
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective chemical labeling of proteins
Xi
Chen
ab and
Yao-Wen
Wu
*ab
aChemical Genomics Centre of the Max Planck Society, Otto-Hahn-Str. 15, 44227 Dortmund, Germany
bMax-Planck Institute for Molecular Physiology, Otto-Hahn-Str. 11, 44227 Dortmund, Germany. E-mail: yaowen.wu@mpi-dortmund.mpg.de
First published on 23rd February 2016
Abstract
Over the years, there have been remarkable efforts in the development of selective protein labeling strategies. In this review, we deliver a comprehensive overview of the currently available bioorthogonal and chemoselective reactions. The ability to introduce bioorthogonal handles to proteins is essential to carry out bioorthogonal reactions for protein labeling in living systems. We therefore summarize the techniques that allow for site-specific “installation” of bioorthogonal handles into proteins. We also highlight the biological applications that have been achieved by selective chemical labeling of proteins.
 Xi Chen | Dr. Xi Chen was born in Sicuan, China in 1986. He received his MSc in Chemistry at the National University of Singapore and PhD degree (2012) in Protein Chemistry at Ulm University. Since 2012, he conducted his postdoctoral research under the supervision of Dr. Yao-Wen Wu at the Max-Planck Institute for Molecular Physiology and the Chemical Genomics Centre of the Max-Planck Society. His research focuses on chemical protein labeling and the development of photochemical approaches for controlling protein function in live cells. |
 Yao-Wen Wu | Dr. Yao-Wen Wu was born in Jiangxi, China in 1980. He received his BSc in Chemistry from Sun Yat-sen University in 2001 and his MSc in Organic Chemistry from Tsinghua University in 2004. After obtaining Dr rer. nat. (summa cum laude) in 2008 at TU Dortmund working at the Max Planck Institute of Molecular Physiology in Dortmund, he then conducted his postdoctoral research in cell biology at King's College London. Since 2010 he has been leader of an Otto Hahn group at the Max Planck Institute in Dortmund. Since 2012 he has been group leader of the Chemical Genomics Centre of the Max Planck Society. His research interests include molecular mechanism of membrane trafficking and autophagy, protein chemical modification, chemical probes for visualizing cellular events, chemical and optogenetics. |
1. Introduction
Selective chemical labeling of proteins is highly useful for probing and controlling protein functions in vitro and in living systems.1–10 Since proteins carry many copies of different functional groups, it is demanding to selectively label a protein at a defined site. Additionally, the labeling conditions have to be mild and reactions should be able to undergo in aqueous solution. Moreover, the labeling reagents should also be orthogonal to other functionalities in living systems when a protein is to be labeled in vivo.11,12 These restrictions make it really challenging to label proteins in a chemoselective and site-specific manner.Recent years have seen tremendous progress in chemical protein labeling. There are already some excellent reviews on bioorthogonal reactions and selective chemical labeling of proteins.2–4,6,12–18 In this review, we compare the features of reported bioorthogonal reactions and chemical tagging approaches, e.g. the reaction rate or time, reaction conditions, reagents, etc. This could be helpful to identify the most suitable labeling reaction for a particular application. Moreover, we highlight the applications of established chemical labeling techniques to tackle biological problems.
2. The toolbox of bioorthogonal chemistry for protein labeling
Bioorthogonal reactions are robust and invaluable tools for chemical protein labeling.4,6 Typically, selective protein labeling is accomplished by incorporation of bioorthogonal groups into a protein, followed by chemoselective modifications.18 This approach is also designated as “tag-and-modify”.19 A variety of bioorthogonal reactions have been developed, which can be classified into: (1) condensation reactions through carbonyls, (2) “click” reactions through azides, (3) inverse electron-demand Diels–Alder cycloadditions (DAINV) and other cycloaddition reactions, (4) transition metal-catalyzed coupling and decaging reactions, and (5) labeling reactions at cysteine residues (Table 1). In parallel, many elegant approaches have been established to selectively equip proteins with bioorthogonal handles (discussed in later sections), fulfilling the requirements for the subsequent bioorthogonal modification.| Reactant A | Reactant B | Product | k (M−1 s−1) or t | Features | Reference(s) |
|---|---|---|---|---|---|
| t: reaction time. (A) RT: room temperature; ABAO: aminobenzamidoxime; KAHA: α-ketoacid-hydroxylamine; ACL: aldehyde capture ligation. (B) PBS: phosphate buffered saline; TBTA: tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine; CuAAC: copper-catalyzed azide alkyne cycloaddition; SPAAC: strain-promoted azide alkyne cycloaddition; BCN: bicyclononyne. (C) TCO: trans-cyclooctene; ED: electron-donating. (D) TQ-ligation: thiovinyl ether o-quinolinone quinone methide ligation; oQQM: o-quinolinone quinone methide; EW: electron-withdrawing. (E) BIAN: bis-imine of acenaphthenequinone and mesitylamine; dba: dibenzylidene acetone. (F) TCEP: tris(2-carboxyethyl)phosphine; Dha: dehydroalanine; MSH: O-mesitylenesulfonylhydroxylamine; CBT: cyanobenzothiazole; NCL: native chemical ligation; MESNA: 2-mercaptoethanesulfonate; MPAA: 4-mercaptophenylacetic acid. | |||||
| A: Condensation through carbonyls | |||||
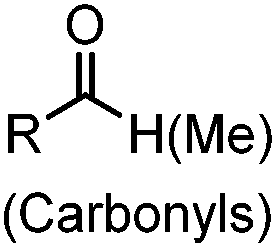
|
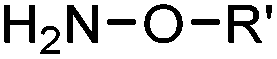
|
|
0.006 (pH 7, RT) | Oxime ligation: p-phenylenediamine (p-PDA) or aniline, etc. as a catalyst | Dirksen (2006)20 |
| 0.29 (pH 7, RT, 10 mM p-PDA) | Wendeler (2014)21 | ||||
|
|
PBS (pH 6.5, RT) 0.0087, or 0.46 (1 mM 5MA) | Hydrazone ligation: aniline or 5-methoxyanthranilic acid (5MA), etc. as a catalyst; ∼85% yield in 5 h | Crisalli (2013)22 | |
|
|
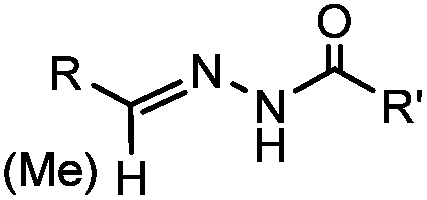
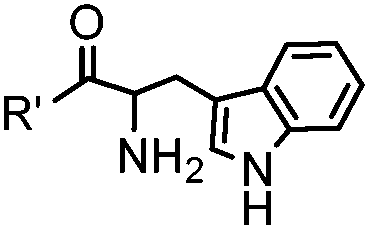
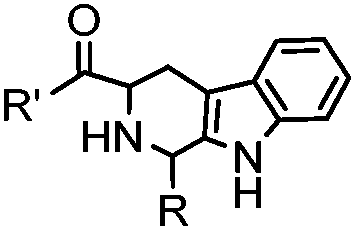
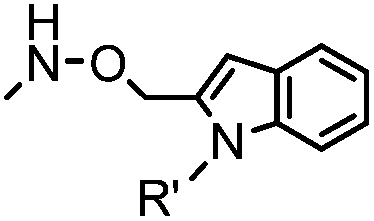
18 h (pH 6.5, 37 °C) | Pictet-Spengler reaction: formation of a stable C–C bond; suitable for N-terminal protein labeling | Sasaki (2008)23 | |
|
|
10 (pD 4.5) | Pictet-Spengler ligation: ligation is faster under lower pH conditions; formation of a stable C–C bond | Agarwal (2013)24 | |
| 2 (pD 6) | |||||
|
|
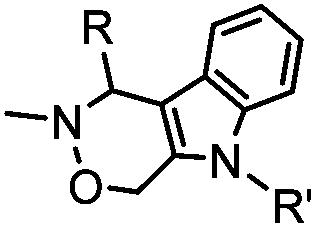
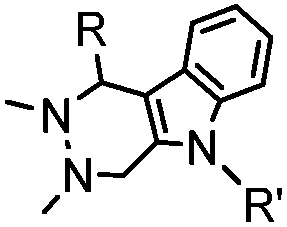
4.17 (pH 6) | Hydrazino-Pictet-Spengler ligation: ligation proceeds fastest under pH 6; formation of a stable C–C bond | Agarwal (2013)25 | |
|
|
40 (pH 4.5) | ABAO ligation: formation of a fluorescent dihydroquinazoline (λmax 490 nm); quantitative labeling in 1 h | Kitov (2014)26 | |
|
|
1.61 (pH 7.4, RT, 10 μM CuSO4, air) | Under air; requires Cu(II) or Zn(II) catalyst; quantitative labeling in 2 h | Ji (2014)27 | |
|
|
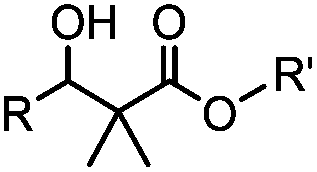
24 h (pH 7, RT) | Mukaiyama-Adol condensation: 11–55% conversion; stable | Alam (2010)28 | |
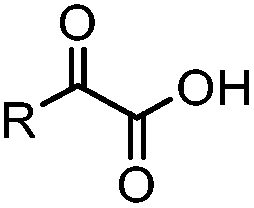
|
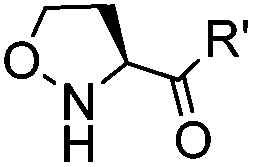
|
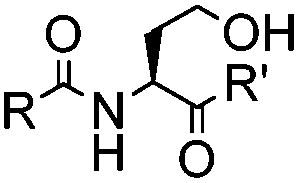
|
4 h (≥10% H2O in DMSO, 60 °C) | KAHA ligation: formation of a native amide bond; 56–76% yield | Bode (2006)29 |
| Pattabiraman (2012)30 | |||||
|
|
|
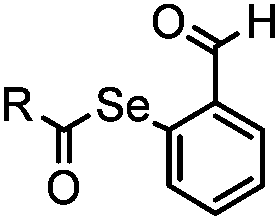
1.65 (DMF, RT); 40 h [10% DMF in PBS (pH 7)] | ACL: selective N-terminal protein labeling is possible at pH 7; ∼70% labeling of ubiquitin | Raj (2015)31 |
| B: Click reactions through azides | |||||
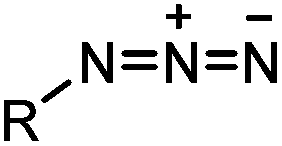
|
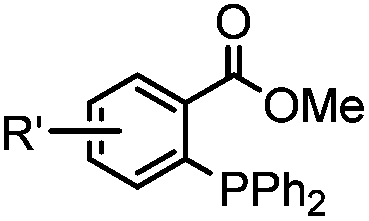
|
|
0.003 (PBS) | Staudinger-Bertozzi ligation: phosphine is prone to oxidation | Saxon (2000)32 |
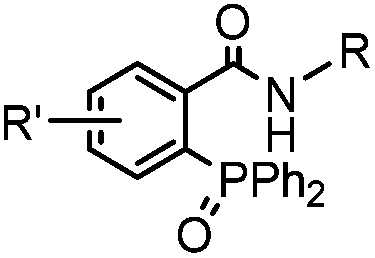
|
|
0.0074 (H2O) | Traceless Staudinger ligation: needs transient protection of phosphine by BH3; 80% yield at pH ≥ 7.5 | Nilsson (2001)33 | |
| Tam (2007)34 | |||||
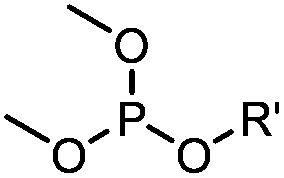
|
|
6–24 h (H2O, RT) | Staudinger-phosphite ligation: phosphite is stable against air oxidation; up to 90% yield | Serwa (2009)35 | |

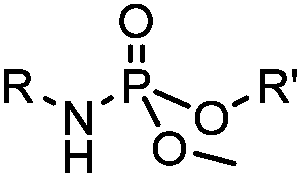
|
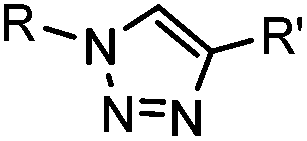
|
∼3 [50 μM Cu(I) + 50 μM TBTA, DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 1:9] :H2O 1:9] |
CuAAC: ligand for copper catalyst is required; up to 94% yield | Rostovtsev (2002)36 | |
| Presolski (2010)37 | |||||

|
|
up to 0.0024 (H2O, 25 °C) | Oxanorbornadiene cycloaddition: metal-free, but is slow | van Berkel (2007)38 | |
|
|
0.0024–0.076 | SPAAC: cyclooctynes are susceptible to thiols | Agard (2004)39 | |
| Baskin (2007)40 | |||||

|
|
0.057–0.96 | SPAAC: cyclooctynes are lipophilic; nonspecifically stick to serum proteins and cellular membranes | Ning (2008)41 | |
| Jewett (2010)42 | |||||
|
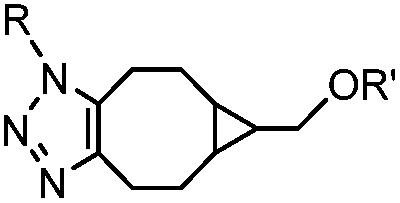
|
0.29; 2–2.9 (with electron-deficient azide) | SPAAC: BCN is readily accessible; the reaction rate is accelerated using an electron-deficient azide | Dommerholt (2010/2014)43,44 | |
| C: Inverse electron-demand Diels–Alder cycloadditions (DA INV ) | |||||
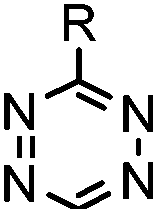
|
|

|
210–∼8.6 × 105 (PBS, 37 °C) | TCO is lipophilic and isomerizes over time; 100% conversion in 5 min | Blackman (2008)45 |
|
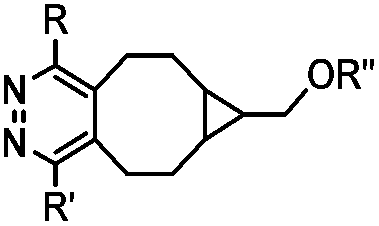
|
437–2672 | BCN is readily accessible; almost quantitative conversion | Lang (2012)46 | |
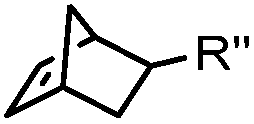
|
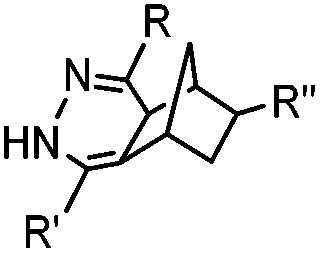
|
0.12–9.46 | Norbornene is stable | Devaraj (2008)47 | |
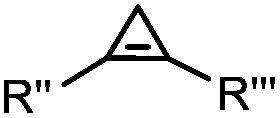
|
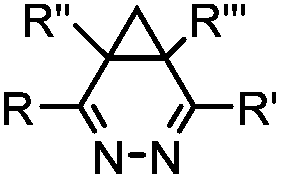
|
0.03–27 | Cyclopropene is small and stable; ED groups increase reactivity; quantitative labeling | Patterson (2012)48 | |
| Elliott (2014)49 | |||||
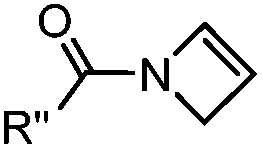
|
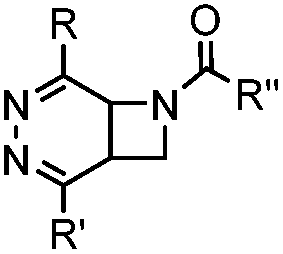
|
0.39 (DMSO:H2O 12:88, 20 °C) |
Used for affinity based protein profiling | Engelsma (2014)50 | |
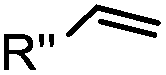
|
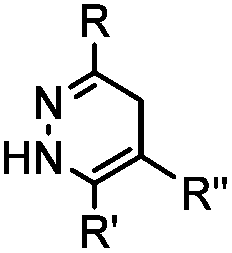
|
0.017–0.041 | Terminal olefin only reacts with highly reactive tetrazines | Niederwieser (2013)51 | |
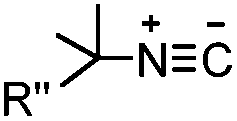
|
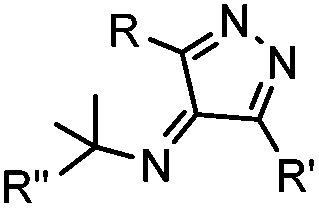
|
0.12–0.58 (THF:H2O 1:1) |
Tertiary isonitrile is required; the reaction is slow | Stöckmann (2011)52 | |
| D: Other dipolar cycloadditions | |||||
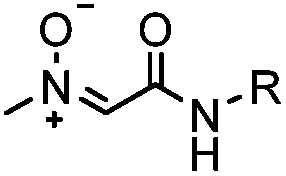
|
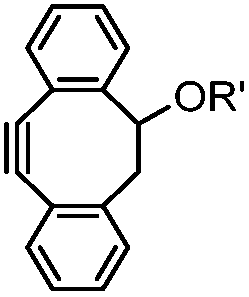
|
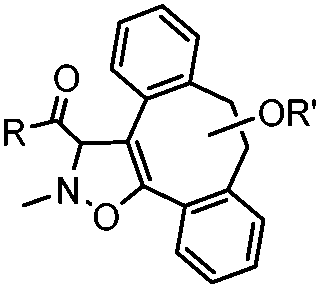
|
39 (MeCN:H2O 1:9, 25 °C) |
Strain-promoted alkyne-nitrone cycloaddition (SPANC): some nitrones are prone to hydrolysis; up to 95% yield | Ning (2010)53 |
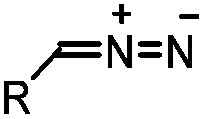
|
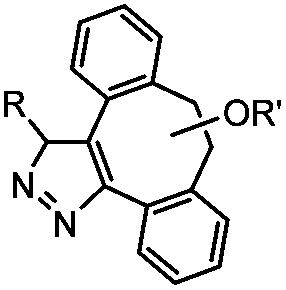
|
0.043 (CDCl3, RT) | Diazo-strained alkyne cycloaddition: the reaction rates are similar to the equivalent reactions of azide | McGrath (2012)54 | |
| Josa-Culleré (2014)55 | |||||
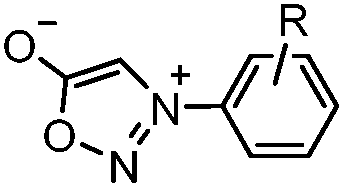
|
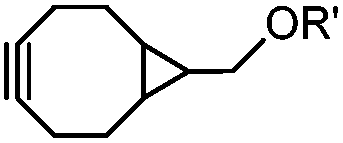
|
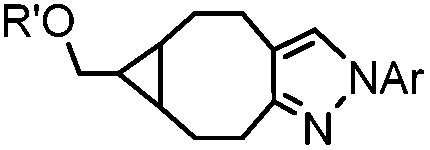
|
0.054 (21 °C, 55% MeOH in H2O) | Strain-promoted sydnone-BCN cycloaddition: quantitative reaction in organic solvent and in aqueous buffer | Wallace (2014)56 |
|
|
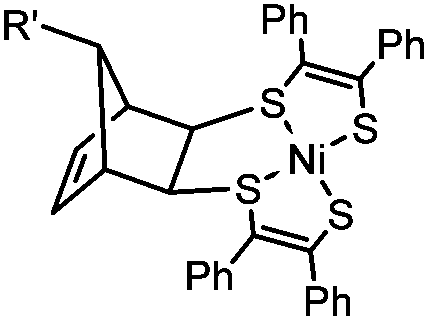
|
0.25 (PBS/EtOH) | Quadricylane ligation: quadricyclane is stable; the product is light sensitive | Sletten (2011)57 |
|

|
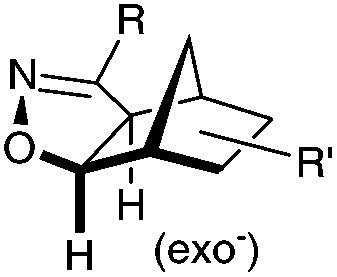
|
N.A. | Nitrile oxide-norbornene cycloaddition: nitrile oxide may cross react with nucleophiles; mostly used for labeling of nucleic acids | Gutsmiedl (2009)58 |
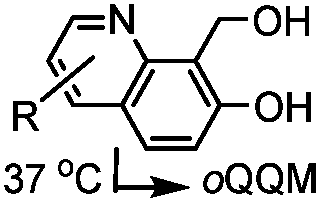
|

|

|
0.0015–0.028 (37 °C, 1:5 MeCN:H2O) |
TQ ligation: oQQM generated in situ; EW-groups on oQQM accelerate the reaction | Li/Dong (2013)59 |
| Zhang (2015)60 | |||||
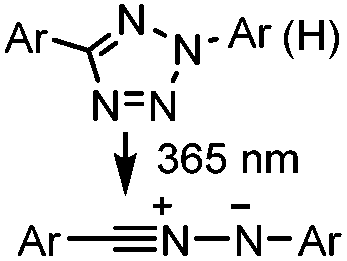
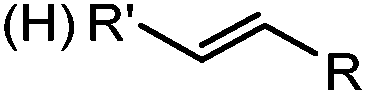
|
|
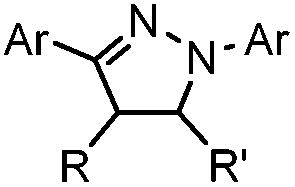
|
0.79 (MeCN:PBS 1:1) |
Tetrazole-alkene photo-click reaction: irradiation wavelength is tetrazole dependent; nitrile imine generated in situ; up to quantitative yield | Wang (2008/2009)61,62 |
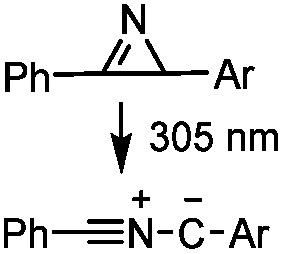
|
|
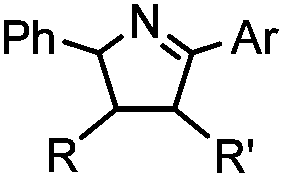
2 min (PBS, RT): give 41% labeling of lysozyme | Azirine ligation: azirine generates a reactive nitrile ylide intermediate in situ | Lim (2010)63 | |
| E: Transition metal-catalyzed coupling/decaging reactions | |||||

|
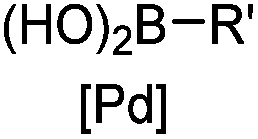
|
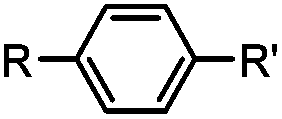
|
30 min (ligand, pH8, 37 °C) | Suzuki–Miyaura coupling: boronic acids are moderately toxic; >95% conversion | Chalker (2009)64 |
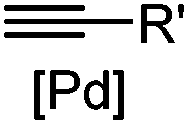
|
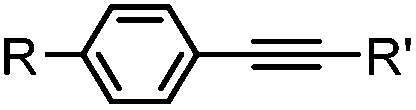
|
30 min (Cu ion, ligand, RT) | Sonogashira coupling: Mg(II) inhibits the binding of Pd(II) to the protein | Kodama (2007)65 | |
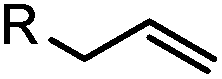
|
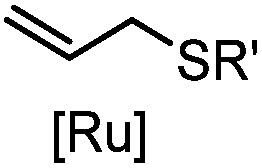
|

|
2–5 h (pH 8, 30% tBuOH/H2O, RT to 37 °C) | Olefin cross metathesis: requires ruthenium catalyst; up to 90% yield | Lin (2008)66 |
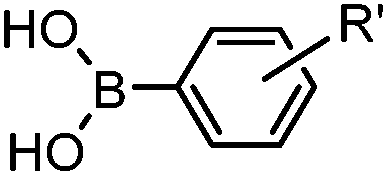
|
|
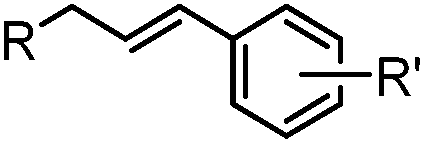
24 h (Pd(OAc)2, BIAN, O2, pH 7, RT) | Aqueous oxidative Heck reaction: reaction opens to air; up to full conversion if excessive catalyst and boronic acid are used | Ourailidou (2014)67 | |
|
allyl2Pd2Cl2 or Pd(dba)2 |

|
180 min (10 μM Pd(dba)2, 37 °C) | Useful for in vivo protein activation | Li (2014)68 |
| F: Labeling at cysteine residue(s) | |||||
|
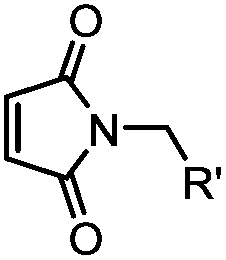
|
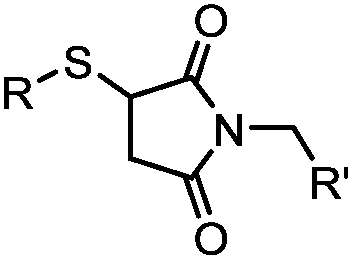
|
<30 min (PBS, RT) | TCEP should be added to maintain cysteine resides in a reduced form | Kim (2008)69 |
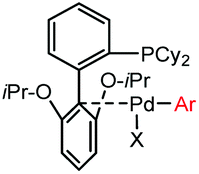
|
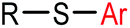
|
5 min (pH 7.5, RT, 5:95 MeCN:H2O) |
X = I, Br, Cl, OTf; quantitative labeling. Cy = cyclohexyl- | Vinogradova (2015)70 | |
|
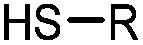
|
|
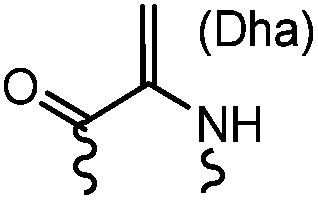
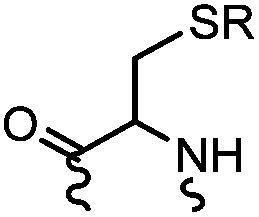
90 min (pH 9.0, 4 °C or RT) | Dha can be derived from cysteine by MSH (pH8.0, 4 °C, 20 min); >95% labeling | Bernardes (2008)71 |
|
|
|
9.19 (PBS) | CBT ligation: side reactions with thiols of CBT | Ren (2009)72 |
|

|
pH 7.0 | NCL: thiol catalysts are used, e.g., MESNA, MPAA | Dawson (1994)73 | |
|
|
|
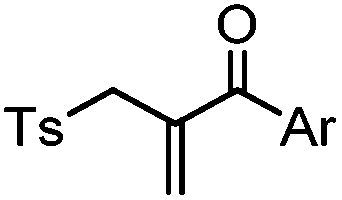
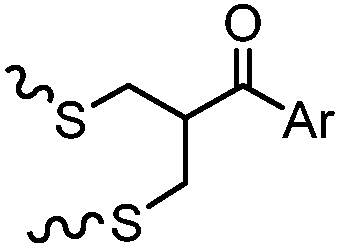
After reduction of disulfide: 2 h (pH 7.8, 4 °C) | S–S intercalation: reaction under mildly basic pH; formation of a stable product | Shaunak (2006)74 |
|

|
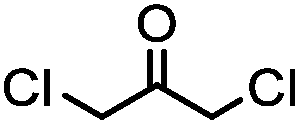
TCEP, 1 h; then pH 8, RT, 3 h | Acetone crosslinking: the introduced ketone allows oxime ligation; cross linking also stabilizes helical structures | Assem (2015)75 | |
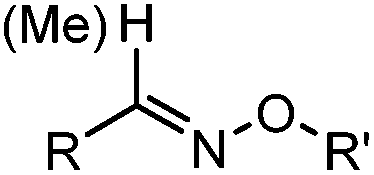
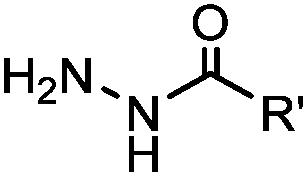
2.1. Condensation through carbonyls
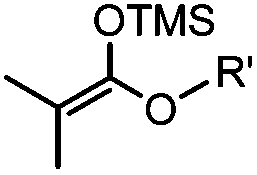
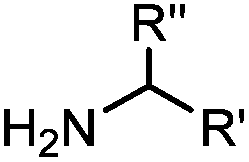
Ketones and aldehydes can react with hydroxyamine and hydrazide compounds under aqueous conditions to form stable oxime and hydrazone linkages, respectively. Oxime ligation76 is slow under neutral pH conditions and therefore an aniline catalyst is required.20 Recently, an improved catalyst p-phenylenediamine (p-PDA) was reported, which displays a higher water solubility and a better catalytic efficacy (10–120 times faster) than aniline.21 With respect to the condensation using hydrazide (hydrazone ligation), electronic and acid/base effects strongly influence the reaction efficiency at pH 7.4. For instance, carbonyl compounds with neighboring acid/base groups (e.g. carboxylate) form hydrazones at accelerated rates of up to 2–20 M−1 s−1.77 In addition, in the presence of a 5-methoxyanthranilic acid (5MA) catalyst, the condensation rate can be substantially enhanced 84-fold (6.6 M−1 s−1, 1 mM 5MA) compared to the reaction without a catalyst (0.08 M−1 s−1) at pH 6.5 (Table 1A).22Aside from oxime ligation and hydrazone ligation, aldehydes and ketones can undergo a Pictet-Spengler reaction with β-arylethylamines,78 which has been used for protein labeling at the N-terminus.23 To date, several modified versions have been introduced, including Pictet-Spengler ligation24 and hydrazino-Pictet-Spengler ligation.25 Additionally, proteins engineered with an N-terminal aldehyde tag can be labeled via the Mukaiyama-Adol condensation using silyl ketene reagents with the formation of a stable C–C bond.28 KAHA (α-ketoacid-hydroxylamine) ligation allows the condensation between an α-ketoacid and hydroxylamine or 5-oxaproline to form a native amide bond. This ligation has been a valuable alternative to native chemical ligation (NCL)73 to join two unprotected peptide fragments in peptide synthesis.29,30 Owing to the rapid association between aldehydes and amines, an aldehyde has been elegantly employed as an amine-capture auxiliary in aldehyde capture ligation (ACL). In ACL, a C-terminal selenobenzaldehyde ester can interact with the N-terminus of a peptide/protein. A native amide bond linkage is formed following a Se→N acyl shift. The ACL has been used for site-specific N-terminal modification of ubiquitin (Table 1A).31
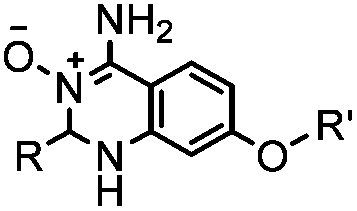
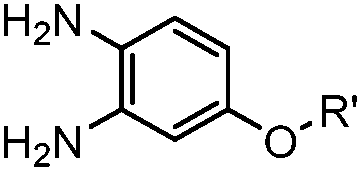
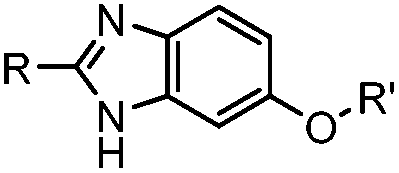
A recently introduced reaction is ABAO (2-aminobenzamidoxime) ligation. ABAO combines an aniline moiety for iminium-based activation of the aldehyde with a nucleophilic group at the ortho-position to the amine for intramolecular ring closure. In addition to the rapid condensation reaction kinetics (up to 40 M−1 s−1), the condensation forms a fluorescent dihydroquinazoline derivative, making it possible to develop fluorogenic aldehyde-reactive probes.26 Alkyl aldehydes can also efficiently couple with aryl diamines under mild conditions (RT, neutral aqueous solution) in the presence of Cu(II) or Zn(II) ions via an oxidative condensation process.27 This reaction forms stable benzimidazole linkages and has been utilized to label the T4 lysozyme protein with an aldehyde dye (Table 1A).
With respect to labeling biomolecules in live cells or organisms, carbonyl compound-related condensations have not been widely used. This is because the catalysts are usually toxic, and endogenous ketones and aldehydes, e.g. glucose and pyruvate, would interfere with the labeling reaction. Nevertheless, ketones and aldehydes are generally not present on the cell surface. Therefore, carbonyls serve as useful chemical handles for labeling biomolecules on the cell surface using hydrazide or aminooxy probes.20,79–83
2.2. “Click” through azides
Azide is a small and stable group, which has a unique dipole for a variety of bioorthogonal reactions.84 Click reactions using azides include Staudinger ligation,32 traceless Staudinger ligation,33,34 Staudinger-phosphite ligation,35 copper-catalyzed azide alkyne cycloaddition (CuAAC),36 strain-promoted azide alkyne cycloaddition (SPAAC) and oxanorbornadiene cycloaddition (Table 1B).38 Among these reactions, CuAAC and SPAAC appear to be the most popular and are discussed in detail in this section.4CuAAC is a hallmark of bioorthogonal chemistry that was reported independently by Sharpless36 and Meldal85 in 2002. Its application as a bioorthogonal reaction revolutionized our ability to modify and manipulate proteins.86,87 CuAAC becomes popular mainly due to the following reasons: (1) the azide and alkyne groups are highly specific toward each other but remain inert to other chemically active molecules in live systems; (2) the reaction produces a regioselective 1,4-triazole product which is stable and inert; (3) CuAAC exhibits fast reaction kinetics (∼3 M−1 s−1 in the presence of 50 μM Cu(I) and 50 μM TBTA)37 and various ligands have been developed to stabilize Cu(I) and further increase the reaction speed.
Cu ions catalyze the production of reactive oxygen species, leading to cytotoxicity.88 This limits the application of CuAAC in living systems. By choosing an appropriate ligand, CuAAC can be biocompatible with minimal cytotoxicity while showing an increased reaction rate.89 A panel of these ligands is summarized in Scheme 1A. Copper-chelating azides bring the Cu(I) ion into close proximity and thereby significantly increase the reaction rate (Scheme 1B).90 The ligand BTTP (3-[4-({bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}methyl)-1H-1,2,3-triazol-1-yl]propanol) stabilizes the Cu(I) ion while speeding up the click reaction (Scheme 1A). In addition, its complex with Cu(I) is cell permeable and non-cytotoxic, facilitating CuAAC in live E. coli cells.91 Using this reaction, an environment-sensitive fluorogenic fluorophore (alky-4DMN) was site-specifically introduced into HdeA in both the periplasm and cytoplasm of E. coli. HdeA, an acid-stress chaperone that adopts pH-dependent conformational changes, was genetically encoded with an azide-carrying unnatural amino acid ACPK at residue 58 within its pH-responsive region. The resulting hybrid pH indicator enables compartment-specific pH measurement to determine the pH gradient across the E. coli cytoplasmic membrane (Scheme 1C).91
 | ||
| Scheme 1 Typical CuAAC ligands (A) and variants of improved CuAAC (B and C). | ||
Fluorogenic azide probes display substantial fluorescence enhancement upon cycloaddition reaction and therefore confer labeling with minimal background (Fig. 1A). Based on photo-induced electron transfer (PET) mechanism, highly fluorogenic blue-emissive azidomethyl substituted anthracene (A) and its analogues have been generated.92 Substitution at the 3 or 7 position of coumarin has a strong impact on its fluorescence properties. Guided by this principle, 3-azido substituted coumarins (B) show an 80-fold increase in fluorescence intensity upon the cycloaddition reaction.93 The quantum yield (Φ, QY) of a probe after a click reaction can be calculated by density function theory (DPF), allowing rational design of fluorogenic azide probes.94 Using this approach, the green-emission azido-fluorescein (C) has been designed, which exhibits a fluorescence enhancement of 29 to 34-fold upon cycloaddition with different alkynes.95 Fluorogenic green- to far red-emitting CalFluors enable sensitive detection of biomolecules under no-wash conditions (D, E, F).96
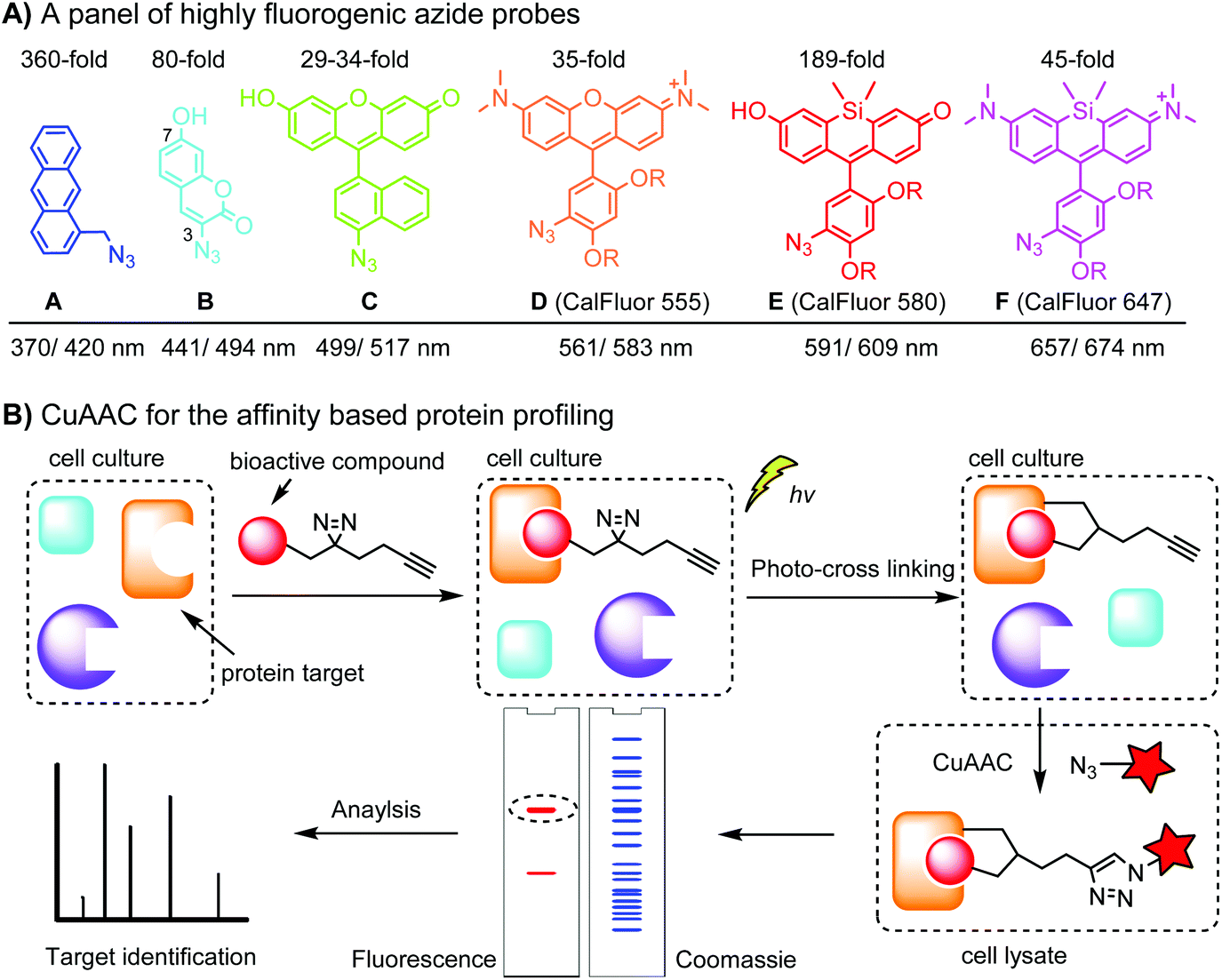 | ||
| Fig. 1 (A) A panel of highly fluorogenic azide probes including a selection of CalFluors; the maximal excitation/emission wavelengths refer to the value after the click reaction; R = –(CH2)2O(CH2)2N+Me2(CH2)3SO3−. (B) Target identification using an affinity-based probe (AfBP) by CuAAC. | ||
An important application of CuAAC lies in the target identification of biologically active small molecules in biomedical research and drug discovery.97 A bioactive compound is typically derivatized with a photo-crosslinking moiety (e.g. diazirine) and a terminal alkyne, which is denoted as an affinity-based probe (AfBP) (Fig. 1B). By photo-crosslinking, the AfBP probe in situ captures the protein target and forms a stable protein–ligand complex in the cell. Subsequent labeling of the target proteins using azide probes is followed by separation via gel-electrophoresis and determination via mass spectroscopy.97 Among many bioorthogonal groups, a terminal alkyne is a well suited tag as it is small with minimal interference of protein–ligand interactions, chemically inert, and can be easily modified via CuAAC.
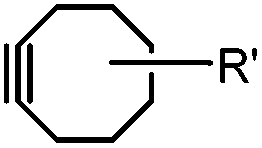
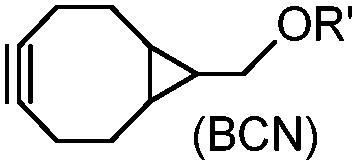
Although the cytotoxicity of Cu(I) can be reduced by using ligands, copper-free click chemistry is more straightforward. Strain-promoted azide–alkyne cycloaddition (SPAAC) and other copper-free click reactions38 without the requirement for a metal catalyst have been developed (Table 2A). The first SPAAC reaction for protein labeling was reported by Bertozzi and coworkers in 2004 using a strained cyclooctyne.39 However, the reaction rate is slow (k = 2.4 × 10−3 M−1 s−1), which is comparable to Staudinger ligation (k = 3 × 10−3 M−1 s−1). To date, new variants of strained alkynes have been developed with improved properties, such as enhanced specificity, reduced lipophilicity98 and increased reaction rates (Table 2B).
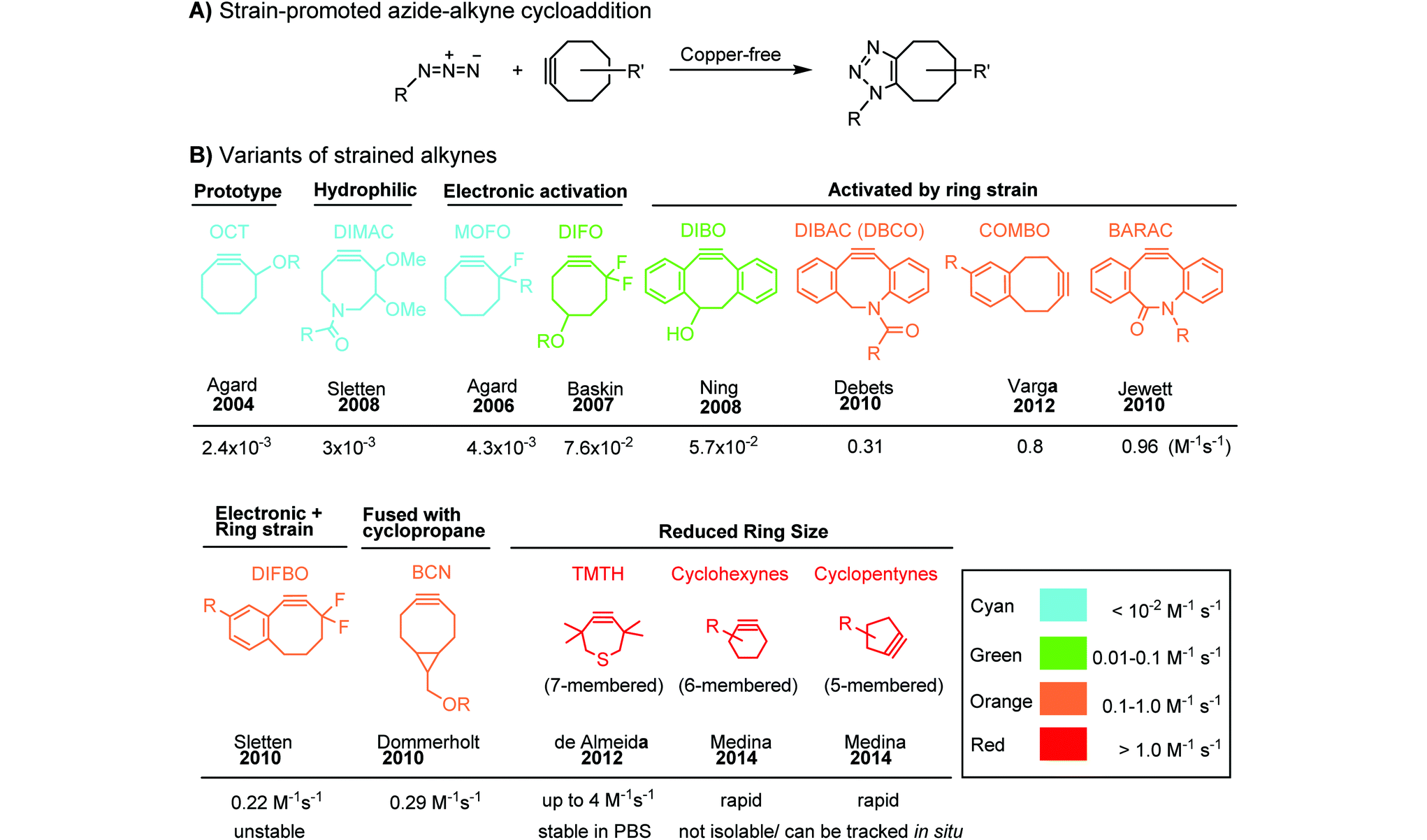
|
There are several ways to improve strained alkynes.99 The first strategy is to modulate the electronic properties by introducing electron-withdrawing (EW) groups, e.g. fluorine, near the triple bond.100 Examples include MOFO101 and DIFO.40 The second approach is to fuse the cyclooctynes with rigid aromatic rings, leading to an enhancement of reactivity by increasing ring strain. The ring-fused cyclooctynes, including DIBO,41,102 DIBAC,103 COMBO104 and BARAC,42 show a 25–400-fold increase in the reaction rate.105 However, synthesis of these cyclooctynes is usually laborious. Bicyclononyne (BCN), a cyclopropane-fused cyclooctyne, shows relatively fast reaction kinetics (0.3–1 M−1 s−1) toward azides and can be facilely prepared in only three steps.43 Using an electron-deficient azide, e.g. 4-azido-1-methylpyridin-1-ium iodide, the cycloaddition reaction rate with BCN can be further increased to 2–2.9 M−1 s−1.44 By introducing both EW groups and ring strain, difluorobenzocyclooctyne (DIFBO) shows only a moderate increase of reactivity (0.22 M−1 s−1) with a significant reduction in stability.106 The third strategy is to shorten the ring size as exemplified by the 7-membered tetramethylthiazacycloheptyne (TMTH),107 cyclohexyne and cyclopentyne.108 However, these compounds have poor stability (Table 2B). The copper-free click reactions have been used for antibody-free western blot analysis,109 visualization of glycosylation on cell surfaces110 and protein labeling inside live cells.111
2.3. Inverse electron-demand Diels–Alder cycloaddition (DAINV) and other cycloadditions
The inverse electron-demand Diels–Alder cycloaddition (DAINV) occurs between an electron-rich dienophile (e.g. strained alkene/alkyne) and an electron-poor diene, typically 1,2,4,5-tetrazine (Table 3A). DAINV represents so far the most rapid bioorthogonal reaction (up to 8.6 × 105 M−1 s−1).45,112 In contrast, the reaction rate of SPAAC is only up to 1 M−1 s−1. Theoretical calculation suggests that the rapid reaction rate can be attributed to the much higher interaction energy between tetrazine and trans-cyclooctene (TCO) in comparison to the interaction energy between an azide and a strained alkyne.113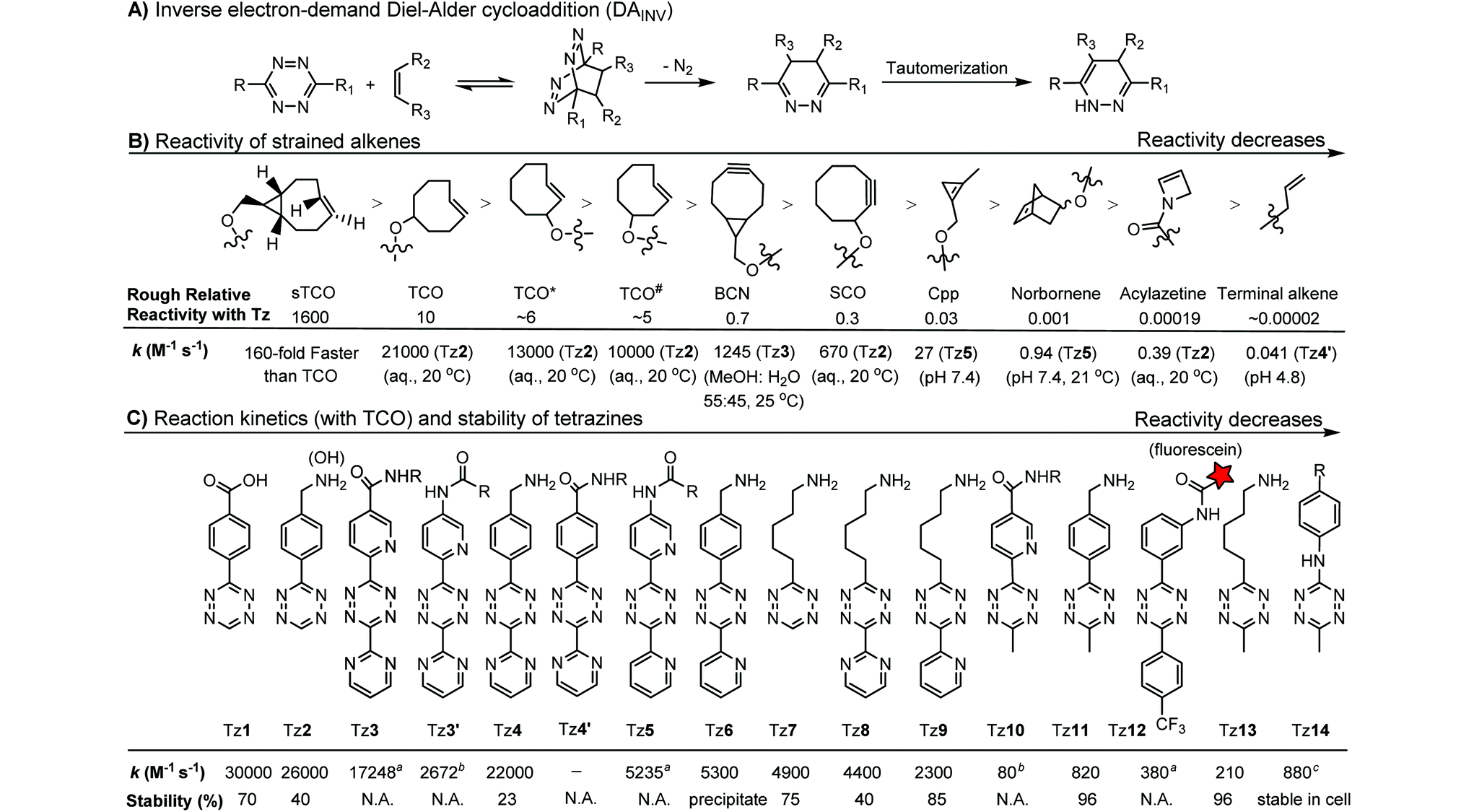
Tetrazines are highly reactive so that they can also readily react with strained alkynes, isonitriles52 and even terminal alkenes. Among these reactions, the reaction between tetrazine and BCN or cyclopropene has proven to be the most useful. Tetrazine reacts with BCN in a rapid fashion with a reaction rate constant of 3000 M−1 s−1.46 In comparison to TCO that requires complicated synthetic procedures, BCN is readily available through organic synthesis. Compared to BCN and TCO groups, cyclopropene (Cpp) is much smaller, and therefore has been employed as a “minimalist” tag for live-cell imaging and affinity-based protein labeling.114
Alkenes and alkynes display different reactivity toward tetrazine (Table 3B).116 sTCO features a cyclopropane ring, which brings additional strain and increases the reaction rate of the cycloaddition up to 160-fold in comparison to TCO.116 The carbamate bond near the trans-double bond in TCO* reduces the chance of nucleophilic attack. Thus, TCO* shows better stability than TCO with only 2-fold reduction in reactivity toward tetrazine.117 The reaction rate of BCN with tetrazine is about 10 times slower than that of TCO.46 Norbornene is ca. 10000 times less reactive than TCO, as it is more bulky and exhibits less ring strain than TCO.46,47 Other alkenes, including Cpp,48,49,114 acylazetine50 and terminal alkene,51 are less reactive, and therefore can react only with highly reactive tetrazines.
Different tetrazines show varied reactivity toward strained alkenes (Table 3C). Studies have been conducted to evaluate the reactivity and stability of tetrazines.115 The introduction of EW substituents can substantially increase the reaction rate. However, it is a double-edged sword. Increase in the reactivity of tetrazine often results in the reduction of stability and lifetime in the serum. ED substituents on the other hand decrease the reactivity of tetrazine.115 Besides electronic effects, steric effects also play a crucial role. In general, there is a trade-off between reactivity and stability. The rate constants of tetrazine reactions with TCO as well as their stability are summarized in Table 3C.
Green- and red-emitting fluorophores display electronic interactions with tetrazine chromophores that have absorption maxima at 500–525 nm. As a consequence, tetrazine-conjugated fluorophores often show reduced fluorescence. After a cycloaddition reaction, tetrazine is deconjugated and loses its quenching capability. Hence, many tetrazine-fluorophores feature fluorogenic properties, such as BODIPY-FL, Oregon Green 488, BODIPY-TMR, VT680,118 TAMRA, and fluorescein.119 These probes show a moderate turn-on ratio (up to 20-fold) (Fig. 2A). By adapting through-bond energy transfer (TBET) for fluorescence quenching, Weissleder and coworkers developed green-emitting BODIPY-tetrazine probes with up to 1600-fold turn-on ratio120 and blue-emitting coumarin-tetrazine probes with up to 11000-fold fluorescence enhancement.121 The tetrazine moiety is attached at the meta-position to the fluorophore on a rigid phenyl ring (Fig. 2A). Under these conditions, the tetrazine group is perpendicular to the fluorophore moiety, leading to the collinear alignment of two dipoles.121
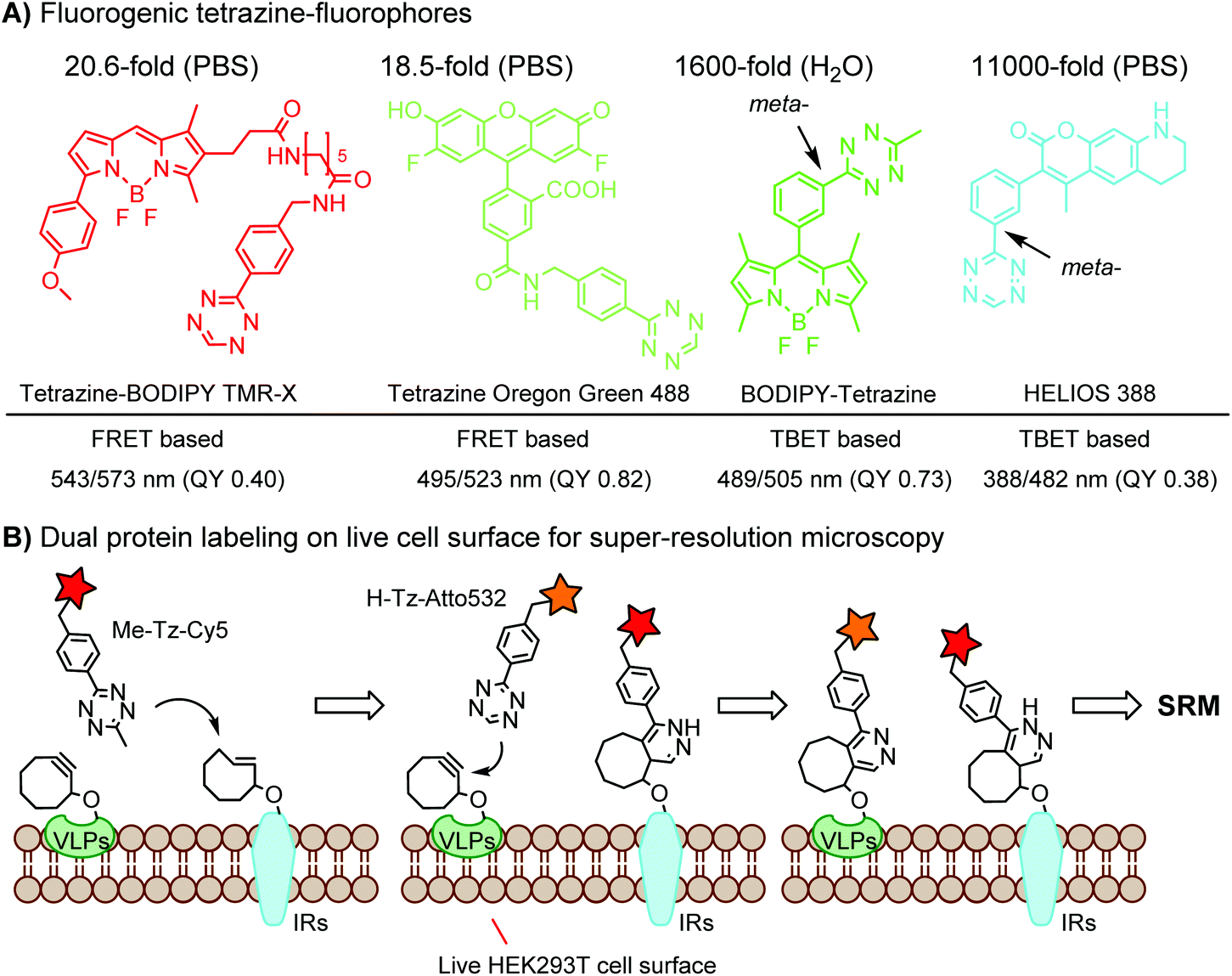 | ||
| Fig. 2 (A) Fluorogenic tetrazine-fluorophores; (B) dual protein labeling using orthogonal DAINV pairs for super-resolution microscopy (SRM). | ||
To achieve dual- or multi-labeling of proteins, mutually orthogonal reactions are desirable.122 By fine-tuning the DAINV reactions, “selectivity enhanced” DAINV reactions have been applied to sequential dual-color labeling of insulin receptors (IRs) and virus-like particles (VLPs) on the cell surface of HEK293T cells, facilitating dual-color super-resolution microscopy (Fig. 2B).117
In addition to CuAAC, SPAAC and DAINV, which are popularly used for protein labeling, other dipolar cycloaddition reactions are summarized in Table 1D, including strain-promoted alkyne-nitrone cycloaddition (SPANC),53 diazo-strained alkyne cycloaddition,54,55 nitrile oxide-norbornene cycloaddition,58 quadricylane ligation,57 TQ-ligation,59,60 strain-promoted sydnone-BCN cycloaddition,56 and plenty of photo-triggered cycloaddition reactions, such as the tetrazole-alkene photo-click reaction61,62 and azirine ligation.63
2.4. Transition metal-catalyzed couplings and decaging reactions
Biocompatible transition metal-catalyzed couplings include the Suzuki–Miyaura coupling,64 Sonogashira coupling, and olefin metathesis. These reactions can be performed under mild and aqueous conditions, despite the necessity to use a transition metal catalyst (Table 1E).15 Suzuki–Miyaura coupling requires an iodophenyl group, a water soluble palladium (0) catalyst and an appropriate ligand. Recently, an improved ligand, 1,1-dimethylguanidine, has been employed for aqueous Suzuki–Miyaura coupling.123 Suzuki–Miyaura coupling has been successfully used for protein glycosylation and labeling at the cell surface.124 Sonogashira coupling is another Pd-catalyzed coupling reaction.65 An alkyne group is incorporated into the protein and subsequently reacts with an iodophenyl probe. Sonogashira coupling has been used for the modification of ubiquitin in live cells.11,15,125Terminal olefins can undergo an oxidative Heck reaction with boronic acid in the presence of Pd(OAc)2/BIAN catalysts. This reaction has been used for the site-specific labeling of 4-oxalocrotonate tautomerase (4-OT).67 Olefin metathesis is the redistribution of fragments of alkenes by scission and regeneration of carbon–carbon double bonds.117,118 Hence, in order to avoid undesired cross coupling products, a terminal olefin and a more reactive thiovinyl ether are required.126 Water soluble ruthenium catalysts have been developed to mediate the reaction in aqueous solution.126,127
Chemical protein labeling not only makes proteins visible but also renders them controllable. Recently, palladium catalysts have been used to manipulate protein function in cells. Pd-mediated cleavage of the propargyl carbamate group leads to the generation of a free lysine residue. The protected lysine analogue can be genetically and site-specifically incorporated into a protein using an unnatural amino acid (UAA) mutagenesis technique (discussed in the later section). This strategy enables protein activation in living cells by decaging the lysine residue located at the active site of a protein, and has been utilized to elucidate the virulence mechanism of a bacterial type III effector protein in its host cells (Fig. 3A).68 A ruthenium-catalyzed cleavage reaction has been used for the cleavage of allyl carbamate to unmask caged Rhodamine 110 (R110) inside living cells (Fig. 3B).128
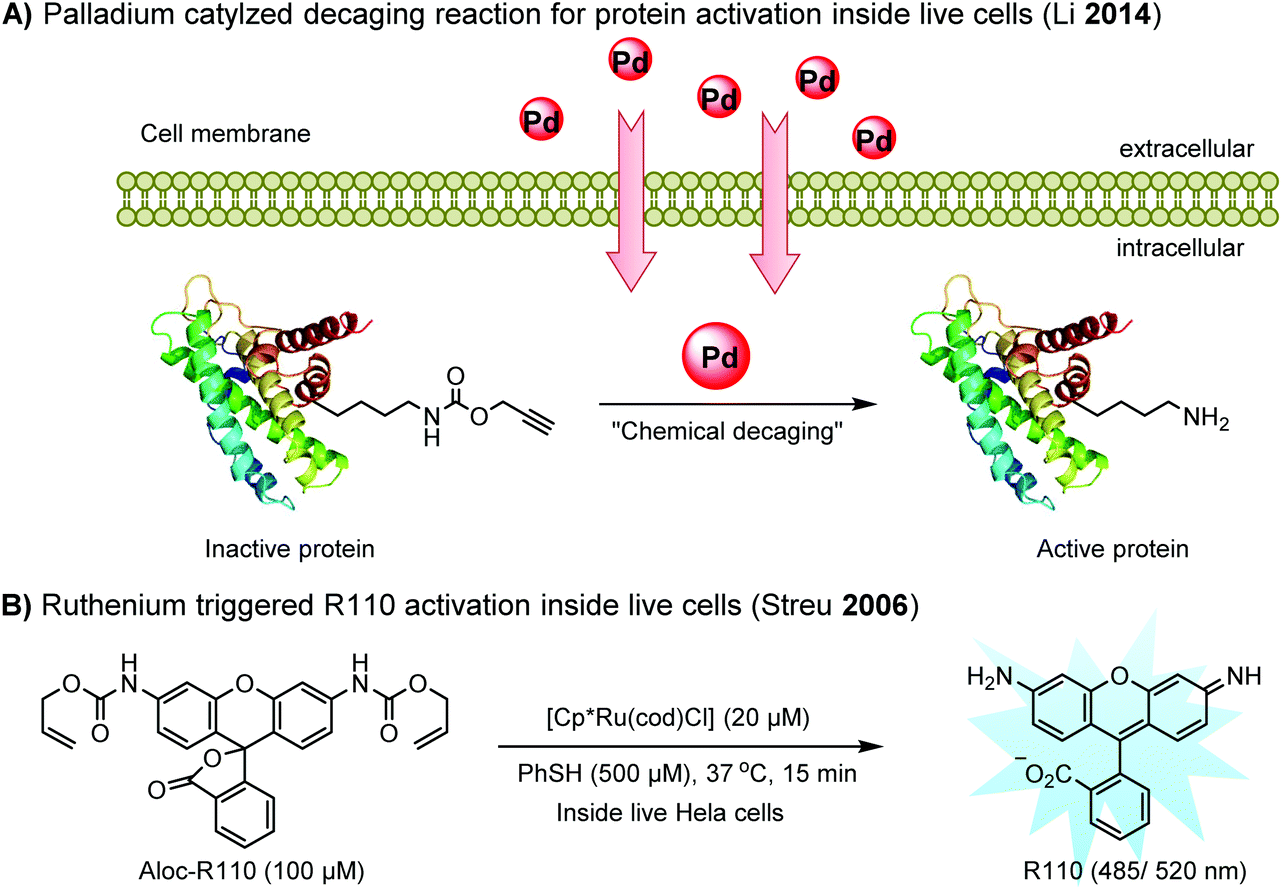 | ||
| Fig. 3 Transition metal-catalyzed decaging reactions under living conditions. | ||
2.5. Selective labeling at cysteine residue
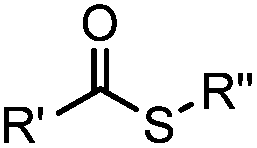
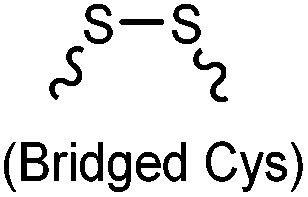
Cysteine is characterized by its remarkable nucleophilicity among 20 common amino acid residues. Traditionally, proteins can be site-specifically labeled at their solvent accessible cysteine residues by thiol-reactive alkylation reagents, e.g., maleimides and iodoacetamides.69,129 However, excess maleimide-based reagents lead to the modification of the side chains of histidine, lysine and α-amino groups. By converting cysteine residues to dehydroalanine (Dha), the Dha residue can then be rapidly and specifically labeled by various thiol reagents.71 This approach has been used for the preparation of post-translationally modified proteins, including phosphorylation, glycosylation, methylation, acetylation, and lipidation (Table 1F).130,131Adjacent cysteines are usually oxidized to form disulfide bonds under non-reducing conditions. In these cases, the solvent accessible disulfide bond can be first gently reduced and subsequently “intercalated” by mono-sulfone reagents. This approach permits the site-specific PEGylation of a variety of therapeutic proteins, including human interferon α-2b and antibody fragments.74 A more recent approach employed 1,3-dichloroacetone (DCA) to introduce a reactive ketone tag, enabling subsequent oxime ligation (Table 1F).75
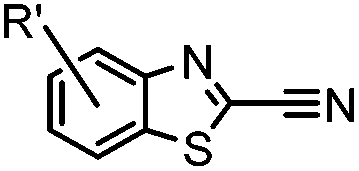
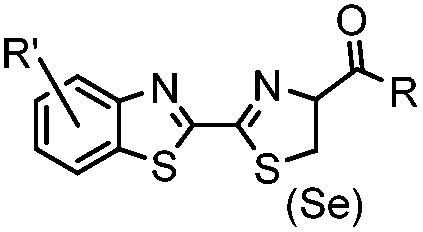
N-terminal cysteine displays unique reactivity. Proteins carrying an N-terminal cysteine can undergo native chemical ligation with thioester probes and chemoselective ligation with aldehydes to form thiazolidines.132 N-terminal cysteine can also specifically react with cyanobenzothiazole (CBT) derivatives at a fast reaction rate (9 M−1 s−1).72 The reaction of CBT compounds with D-cysteine is highly biocompatible and has been used for bioluminescent imaging of protease activity in live mice.133
Palladium-tolyl complexes using 2-dicyclohexylphosphino-2′,6′-diisopropylbiphenyl (RuPhos) as the ligand have been developed to mediate efficient and highly selective cysteine conjugation reactions under biocompatible reaction conditions.70 At pH 7.5, the rate of a palladium-medicated reaction is comparable to that of the maleimide reaction. This bioconjugation strategy has demonstrated its broad utility for making stapled peptides, site-specific labeling of proteins with a coumarin fluorophore, and the preparation of antibody-drug conjugates (ADCs) (Scheme 2).70
 | ||
| Scheme 2 Organometallic palladium reagents for cysteine bioconjugation on proteins. | ||
3. Chemoselective labeling of native proteins
Chemoselective labeling of native proteins is useful for the preparation of protein conjugates such as ADCs134 and the labeling of endogenous proteins in living cells.135 In addition to selective labeling at cysteine residues as discussed in the previous section, there are a few other approaches available (Scheme 3).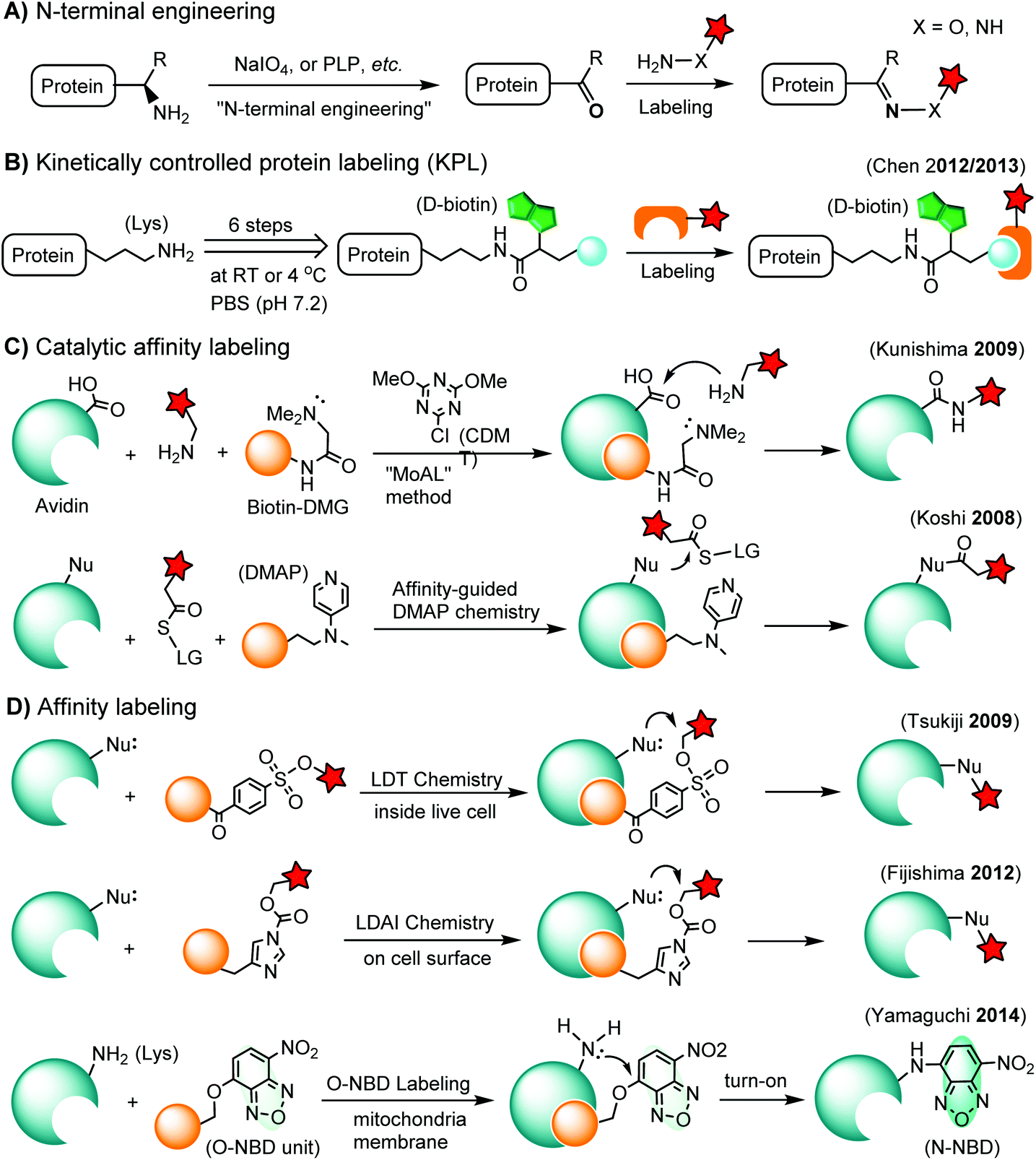 | ||
| Scheme 3 Methods for chemoselective labeling of native proteins. LG: leaving group. | ||
3.1. N-terminal labeling
The N-terminus of a protein shows unique reactivity and can be site-specifically labeled or converted to a bioorthogonal handle. The N-terminal α-amino group features a lower pKa value (ca. 8) than that of the ε-amino group of lysine (ca. 10). Transamination methods have been used to convert the N-terminal α-amino group to an aldehyde or a ketone using glyoxylate in the presence of a divalent metal ion and a base136 or using pyridoxal-5-phosphate (PLP).137 N-terminal glycine shows the highest reactivity in the transamination reaction.137 A ketene compound has been reported to selectively react with an N-terminal α-amino group at pH 6.3.138 Another approach is the oxidation of an N-terminal serine or threonine to an aldehyde or ketone by sodium m-periodate at neutral pH.139 Afterwards, the N-terminal aldehyde or ketone tag is ready for oxime ligation, Mukaiyama-Adol condensation, Pictet-Spengler reaction, etc., as discussed in previous sections (Scheme 3A).3.2. Kinetically-controlled protein labeling (KPL)
Under normal bioconjugation conditions for protein labeling at lysine side chains, it is usually difficult to achieve site-specificity using regular amine-reactive reagents, such as an N-hydroxysuccinimidyl (NHS) ester. Nonetheless, lysine residues on a protein’s surface display subtle differences regarding their individual reactivity. This difference may originate from the different solvent accessibility of lysine, or the interaction of lysine with neighboring residues, or the combination of both.140 Through kinetically-controlled protein labeling (KPL), proteins can be mono-modified at a specific lysine side chain.140 This approach enables the site-selective introduction of a terminal alkyne, or azide into native proteins for a click reaction140 or Staudinger ligation (Scheme 3B).141 KPL has been utilized for site-specific biotinylation of pharmaceutically relevant proteins for delivery into macrophages142 and dual labeling of the peptide hormone, somatostatin, for the visualization of targeted drug delivery in tumor cells.1433.3. Catalytic affinity labeling
Catalytic modules tethered with a ligand can be targeted to the binding pocket of a protein to locally catalyze the labeling reaction on the protein. For instance, a biotin-conjugated dimethylglycine (DMG) group is brought to the proximity of the avidin binding pocket, where it specifically catalyzes the acylation of amine probes with Asp108.144 In the so-called modular affinity labeling (MoAL) approach, three modules are required: (1) the catalytic ligand module (biotin-DMG), (2) the labeling module (amine probe) and (3) the reactive module (CDMT). Another catalytic affinity labeling method is termed affinity-guided DMAP (4-dimethylaminopyridine) chemistry. DMAP is an effective acyl transfer catalyst, which can activate an acyl ester for its transfer to a nucleophilic residue. Therefore, a ligand tethered with DMAP allows specific labeling of native proteins by acyl ester probes (Scheme 3C).1453.4. Affinity labeling of endogenous proteins
In ligand-directed tosyl (LDT) chemistry, ligand-tethered tosyl ester probes are used to label endogenous proteins in cells. Upon labeling, the ligand is cleaved off at the tosyl ester linkage. Therefore, the protein is still active after labeling because the active center is no longer occupied by its ligand.146 The concept was employed to construct a turn-on probe based on the release of the quencher upon labeling.147 The LDT chemistry has been applied to the labeling of cell surface receptors148 and native FKBP12 in live cells.149 LDT chemistry is slow and typically requires over 12 hours of incubation time. Recently, a faster affinity labeling approach, known as ligand-directed acyl imidazole (LDAI) chemistry, was used to selectively modify the endogenous folate receptor at the cell surface (Scheme 3D). The experiments showed that LDAI labeling is 12-fold more efficient than LDT labeling.150 New emerging affinity labeling reagents include chemical probes bearing an O-nitrobenzoxadiazole (O-NBD) unit. Upon the ligand-directed reaction with a lysine side chain, the non-fluorescent O-NBD is converted to fluorescent N-NBD. Translocator protein (TSPO) ligands carrying an O-NBD unit enabled proteomic identification of a partner protein of TSPO, a voltage-dependent anion channel (VDAC). The affinity labeling reaction using O-NBD is quite efficient with yields of 41% and 76% after 1 h and 12 h, respectively.1514. Chemical labeling of proteins in living cells
Proteins function in signaling pathways and interacting networks in the complicated surroundings of cells and organisms. Fluorescent proteins (FPs) have revolutionized our ability to visualize and investigate protein function directly in living cells and organisms by fusion of individual proteins with FPs. Chemical probes, including organic dyes, are able to achieve properties that are not readily possible when using FPs. For instance, many organic dyes are superior to FPs in terms of brightness, photostability, far red-emission, environmental sensitivity, pulse-chase labeling and the flexibility for modifications to their spectral and biochemical properties. In the chemical tagging approach, a protein of interest (POI) is fused with a polypeptide tag, which is subsequently labeled with chemical probes. In general, these tags can be classified in the following categories: (1) metal chelation based peptide tags; (2) self-labeling peptide tags; (3) ligand binding domains (LBDs); (4) self-labeling enzymatic domains; (5) peptide sequences for enzymatic modifications; and (6) genetically encoded unnatural amino acids (UAAs) as “minimalist” tags (Table 4).| Tag name (size in AA) | Probe | Enzyme | Cellular labeling | Features | Reference(s) |
|---|---|---|---|---|---|
| AA: amino acids. (A) BG: O6-benzylguanine; BC: O2-benzylcytosine; BG-FL: BG-fluorescein conjugate; BC-FL: BC-fluorescein conjugate. (E) ACP: acyl carrier protein; PPTase: 4′-phosphopantetheinyl transferase; CoA: coenzyme A; PCP: peptide carrier proteins. | |||||
| A. Metal chelation based peptide tags | |||||
| His-tag (6): HHHHHH |

|
No | Cell surface/intracellular | Ni(II) is toxic to cells and quenches fluorescence; intracellular labeling is possible using cell-penetrating multivalent chelator carrier complexes | Guignet (2004)152 |
| Wieneke (2014)153 | |||||
| His-tag (6): HHHHHH | Zn(II) complex: HisZiFit | No | Cell surface | Zn(II) is non-toxic; the zinc complex is membrane-impermeant | Hauser (2007)154 |
| D4 tag (4): (DDDD)n, n = 1–3 | Multinuclear Zn(II) complexes: Zn(II)-DpaTyrs probe | No | Cell surface | Spectroscopic change upon chelation; D4 tag/Zn(II)-DpaTyrs pair is orthogonal to the His tag/Ni(II)-NTA pair | Ojida (2006)155 |
| B. Self-labeling peptide tags | |||||
| CCXXCC (6) (tetracysteine) |
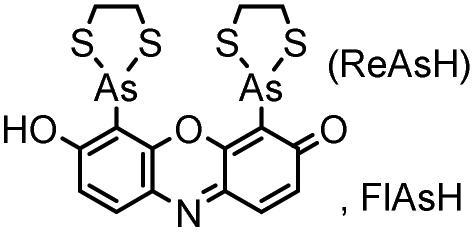
|
No | Intracellular | Fluorogenic; non-specific reactions and interactions to endogenous thiols; biarsenical probes may be cytotoxic | Griffin (1998)156 |
| Gaietta (2002)157 | |||||
| SSPGSS (6) (tetraserine) |

|
No | Cell surface | Fluorogenic; reduced background staining; cytotoxic; off-target labeling of endogenous tetraserine motif; RhoBo is cell permeable | Halo (2009)158 |
| HyRe tag (11): HKSNHSSKNRE |
|
No | Cell surface | Neutral pH; imine byproduct formation at pH <6 | Eldridge (2011)159 |
| SpyTag (13) | SpyCatcher protein domain (138 AA) | No | Cell surface | k = 1.4 × 103 M−1 s−1; high yield under diverse conditions of pH, temperature and buffer; formation of a stable amide bond linkage; not traceless | Zakeri (2012)160 |
| E3 tag (21): (EIAALEK)3 | K3 peptide (21 AA): (KIAALKE)3 | No | Cell surface | E3/K3 coiled-coil template induces proximity-driven rhodium(II)-catalyzed modification or acyl transfer reaction (t1/2 = 10 min) | Chen (2011)161 |
| Reinhardt (2014)162 | |||||
| C. Ligand binding domains (LBDs) | |||||
| FKBP’ (108) (FKBP12_F36 V) |
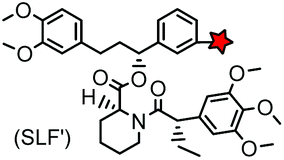
|
No | Intracellular | Specific binding between FKBP’ and SLF’; non-covalent | Clackson (1998)163 |
| Marks (2004)164 | |||||
| eDHFR (159) |
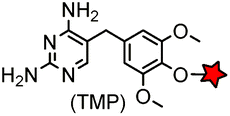
|
No | Intracellular | Tight and specific binding between eDHFR and TMP; non-covalent/covalent; non-cytotoxic; affinity conjugation is rapid (t1/2 = 1.8 min) | Miller (2005)165 |
| Liu (2014)166 | |||||
| PYP-tag (125) |
|
No; PYP tag is derived from a purple bacteria | Intracellular | PYP-tag covalently binds to the thioester derivative of cinnamic acid/coumarin; fluorogenic; up to k = 3950 M−1 s−1 | Hori (2009/2013)167,168 |
| D. Self-labeling enzymatic domains | |||||
| SNAP tag (182) |
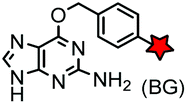
|
hAGT (mutated human O6-alkylguanine-DNA alkyltransferase) | Intracellular | BG: k = 0.3 × 104 M−1 s−1; BG-FL: k = 2.8 × 104 M−1 s−1; removal of guanine after labeling | Keppler (2003)169 |
| CLIP tag (182) |
|
Intracellular | BC-FL: k = 0.1 × 104 M−1 s−1. SNAP-tag and CLIP-tag possess orthogonal substrate specificities; removal of cytosine after labeling | Gautier (2008)157 | |
| BL-tag (263) |
|
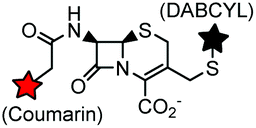
Non-catalytic β-lactamase variant | Cell surface | k = 7.8 × 104 M−1 s−1, E166N mutant of β-lactamase; fluorogenic labeling | Mizukami (2009)170 |
| Halo tag (290) |

|
Dha (mutated haloalkane dehalogenase) | Intracellular | k = 2.7 × 106 M−1 s−1; covalent; probe easily accessible by synthesis | Los (2008)171 |
| Cutinase (197) |

|
A small globular serine esterase of 22 kDa | Cell surface | Cutinase is labeled by the covalent inhibitor, p-nitrophenyl phosphonate (pNPP) | Bonasio (2007)172 |
| E. Enzymatic modifications | |||||
| ACP (77)/S6(12)/A1(12)/PCP(80)/ybbR tag(11) | Coenzyme A (CoA) derivatives | PPTase (AcpS or Sfp) | Cell surface | AcpS/Sfp catalyzes the transfer of a CoA-activated probe to the Ser residue of the tag. kcat = 5.5 × 10−4–0.32 s−1, kcat/Km = 5.3–4550 M−1 s−1 | George (2004)173 |
| Yin (2005)174 | |||||
| Zhou (2007)175 | |||||
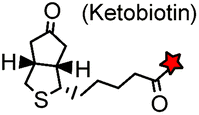
AP tag (15): GLNDIFEAQ![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) IEWHE IEWHE |
|
Biotin ligase (BirA) | Cell surface | Ketobiotin is a substrate for BirA and is incorporated into the Lys residue of the peptide tag; labeling time >20 min | Chen (2005)83 |
| Q-tag (7): PKPQQFM | Cadaverine derivatives | Transglutaminase (TGase) | Cell surface | TGase mediates the formation of an amide bond between the amine of cadaverine and the Glu residue of the Q-tag | Sato (1996)176 |
| Lin (2006)177 | |||||
| LAP tag (13–22) |
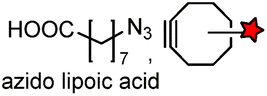
|
Lipoic acid ligase (LplA)/LplA mutant | Cell surface/intracellular | k cat = 0.048 s−1 for the conjugation of azido lipoic acid; coumarin addition by W37VLplA: kcat = 0.019 s−1, kcat/Km = 3.8 × 102 M−1 s−1; W37ILplA: kcat = 0.016 s−1, kcat/Km = 0.6 × 102 M−1 s−1 | Fernandez-Suarez (2007)178 |
L![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) XPXR (6) XPXR (6) |
|
Formylglycine generating enzyme (FGE) | Cell surface | FGE catalyzes the transformation of a Cys of the peptide tag to a formylglycine | Carrico (2007)79 |
LPX![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) G (5) G (5) |

|
Sortase A (SrtA) | Cell surface | SrtA cleaves the peptide at the Thr-Gly site, and attaches a labeled polyglycine peptide | Popp (2007)179 |
| CAAX (4) |
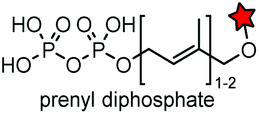
|
Protein farnesyltransferase (PTFase) | Cell surface | CAAX is also present in endogenous proteins; an alkyne group is introduced for click labeling | Wollack (2009)180 |
| Split intein [IntC (35) or IntN (25)] | Another half of split intein [IntN (100) or IntC (104)] | Reconstituted split inteins | Cell surface/intracellular | Intein-mediated protein trans splicing removes intein after reaction (traceless); yield 55–90% and k = 0.9–4.7 s−1 using Npu DnaE intein and AceL-TerL intein | Giriat (2003)181 |
| Schütz (2014)182 | |||||
TITS![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) YYR (8) YYR (8) |
|
AnkX from Legionella pneumophila | In vitro | k cat/Km = 0.9–5 × 102 M−1 s−1, covalent labeling can be removed by Lem1 | Heller (2015)183 |
| Tub-tag (14): VDSVEGEGEEEGEE |
|
Recombinant tubulin tyrosine ligase (TTL) | In fixed cell | Yield up to 99% with moderate enzyme concentrations and short reaction times | Schumacher (2015)184 |
| F. Incorporation of UAA as a “minimalist” tag | |||||
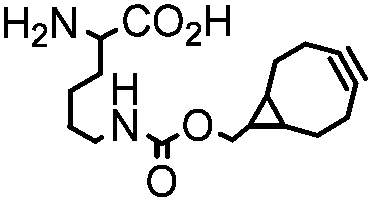
|
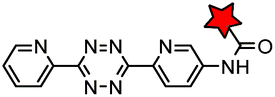
|
BCN-RS/tRNACUA | Cell surface/intracellular | UAA bearing BCN or TCO can be site-specifically incorporated to proteins in live cells for labeling with tetrazine dyes | Lang (2012)46 |
4.1. Metal chelation based peptide tags
Metal chelation methods have been adopted for affinity chromatography in protein purification. The principle has also been utilized for protein labeling, either by non-covalent complex formation or by chelation-driven affinity conjugation.152,185 Examples include the poly-histidine tag (His-tag, HHHHHH) and the tetra-aspartate tag [D4 tag, (DDDD)n, n = 1–3], which can be labeled using Ni-NTA probes and zinc complexes, respectively.152,154,155,186–189 Specific intracellular labeling of His-tagged proteins was achieved using cell-penetrating multivalent N-nitrilotriacetic acid (NTA) carrier complexes.153 The advantages of metal chelation based labeling can be attributed to the small size of the tag which confers minimal disturbance to protein function, the high labeling efficiency, selectivity and accessibility of various functional probes (Table 4A).1904.2. Self-labeling peptide tags
Tsien and coworkers reported the biarsenical FlAsH (fluorescein arsenical hairpin) as the first chemical surrogate to FPs for labeling proteins in live cells.156 A red version of FlAsH, a resorufin-based biarsenical (ReAsH), was developed later. FlAsH and ReAsH probes allow specific and fluorogenic labeling of proteins fused to a tetracysteine motif (CCXXCC).191–193 However, this labeling technology suffers practically from nonspecific labeling of thiol-rich biomolecules in the cell and the toxicity of the biarsenical ligands.194 Nevertheless, the tetracysteine motif (CCXXCC) is much smaller than FPs. Other self-labeling peptide tags include the bisboronic RhoBo-tetraserine tagging system,158 hydrazide-reactive (HyRe) tag,159 SpyTag160 and E3 tag161,162 (Table 4B).Prominent applications of self-labeling peptide tag approaches involve visualization of newly synthesized proteins and tracking of protein trafficking in live cells. This is readily achieved via the “pulse-chase” technique. The old populations of proteins were pulse-labeled by green-emitting FlAsH, while the newly synthesized proteins were chased by red-emitting ReAsH. Consequently, old and new copies of an individual protein were labeled using two colors. In one example, this approach was used to elucidate the mechanism of connexin assembly and turnover in HeLa cells.191 In another example, the approach was employed to study AMPA receptor (AMPAR) trafficking. Regulation of AMPA receptor (AMPAR) trafficking is important for neural plasticity. GluR1 and GluR2 are two AMPAR subunits that play a key role in the activity-dependent trafficking of the AMPARs during long-term potentiation (LTP) and depression (LDT). In order to examine the trafficking and synthesis of GluR1 and GluR2, a tetracysteine motif (EAAAREACCRECCARA) was attached at the C-termini. ReAsH-EDT2 was first applied and after 6–8 h, FlAsH-EDT2 was applied to cells expressing tetracysteine-tagged GluR1 or GluR2 (Scheme 4B). In this case, the red ReAsH-EDT2 labels all preexisting GluR1/2 subunits, while the green FlAsH-EDT2 labels those AMPRAR subunits synthesized during the 6–8 h chase period. The measurements suggested that both GluR1 and GluR2 are synthesized in dendrites and that an activity blockade enhances the dendritic synthesis of GluR1 but not GluR2.195
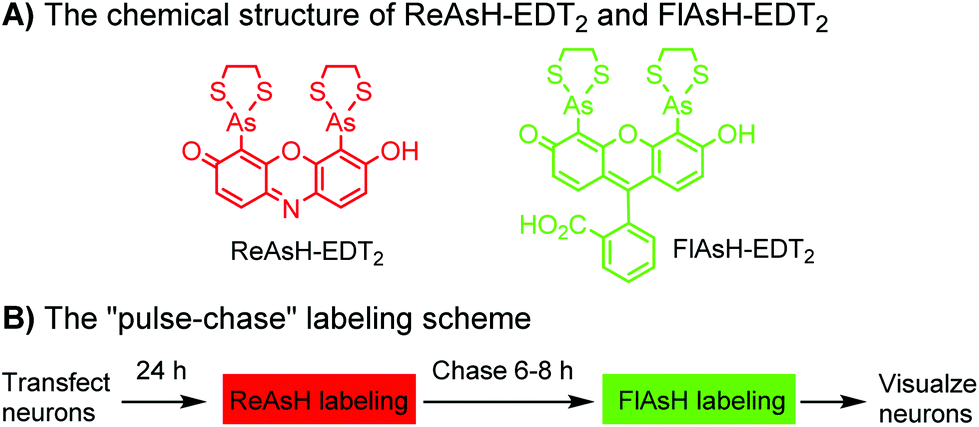 | ||
| Scheme 4 Dual-color “pulse-chase” labeling of neurons using ReAsH and FlAsH for the visualization of the synthesis and trafficking of AMPA receptors. | ||
4.3. Ligand binding domains (LBDs)
The specific interaction between the ligand binding domain (LBD) and its small-molecule ligand confers specificity of labeling in cells. SLF’, a derivative of the synthetic ligand of FKBP12 (FK506 binding protein), binds to FKBP’ (FKBP12_F36 V mutant) with more than 1000-fold selectivity over the wild type FKBP12 protein.163 Based on this specific binding, proteins fused with FKBP’ have been selectively labeled by SLF’-conjugated probes in live cells.164 The antibiotic trimethoprim (TMP) is a specific inhibitor of E. coli dihydrofolate reductase (eDHFR). TMP binds to eDHFR with nanomolar affinity, which is over 1000-fold higher than the interaction with mammalian DHFR. As a result, the off-target labeling by TMP probes is minimal under certain conditions.165Because of the non-covalent binding, the labeling via the FKBP’ or eDHFR tag is reversible. In order to achieve a stable labeling, the affinity conjugation approach has been introduced. A cysteine mutation is introduced in the proximity of the TMP binding site on eDHFR, which can be specifically labeled by TMP-acryloyl probes due to a proximity-induced effect.196 The reaction of a mildly reactive acryloyl group with other thiols in the cell is minimal under certain conditions.197 Based on the affinity conjugation principle, a rapid and fluorogenic TMP-AcBOPDIPY probe is developed with a half-life of less than 2 min for covalent labeling (Fig. 4A).166 Intracellular proteins fused with eDHFR_N23C were rapidly labeled by the TMP-AcBOPDIPY probe under no-wash conditions (Fig. 4B). In addition, the chemical probe displays a superior dynamic range in fluorescence lifetime imaging microscopy (FLIM) for intracellular FRET studies.
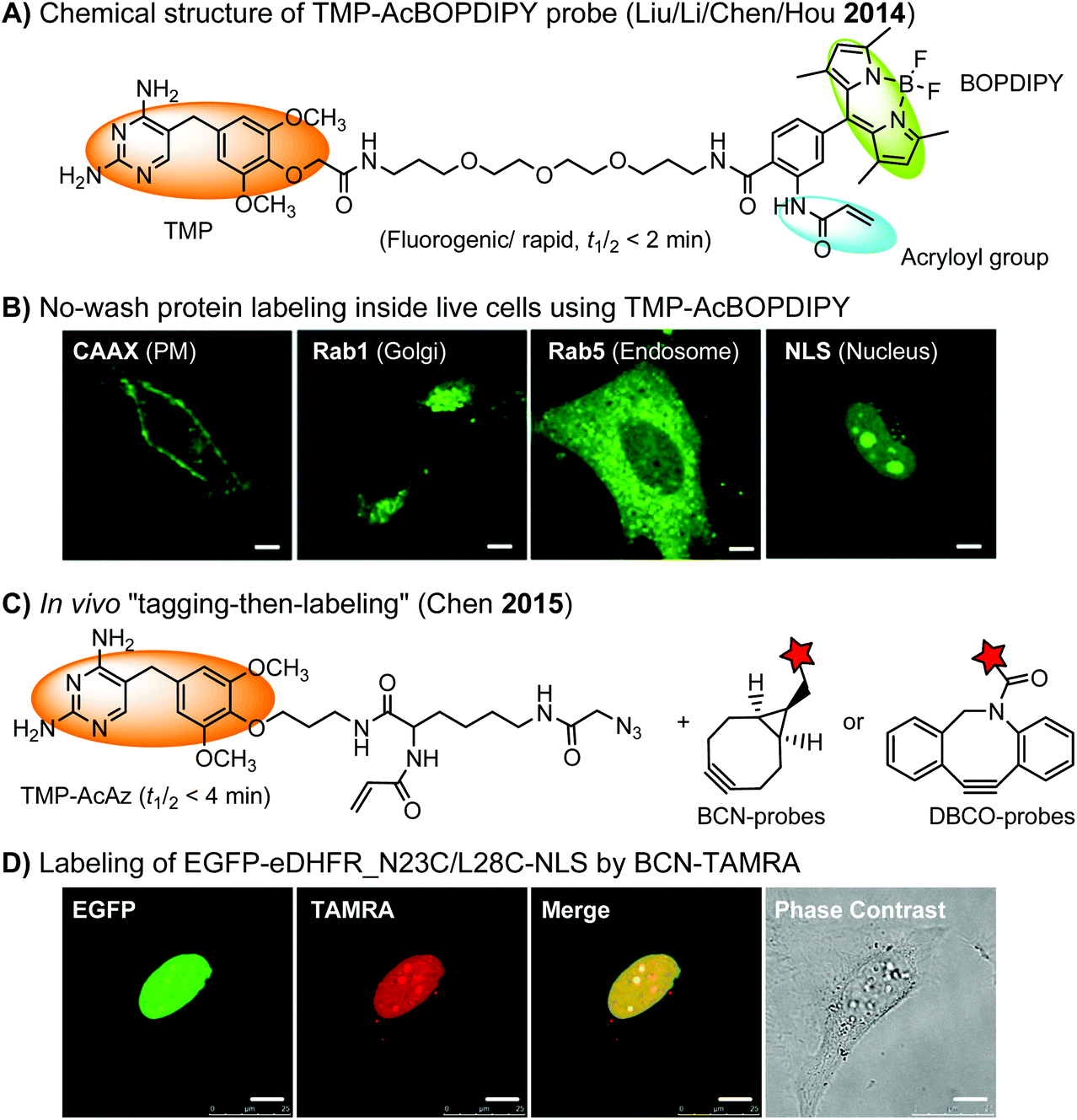 | ||
| Fig. 4 (A) The chemical structure of the TMP-AcBOPDIPY probe (BOPDIPY: boron phenyldipyrrolemethene). (B) Cellular labeling of eDHFR_N23C fused with a K-Ras C-terminal sequence (CAAX), Rab1, Rab5, and nucleus localizing sequence (NLS) at the plasma membrane (PM), the Golgi body, the endosomes, and the nucleus, respectively, using the TMP-AcBOPDIPY probe under no-wash conditions. (C) The “tagging-then-labeling” approach using the TMP-AcAz ligand and BCN or DBCO probes for labeling intracellular proteins in live cells. (D) Specific labeling of EGFP-eDHFR_N23C/L28C-NLS at the nucleus in living HeLa cells by the BCN-TAMRA probe. Scale bar: 10 μm. | ||
Recently, a more versatile “tagging-then-labeling” approach has been realized, enabling efficient introduction of bioorthogonal groups into proteins for bioorthogonal labeling in live cells. The TMP-AcAz ligand incorporates an azido group to proteins fused with the eDHFR tag (Fig. 4C). Subsequently, strain promoted cycloaddition reactions using DBCO- or BCN-conjugates facilitate protein labeling with various probes inside live cells (Fig. 4D).111
The eDHFR tag has been used for live-cell imaging of protein–protein interactions (PPIs) between the first PDZ domain of ZO-1 (fused with eDHFR) and the C-terminal YV motif of claudin-1 (fused with GFP) using time resolved luminescence resonance energy transfer (LRET) technique (Fig. 5).198 Conventional FRET imaging suffers from fluorescence breed-through, leading to high background. In the LRET approach, background signals from cellular auto-fluorescence and direct excitation of GFP were effectively eliminated by imposing a time delay of 10 μs between excitation and detection.
 | ||
| Fig. 5 Time resolved LRET for live-cell imaging of protein–protein interactions using the eDHFR tag. | ||
Photoactive yellow protein (PYP) is a small (14 kDa) soluble protein found in several purple bacteria. It binds to a natural cofactor, the CoA thioester of 4-hydroxycinnamic acid through transthioesterification with Cys69. It also binds to the thioester derivative of coumarin-3-carboxylic acid. Since PYP and its ligands do not exist in animal cells, they can therefore be employed for bioorthogonal labeling of proteins (k = 1.1–124 M−1 s−1).167,168
4.4. Self-labeling enzymatic domains
Enzyme-catalyzed reactions that proceed via irreversible conjugation with “suicide substrates” have been used for protein labeling in live cells. A variety of enzyme–substrate pairs are available. Many of these enzymes are able to tolerate modifications to their substrates. By generating fusion constructs with a protein, it is possible to covalently link the modified substrate to the protein. Generally, these reactions are specific, rapid and irreversible.O 6-Alkylguanine transferase (AGT), a human DNA repair protein, has been used as a self-labeling tag.169,199 The reaction involves the irreversible transfer of the alkyl group of O6-benzylguanine (BG) derivatives to the reactive cysteine residue within the enzyme to generate a covalently modified protein. More efficient AGT mutants, termed SNAP-tags (19 kDa), have been developed.169 An orthogonal AGT-based tag, termed a CLIP-tag, reacts specifically with O2-benzylcytosine derivatives.157
Single-molecule imaging often requires photo-stable and bright organic dyes, which are made possible using chemical protein labeling approaches. The spliceosome is a complex machine responsible for removing introns from the precursors of messenger RNAs (pre-mRNAs). The SNAP-tag has been exploited in combination with the eDHFR-tag to enable single-molecule imaging of the spliceosome in yeast cell extracts (Fig. 6A). The SNAP- and eDHFR-tags facilitate labeling pairs of the small nuclear ribonucleoprotein (snRNP) components of the spliceosome in cell extracts with bright organic dyes (TMP-Cy5 and BG-DY549), thereby enabling imaging of their assembly on individual pre-mRNAs. The measurements revealed that individual spliceosomal subcomplexes associate with pre-mRNA sequentially through an ordered pathway and that subcomplex binding is reversible.200
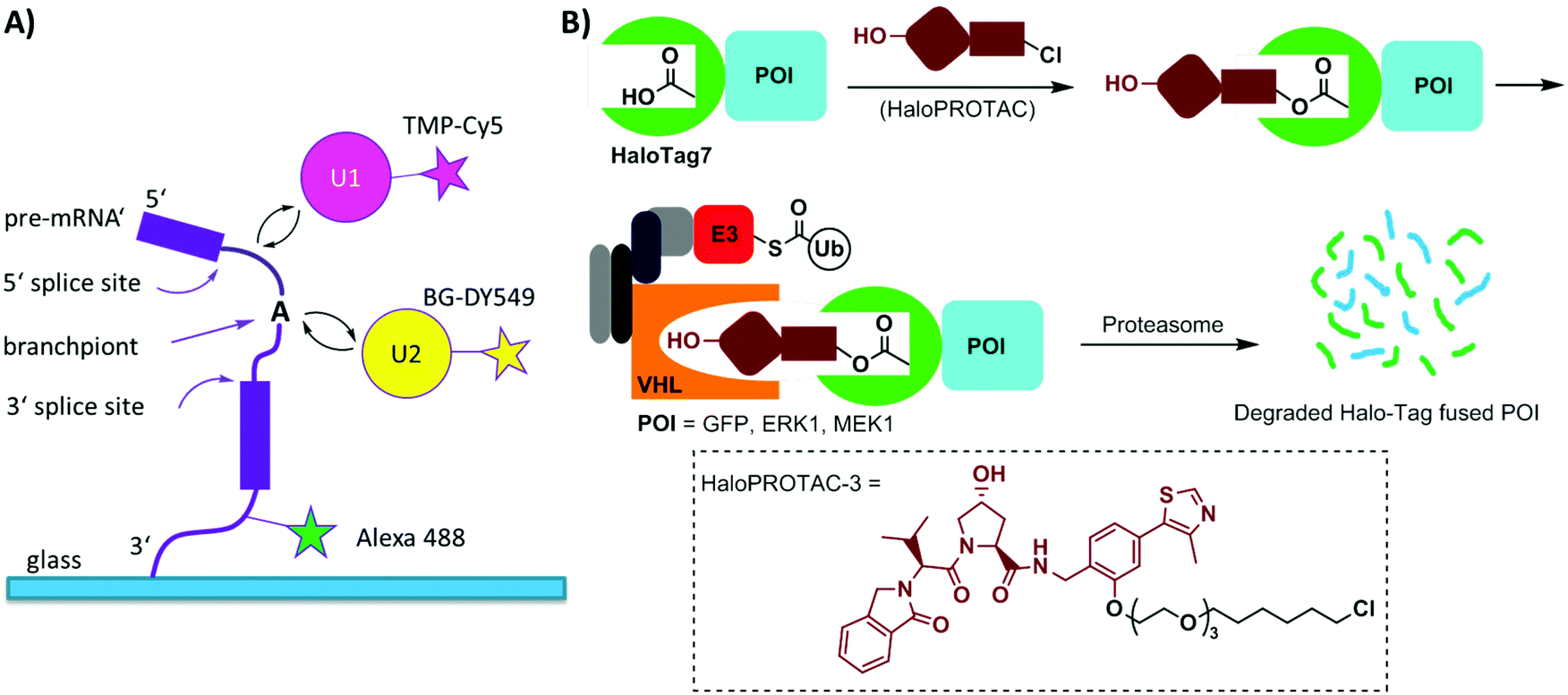 | ||
| Fig. 6 (A) Experimental setup for single-molecule tracing of pre-mRNA splicing in yeast cell extracts using SNAP tagging and TMP tagging technologies. (B) Schematic depiction of HaloPROTAC in inducing degradation of HaloTag7 fused proteins. The chemical structure of one representative HaloPROTAC, HaloPROTAC-3 is given. Ub = ubiquitin; E3 = E3 ligase. | ||
Halo-tag is a modified haloalkane dehalogenase that covalently binds to synthetic chloroalkane derivatives.171,201 Halo-tag is commercially available and has been widely used to label proteins in cells. This approach has been applied to small molecule-induced protein degradation, namely, HaloPROTACs. PROTACs are a class of heterobifunctional molecules that link a ligand of E3 ligase to a ligand for a protein of interest (POI).202 PROTACs recruit the E3 ligase to the POI, resulting in its ubiquitination and subsequent degradation by the proteasome. A bifunctional HaloPROTAC contains a chloroalkane and a hydroxyproline derivative which binds an E3 ligase VHL. The compound induces binding between the HaloTag7 fusion protein and the E3 ligase, leading to the degradation of the HaloTag7 fusion proteins by the proteasome (Fig. 6B).203
β-Lactamase is a small bacterial enzyme (29 kDa) that hydrolyzes β-lactam antibiotics. The E166N mutation of β-lactamase leads to the accumulation of acyl-enzyme intermediates due to the dramatic suppression of deacylation. The mutant β-lactamase, termed BL-tag, and β-lactam probes have been used for the covalent labeling of proteins in cells.170 Enormous efforts that have been made on β-lactam antibiotics render it possible to design various β-lactam probes.204 Cephalosporin-based probes featuring substituent elimination facilitate the development of fluorogenic probes.205 Other examples in this category include cutinase172 and catalytic antibodies (Abs)206 (Table 4D).
4.5. Enzymatic modifications
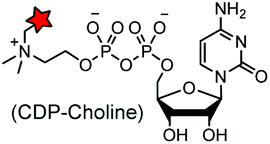
A number of post-translational modifications have been harnessed to specifically incorporate chemical probes or bioorthogonal handles into proteins (Table 4E). The first example is the modification of cell surface proteins using E. coli biotin ligase (BirA). The enzyme recognizes and biotinylates a 15-mer acceptor peptide (AP) sequence. Using a synthetic ketone-containing biotin isostere (keto-biotin) as a substrate, BirA can be used as a “ketone ligase” to introduce a ketone handle for oxime ligation.83 Another example is E. coli lipoic acid ligase, which is able to transfer a lipoic acid derivative carrying an azide178 or a TCO moiety to proteins.119 The introduced azide and TCO moieties facilitate subsequent labeling using strained cyclooctyne and terazine probes, respectively.119,178 The formylglycine-generating enzyme (FGE) specifically oxidizes the cysteine in the consensus LCXPXR motif to the formyl glycine. Subsequently, the aldehyde tag generated by the FGE on the target protein can selectively react with aminooxy and hydrazide probes.79 Reconstitution of the split inteins mediates protein trans splicing (PTS). After ligation of the N- and C-peptide fragments, the intein is eventually removed.181 By choosing appropriate naturally split inteins, PTS reactions can proceed efficiently in live cells.182,207 The bacterial enzyme AnkX from Legionella pneumophila has been used to transfer phosphocholine moieties from synthetically produced CDP-choline derivatives to a consensus sequence, TITSSYYR, at the termini or internal loop regions of a POI. The covalent labeling can be removed by another Legionella dephosphocholination enzyme Lem3.183Many other enzymes have also been used for the selective modification of proteins, including phosphopantetheine transferase (AcpS or Sfp),173–175,208 transglutaminases (TGases),176,177 sortase (SrtA),179 protein farnesyl transferase (PFTase),180 glycosyltransferase,209N-myristoyl transferase (NMT)210 and tubulin tyrosine ligase (TTL)184 (Table 4E). The advantages of enzymatic modifications lie in the small size of the tag and the highly efficient reactions. However, many of the substrates are not cell permeable and therefore are not suited for intracellular labeling.
The enzymatic modification approach has been elegantly applied to spatially-resolved proteomic mapping in living cells.211 An ascorbate peroxidase (APEX) fused with a “mito” sequence was targeted to the mitochondrial matrix. Labeling was initiated by the addition of biotin–phenol and H2O2 to live cells. The resulting phenoxyl radicals are short-lived and membrane-impermeant and therefore only label neighboring endogenous proteins. The biotinylated proteins were recovered with streptavidin-coated beads and identified using mass spectrometry (Fig. 7). This approach led to the identification of 495 proteins within the human mitochondrial matrix, including 31 proteins that were not previously linked to mitochondria.
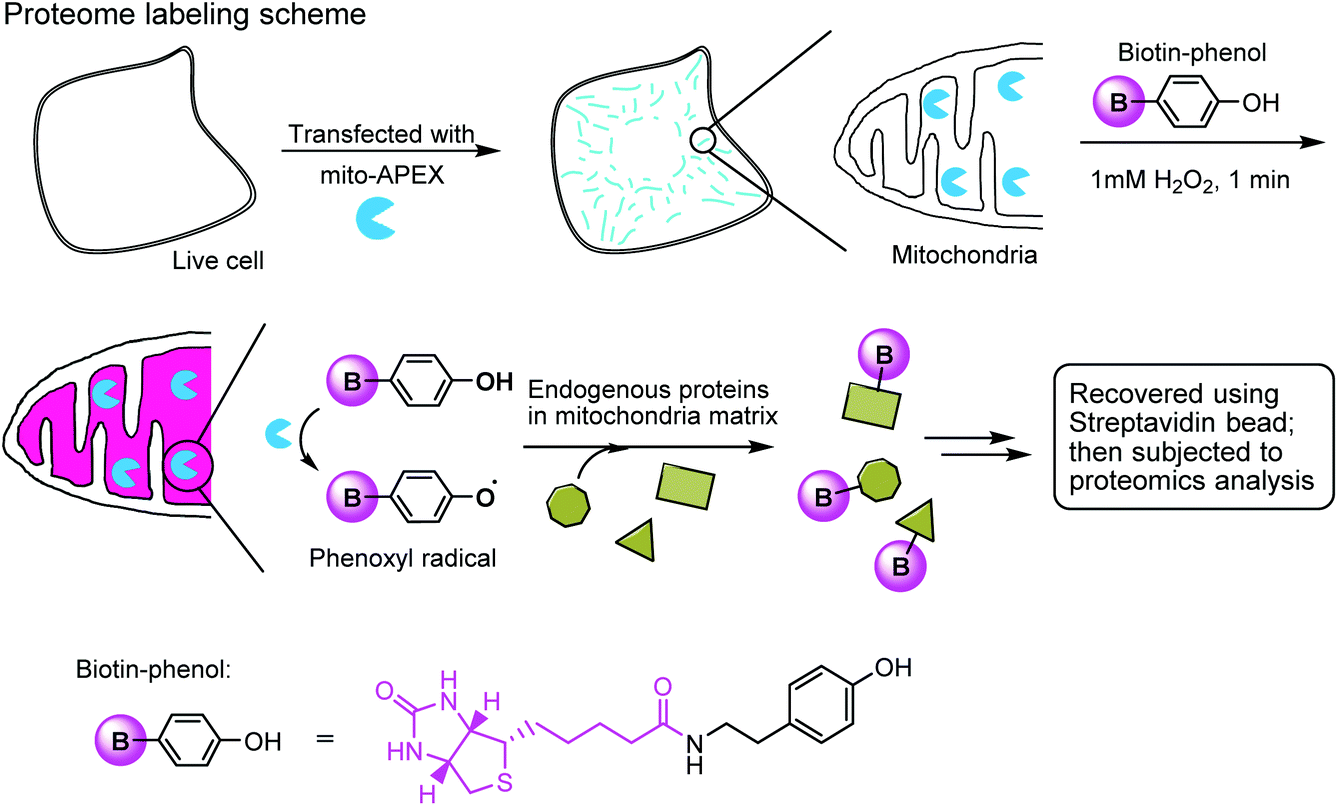 | ||
| Fig. 7 Labeling the mitochondrial matrix proteome in living cells using mito-APEX and biotin-phenol. | ||
4.6. Incorporation of UAA as a “minimalist” tag
Unnatural amino acid (UAA) mutagenesis has emerged as a powerful tool for the site-specific modification of proteins.212 The incorporation of UAAs into a protein sequence can be considered as the introduction of a “minimalist tag” (e.g. only an individual amino acid residue as opposed to a peptide sequence or a protein domain). Unnatural amino acids can be co-translationally incorporated into proteins in either a residue- or a site-specific fashion. The incorporation of UAAs carrying small bioorthogonal groups followed by chemoselective reactions makes it possible to label proteins with a diverse range of probes. A pool of UAAs featuring a variety of bioorthogonal handles has been added to the genetic codes of E. coli, yeast, and mammalian cells.3 This approach has been highly useful for super-resolution imaging, preparation of post-translationally modified proteins, and controlling protein dynamics and functions in living cells.46,68,117,213–215 This approach has also been used to make therapeutic proteins and to prepare a new generation of ADCs.134 More details about this topic are summarized in a recent review.35. Concluding remarks
Selective protein labeling techniques have provided unprecedented views of protein structures, dynamics and functions in vitro, in live cells, and in whole organisms. These approaches have demonstrated the power of chemistry as useful tools in a wide range of biological research areas, including post-translational modifications of proteins, preparation of protein-based pharmaceuticals, super-resolution imaging,117,216 visualization of intracellular protein–protein interactions, modulation of protein function in live cells, proteome labeling49 and affinity-based protein profiling.114Despite successful applications in selective protein labeling, numerous challenges remain in bioorthogonal chemistry. Firstly, many bioorthogonal functional groups are not truly “bioorthogonal”. For instance, strained alkynes may react with free thiols in live systems. Aldehyde and ketone functionalities are also present in many metabolites in living systems. Secondly, some of the bioorthogonal groups are too large and lipophilic, such as cyclooctynes and cyclooctenes, causing non-specific staining and reduction of effective reactants in live cells. Thirdly, the instability of tetrazine, phosphine and other groups in live cells during a prolonged incubation time leads to the deactivation of the bioorthogonal moieties. Fourthly, some metal-catalyzed reactions are incompatible with living conditions, as exemplified by the cytotoxicity of the copper ion used for CuAAC. To address these issues, the development of new bioorthogonal chemistry is definitely required. Since there are few “perfect” bioorthogonal reactions available, one has to carefully consider both the pros and cons of each reaction in order to identify the most suited ones for a particular application.
Chemical tagging approaches allow for the incorporation of probes or bioorthogonal handles into proteins in cells and organisms. However, most of these approaches rely on exogenous expression of the POI with tags or UAAs. Although the investigation of exogenous proteins is useful for unraveling biological processes, the advance in selective labeling of endogenous proteins should facilitate the proteomic analysis of cellular organelles and protein complexes, target identification and diagnosis. An advantage of chemical probes over FPs lies in the flexibility of modifications on organic dyes. Therefore, the development of new organic dyes with special properties, e.g. photo-switchable, activatable, highly fluorogenic, bright, far-red emissive, etc., should substantially help to understand the biological mechanisms of proteins in the context of living systems.
Acknowledgements
This work was supported by DFG grants (grant no. SPP 1623 and SFB 642 to Y. W. W.).References
- L. Spasser and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 6840–6862 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- K. M. Marks and G. P. Nolan, Nat. Methods, 2006, 3, 591–596 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- M. Vila-Perello and T. W. Muir, Cell, 2010, 143, 191–200 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- C. R. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS PubMed.
- Y. W. Wu and R. S. Goody, J. Pept. Sci., 2010, 16, 514–523 CrossRef CAS PubMed.
- J. Li and P. R. Chen, ChemBioChem, 2012, 13, 1728–1731 CrossRef CAS PubMed.
- H. W. Shih, D. N. Kamber and J. A. Prescher, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed.
- M. F. Debets, J. C. M. van Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2013, 11, 6439–6455 CAS.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- Y. K. Gong and L. F. Pan, Tetrahedron Lett., 2015, 56, 2123–2132 CrossRef CAS.
- R. K. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589–1600 RSC.
- J. M. Chalker, G. J. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed.
- P. Crisalli and E. T. Kool, J. Org. Chem., 2013, 78, 1184–1189 CrossRef CAS PubMed.
- T. Sasaki, K. Kodama, H. Suzuki, S. Fukuzawa and K. Tachibana, Bioorg. Med. Chem. Lett., 2008, 18, 4550–4553 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS PubMed.
- P. I. Kitov, D. F. Vinals, S. Ng, K. F. Tjhung and R. Derda, J. Am. Chem. Soc., 2014, 136, 8149–8152 CrossRef CAS PubMed.
- A. Ji, W. Ren and H. W. Ai, Chem. Commun., 2014, 50, 7469–7472 RSC.
- J. Alam, T. H. Keller and T. P. Loh, J. Am. Chem. Soc., 2010, 132, 9546–9548 CrossRef CAS PubMed.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- V. R. Pattabiraman, A. O. Ogunkoya and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5114–5118 CrossRef CAS PubMed.
- M. Raj, H. B. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 6932–6940 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2001, 3, 9–12 CrossRef CAS PubMed.
- A. Tam, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 11421–11430 CrossRef CAS PubMed.
- R. Serwa, I. Wilkening, G. Del Signore, M. Muhlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234–8239 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- S. S. van Berkel, A. T. J. Dirks, M. F. Debets, F. L. van Delft, J. J. L. M. Cornelissen, R. J. M. Nolte and F. P. J. T. Rutjes, ChemBioChem, 2007, 8, 1504–1508 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- X. H. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. C. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378–5384 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- T. S. Elliott, F. M. Townsley, A. Bianco, R. J. Ernst, A. Sachdeva, S. J. Elsasser, L. Davis, K. Lang, R. Pisa, S. Greiss, K. S. Lilley and J. W. Chin, Nat. Biotechnol., 2014, 32, 465–472 CrossRef CAS PubMed.
- S. B. Engelsma, L. I. Willems, C. E. van Paaschen, S. I. van Kasteren, G. A. van der Marel, H. S. Overkleeft and D. V. Filippov, Org. Lett., 2014, 16, 2744–2747 CrossRef CAS PubMed.
- A. Niederwieser, A. K. Spate, L. D. Nguyen, C. Jungst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- H. Stockmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Org. Biomol. Chem., 2011, 9, 7303–7305 Search PubMed.
- X. H. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G. J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065–3068 CrossRef CAS PubMed.
- N. A. McGrath and R. T. Raines, Chem. Sci., 2012, 3, 3237–3240 RSC.
- L. Josa-Cullere, Y. A. Wainman, K. M. Brindle and F. J. Leeper, RSC Adv., 2014, 4, 52241–52244 RSC.
- S. Wallace and J. W. Chin, Chem. Sci., 2014, 5, 1742–1744 RSC.
- E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2011, 133, 17570–17573 CrossRef CAS PubMed.
- K. Gutsmiedl, C. T. Wirges, V. Ehmke and T. Carell, Org. Lett., 2009, 11, 2405–2408 CrossRef CAS PubMed.
- Q. Li, T. Dong, X. H. Liu and X. G. Lei, J. Am. Chem. Soc., 2013, 135, 4996–4999 CrossRef CAS PubMed.
- X. Y. Zhang, T. Dong, Q. Li, X. H. Liu, L. Li, S. Chen and X. G. Lei, ACS Chem. Biol., 2015, 10, 1676–1683 CrossRef CAS PubMed.
- Y. Z. Wang, W. J. Hu, W. J. Song, R. K. V. Lint and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS PubMed.
- Y. Z. Wang, W. J. Song, W. J. Hu and Q. Lin, Angew. Chem., Int. Ed., 2009, 48, 5330–5333 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 7993–7995 RSC.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- K. Kodama, S. Fukuzawa, H. Nakayama, K. Sakamoto, T. Kigawa, T. Yabuki, N. Matsuda, M. Shirouzu, K. Takio, S. Yokoyama and K. Tachibana, ChemBioChem, 2007, 8, 232–238 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- M. E. Ourailidou, J. Y. van der Meer, B. J. Baas, M. Jeronimus-Stratingh, A. L. Gottumukkala, G. J. Poelarends, A. J. Minnaard and F. J. Dekker, ChemBioChem, 2014, 15, 209–212 CrossRef CAS PubMed.
- J. Li, J. T. Yu, J. Y. Zhao, J. Wang, S. Q. Zheng, S. X. Lin, L. Chen, M. Y. Yang, S. Jia, X. Y. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- Y. G. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- H. J. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. X. Xie, Z. Y. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- S. Shaunak, A. Godwin, J. W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS PubMed.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- E. T. Kool, D. H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- J. Stockigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS PubMed.
- H. C. Hang and C. R. Bertozzi, J. Am. Chem. Soc., 2001, 123, 1242–1243 CrossRef CAS PubMed.
- Y. Zeng, T. N. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, Nat. Methods, 2009, 6, 207–209 CrossRef CAS PubMed.
- I. Chen, M. Howarth, W. Y. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- M. F. Debets, C. W. van der Doelen, F. P. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- M. D. Best, Biochemistry, 2009, 48, 6571–6584 CrossRef CAS PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- D. C. Kennedy, C. S. McKay, M. C. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852–5856 CrossRef CAS PubMed.
- M. Y. Yang, A. S. Jalloh, W. Wei, J. Zhao, P. Wu and P. R. Chen, Nat. Commun., 2014, 5 Search PubMed , 4981.
- F. Xie, K. Sivakumar, Q. B. Zeng, M. A. Bruckman, B. Hodges and Q. Wang, Tetrahedron, 2008, 64, 2906–2914 CrossRef CAS.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed.
- Y. Urano, M. Kamiya, K. Kanda, T. Ueno, K. Hirose and T. Nagano, J. Am. Chem. Soc., 2005, 127, 4888–4894 CrossRef CAS PubMed.
- P. Shieh, M. J. Hangauer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 17428–17431 CrossRef CAS PubMed.
- P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137, 7145–7151 CrossRef CAS PubMed.
- Y. Su, J. Y. Ge, B. W. Zhu, Y. G. Zheng, Q. Zhu and S. Q. Yao, Curr. Opin. Chem. Biol., 2013, 17, 768–775 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097–3099 CrossRef CAS PubMed.
- B. Gold, N. E. Shevchenko, N. Bonus, G. B. Dudley and I. V. Alabugin, J. Org. Chem., 2012, 77, 75–89 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. T. Dirks, F. P. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. Rutjes, J. C. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- B. R. Varga, M. Kallay, K. Hegyi, S. Beni and P. Kele, Chem. – Eur. J., 2012, 18, 822–828 CrossRef CAS PubMed.
- C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed.
- E. M. Sletten, H. Nakamura, J. C. Jewett and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 11799–11805 CrossRef CAS PubMed.
- G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed.
- J. M. Medina, T. C. McMahon, G. Jimenez-Oses, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 14706–14709 CrossRef CAS PubMed.
- S. Jang, K. Sachin, H. J. Lee, D. W. Kim and H. S. Lee, Bioconjugate Chem., 2012, 23, 2256–2261 CrossRef CAS PubMed.
- B. Belardi, A. de la Zerda, D. R. Spiciarich, S. L. Maund, D. M. Peehl and C. R. Bertozzi, Angew. Chem., Int. Ed., 2013, 52, 14045–14049 CrossRef CAS PubMed.
- X. Chen, F. Li and Y. W. Wu, Chem. Commun., 2015, 51, 16537–16540 RSC.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904–17907 CrossRef CAS PubMed.
- Z. Li, D. Wang, L. Li, S. Pan, Z. Na, C. Y. Tan and S. Q. Yao, J. Am. Chem. Soc., 2014, 136, 9990–9998 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS PubMed.
- I. Nikic, T. Plass, O. Schraidt, J. Szymanski, J. A. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 CrossRef CAS PubMed.
- J. C. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920–922 CrossRef CAS PubMed.
- Z. Gao, V. Gouverneur and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 13612–13615 CrossRef CAS PubMed.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed.
- N. Li, R. K. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- Y. A. Lin and B. G. Davis, Beilstein J. Org. Chem., 2010, 6, 1219–1228 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805–16811 CrossRef CAS PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- S. Sechi and B. T. Chait, Anal. Chem., 1998, 70, 5150–5158 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernandez-Gonzalez, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835–1839 CrossRef CAS PubMed.
- T. J. Tolbert and C. H. Wong, Angew. Chem., Int. Ed., 2002, 41, 2171–2174 CrossRef CAS.
- A. Godinat, H. M. Park, S. C. Miller, K. Cheng, D. Hanahan, L. E. Sanman, M. Bogyo, A. Yu, G. F. Nikitin, A. Stahl and E. A. Dubikovskaya, ACS Chem. Biol., 2013, 8, 987–999 CrossRef CAS PubMed.
- P. M. Drake and D. Rabuka, Curr. Opin. Chem. Biol., 2015, 28, 174–180 CrossRef CAS PubMed.
- Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2013, 52, 4088–4106 CrossRef CAS PubMed.
- H. B. F. Dixon, J. Protein Chem., 1984, 3, 99–108 CrossRef CAS.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- A. O. Y. Chan, C. M. Ho, H. C. Chong, Y. C. Leung, J. S. Huang, M. K. Wong and C. M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS PubMed.
- X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjugate Chem., 2012, 23, 500–508 CrossRef CAS PubMed.
- X. Chen, L. Henschke, Q. Wu, K. Muthoosamy, B. Neumann and T. Weil, Org. Biomol. Chem., 2013, 11, 353–361 CAS.
- M. Lillich, X. Chen, T. Weil, H. Barth and J. Fahrer, Bioconjugate Chem., 2012, 23, 1426–1436 CrossRef CAS PubMed.
- T. Wang, D. Y. W. Ng, Y. Z. Wu, J. Thomas, T. TamTran and T. Weil, Chem. Commun., 2014, 50, 1116–1118 RSC.
- M. Kunishima, S. Nakanishi, J. Nishida, H. Tanaka, D. Morisaki, K. Hioki and H. Nomoto, Chem. Commun., 2009, 5597–5599 RSC.
- Y. Koshi, E. Nakata, M. Miyagawa, S. Tsukiji, T. Ogawa and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 245–251 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed.
- S. Tsukiji, H. X. Wang, M. Miyagawa, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 9046–9054 CrossRef CAS PubMed.
- H. X. Wang, Y. Koshi, D. Minato, H. Nonaka, S. Kiyonaka, Y. Mori, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2011, 133, 12220–12228 CrossRef CAS PubMed.
- T. Tamura, Y. Kioi, T. Miki, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2013, 135, 6782–6785 CrossRef CAS PubMed.
- S. H. Fujishima, R. Yasui, T. Miki, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 3961–3964 CrossRef CAS PubMed.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- E. G. Guignet, R. Hovius and H. Vogel, Nat. Biotechnol., 2004, 22, 440–444 CrossRef CAS PubMed.
- R. Wieneke, N. Laboria, M. Rajan, A. Kollmannsperger, F. Natale, M. C. Cardoso and R. Tampe, J. Am. Chem. Soc., 2014, 136, 13975–13978 CrossRef CAS PubMed.
- C. T. Hauser and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3693–3697 CrossRef CAS PubMed.
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452–10459 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa Jr., M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2009, 131, 438–439 CrossRef CAS PubMed.
- G. M. Eldridge and G. A. Weiss, Bioconjugate Chem., 2011, 22, 2143–2153 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- Z. Chen, B. V. Popp, C. L. Bovet and Z. T. Ball, ACS Chem. Biol., 2011, 6, 920–925 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, S. Zernia, K. Morl, A. G. Beck-Sickinger and O. Seitz, Angew. Chem., Int. Ed., 2014, 53, 10237–10241 CrossRef CAS PubMed.
- T. Clackson, W. Yang, L. W. Rozamus, M. Hatada, J. F. Amara, C. T. Rollins, L. F. Stevenson, S. R. Magari, S. A. Wood, N. L. Courage, X. Lu, F. Cerasoli Jr., M. Gilman and D. A. Holt, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10437–10442 CrossRef CAS.
- K. M. Marks, P. D. Braun and G. P. Nolan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9982–9987 CrossRef CAS PubMed.
- L. W. Miller, Y. Cai, M. P. Sheetz and V. W. Cornish, Nat. Methods, 2005, 2, 255–257 CrossRef CAS PubMed.
- W. Liu, F. Li, X. Chen, J. Hou, L. Yi and Y. W. Wu, J. Am. Chem. Soc., 2014, 136, 4468–4471 CrossRef CAS PubMed.
- Y. Hori, H. Ueno, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 16610–16611 CrossRef CAS PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360–12365 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- S. Mizukami, S. Watanabe, Y. Hori and K. Kikuchi, J. Am. Chem. Soc., 2009, 131, 5016–5017 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- R. Bonasio, C. V. Carman, E. Kim, P. T. Sage, K. R. Love, T. R. Mempel, T. A. Springer and U. H. von Andrian, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14753–14758 CrossRef CAS PubMed.
- N. George, H. Pick, H. Vogel, N. Johnsson and K. Johnsson, J. Am. Chem. Soc., 2004, 126, 8896–8897 CrossRef CAS PubMed.
- J. Yin, P. D. Straight, S. M. McLoughlin, Z. Zhou, A. J. Lin, D. E. Golan, N. L. Kelleher, R. Kolter and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820 CrossRef CAS PubMed.
- Z. Zhou, P. Cironi, A. J. Lin, Y. Q. Xu, S. Hrvatin, D. E. Golan, P. A. Silver, C. T. Walsh and J. Yin, ACS Chem. Biol., 2007, 2, 337–346 CrossRef CAS PubMed.
- H. Sato, M. Ikeda, K. Suzuki and K. Hirayama, Biochemistry, 1996, 35, 13072–13080 CrossRef CAS PubMed.
- C. W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542–4543 CrossRef CAS PubMed.
- M. Fernandez-Suarez, H. Baruah, L. Martinez-Hernandez, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef CAS PubMed.
- M. W. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707–708 CrossRef CAS PubMed.
- J. W. Wollack, J. M. Silverman, C. J. Petzold, J. D. Mougous and M. D. Distefano, ChemBioChem, 2009, 10, 2934–2943 CrossRef CAS PubMed.
- I. Giriat and T. W. Muir, J. Am. Chem. Soc., 2003, 125, 7180–7181 CrossRef CAS PubMed.
- V. Schutz and H. D. Mootz, Angew. Chem., Int. Ed., 2014, 53, 4113–4117 CrossRef PubMed.
- K. Heller, P. Ochtrop, M. F. Albers, F. B. Zauner, A. Itzen and C. Hedberg, Angew. Chem., Int. Ed., 2015, 54, 10327–10330 CrossRef CAS PubMed.
- D. Schumacher, J. Helma, F. A. Mann, G. Pichler, F. Natale, E. Krause, M. C. Cardoso, C. P. R. Hackenberger and H. Leonhardt, Angew. Chem., Int. Ed., 2015, 54, 13787–13791 CrossRef CAS PubMed.
- S. H. Uchinomiya, H. Nonaka, S. H. Fujishima, S. Tsukiji, A. Ojida and I. Hamachi, Chem. Commun., 2009, 5880–5882 RSC.
- K. Honda, S. H. Fujishima, A. Ojida and I. Hamachi, ChemBioChem, 2007, 8, 1370–1372 CrossRef CAS PubMed.
- K. Honda, E. Nakata, A. Ojida and I. Hamachi, Chem. Commun., 2006, 4024–4026 RSC.
- H. Nonaka, S. Tsukiji, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 15777–15779 CrossRef CAS PubMed.
- C. R. Goldsmith, J. Jaworski, M. Sheng and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 418–419 CrossRef CAS PubMed.
- S. Uchinomiya, H. Nonaka, S. Wakayama, A. Ojida and I. Hamachi, Chem. Commun., 2013, 49, 5022–5024 RSC.
- G. Gaietta, T. J. Deerinck, S. R. Adams, J. Bouwer, O. Tour, D. W. Laird, G. E. Sosinsky, R. Y. Tsien and M. H. Ellisman, Science, 2002, 296, 503–507 CrossRef CAS PubMed.
- B. R. Martin, B. N. Giepmans, S. R. Adams and R. Y. Tsien, Nat. Biotechnol., 2005, 23, 1308–1314 CrossRef CAS PubMed.
- M. Andresen, R. Schmitz-Salue and S. Jakobs, Mol. Biol. Cell, 2004, 15, 5616–5622 CrossRef CAS PubMed.
- C. Hoffmann, G. Gaietta, A. Zurn, S. R. Adams, S. Terrillon, M. H. Ellisman, R. Y. Tsien and M. J. Lohse, Nat. Protoc., 2010, 5, 1666–1677 CrossRef CAS PubMed.
- W. Ju, W. Morishita, J. Tsui, G. Gaietta, T. J. Deerinck, S. R. Adams, C. C. Garner, R. Y. Tsien, M. H. Ellisman and R. C. Malenka, Nat. Neurosci., 2004, 7, 244–253 CrossRef CAS PubMed.
- S. S. Gallagher, J. E. Sable, M. P. Sheetz and V. W. Cornish, ACS Chem. Biol., 2009, 4, 547–556 CrossRef CAS PubMed.
- Z. X. Chen, C. R. Jing, S. S. Gallagher, M. P. Sheetz and V. W. Cornish, J. Am. Chem. Soc., 2012, 134, 13692–13699 CrossRef CAS PubMed.
- H. E. Rajapakse, N. Gahlaut, S. Mohandessi, D. Yu, J. R. Turner and L. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13582–13587 CrossRef CAS PubMed.
- A. Juillerat, T. Gronemeyer, A. Keppler, S. Gendreizig, H. Pick, H. Vogel and K. Johnsson, Chem. Biol., 2003, 10, 313–317 CrossRef CAS PubMed.
- A. A. Hoskins, L. J. Friedman, S. S. Gallagher, D. J. Crawford, E. G. Anderson, R. Wombacher, N. Ramirez, V. W. Cornish, J. Gelles and M. J. Moore, Science, 2011, 331, 1289–1295 CrossRef CAS PubMed.
- Y. Zhang, M. K. So, A. M. Loening, H. Yao, S. S. Gambhir and J. Rao, Angew. Chem., Int. Ed., 2006, 45, 4936–4940 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, ACS Chem. Biol., 2015, 10, 1831–1837 CrossRef CAS PubMed.
- S. Mizukami, Y. Hori and K. Kikuchi, Acc. Chem. Res., 2014, 47, 247–256 CrossRef CAS PubMed.
- S. Mizukami, S. Watanabe, Y. Akimoto and K. Kikuchi, J. Am. Chem. Soc., 2012, 134, 1623–1629 CrossRef CAS PubMed.
- R. K. Goswami, Z. Z. Huang, J. S. Forsyth, B. Felding-Habermann and S. C. Sinha, Bioorg. Med. Chem. Lett., 2009, 19, 3821–3824 CrossRef CAS PubMed.
- I. V. Thiel, G. Volkmann, S. Pietrokovski and H. D. Mootz, Angew. Chem., Int. Ed., 2014, 53, 1306–1310 CrossRef CAS PubMed.
- J. Yin, F. Liu, X. H. Li and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 7754–7755 CrossRef CAS PubMed.
- S. DeFrees, Z. G. Wang, R. Xing, A. E. Scott, J. Wang, D. Zopf, D. L. Gouty, E. R. Sjoberg, K. Panneerselvam, E. C. Brinkman-Van der Linden, R. J. Bayer, M. A. Tarp and H. Clausen, Glycobiology, 2006, 16, 833–843 CrossRef CAS PubMed.
- H. C. Hang, E. J. Geutjes, G. Grotenbreg, A. M. Pollington, M. J. Bijlmakers and H. L. Ploegh, J. Am. Chem. Soc., 2007, 129, 2744–2745 CrossRef CAS PubMed.
- H. W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328–1331 CrossRef CAS PubMed.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS PubMed.
- H. Xiao, A. Chatterjee, S. H. Choi, K. M. Bajjuri, S. C. Sinha and P. G. Schultz, Angew. Chem., Int. Ed., 2013, 52, 14080–14083 CrossRef CAS PubMed.
- D. T. Rogerson, A. Sachdeva, K. H. Wang, T. Haq, A. Kazlauskaite, S. M. Hancock, N. Huguenin-Dezot, M. M. K. Muqit, A. M. Fry, R. Bayliss and J. W. Chin, Nat. Chem. Biol., 2015, 11, 496–503 CrossRef CAS PubMed.
- Y. H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- G. Lukinavicius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Correa Jr., Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |