH3), 2.01 (1H, t, J 2.5 Hz, 7-H), 2.34–2.39 (2H, m, 5-H2), 2.41–2.48 (2H, m, 4-H2), 4.20 (2H, q, J 7.1 Hz, OCH2CH3), 5.90 (1H, dt, J 15.7, 1.5 Hz, 2-H), 6.97 (1H, dt, J 15.7, 6.7 Hz, 3-H); δC (126 MHz, CDCl3) 14.3 (CH3), 17.4 (CH2), 31.0 (CH2), 60.3 (CH2), 69.4 (CH), 82.7 (C), 122.6 (CH), 146.3 (CH), 166.4 (C); m/z (CI) 153 (MH+. 100%), 139 (5), 113 (10), 97 (5), 81 (15), 69 (15).
Ethyl (2E)-oct-2-en-6-ynoate (11)
Ethyl (2E)-oct-2-en-6-ynoate (11) was synthesised as described for ethyl (2E)-hept-2-en-6-ynoate (10) using hex-4-yne-1-ol (9) (0.17 g, 1.68 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 8:2) gave ethyl (2E)-oct-2-en-6-ynoate (11) (0.27 g, 95%) as a colourless oil. νmax/cm−1 (neat) 2921 (CH), 1721 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1657, 1368, 1265, 1157, 1039, 975; δH (400 MHz, CDCl3) 1.29 (3H, t, J 7.1 Hz, OCH2CH3), 1.77 (3H, t, J 2.3, 8-H3), 2.25–2.32 (2H, m, 5-H2), 2.34–2.41 (2H, m, 4-H2), 4.19 (2H, q, J 7.1 Hz, OCH2CH3), 5.87 (1H, dt, J 15.7, 1.5 Hz, 2-H), 6.98 (1H, dt, J 15.7, 6.6 Hz, 3-H); δC (126 MHz, CDCl3) 3.4 (CH3), 14.2 (CH3), 17.7 (CH2), 31.6 (CH2), 60.2 (CH2), 76.6 (C), 77.5 (C), 122.1 (CH), 147.1 (CH), 166.5 (C); m/z (ESI) 189.0883 (MNa+. C10H14NaO2 requires 189.0886).
O), 1657, 1368, 1265, 1157, 1039, 975; δH (400 MHz, CDCl3) 1.29 (3H, t, J 7.1 Hz, OCH2CH3), 1.77 (3H, t, J 2.3, 8-H3), 2.25–2.32 (2H, m, 5-H2), 2.34–2.41 (2H, m, 4-H2), 4.19 (2H, q, J 7.1 Hz, OCH2CH3), 5.87 (1H, dt, J 15.7, 1.5 Hz, 2-H), 6.98 (1H, dt, J 15.7, 6.6 Hz, 3-H); δC (126 MHz, CDCl3) 3.4 (CH3), 14.2 (CH3), 17.7 (CH2), 31.6 (CH2), 60.2 (CH2), 76.6 (C), 77.5 (C), 122.1 (CH), 147.1 (CH), 166.5 (C); m/z (ESI) 189.0883 (MNa+. C10H14NaO2 requires 189.0886).
Ethyl (2E)-7-phenylhept-2-en-6-ynoate (12)36
Ethyl (2E)-7-phenylhept-2-en-6-ynoate (12) was synthesised as described for ethyl (2E)-hept-2-en-6-ynoate (10) using 5-phenylpent-4-yn-1-ol (5) (0.64 g, 3.96 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 17:3) gave ethyl (2E)-7-phenylhept-2-en-6-ynoate (12) (0.86 g, 95%) as a yellow oil. Spectroscopic data was consistent with the literature.36δH (400 MHz, CDCl3) 1.30 (3H, t, J 7.1 Hz, OCH2CH3), 2.47–2.61 (4H, m, 4-H2 and 5-H2), 4.20 (2H, q, J 7.1 Hz, OCH2CH3), 5.93 (1H, dt, J 15.7, 1.5 Hz, 2-H), 7.04 (1H, dt, J 15.7, 6.6 Hz, 3-H), 7.26–7.31 (3H, m, 3 × ArH), 7.36–7.41 (2H, m, 2 × ArH); δC (101 MHz, CDCl3) 14.3 (CH3), 18.4 (CH2), 31.4 (CH2), 60.2 (CH2), 81.7 (C), 88.3 (C), 122.5 (CH), 123.6 (C), 127.8 (CH), 128.2 (2 × CH), 131.6 (2 × CH), 146.6 (CH), 166.3 (C); m/z (CI) 229 (MH+. 100%), 155 (7), 113 (13), 81 (25), 69 (34).
Ethyl (2E)-7-(4′-nitrophenyl)hept-2-en-6-ynoate (13)
Ethyl (2E)-7-(4′-nitrophenyl)hept-2-en-6-ynoate (13) was synthesised as described for ethyl (2E)-hept-2-en-6-ynoate (10) using 5-(4′-nitrophenyl)pent-4-yn-1-ol (6) (0.46 g, 2.24 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 8:2) gave ethyl (2E)-7-(4′-nitrophenyl)hept-2-en-6-ynoate (13) (0.86 g, 95%) as a yellow solid. Mp 56–58 °C; νmax/cm−1 (neat) 2960 (CH), 1714 (CO), 1591 (CC), 1509, 1340, 1154, 854, 750; δH (400 MHz, CDCl3) 1.30 (3H, t, J 7.1 Hz, OCH2CH3), 2.50–2.57 (2H, m, 4-H2), 2.60–2.66 (2H, m, 5-H2), 4.21 (2H, q, J 7.1 Hz, OCH2CH3), 5.94 (1H, dt, J 15.7, 1.5 Hz, 2-H), 7.02 (1H, dt, J 15.7, 6.7 Hz, 3-H), 7.49–7.54 (2H, m, 2′-H and 6′-H), 8.14–8.18 (2H, m, 3′-H and 5′-H); δC (101 MHz, CDCl3) 14.3 (CH3), 18.6 (CH2), 30.9 (CH2), 60.4 (CH2), 80.3 (C), 94.3 (C), 122.8 (CH), 123.5 (2 × CH), 130.6 (C), 132.3 (2 × CH), 146.0 (CH), 146.8 (C), 166.3 (C); m/z (ESI) 296.0881 (MNa+. C15H15NNaO4 requires 296.0893).
Ethyl (2E)-7-(4′-methoxyphenyl)hept-2-en-6-ynoate (14)36
Ethyl (2E)-7-(4′-methoxyphenyl)hept-2-en-6-ynoate (14) was synthesised as described for ethyl (2E)-hept-2-en-6-ynoate (10) using 5-(4′-methoxyphenyl)pent-4-yn-1-ol (7) (0.55 g, 2.89 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 8:2) gave ethyl (2E)-7-(4′-methoxyphenyl)hept-2-en-6-ynoate (14) (0.70 g, 94%) as a yellow oil. Spectroscopic data was consistent with the literature.36δH (400 MHz, CDCl3) 1.29 (3H, t, J 7.1 Hz, OCH2CH3), 2.46–2.59 (4H, m, 4-H2and 5-H2), 3.80 (3H, s, OCH3), 4.20 (2H, q, J 7.1 Hz, OCH2CH3), 5.92 (1H, dt, J 15.7, 1.5 Hz, 2-H), 6.79–6.84 (2H, m, 3′-H and 5′-H), 7.03 (1H, dt, J 15.7, 6.5 Hz, 3-H), 7.30–7.35 (2H, m, 2′-H and 6′-H); δC (126 MHz, CDCl3) 14.3 (CH3), 18.4 (CH2), 31.5 (CH2), 55.2 (CH3), 60.3 (CH2), 81.4 (C), 86.7 (C), 113.8 (2 × CH), 115.7 (C), 122.4 (CH), 132.9 (2 × CH), 146.8 (CH), 159.2 (C), 166.4 (C); m/z (EI) 258 (M+. 22%), 230 (20), 185 (27), 145 (100), 130 (6), 102 (13), 83 (11).
(2E)-Hept-2-en-6-yn-1-ol (15)37
Ethyl (2E)-hept-2-en-6-ynoate (10) (2.28 g, 15.0 mmol) was dissolved in diethyl ether (80 mL) and cooled to −78 °C. DIBAL-H (1 M in hexane) (33.0 mL, 33.0 mmol) was added dropwise and the reaction mixture was stirred at −78 °C for 3 h, before warming to room temperature overnight. The solution was cooled to 0 °C and quenched by the addition of a saturated solution of Rochelle salt (50 mL) and warmed to room temperature with vigorous stirring for 1 h, producing a white precipitate that was filtered through a pad of Celite® and washed with diethyl ether (3 × 75 mL). The filtrate was then dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (petroleum ether/diethyl ether, 1:1) gave (2E)-hept-2-en-6-yn-1-ol (15) (1.44 g, 87%) as a pale yellow oil. Spectroscopic data was consistent with the literature.37δH (500 MHz, CDCl3) 1.42 (1H, br s, OH), 1.99 (1H, t, J 2.5 Hz, 7-H), 2.28–2.33 (4H, m, 4-H2 and 5-H2), 4.14 (2H, d, J 4.0 Hz, 1-H2), 5.70–5.81 (2H, m, 2-H and 3-H); δC (126 MHz, CDCl3) 18.5 (CH2), 31.1 (CH2), 63.5 (CH2), 68.8 (CH), 83.7 (C), 130.5 (CH), 130.6 (CH); m/z (CI) 111 (MH+. 3%), 107 (15), 93 (100), 81 (10), 69 (10).
(2E)-Oct-2-en-6-yn-1-ol (16)
(2E)-Oct-2-en-6-yn-1-ol (16) was synthesised as described for (2E)-hept-2-en-6-yn-1-ol (15) using ethyl (2E)-oct-2-en-6-ynoate (11) (0.56 g, 3.36 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 1:1) gave (2E)-oct-2-en-6-yn-1-ol (16) (0.36 g, 87%) as a colourless oil. νmax/cm−1 (neat) 3337 (OH), 2919 (CH), 1436, 1082, 1000, 968; δH (500 MHz, CDCl3) 1.24–1.29 (1H, m, OH), 1.78 (3H, t, J 2.4 Hz, 8-H3), 2.18–2.27 (4H, m, 4-H2 and 5-H2), 4.11 (2H, t, J 5.3 Hz, 1-H2), 5.66–5.79 (2H, m, 2-H and 3-H); δC (126 MHz, CDCl3) 3.4 (CH3), 18.8 (CH2), 31.7 (CH2), 63.5 (CH2), 76.0 (C), 78.5 (C), 130.1 (CH), 131.3 (CH); m/z (ESI) 147.0782 (MNa+. C8H12NaO requires 147.0780), 135 (13%), 91 (22).
(2E)-7-Phenylhept-2-en-6-yn-1-ol (17)15
(2E)-7-Phenylhept-2-en-6-yn-1-ol (17) was synthesised as described for (2E)-hept-2-en-6-yn-1-ol (15) using ethyl (2E)-7-phenylhept-2-en-6-ynoate (12) (0.67 g, 3.00 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 13:7) gave (2E)-7-phenylhept-2-en-6-yn-1-ol (17) (0.54 g, 98%) as a colourless oil. Spectroscopic data was consistent with the literature.15δH (400 MHz, CDCl3) 1.29 (1H, br s, OH), 2.33–2.40 (2H, m, 4-H2), 2.50 (2H, t, J 6.8 Hz, 5-H2), 4.13 (2H, br s, 1-H2), 5.72–5.87 (2H, m, 2-H and 3-H), 7.25–7.32 (3H, m, 3 × ArH), 7.36–7.42 (2H, m, 2 × ArH); δC (101 MHz, CDCl3) 19.5 (CH2), 31.5 (CH2), 63.5 (CH2), 81.2 (C), 89.4 (C), 123.9 (C), 127.7 (CH), 128.2 (2 × CH), 130.5 (CH), 130.9 (CH), 131.6 (2 × CH); m/z (EI) 186 (M+. 13%), 167 (12), 155 (11), 142 (16), 128 (9), 115 (100), 105 (10), 84 (14).
(2E)-7-(4′-Nitrophenyl)hept-2-en-6-yn-1-ol (18)
(2E)-7-(4′-Nitrophenyl)hept-2-en-6-yn-1-ol (18) was synthesised as described for (2E)-hept-2-en-6-yn-1-ol (15) using ethyl (2E)-7-(4′-nitrophenyl)hept-2-en-6-ynoate (13) (0.67 g, 2.45 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 6:4) gave (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.47 g, 83%) as a dark green solid. Mp 64–66 °C; νmax/cm−1 (neat) 3374 (OH), 2924 (CH), 1593 (CC), 1516, 1341, 855, 750; δH (400 MHz, CDCl3) 1.35 (1H, br s, OH), 2.35–2.42 (2H, m, 4-H2), 2.54 (2H, t, J 7.1 Hz, 5-H2), 4.14 (2H, d, J 3.2 Hz, 1-H2), 5.72–5.85 (2H, m, 2-H and 3-H), 7.49–7.54 (2H, m, 2′-H and 6′-H), 8.13–8.18 (2H, m, 3′-H and 5′-H); δC (101 MHz, CDCl3) 19.6 (CH2), 31.1 (CH2), 63.5 (CH2), 79.8 (C), 95.6 (C), 123.5 (2 × CH), 130.4 (CH), 130.8 (CH), 130.9 (C), 132.3 (2 × CH), 146.7 (C); m/z (ESI) 254.0784 (MNa+. C13H13NNaO3 requires 254.0788), 227 (9%), 199 (9).
(2E)-7-(4′-Methoxyphenyl)hept-2-en-6-yn-1-ol (19)
(2E)-7-(4-Methoxyphenyl)hept-2-en-6-yn-1-ol (19) was synthesised as described for (2E)-hept-2-en-6-yn-1-ol (15) using ethyl (2E)-7-(4′-methoxyphenyl)hept-2-en-6-ynoate (14) (0.44 g, 1.68 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 1:1) gave (2E)-7-(4′-methoxyphenyl)hept-2-en-6-yn-1-ol (19) (0.34 g, 94%) as a yellow oil. νmax/cm−1 (neat) 3368 (OH), 2916 (CH), 1607 (CC), 1508, 1244, 831; δH (400 MHz, CDCl3) 1.43 (1H, br s, OH), 2.30–2.41 (2H, m, 4-H2), 2.47 (2H, t, J 7.1 Hz, 5-H2), 3.79 (3H, s, OCH3), 4.12 (2H, d, J 4.5 Hz, 1-H2), 5.69–5.85 (2H, m, 2-H and 3-H), 6.78–6.83 (2H, m, 3′-H and 5′-H), 7.29–7.34 (2H, m, 2′-H and 6′-H); δC (101 MHz, CDCl3) 19.5 (CH2), 31.6 (CH2), 55.2 (CH3), 63.2 (CH2), 80.9 (C), 87.9 (C), 113.9 (2 × CH), 116.0 (C), 130.4 (CH), 130.7 (CH), 132.9 (2 × CH), 159.1 (C); m/z (EI) 216.1153 (M+. C14H16O2 requires 216.1150), 172 (17), 145 (100), 130 (7), 102 (15).
3-(2′,2′,2′-Trichloromethylcarbonylamino)hept-1-en-6-yne (20)15
(2E)-Hept-2-en-6-yn-1-ol (15) (0.11 g, 1.00 mmol) was dissolved in dichloromethane (20 mL) and cooled to 0 °C. To the solution was added 1,8-diazabicyclo[5.4.0]undec-7-ene (0.03 mL, 0.20 mmol) and trichloroacetonitrile (0.15 mL, 1.50 mmol). The reaction mixture was allowed to warm to room temperature before stirring for 3 h. The reaction mixture was filtered through a short pad of silica gel and the filtrate concentrated in vacuo to give the allylic trichloroacetimidate, which was used without further purification. The allylic trichloroacetimidate was dissolved in toluene (21 mL) under an argon atmosphere. Bis(acetonitrile)palladium chloride (0.026 g, 0.10 mmol) was then added to the solution and the reaction mixture was stirred at 40 °C for 24 h. To the reaction mixture, an additional portion of bis(acetonitrile)palladium chloride (0.026 g, 0.10 mmol) was added and the reaction was stirred at 40 °C for 24 h and the solvent was evaporated. Flash column chromatography using silica (petroleum ether/diethyl ether 10:1) gave 3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (20) (0.086 g, 34%) as a white solid. Mp 35–37 °C; spectroscopic data was consistent with the literature.15δH (500 MHz, CDCl3) 1.84–2.00 (2H, m, 4-H2), 2.05 (1H, t, J 2.7 Hz, 7-H), 2.26–2.39 (2H, m, 5-H2), 4.56–4.63 (1H, m, 3-H), 5.27 (1H, d, J 10.5 Hz, 1-HH), 5.30 (1H, d, J 17.2 Hz, 1-HH), 5.82 (1H, ddd, J 17.2, 10.5, 5.6 Hz, 2-H), 6.93 (1H, br s, NH); δC (126 MHz, CDCl3) 14.8 (CH2), 32.5 (CH2), 53.0 (CH), 69.9 (CH), 83.1 (C), 92.7 (C), 116.9 (CH2), 135.4 (CH), 161.3 (C); m/z (CI) 254 (MH+. 72%), 220 (55), 186 (42), 184 (37), 132 (12), 89 (100), 69 (27).
3-(2′,2′,2′-Trichloromethylcarbonylamino)oct-1-en-6-yne (21)
3-(2′,2′,2′-Trichloromethylcarbonylamino)oct-1-en-6-yne (21) was synthesised as described for compound 20, except using (2E)-oct-2-en-6-yn-1-ol (16) (0.04 g, 0.34 mmol) and a single portion of bis(acetonitrile)palladium chloride (0.009 g, 0.034 mmol). The reaction was performed at 20 °C for 24 h. Purification by flash column chromatography (petroleum ether/diethyl ether, 7:3) gave 3-(2′,2′,2′-trichloromethylcarbonylamino)oct-1-en-6-yne (21) (0.050 g, 55%) as a colourless oil. νmax/cm−1 (neat) 3331 (NH), 2920 (CH), 1694 (CO), 1516 (CC), 1441, 1250, 926, 822; δH (400 MHz, CDCl3) 1.73–1.95 (5H, m, 4-H2 and 8-H3), 2.20–2.30 (2H, m, 5-H2), 4.51–4.62 (1H, m, 3-H), 5.19–5.30 (2H, m, 1-H2), 5.79 (1H, ddd, J 17.2, 10.4, 5.4 Hz, 2-H), 7.14 (1H, d, J 5.4, NH); δC (101 MHz, CDCl3) 3.7 (CH3), 14.9 (CH2), 32.7 (CH2), 53.2 (CH), 77.3 (C), 78.0 (C), 92.8 (C), 116.4 (CH2), 135.6 (CH), 161.2 (C); m/z (ESI) 289.9865 (MNa+. C10H1235Cl3NNaO requires 289.9877).
7-Phenyl-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (22)
7-Phenyl-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (22) was synthesised as described for compound 20, except using (2E)-7-phenylhept-2-en-6-yn-1-ol (17) (0.08 g, 0.44 mmol) and a single portion of bis(acetonitrile)palladium chloride (0.012 g, 0.044 mmol). The reaction was performed at 20 °C for 12 h. Flash column chromatography using silica (petroleum ether/diethyl ether, 1:1) gave 7-phenyl-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (22) (0.12 g, 81%) as a colourless oil. νmax/cm−1 (neat) 3304 (NH), 2955 (CH), 2362, 1714 (CO), 1511, 1265, 1175; δH (400 MHz, CDCl3) 1.90–2.09 (2H, m, 4-H2), 2.48–2.61 (2H, m, 5-H2), 4.60–4.69 (1H, m, 3-H), 5.27 (1H, d, J 10.4 Hz, 1-HH), 5.32 (1H, d, J 17.2 Hz, 1-HH), 5.85 (1H, ddd, J 17.2, 10.4, 5.6 Hz, 2-H), 6.98 (1H, d, J 7.4 Hz, NH), 7.26–7.32 (3H, m, 3 × ArH), 7.37–7.44 (2H, m, 2 × ArH); δC (101 MHz, CDCl3) 15.9 (CH2), 32.8 (CH2), 53.2 (CH), 82.0 (C), 88.4 (C), 92.7 (C), 116.9 (CH2), 123.4 (C), 128.0 (CH), 128.3 (2 × CH), 131.7 (2 × CH), 135.6 (CH), 161.4 (C); m/z (ESI) 352.0019 (MNa+. C15H1435Cl3NNaO requires 352.0033).
7-(4′′-Nitrophenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (23)
7-(4′′-Nitrophenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (23) was synthesised as described for compound 20, except using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.06 g, 0.26 mmol) and a single portion of bis(acetonitrile)palladium chloride (0.008 g, 0.026 mmol). The reaction was performed at 20 °C for 12 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 8:2) gave 7-(4′′-nitrophenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (23) (0.07 g, 76%) as a yellow oil. νmax/cm−1 (neat) 3339 (NH), 2932 (CH), 1697 (CO), 1514 (CC), 1341, 1107, 852, 820; δH (400 MHz, CDCl3) 1.93–2.08 (2H, m, 4-H2), 2.50–2.64 (2H, m, 5-H2), 4.58–4.68 (1H, m, 3-H), 5.25–5.36 (2H, m, 1-H2), 5.85 (1H, ddd, J 17.2, 10.4, 5.7 Hz, 2-H), 6.80 (1H, d, J 7.9, NH), 7.50–7.55 (2H, m, 2′′-H and 6′′-H), 8.12–8.17 (2H, m, 3′′-H and 5′′-H); δC (101 MHz, CDCl3) 16.2 (CH2), 32.7 (CH2), 53.0 (CH), 80.4 (C), 92.7 (C), 94.3 (C), 117.2 (CH2), 123.5 (2 × CH), 130.5 (C), 132.4 (2 × CH), 135.5 (CH), 146.8 (C), 161.4 (C); m/z (ESI) 396.9873 (MNa+. C15H1335Cl3N2NaO3 requires 396.9884).
7-(4′′-Methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24)
7-(4′′-Methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) was synthesised as described for compound 20, except using (2E)-7-(4′-methoxyphenyl)hept-2-en-6-yn-1-ol (19) (0.11 g, 0.49 mmol) and a single portion of bis(acetonitrile)palladium chloride (0.014 g, 0.049 mmol). The reaction was performed at 20 °C for 12 h. Purification by flash column chromatography (petroleum ether/ethyl acetate, 8:2) gave 7-(4′′-methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) (0.15 g, 83%) as a colourless oil. νmax/cm−1 (neat) 3340 (NH), 2925 (CH), 1697 (CO), 1509 (CC), 1246, 1173, 831; δH (400 MHz, CDCl3) 1.89–2.08 (2H, m, 4-H2), 2.45–2.62 (2H, m, 5-H2), 3.80 (3H, s, OCH3), 4.59–4.69 (1H, m, 3-H), 5.24–5.35 (2H, m, 1-H2), 5.85 (1H, ddd, J 17.1, 10.4, 5.5 Hz, 2-H), 6.79–6.84 (2H, m, 3′′-H and 5′′-H), 7.02 (1H, d, J 8.0 Hz, NH), 7.30–7.36 (2H, m, 2′′-H and 6′′-H); δC (126 MHz, CDCl3) 15.8 (CH2), 32.8 (CH2), 53.2 (CH), 55.3 (CH3), 81.9 (C), 86.8 (C), 92.7 (C), 113.9 (2 × CH), 115.4 (C), 116.7 (CH2), 133.0 (2 × CH), 135.6 (CH), 159.3 (C), 161.4 (C); m/z (ESI) 382.0120 (MNa+. C16H1635Cl3NNaO2 requires 382.0139).
(3aS*,8R*,8aS*,8bR*)-2,5-Diphenyl-3a,4,6,7,8a,8b-hexahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (25)15
(2E)-7-Phenylhept-2-en-6-yn-1-ol (17) (0.12 g, 0.64 mmol) was dissolved in dichloromethane (16 mL) and cooled to 0 °C. To the solution, 1,8-diazabicyclo[5.4.0]undec-7-ene (0.02 mL, 0.013 mmol) and trichloroacetonitrile (0.10 mL, 0.966 mmol) were added. The reaction mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was filtered through a short pad of silica gel with diethyl ether (300 mL) and the filtrate concentrated in vacuo to give the allylic trichloroacetimidate, which was used without further purification. The allylic trichloroacetimidate was dissolved in toluene (16 mL) and bis(acetonitrile)palladium chloride (0.018 g, 0.064 mmol) was then added and the reaction mixture was stirred at room temperature for 12 h. Grubbs second generation catalyst (0.04 g, 0.05 mmol) was added with 1,7-octadiene (0.39 mL, 2.58 mmol) and the reaction mixture was stirred for 18 h at 90 °C. N-Phenyl maleimide (0.17 g, 0.96 mmol) was added with hydroquinone (0.008 g, 0.008 mmol). The reaction mixture was stirred for 18 h at 75 °C. The reaction mixture was then cooled and the solvent was evaporated. Flash column chromatography (petroleum ether/diethyl ether, 7:3) gave compound 25 (0.17 g, 51%) as a yellow solid. Mp 151–153 °C; spectroscopic data was consistent with the literature.15δH (400 MHz, CDCl3) 1.75 (1H, dq, J 12.3, 10.2 Hz, 7-HH), 2.10–2.20 (1H, m, 7-HH), 2.53–2.66 (3H, m, 4-HH and 6-H2), 3.12 (1H, dd, J 9.1, 5.8 Hz, 8a-H), 3.30 (1H, dd, J 15.2, 1.4 Hz, 4-HH), 3.46–3.56 (2H, m, 3a-H and 8b-H), 4.88–5.01 (1H, m, 8-H), 7.06–7.10 (2H, m, 2 × ArH), 7.23–7.47 (8H, m, 8 × ArH), 8.96 (1H, d, J 9.6 Hz, NH); δC (126 MHz, CDCl3) 28.4 (CH2), 29.9 (CH2), 31.6 (CH2), 40.3 (CH), 41.7 (CH), 43.7 (CH), 52.8 (CH), 92.9 (C), 126.5 (2 × CH), 127.2 (CH), 127.5 (2 × CH), 128.5 (2 × CH), 129.2 (CH), 129.4 (2 × CH), 130.3 (C), 131.4 (C), 139.0 (C), 139.6 (C), 162.3 (C), 178.5 (C), 179.7 (C); m/z (ESI) 525 (MNa+. 100%), 481 (18%), 454 (7), 413 (7), 345 (24), 323 (21), 297 (9), 236 (11), 218 (7).
(3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-Hexahydro-5-methyl-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (26)
(3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-Hexahydro-5-methyl-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (26) was synthesised as described for compound 25 using (2E)-oct-2-en-6-yn-1-ol (16) (0.05 g, 0.36 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 7:3) gave compound 26 (0.04 g, 25%) as a white solid. Mp 126–128 °C; νmax/cm−1 (neat) 3303 (NH), 2928 (CH), 1696 (CO), 1518 (CC), 1389, 1188, 736; δH (400 MHz, CDCl3) 1.69–1.81 (4H, m, 7-HH and 5-CH3), 2.09–2.22 (2H, m, 6-HH and 7-HH), 2.26–2.35 (1H, m, 4-HH), 2.45–2.55 (1H, m, 6-HH), 2.69 (1H, dd, J 14.8, 1.4 Hz, 4-HH), 2.84–2.91 (1H, m, 8a-H), 3.31 (1H, ddd, J 8.6, 7.1, 1.4 Hz, 3a-H), 3.38 (1H, dd, J 8.6, 6.2 Hz, 8b-H), 4.76–4.89 (1H, m, 8-H), 7.11–7.15 (2H, m, 2 × ArH), 7.38–7.50 (3H, m, 3 × ArH), 8.97 (1H, d, J 9.6 Hz, NH); δC (101 MHz, CDCl3) 19.3 (CH3), 26.1 (CH2), 31.6 (CH2), 31.9 (CH2), 39.9 (CH), 41.4 (CH), 42.5 (CH), 53.1 (CH), 92.9 (C), 126.2 (C), 126.4 (2 × CH), 129.1 (CH), 129.3 (2 × CH), 131.6 (C), 136.3 (C), 162.2 (C), 178.5 (C), 179.9 (C); m/z (ESI) 463.0350 (MNa+. C20H1935Cl3N2NaO3 requires 463.0353).
(9R*,9aS*)-2,6-Diphenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (27)
(9R*,9aS*)-2,6-Diphenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (27) was synthesised as described for compound 25 using (2E)-7-phenylhept-2-en-6-yn-1-ol (17) (0.08 g, 0.40 mmol) and 4-phenyl-1,2,4-triazole-3,5-dione (0.13 g, 0.72 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 1:1) gave compound 27 (0.08 g, 40%) as a dark yellow solid. Mp 176–178 °C; νmax/cm−1 (neat) 3405 (NH), 2925 (CH), 1714 (CO), 1704 (CO) 1503 (CC), 1420, 752; δH (400 MHz, CDCl3) 2.14–2.29 (2H, m, 8-H2), 2.44–2.54 (1H, m, 7-HH), 2.60–2.71 (1H, m, 7-HH), 4.40 (1H, ddd, J 16.6, 5.3, 2.3 Hz, 5-HH), 4.54 (1H, ddd, J 16.6, 5.3, 2.9 Hz, 5-HH), 4.57–4.61 (1H, m, 9a-H), 4.90–4.95 (1H, m, 9-H), 6.73 (1H, d, J 6.0 Hz, NH), 7.28–7.56 (10H, m, 10 × ArH); δC (101 MHz, CDCl3) 24.4 (CH2), 27.8 (CH2), 45.6 (CH2), 52.5 (CH), 59.9 (CH), 92.7 (C), 125.5 (2 × CH), 127.7 (2 × CH), 128.4 (CH), 128.7 (CH), 128.7 (C), 128.9 (2 × CH), 129.2 (2 × CH), 130.9 (C), 132.3 (C), 136.3 (C), 151.7 (C), 152.7 (C), 161.3 (C); m/z (ESI) 527.0395 (MNa+. C23H1935Cl3N4NaO3 requires 527.0415).
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-phenyl-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (28)
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-phenyl-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (28) was synthesised as described for compound 25 using (2E)-7-phenylhept-2-en-6-yn-1-ol (17) (0.06 g, 0.32 mmol) and tetracyanoethylene (0.25 g, 1.93 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 10:1) gave compound 28 (0.08 g, 56%) as a yellow solid. Mp 136–138 °C; νmax/cm−1 (neat) 3347 (NH), 2927 (CH), 1705 (CO), 1517 (CC), 1218, 823, 769; δH (400 MHz, CDCl3) 1.90–2.03 (1H, m, 2-HH), 2.33–2.47 (2H, m, 2-HH and 3-HH), 2.63–2.74 (1H, m, 3-HH), 3.31 (1H, dt, J 16.4, 1.7 Hz, 5-HH), 3.46–3.56 (2H, m, 5-HH and 7a-H), 4.41–4.53 (1H, m, 1-H), 6.92 (1H, J 7.6 Hz, NH), 7.17–7.22 (2H, m, 2 × ArH), 7.35–7.47 (3H, m, 3 × ArH); δC (126 MHz, CDCl3) 26.3 (CH2), 29.0 (CH2), 37.5 (CH2), 39.7 (C), 41.4 (C), 49.7 (CH), 54.9 (CH), 91.8 (C), 108.2 (C), 110.5 (C), 110.8 (C), 110.8 (C), 127.1 (2 × CH), 127.8 (C), 129.0 (CH), 129.2 (2 × CH), 131.4 (C), 136.4 (C), 162.5 (C); m/z (ESI) 480.0133 (MNa+. C21H1435Cl3N5NaO requires 480.0156).
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-phenyl-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (29)
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-phenyl-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (29) was synthesised as described for compound 25 using (2E)-7-phenylhept-2-en-6-yn-1-ol (17) (0.09 g, 0.48 mmol) and methyl acrylate (0.13 mL, 1.44 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 8:2) gave compound 29 (0.09 g, 46%) as a yellow oil; νmax/cm−1 (neat) 3414 (NH), 2952 (CH), 1712 (CO), 1511 (CC), 1200, 822, 757; δH (500 MHz, CDCl3) 1.65–1.77 (1H, m, 2-HH), 1.95–2.10 (2H, m, 2-HH and 6-HH) 2.22–2.38 (3H, m, 3-HH, 5-HH and 6-HH), 2.40–2.49 (1H, m, 3-HH), 2.50–2.62 (1H, m, 5-HH), 3.00 (1H, q, J 4.5 Hz, 7-H), 3.04–3.12 (1H, m, 7a-H), 3.72 (1H, s, OCH3), 4.63 (1H, qd, J 8.6, 5.0 Hz, 1-H), 7.16–7.26 (3H, m, 3 × ArH), 7.29–7.36 (2H, m, 2 × ArH), 7.68 (1H, d, J 8.6 Hz, NH); δC (126 MHz, CDCl3) 26.4 (CH2), 27.8 (CH2), 28.9 (CH2), 31.7 (CH2), 39.5 (CH), 43.9 (CH), 52.1 (CH), 53.2 (CH3), 92.9 (C), 126.6 (CH), 127.6 (2 × CH), 128.1 (2 × CH), 130.0 (C), 136.0 (C), 141.9 (C), 161.7 (C), 175.5 (C); m/z (ESI) 438.0381 (MNa+. C19H2035Cl3NNaO3 requires 438.0401).
(3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-Hexahydro-5-(4′′-nitrophenyl)-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (30)
(3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-Hexahydro-5-(4′′-nitrophenyl)-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (30) was synthesised as described for compound 25 using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.05 g, 0.20 mmol). Flash column chromatography (petroleum ether/ethyl acetate, 7:3) gave compound 30 (0.08 g, 69%) as a yellow solid. Mp 160–162 °C; νmax/cm−1 (neat) 3306 (NH), 2956 (CH), 1695 (CO), 1513 (CC), 1344, 1191, 821, 753; δH (400 MHz, CDCl3) 1.79 (1H, qd, J 12.4, 8.0 Hz, 7-HH), 2.16–2.26 (1H, m, 7-HH), 2.50–2.70 (3H, m, 4-HH and 6-H2), 3.17 (1H, dd, J 9.5, 6.1 Hz, 8a-H), 3.33 (1H, dd, J 14.8, 1.0 Hz, 4-HH), 3.53–3.59 (2H, m, 3a-H and 8b-H), 4.90–5.02 (1H, m, 8-H), 7.02–7.08 (2H, m, 2′′-H and 6′′-H), 7.37–7.49 (5H, m, 5 × ArH), 8.16–8.27 (2H, m, 3′′-H and 5′′-H), 8.93 (1H, d, J 9.5 Hz, NH); δC (126 MHz, CDCl3) 28.6 (CH2), 31.3 (CH2), 31.7 (CH2), 40.2 (CH), 41.4 (CH), 44.2 (CH), 52.6 (CH), 92.8 (C), 123.8 (2 × CH), 126.4 (2 × CH), 128.2 (2 × CH), 128.9 (C), 129.4 (CH), 129.5 (2 × CH), 131.2 (C), 143.7 (C), 145.4 (C), 146.7 (C), 162.4 (C), 178.3 (C), 179.3 (C); m/z (ESI) 570.0347 (MNa+. C25H2035Cl3N3NaO5 requires 570.0361).
(9R*,9aS*)-6-(4′′-Nitrophenyl)-2-phenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (31)
(9R*,9aS*)-6-(4′′-Nitrophenyl)-2-phenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (31) was synthesised as described for compound 25 using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.07 g, 0.30 mmol) and 4-phenyl-1,2,4-triazole-3,5-dione (0.06 g, 0.36 mmol). Purification by flash column chromatography (petroleum ether/ethyl acetate, 1:1) gave compound 31 (0.11 g, 63%) as a yellow solid. Mp 160–162 °C; νmax/cm−1 (neat) 3398 (NH), 2925 (CH), 1711 (CO), 1515 (CC), 1420, 1343, 854, 751; δH (400 MHz, CDCl3) 2.22–2.33 (2H, m, 8-H2), 2.45–2.56 (1H, m, 7-HH), 2.60–2.72 (1H, m, 7-HH), 4.41 (1H, dq, J 16.0, 4.0 Hz, 5-HH), 4.52 (1H, dq, J 16.0, 4.0 Hz, 5-HH), 4.57–4.62 (1H, m, 9a-H), 4.86–4.93 (1H, m, 9-H), 6.77 (1H, d, J 5.8 Hz, NH), 7.33–7.54 (7H, m, 7 × ArH), 8.24–8.31 (2H, m, 3′′-H and 5′′-H); δC (101 MHz, CDCl3) 24.9 (CH2), 27.6 (CH2), 45.3 (CH2), 52.6 (CH), 60.4 (CH), 92.6 (C), 124.2 (2 × CH), 125.5 (2 × CH), 126.5 (C), 128.6 (CH), 128.7 (2 × CH), 129.3 (2 × CH), 130.7 (C), 136.0 (C), 142.9 (C), 147.6 (C), 151.8 (C), 152.8 (C), 161.5 (C); m/z (ESI) 572.0239 (MNa+. C23H1835Cl3N5NaO5 requires 572.0266).
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-(4′′-nitrophenyl)-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (32)
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-(4′′-nitrophenyl)-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (32) was synthesised as described for compound 25 using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.07 g, 0.30 mmol) and tetracyanoethylene (0.23 g, 1.8 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 6:4) gave compound 32 (0.11 g, 73%) as a yellow solid. Mp 128–130 °C; νmax/cm−1 (neat) 3334 (NH), 2924 (CH), 1709 (CO), 1520 (CC), 1347, 1218, 855, 757; δH (400 MHz, CDCl3) 1.98–2.09 (1H, m, 2-HH), 2.31–2.46 (2H, m, 2-HH and 3-HH), 2.65–2.74 (1H, m, 3-HH), 3.29–3.37 (1H, m, 5-HH), 3.50–3.58 (2H, m, 5-HH and 7a-H), 4.43–4.52 (1H, m, 1-H), 6.95 (1H, d, J 8.3 Hz, NH), 7.39–7.44 (2H, m, 2′′-H and 6′′-H), 8.29–8.34 (2H, m, 3′′-H and 5′′-H); δC (101 MHz, CDCl3) 26.4 (CH2), 28.8 (CH2), 37.0 (CH2), 39.6 (C), 41.3 (C), 49.8 (CH), 54.8 (CH), 91.7 (C), 108.1 (C), 110.2 (C), 110.5 (C), 110.5 (C), 124.5 (2 × CH), 126.0 (C), 128.6 (2 × CH), 134.0 (C), 142.8 (C), 148.0 (C), 162.7 (C); m/z (ESI) 524.9983 (MNa+. C21H1335Cl3N6NaO3 requires 525.0007), 357 (21%), 303 (22), 289 (29), 253 (25), 235 (8).
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-(4′′-nitrophenyl)-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (33)
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-(4′′-nitrophenyl)-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (33) was synthesised as described for compound 25 using (2E)-7-(4′-nitrophenyl)hept-2-en-6-yn-1-ol (18) (0.07 g, 0.29 mmol) and methyl acrylate (0.08 mL, 0.87 mmol). Purification by flash column chromatography (petroleum ether/diethyl ether, 8:2) gave compound 33 (0.06 g, 46%) as a colourless oil; νmax/cm−1 (neat) 3389 (NH), 2930 (CH), 1710 (CO), 1514 (CC), 1344, 821, 752; δH (400 MHz, CDCl3) 1.71–1.84 (1H, m, 2-HH), 1.95–2.15 (2H, m, 2-HH and 3-HH), 2.24–2.61 (5H, m, 3-HH, 5-H2 and 6-H2), 3.04 (1H, q, J 4.2 Hz, 7-H), 3.09–3.15 (1H, m, 7a-H), 3.72 (1H, s, OCH3), 4.66 (1H, qd, J 9.0, 7.2 Hz, 1-H), 7.32–7.38 (2H, m, 2′′-H and 6′′-H), 7.58 (1H, d, J 9.0 Hz, NH), 8.16–8.22 (2H, m, 3′′-H and 5′′-H); δC (101 MHz, CDCl3) 26.5 (CH2), 27.3 (CH2), 29.4 (CH2), 31.8 (CH2), 39.2 (CH), 44.2 (CH), 52.2 (CH), 52.9 (CH3), 92.7 (C), 123.5 (2 × CH), 127.8 (C), 128.4 (2 × CH), 139.5 (C), 146.4 (C), 148.7 (C), 161.8 (C), 175.5 (C); m/z (EI) 460.0359 (M+. C19H1935Cl3N2O5 requires 460.0360), 299 (100%), 240 (71), 194 (16), 165 (12), 83 (17).
(3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-Hexahydro-5-(4′′-methoxyphenyl)-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (34)
7-(4′′-Methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) (0.04 g, 0.12 mmol) was dissolved in toluene (3 mL) and Grubbs second generation catalyst (0.007 g, 0.008 mmol) was added with 1,7-octadiene (0.07 mL, 0.48 mmol) and the reaction mixture was stirred for 20 h at 40 °C. N-Phenyl maleimide (0.03 g, 0.18 mmol) was added with hydroquinone (0.003 g, 0.003 mmol). The reaction mixture was stirred for 24 h at 75 °C. The reaction mixture was then cooled and the solvent was evaporated. Flash column chromatography (petroleum ether/diethyl ether, 8:2) gave (3aS*,8R*,8aS*,8bR*)-3a,4,6,7,8a,8b-hexahydro-5-(4′′-methoxyphenyl)-2-phenyl-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (34) (0.042 g, 65%) as a yellow solid. Mp 115–117 °C; νmax/cm−1 (neat) 3308 (NH), 2959 (CH), 1698 (CO), 1512 (CC), 1391, 1247, 823; δH (400 MHz, CDCl3) 1.66–1.81 (1H, m, 7-HH), 2.09–2.19 (1H, m, 7-HH), 2.52–2.62 (3H, m, 4-HH and 6-H2), 3.10 (1H, dd, J 8.9, 6.3 Hz, 8a-H), 3.27 (1H, dd, J 15.2, 1.2 Hz, 4-HH), 3.44–3.54 (2H, m, 3a-H and 8b-H), 3.81 (3H, s, OCH3), 4.87–4.99 (1H, m, 8-H), 6.85–6.92 (2H, m, 3′′-H and 5′′-H), 7.04–7.09 (2H, m, 2 × ArH), 7.17–7.23 (2H, m, 2′′-H and 6′′-H), 7.34–7.46 (3H, m, 3 × ArH), 8.97 (1H, d, J 9.6 Hz, NH); δC (101 MHz, CDCl3) 28.3 (CH2), 31.6 (CH2), 31.7 (CH2), 40.3 (CH), 41.7 (CH), 43.7 (CH), 52.9 (CH), 55.3 (CH3), 92.9 (C), 113.8 (2 × CH), 126.5 (2 × CH), 128.7 (2 × CH), 129.1 (CH), 129.4 (2 × CH), 129.8 (C), 131.4 (C), 131.5 (C), 138.1 (C), 158.7 (C), 162.3 (C), 178.6 (C), 179.8 (C); m/z (ESI) 555.0599 (MNa+. C26H2335Cl3N2NaO4 requires 555.0616).
(9R*,9aS*)-6-(4′′-Methoxyphenyl)-2-phenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (35)
(9R*,9aS*)-6-(4′′-Methoxyphenyl)-2-phenyl-7,8,9,9a-tetrahydro-9-(2′,2′,2′-trichloromethylcarbonylamino)-1H,5H-cyclopent[c][2,4,10]triazolo[1,2-a]pyridazine-1,3(2H)-dione (35) was synthesised as described for compound 34 using 7-(4′′-methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) (0.03 g, 0.09 mmol) and 4-phenyl-1,2,4-triazole-3,5-dione (0.02 g, 0.11 mmol). The Diels–Alder reaction was stirred for 24 h at 75 °C. Purification by flash column chromatography (petroleum ether/ethyl acetate, 6:4) gave compound 35 (0.04 g, 76%) as a dark yellow solid. Mp 154–156 °C; νmax/cm−1 (neat) 3406 (NH), 2932 (CH), 1774 (CO), 1715 (CO), 1703 (CO), 1510 (CC), 1420, 1250, 821, 734; δH (400 MHz, CDCl3) 2.12–2.30 (2H, m, 8-H2), 2.44–2.56 (1H, m, 7-HH), 2.61–2.71 (1H, m, 7-HH), 3.85 (3H, s, OCH3), 4.36 (1H, ddd, J 16.0, 4.8, 2.6 Hz, 5-HH), 4.49–4.61 (2H, m, 5-HH and 9a-H), 4.92 (1H, q, J 5.8 Hz, 9-H), 6.70 (1H, d, J 5.8 Hz, NH), 6.93–6.99 (2H, m, 3′′-H and 5′′-H), 7.22–7.56 (7H, m, 7 × ArH); δC (101 MHz, CDCl3) 24.5 (CH2), 27.9 (CH2), 45.5 (CH2), 52.3 (CH), 55.4 (CH3), 59.8 (CH), 92.6 (C), 114.3 (2 × CH), 125.5 (2 × CH), 128.2 (C), 128.4 (CH), 128.4 (C), 128.9 (2 × CH), 129.2 (2 × CH), 130.9 (C), 131.1 (C), 151.7 (C), 152.6 (C), 159.8 (C), 161.2 (C); m/z (ESI) 557.0510 (MNa+. C24H2135Cl3N4NaO4 requires 557.0521).
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-(4′′-methoxyphenyl)-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (36)
(1R*,7aR*)-2,3,5,6,7,7a-Hexahydro-4-(4′′-methoxyphenyl)-6,6,7,7-tetracyano-1-(2′,2′,2′-trichloromethylcarbonylamino)indene (36) was synthesised as described for compound 34 using 7-(4′′-methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) (0.04 g, 0.10 mmol) and tetracyanoethylene (0.08 g, 0.60 mmol). The Diels–Alder reaction was stirred for 24 h at 75 °C. Purification by flash column chromatography (petroleum ether/ethyl acetate, 6:4) gave compound 36 (0.03 g, 57%) as a yellow solid. Mp 116–118 °C; νmax/cm−1 (neat) 3370 (NH), 2935 (CH), 1711 (CO), 1513 (CC), 1248, 1178, 824; δH (400 MHz, CDCl3) 1.88–2.02 (1H, m, 2-HH), 2.33–2.47 (2H, m, 2-HH and 3-HH), 2.64–2.77 (1H, m, 3-HH), 3.24–3.33 (1H, m, 5-HH), 3.44–3.54 (2H, m, 5-HH and 7a-H), 3.84 (3H, s, OCH3), 4.39–4.51 (1H, m, 1-H), 6.91–6.97 (3H, m, 3′′-H, 5′′-H and NH), 7.11–7.15 (2H, m, 2′′-H and 6′′-H); δC (101 MHz, CDCl3) 26.4 (CH2), 29.1 (CH2), 37.6 (CH2), 39.7 (C), 41.4 (C), 50.0 (CH), 54.9 (CH), 55.4 (CH3), 91.7 (C), 108.2 (C), 110.4 (C), 110.7 (C), 110.8 (C), 114.5 (2 × CH), 127.5 (C), 128.4 (2 × CH), 128.6 (C), 130.5 (C), 159.9 (C), 162.5 (C); m/z (ESI) 510.0253 (MNa+. C22H1635Cl3N5NaO2 requires 510.0262).
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-(4′′-methoxyphenyl)-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (37)
Methyl (1R*,7S*,7aS*)-2,3,5,6,7,7a-hexahydro-4-(4′′-methoxyphenyl)-1-(2′,2′,2′-trichloromethylcarbonylamino)indene-7-carboxylate (37) was synthesised as described for compound 34 using 7-(4′′-methoxyphenyl)-3-(2′,2′,2′-trichloromethylcarbonylamino)hept-1-en-6-yne (24) (0.04 g, 0.11 mmol) and methyl acrylate (0.03 mL, 0.33 mmol). The Diels–Alder reaction was stirred for 5 days at 111 °C. Purification by flash column chromatography (petroleum ether/diethyl ether, 7:3) gave compound 37 (0.03 g, 59%) as a pale yellow oil; νmax/cm−1 (neat) 3350 (NH), 2935 (CH), 1712 (CO), 1608, 1510 (CC), 1246, 1175, 1035, 822, 737; δH (400 MHz, CDCl3) 1.64–1.76 (1H, m, 2-HH), 1.93–2.08 (2H, m, 2-HH, and 6-HH), 2.21–2.38 (3H, m, 3-HH, 5-HH and 6-HH), 2.40–2.59 (2H, m, 3-HH and 5-HH), 2.99 (1H, q, J 4.6 Hz, 7-H), 3.03–3.11 (1H, m, 7a-H), 3.71 (1H, s, OCH3), 3.81 (1H, s, OCH3), 4.63 (1H, qd, J 9.0, 6.9 Hz, 1-H), 6.84–6.89 (2H, m, 3′′-H and 5′′-H), 7.10–7.16 (2H, m, 2′′-H and 6′′-H), 7.68 (1H, d, J 9.0 Hz, NH); δC (101 MHz, CDCl3) 26.4 (CH2), 27.8 (CH2), 29.0 (CH2), 31.6 (CH2), 39.5 (CH), 43.9 (CH), 52.1 (CH), 53.2 (CH3), 55.3 (CH3), 92.9 (C), 113.4 (2 × CH), 128.7 (2 × CH), 129.4 (C), 134.3 (C), 135.1 (C), 158.3 (C), 161.7 (C), 175.5 (C); m/z (ESI) 468.0486 (MNa+. C20H2235Cl3NNaO4 requires 468.0507).
(3aS*,8R*,8aS*,8bR*)-2,5-Diphenyl-3a,4,6,7,8a,8b-hexahydro-8-(2′,2′-dichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (38)
To a solution of (3aS*,8R*,8aS*,8bR*)-2,5-diphenyl-3a,4,6,7,8a,8b-hexahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (25) (0.08 g, 0.16 mmol) in ethyl acetate (4 mL) was added 10% palladium on charcoal (0.02 g). The mixture was stirred under an atmosphere of hydrogen at room temperature for 48 h. The reaction mixture was filtered through a short pad of Celite® with diethyl ether (80 mL) and the solvent was evaporated. Purification by flash column chromatography (petroleum ether/ethyl acetate, 7:3) gave compound 38 (0.04 g, 48%) as a white solid. Mp 138–140 °C; νmax/cm−1 (neat) 3323 (NH), 2922 (CH), 1690 (CO), 1524 (CC), 1497, 1389, 1196, 808, 734; δH (400 MHz, CDCl3) 1.66–1.80 (1H, m, 7-HH), 2.05–2.15 (1H, m, 7-HH), 2.52–2.64 (3H, m, 4-HH and 6-H2), 3.07 (1H, dd, J 8.7, 6.4 Hz, 8a-H), 3.29 (1H, dd, J 15.3, 2.9 Hz, 4-HH), 3.44–3.54 (2H, m, 3a-H and 8b-H), 4.89–5.01 (1H, m, 8-H), 5.99 (1H, s, CHCl2), 7.06–7.12 (2H, m, 2 × ArH), 7.22–7.53 (8H, m, 8 × ArH), 8.66 (1H, d, J 9.7 Hz, NH); δC (126 MHz, CDCl3) 28.4 (CH2), 31.7 (2 × CH2), 40.3 (CH), 41.7 (CH), 43.7 (CH), 51.6 (CH), 66.7 (CH), 126.6 (2 × CH), 127.2 (CH), 127.5 (2 × CH), 128.4 (2 × CH), 129.1 (CH), 129.4 (2 × CH), 130.1 (C), 131.5 (C), 139.0 (C), 139.9 (C), 164.6 (C), 178.6 (C), 179.7 (C); m/z (EI) 468.1012 (M+. C25H2235Cl2N2O3 requires 468.1007), 341 (100%), 194 (71), 167 (34), 152 (11), 77 (11).
(3aS*,5R*,5aR*,8R*,8aR*,8bR*)-5,5a-Dihydroxy-2,5-diphenyl-3a,4,5,5a,6,7,8a,8b-octahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (39)
(3aS*,8R*,8aS*,8bR*)-2,5-Diphenyl-3a,4,6,7,8a,8b-hexahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (25) (0.04 g, 0.08 mmol) was dissolved in dichloromethane (2 mL) at −78 °C. Tetramethylethylenediamine (0.013 mL, 0.09 mmol) was added and the reaction mixture stirred for 0.1 h, before the addition of osmium tetroxide (0.02 g, 0.09 mmol). The dark coloured solution was stirred for 1 h at −78 °C before warming to room temperature and then stirred for 2 h. The solvent was removed in vacuo and the dark coloured solid was dissolved in methanol (2 mL). 12 M Hydrochloric acid (0.5 mL) was added and the mixture stirred for a further 2 h. The solvent was removed in vacuo to afford a dark solid. Flash column chromatography (petroleum ether/ethyl acetate, 4:1) gave compound 39 (0.04 g, 100%) as a white solid. Mp 164–166 °C; νmax/cm–1 (neat) 3475 (NH/OH), 2931 (CH), 1698 (CO), 1500 (CC), 1380, 1216, 818, 753; δH (400 MHz, CD3OD) 1.13–1.23 (1H, m, 6-HH), 1.86–2.06 (2H, m, 6-HH and 7-HH), 2.14–2.30 (2H, m, 4-HH and 7-HH), 2.58 (1H, dd, J 14.3, 12.5 Hz, 4-HH), 2.87–2.93 (1H, m, 8a-H), 3.41–3.49 (1H, m, 3a-H), 3.59 (1H, t, J 7.6 Hz, 8b-H), 5.10 (1H, dt, J 10.8, 7.6 Hz, 8-H), 7.21–7.27 (1H, m, ArH), 7.29–7.38 (4H, m, 4 × ArH), 7.40–7.46 (1H, m, ArH), 7.47–7.58 (4H, m, 4 × ArH); δC (126 MHz, CD3OD) 27.8 (CH2), 30.6 (CH2), 36.2 (CH2), 39.9 (CH), 41.6 (CH), 46.0 (CH), 52.0 (CH), 74.5 (C), 83.7 (C), 92.7 (C), 126.4 (2 × CH), 126.8 (CH), 126.8 (2 × CH), 127.3 (2 × CH), 128.4 (CH), 128.7 (2 × CH), 132.0 (C), 143.5 (C), 162.0 (C), 178.8 (C), 178.8 (C); m/z (ESI) 559.0564 (MNa+. C25H2335Cl3N2NaO5 requires 559.0565).
(3aS*,5R*,5aR*,8R*,8aR*,8bR*)-5,5a-Epoxy-2,5-diphenyl-3a,4,5,5a,6,7,8a,8b-octahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (40)
3-Chloroperbenzoic acid (0.03 g, 0.18 mmol) was added to a stirred solution of (3aS*,8R*,8aS*,8bR*)-2,5-diphenyl-3a,4,6,7,8a,8b-hexahydro-8-(2′,2′,2′-trichloromethylcarbonylamino)cyclopent[e]isoindole-1,3(2H,3aH)-dione (25) (0.02 g, 0.044 mmol) in dichloromethane (2 mL) at 0 °C. The reaction mixture was warmed from 0 °C to room temperature over 18 h then cooled to 0 °C before 3-chloroperbenzoic acid (0.03 g, 0.18 mmol) was added. The reaction mixture was stirred for a further 24 h at room temperature, quenched by the addition of a saturated solution of sodium sulfite (3 mL) and extracted with dichloromethane (2 × 3 mL). The combined organic layers were washed with a saturated solution of sodium hydrogen carbonate (3 × 6 mL), water (6 mL), brine (6 mL), then dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (petroleum ether/diethyl ether, 1:1) gave compound 40 (0.02 g, 65%) as a white solid. Mp 142–144 °C; νmax/cm−1 (neat) 3340 (NH), 2921 (CH), 1701 (CO), 1514 (CC), 1390, 1192, 821, 756; δH (400 MHz, CDCl3) 1.58–1.71 (1H, m, 6-HH), 1.85–1.99 (2H, m, 6-HH and 7-HH), 2.23–2.33 (2H, m, 4-HH and 7-HH), 2.75 (1H, dd, J 9.0, 7.1 Hz, 8a-H), 3.22 (1H, dd, J 15.9, 1.3, 4-HH), 3.40 (1H, dd, J 9.3, 7.1, 8b-H), 3.49 (1H, td, J 9.3, 1.3 Hz, 3a-H), 5.02–5.13 (1H, m, 8-H), 7.11–7.15 (2H, m, 2 × ArH), 7.21–7.37 (5H, m, 5 × ArH), 7.42–7.55 (3H, m, 3 × ArH), 8.52 (1H, d, J 9.3 Hz, NH); δC (101 MHz, CDCl3) 25.1 (CH2), 29.5 (CH2), 33.0 (CH2), 38.3 (CH), 40.0 (CH), 45.5 (CH), 52.0 (CH), 61.6 (C), 71.8 (C), 92.7 (C), 126.0 (2 × CH), 126.5 (2 × CH), 128.1 (CH), 128.5 (2 × CH), 129.4 (CH), 129.5 (2 × CH), 131.2 (C), 136.9 (C), 162.0 (C), 178.5 (C), 178.9 (C); m/z (ESI) 541.0450 (MNa+. C25H2135Cl3N2NaO4 requires 541.0459).
X-ray procedure for 39
Single crystal diffraction data for 39 were collected by the EPSRC UK National Crystallography Service.38 Data reduction was carried out using Crysalis PRO (Agilent Technologies, 2014). The structure was solved by charge-flipping methods using SuperFlip39 and refined against F2 using full-matrix least-squares refinement using SHELX201440 within OLEX2.41 Positional and anisotropic atomic displacement parameters (adps) were refined for all non-hydrogen atoms. Hydrogen atoms bound to carbon and nitrogen atoms were placed at calculated positions and refined as part of a riding model except for the MeOH methyl hydrogen and all hydroxyl hydrogen atoms which located in difference Fourier maps and were refined as a rigid rotor. There are two independent molecules of both the compound and of MeOH in the asymmetric unit although the conformation of the two molecules is essentially the same.Acknowledgements
Financial support from the Ministry of Higher Education and Scientific Research, Omar Al-Mukhtar University, Libya (studentship to M. A. B. M.) and the University of Glasgow (University Scholarship to M. W. G.) is gratefully acknowledged. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data.Notes and references
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) T. J. Ritchie, S. J. F. MacDonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS PubMed; (c) A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799 CrossRef CAS PubMed.
- M. Dow, M. Fisher, T. James, F. Marchetti and A. Nelson, Org. Biomol. Chem., 2012, 10, 17 CAS.
- S. V. Bhat, B. A. Nagasampagi and M. Sivakumar, in Chemistry of Natural Products, Springer, Berlin, Germany, 2005 Search PubMed.
- (a) H. Sorek, A. Rudi, S. Gueta, F. Reyes, M. J. Martin, M. Aknin, E. Gaydou and J. Vacelet, Tetrahedron, 2006, 62, 8838 CrossRef CAS; (b) E. Gros, A. Al-Mourabit, M.-T. Martin, J. Sorres, J. Vacelet, M. Frederich, M. Aknin, Y. Kashman and A. Gauvin-Bialecki, J. Nat. Prod., 2014, 77, 818 CrossRef CAS PubMed.
- A. E. Walts and W. R. Roush, Tetrahedron, 1985, 41, 3463 CrossRef CAS and references therein.
- For recent developments, see: H. Yu, I. J. Kim, J. E. Folk, X. Tian, R. B. Rothman, M. H. Baumann, C. M. Dersch, J. L. Flippen-Anderson, D. Parrish, A. E. Jacobsen and K. C. Rice, J. Med. Chem., 2004, 47, 2624 CrossRef CAS PubMed.
- (a) A. Graul and J. Castaner, Drugs Future, 1998, 23, 903 Search PubMed; (b) J. Sterling, A. Veinberg, D. Lerner, W. Goldenberg, R. Levy, M. Youdim and J. Finberg, J. Neural Transm., Suppl., 1998, 52, 301 CAS.
- Z. Kádár, J. Molnár, G. Schneider, I. Zupkó and É. Frank, Bioorg. Med. Chem., 2012, 20, 1396 CrossRef PubMed.
- J. P. Vacca, B. D. Dorsey, W. A. Schleif, R. B. Levin, S. L. McDaniel, P. L. Darke, J. Zugay, J. C. Quintero, O. M. Blahy, E. Roth, V. V. Sardana, A. J. Schlabach, P. I. Graharm, J. H. Condra, L. Gotlib, M. K. Holloway, J. Lin, I.-W. Chen, K. Vastag, D. Ostovic, P. S. Anderson, E. A. Emini and J. R. Huff, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4096 CrossRef CAS.
- K. M. Brummond, D. Chen, T. O. Painter, S. Mao and D. D. Seifried, Synlett, 2008, 759 CrossRef CAS.
- F. Lhermitte and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 2459 CrossRef.
- For examples of amino-substituted indane synthesis, see: (a) C. J. Roxburgh, Synthesis, 1996, 307 CrossRef CAS; (b) X.-H. Gu, H. Yu, A. E. Jacobsen, R. B. Rothman, C. M. Dersch, C. George, J. F. Flippen-Anderson and K. C. Rice, J. Med. Chem., 2000, 43, 4868 CrossRef CAS PubMed; (c) M. Froimowitz, K.-M. Wu, A. Moussa, R. M. Haidar, J. Jurayj, C. George and E. L. Gardner, J. Med. Chem., 2000, 43, 4981 CrossRef CAS PubMed; (d) S. H. Lee, I. S. Kim, Q. R. Li, G. R. Dong, L. S. Jeong and Y. H. Jung, J. Org. Chem., 2011, 76, 10011 CrossRef CAS PubMed; (e) S. H. Lee, S. J. Park, I. S. Kim and Y. H. Jung, Tetrahedron, 2013, 69, 1877 CrossRef CAS; (f) D. R. White, J. T. Hutt and J. P. Wolfe, J. Am. Chem. Soc., 2015, 137, 11246 CrossRef CAS PubMed.
- M. W. Grafton, L. J. Farrugia and A. Sutherland, J. Org. Chem., 2013, 78, 7199 CrossRef CAS PubMed.
- M. W. Grafton, L. J. Farrugia, H. M. Senn and A. Sutherland, Chem. Commun., 2012, 48, 7994 RSC.
- M. W. Grafton, S. A. Johnston, L. J. Farrugia and A. Sutherland, Tetrahedron, 2014, 70, 7133 CrossRef CAS.
- F. I. McGonagle, L. Brown, A. Cooke and A. Sutherland, Org. Biomol. Chem., 2010, 8, 3418 CAS.
- K. R. Roesch and R. C. Larock, J. Org. Chem., 2001, 66, 412 CrossRef CAS PubMed.
- S. Hötling, B. Haberlag, M. Tamm, J. Collatz, P. Mack, J. L. M. Steidle, M. Vences and S. Schulz, Chem. – Eur. J., 2014, 20, 3183 CrossRef PubMed.
- R. E. Ireland and D. W. Norbeck, J. Org. Chem., 1985, 50, 2198 CrossRef CAS.
- M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183 CrossRef CAS.
- C. E. Anderson, L. E. Overman and M. P. Watson, Org. Synth., 2005, 82, 134 CrossRef CAS.
- L. E. Overman and N. E. Carpenter, in Organic Reactions, ed. L. E. Overman, Wiley, Hoboken, NJ, 2005, vol. 66, pp. 1–107 and references therein Search PubMed.
- (a) C. S. Poulsen and R. Madsen, Synthesis, 2003, 1 CAS; (b) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317 CrossRef CAS PubMed; (c) H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55 RSC.
- (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed; (b) M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543 CrossRef CAS PubMed.
- S. Fustero, P. Bello, J. Miró, A. Simón and C. del Pozo, Chem. – Eur. J., 2012, 18, 10991 CrossRef CAS PubMed.
- A preliminary study describing the synthesis of 25 has been previously published. See ref. 15 for details.
- See ESI† for NOE experiments for all aminobicyclo[4.3.0]nonanes.
- For example, see: (a) A. Fürstner, L. Ackermann, B. Gabor, R. Goddard, C. W. Lehmann, R. Mynott, F. Stelzer and O. R. Thiel, Chem. – Eur. J., 2001, 7, 3236 CrossRef; (b) B. M. Trost, A. C. Gutierrez and E. M. Ferreira, J. Am. Chem. Soc., 2010, 132, 9206 CrossRef CAS PubMed.
- (a) T. J. Donohoe, K. Blades, M. Helliwell, P. R. Moore and J. J. G. Winter, J. Org. Chem., 1999, 64, 2980 CrossRef CAS PubMed; (b) K. Blades, T. J. Donohoe, J. J. G. Winter and G. Stemp, Tetrahedron Lett., 2000, 41, 4701 CrossRef CAS; (c) T. J. Donohoe, K. Blades, P. R. Moore, M. J. Waring, J. J. G. Winter, M. Helliwell, N. J. Newcombe and G. Stemp, J. Org. Chem., 2002, 67, 7946 CrossRef CAS PubMed; (d) T. J. Donohoe, Synlett, 2002, 1223 CrossRef CAS.
- Crystal data for 39. C25H23Cl3N2O5·CH4O, Mr = 569.84, Triclinic, a = 11.7084 (4) Å, b = 14.1127 (6) Å, c = 15.5928 (6) Å, α = 90.096 (3)°, β = 95.408 (3)°, γ = 91.971 (3)°, V = 2563.48 (17) Å3, T = 100 K, space group P (no. 2), Z = 4, 34139 reflections measured, 9008 unique (Rint = 0.073), which were used in all calculations. The final R1 = 0.054 for 6437 observed data [F2 > 2s(F2)] and wR2(F2) = 0.136 (all data).
- Crystallographic data for compound 39 in this paper have been deposited with the Cambridge Crystallographic Data Centre with code CCDC 1429493.
- K. Fuji, T. Morimoto, K. Tsutsumi and K. Kakiuchi, Chem. Commun., 2005, 3295 RSC.
- K. M. Gericke, D. I. Chai and M. Lautens, Tetrahedron, 2008, 64, 6002 CrossRef CAS.
- H.-Y. Hsieh, W.-C. Lee, G. C. Senadi, W.-P. Hu, J.-J. Liang, T.-R. Tsai, Y.-W. Chou, K.-K. Kuo, C.-Y. Chen and J.-J. Wang, J. Med. Chem., 2013, 56, 5422 CrossRef CAS PubMed.
- A. B. Smith, J. W. Leahy, I. Noda, S. W. Remiszewski, N. J. Liverton and R. Zibuck, J. Am. Chem. Soc., 1992, 114, 2995 CrossRef CAS.
- M. Nishida, N. Adachi, K. Onozuka, H. Matsumura and M. Mori, J. Org. Chem., 1998, 63, 9158 CrossRef CAS.
- A. Tsimelzon and R. Braslau, J. Org. Chem., 2005, 70, 10854 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea and J. A. K. Howard, J. Appl. Crystallogr., 2009, 42, 336 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NOE data for compounds 25–37 and, 1H and 13C NMR spectra of all new compounds. CCDC 1429493. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob00165c |
| This journal is © The Royal Society of Chemistry 2016 |