
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative coupling between C(sp2)–H and C(sp3)–H bonds of indoles and cyclic ethers/cycloalkanes†
Qingjing
Yang
a,
Pui Ying
Choy
a,
Yinuo
Wu
*b,
Baomin
Fan
*c and
Fuk Yee
Kwong
*a
aState Key Laboratory of Chirosciences and Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China. E-mail: fuk-yee.kwong@polyu.edu.hk
bSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, People's Republic of China. E-mail: wuyinuo3@mail.sysu.edu.cn
cYMU-HKBU Joint Laboratory of Traditional Natural Medicine, Yunnan Minzu University, Kunming 650500, People's Republic of China. E-mail: adams.bmf@hotmail.com
First published on 28th January 2016
Abstract
Cross-dehydrogenative-coupling (CDC) between C–H/C–H bonds of indoles and cyclic ethers/cycloalkanes is made viable through a simple transition-metal-free pathway. With the aid of only di-tert-butyl peroxide, a number of inactive cyclic ethers and cycloalkanes can be directly coupled with indole derivatives in satisfactory yields.
Direct cross-dehydrogenative-coupling (CDC) reactions of two different C–H bonds under oxidative conditions have emerged as one of the most effective and straightforward strategies for constructing C–C bonds in organic synthesis.1 This approach is highly desirable as the direct engagement of naturally abundant C–H starting materials together can circumvent the prefunctionalization/preactivation of substrates and therefore lead to a better atom economy.2 Yet, the challenges of CDC reactions are low reactivity and selectivity, owing to the high dissociation energy and the ubiquity of C–H bonds, respectively. Transition metal complexes are employed to overcome these difficulties3 and the development of new catalyst systems for more efficient transformations is indeed a central theme in modern organic synthesis.
The indolyl framework is a common sub-unit in various pharmaceutically attractive and naturally occurring products.4 Particularly, the relevant indolyl motifs exhibit unique anti-inflammatory and phosphodiesterase inhibitory activities.5 Therefore, facile convergent approaches for accessing an array of C-3 or C-2 alkylated indole derivatives are in high demand. Nevertheless, the reported procedures are sometimes cumbersome and/or require costly catalysts/substrates (Scheme 1A).6 In fact, the direct C–C bond construction of α-substituted cyclic ethers (e.g. five- or six-membered ring) or cycloalkanes to the C-3 or C-2 position of indoles is still rare by virtue of the low reactivity of these C–H bonds. The transition-metal-catalyzed C–C bond formation of ethers/cycloalkanes with heterocycles via cross-dehydrogenative-coupling (CDC) reactions has received considerable attention and advanced remarkably (Scheme 1B).7 However, C–C coupling of indoles with ethers/cycloalkanes is far less explored.8 In 2015, Cai reported a nickel-catalyzed regioselective CDC of inactive C(sp3)–H bonds with indole derivatives.9 Regiospecific C-3 or C-2 coupling of indoles with 1,4-dioxane was shown with the aid of a Ni(acac)2 or NiF2 complex (Scheme 1B). Apart from the transition metal-catalyzed pathway, the CDC reaction would be further attractive if this reaction can proceed in a metal-free manner.10 Nevertheless, a simple metal-free oxidative coupling between indoles and ethers/cycloalkanes remains sporadically studied. Cai showed that di-tert-butyl peroxide (DTBP) mediated oxidative coupling of isochroman with indole derivatives.11 Tian and Li reported a KOt-Bu-mediated coupling of indoles and [60]fullerene with high regioselectivity at the C-3 position of indoles.12 Continuing our research interest on metal-free coupling reactions,13 C–H functionalization of heteroarene14 and CDC reaction of ortho-acylaniline synthesis,15 herein we demonstrate a metal-free protocol for coupling of cyclic ethers or cycloalkanes with the C-3 or C-2 position of indole derivatives using di-tert-butyl peroxide (DTBP) as the oxidant (Scheme 1C).
 | ||
| Scheme 1 Selected examples of preparation of ethereal azoles. | ||
We first started our investigation by using indole 1a and 1,4-dioxane (2a) as the model substrates (Table 1). A screening of oxidants showed that no reaction occurred when commonly used oxidants such as tert-butyl hydroperoxide (TBHP), benzoyl peroxide (BPO) and potassium persulfate (K2S2O8) were employed (Table 1, entries 3–5). To our delight, di-tert-butyl peroxide (DTBP) and tert-butylperoxybenzoate (TBPB) gave the desired product 3a in 69% and 45% yields, respectively (entries 1 and 2). There were no improvements when CuI, KI, Bu4NI or Pd(OAc)2 was added as the catalyst (entries 7–10). Control experiments revealed that the reaction did not proceed without DTBP, which suggested the crucial importance of peroxide in this transformation (entry 6). Increasing the amount of DTBP or lowering the reaction temperature led to the decrease of the product yield (entries 11 vs. 12 vs. 14). The optimal stoichiometry of DTBP employed was 1.5 equivalents with respect to 2a (entry 1). The best yield was obtained under a nitrogen atmosphere (entry 13).
|
|
|||
|---|---|---|---|
| Entry | [Cat.] (10 mol%) | Oxidants (equiv.) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (2 mL), [Cat.] (10% mol) and oxidant (0.75 mmol) were stirred at 140 °C in air for 20 hours. (DTBP = di-tert-butyl peroxide, TBPB = tert-butylperoxybenzoate, TBHP = tert-butyl hydroperoxide (70% in aqueous solution), BPO = benzoyl peroxide.) b Isolated yield. c Under a nitrogen atmosphere. d 120 °C was used. e 1 mL of 2a was used. | |||
| 1 | — | DTBP (1.5) | 69 |
| 2 | — | TBPB (1.5) | 45 |
| 3 | — | TBHP (1.5) | 0 |
| 4 | — | K2S2O8 (1.5) | 0 |
| 5 | — | BPO (1.5) | 0 |
| 6 | — | DTBP (0) | 0 |
| 7 | Pd(OAc)2 | DTBP (1.5) | 68 |
| 8 | CuI | DTBP (1.5) | 69 |
| 9 | KI | DTBP (1.5) | 65 |
| 10 | Bu4NI | DTBP (1.5) | 35 |
| 11 | — | DTBP (1.0) | 61 |
| 12 | — | DTBP (2.0) | 58 |
| 13 | — | DTBP (1.5) | 78 |
| 14 | — | DTBP (1.5) | 74c,d |
| 15 | — | DTBP (1.5) | 61e |

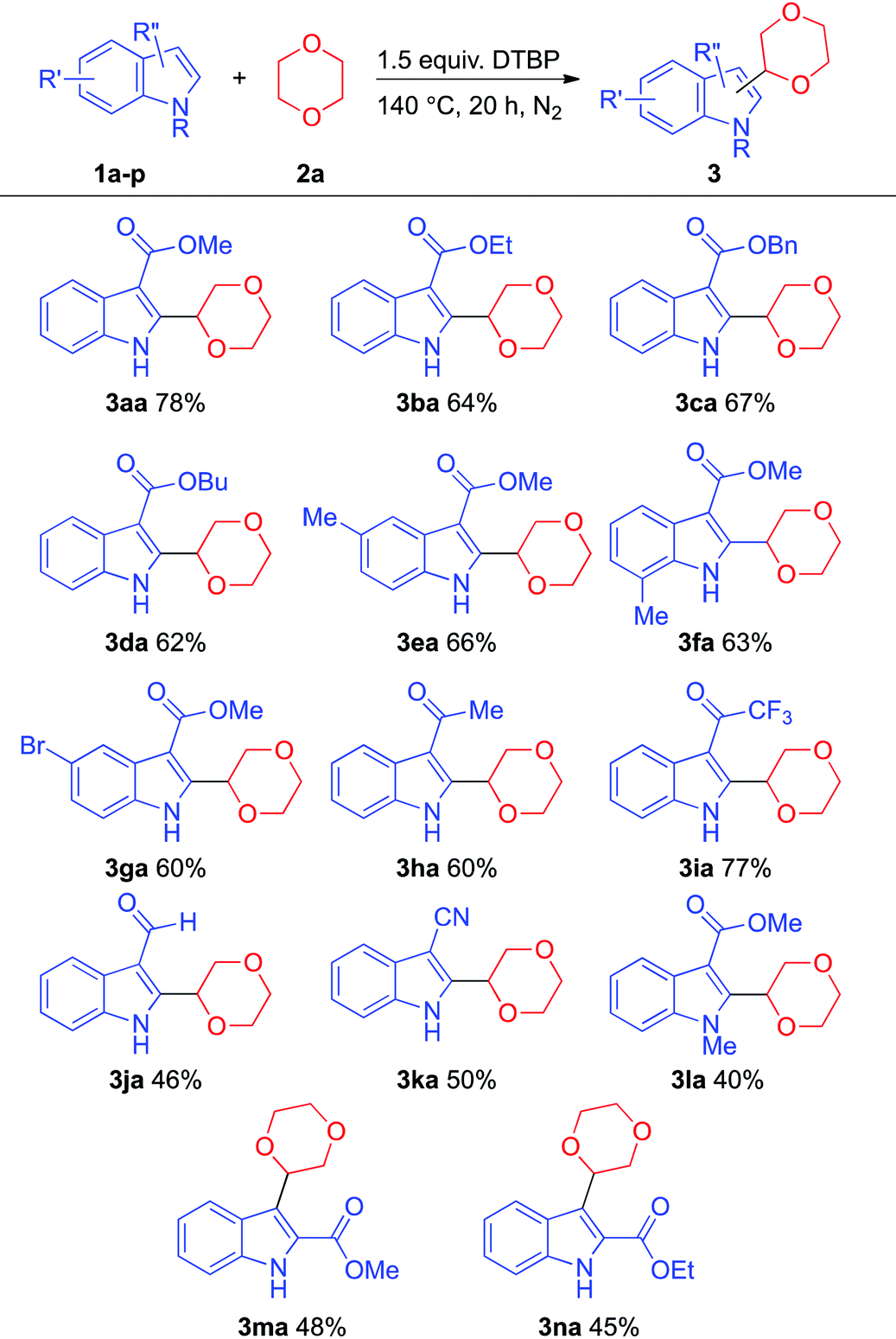
With the optimized reaction conditions in hand, we next tested the scope of this transformation (Table 2). A series of unprotected N–H indole derivatives coupled well with 1,4-dioxane to give the corresponding products in good yields. When the bulkiness of the ester-substituted group was increased, the yield of the desired product decreased (e.g., product 3aavs. product 3da). To the best of our knowledge, there have been no successful examples of indoles containing ketone and cyano groups in the oxidative C–C coupling with cyclic ethers. Gratifyingly, the reaction protocol displayed a good functional group tolerance, and bromo (e.g., product 3ga), keto (e.g., product 3ha, 3ia), and cyano groups (e.g., product 3ka) were found compatible under these conditions. Surprisingly, the aldehyde group remained intact under these reaction conditions with moderate yield (e.g., product 3ja). N-Protected indole derivatives were suitable substrates for this reaction with a lower yield possibly due to the steric effect (e.g., product 3la). Apart from the C–C bond formation at the C-2 position of indoles, C-3 target products could also be afforded under this protocol (e.g., products 3oa and 3pa).
| a Reaction conditions: substituted indoles 1a–1p (0.5 mmol), 1,4-dioxane (2a) (2 mL), and DTBP (0.75 mmol, 1.5 equiv.) were stirred at 140 °C under N2 for 20 hours. Isolated yields are reported. Reaction times are not optimized for each substrate. |
|---|
|
We next turned our attention to extend the substrate scope regarding the ether coupling partner and the results are compiled in Table 3. Apart from 1,4-dioxane, other ethereal entities were tested. Cyclic ethers such as tetrahydrofuran (2b), 1,3-dioxane (2c), and 1,3-dioxolane (2d) coupled smoothly with moderate yields (e.g., products 3ab, 3ac, and 3ad). Acyclic ethers including diethoxymethane (2e) and dibutyl ether (2f) also underwent the coupling and gave the corresponding coupled products 3ae and 3af in 50% and 74% isolated yields, respectively. In order to evaluate the scope of this system, we attempted to use cycloalkanes as the coupling partners instead of cyclic ethers. When cyclohexane was used, the desired coupling product was obtained in 60% yield (e.g., product 3ah). Cycloheptane and cyclooctane were also feasible coupling partners to yield the desired products (e.g., products 3ah and 3ai). It is noteworthy that the bulky adamantane also underwent the target reaction (e.g., product 3aj).
| a Reaction conditions: substituted indole 1a or 1o (0.5 mmol), ether or cycloalkane 2a–j (2 mL), and DTBP (0.75 mmol, 1.5 equiv.) were stirred at 140 °C under N2 for 20 hours. Isolated yields are reported. Reaction times are not optimized for each substrate. b 2.0 mmol of DTBP was used. c 5 equivalents of 1j were used with 2 mL of xylene as a solvent. |
|---|

|
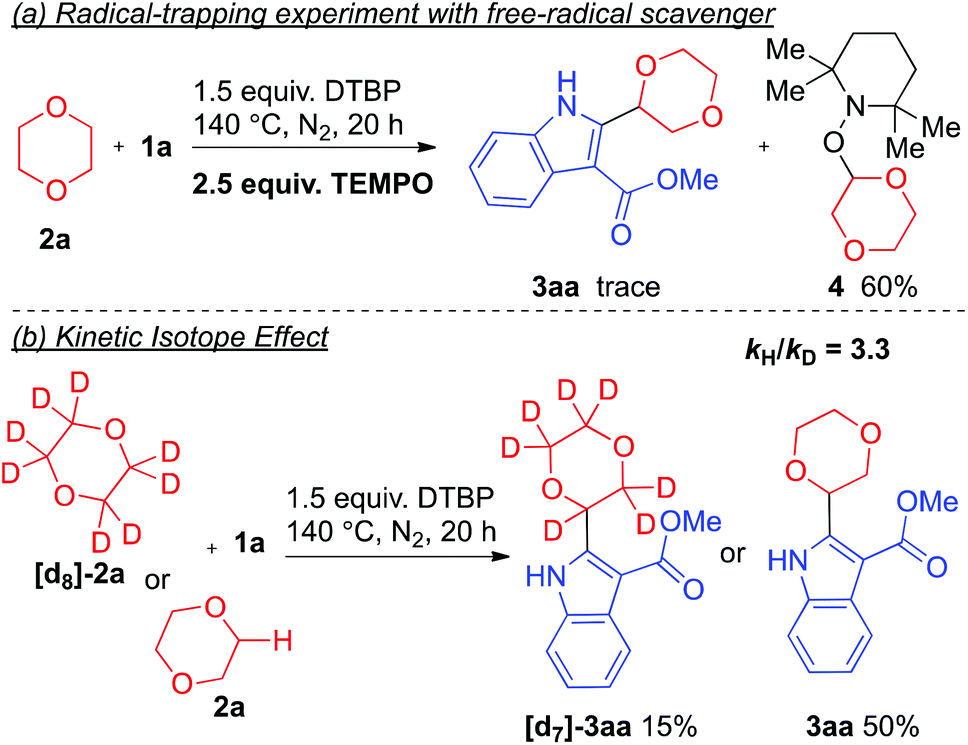
To verify whether the reaction proceeded through a radical pathway, a radical trapping experiment was conducted. When 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) was added, a drastic suppression of the reaction resulted and only a trace amount of the desired product 3aa was detected whereas the 1,4-dioxane-TEMPO coupling product was isolated (Scheme 2a). These results suggested that this reaction likely proceeds via a free-radical intermediate. Meanwhile, a kinetic isotopic effect (KIE) experiment was also performed for probing the dependence of C–H bond cleavage (Scheme 2b). As is depicted, a significant KIE was observed with kH/kD = 3.3. This result indicated that the –C–H bond cleavage of 1,4-dioxane could be the kinetically-dependent rate-limiting step of this reaction. Based on the reported literature,8 it is believed that a tert-butoxyl radical was generated by homolytic cleavage through thermal decomposition of di-tert-butyl peroxide. Then, the tert-butoxyl radical undergoes a hydrogen abstraction of the α-proton adjacent to the oxygen atom of the cyclic ether to afford an ether radical. The radical species further undergoes addition to the indole derivative, followed by SET leading to the oxidative coupling final product.
 | ||
| Scheme 2 Control experiments for the CDC reaction of 1,4-dioxane with indole 1a. | ||
Conclusions
In summary, we have demonstrated an effective metal-free method for a cross-dehydrogenative-coupling reaction between indole derivatives and ethers/cycloalkanes. The usage of rich feedstock starting materials (e.g. non-prefunctionalized simple cyclic ethers and cycloalkanes), good compatibility of functional groups (e.g. bromo, keto, nitrile, ester, N–H amino and aldehyde) and particularly without the need of transition metal catalysts underline the advantage of this straightforward protocol. This also shows the first examples of the CDC reaction between indole derivatives and cycloalkanes under metal-free reaction conditions.Acknowledgements
We thank the Research Grants Council of Hong Kong, Collaborative Research Fund (CRF: C5023-14G), and General Research Fund (GRF: PolyU 153008/14P) for financial support. Grateful to Prof. Albert S. C. Chan's research group (PolyU Hong Kong) for sharing of GC-FID and GC-MS instruments. We especially thank Dr Man-Kin Wong (PolyU Hong Kong) for providing some starting materials and helpful suggestions.Notes and references
- For selected reviews on oxidative C–H functionalization/cross-dehydrogenative-coupling, see: (a) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (b) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459 CrossRef CAS PubMed; (c) Z.-J. Shi and B.-J. Li, Chem. Soc. Rev., 2012, 41, 5588 RSC; (d) C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (e) C. L. Sun, B. J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (f) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC; (g) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (h) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed. For a recent book of cross-dehydrogenative-coupling, see: (i) RSC Green Chemistry Series, From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling, ed. C.-J. Li, The Royal Society of Chemistry, 2015 Search PubMed.
- For the concept atom economy, see: B. M. Trost, Science, 1991, 254, 1471 CAS.
- For recent selected publications/book chapter/review on TM-catalyzed CDC reactions, see: (a) S. Guo, Y. Li, Y. Wang, X. Guo, X. Meng and B. Chen, Adv. Synth. Catal., 2015, 357, 950 CrossRef CAS; (b) I. Marek and Z. Rappoport, Chemistry of Organoiron Compounds, ed. N. Yoshikai, John Wiley & Sons, 2014 Search PubMed; (c) Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem. – Asian J., 2014, 9, 26 CrossRef CAS PubMed.
- (a) G. W. Gribble, Topics in Heterocyclic Chemistry, ed. B. U. W. Maes, Springer, New York, 2010, vol. 26 Search PubMed; (b) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, Wiley-VCH, Weinheim, 2nd edn, 2003 CrossRef.
- (a) G. Dyker, D. Hildebrandt, J. H. Liu and K. Merz, Angew. Chem., Int. Ed., 2003, 42, 4399 CrossRef CAS PubMed; (b) E. M. Ferreira and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 9578 CrossRef CAS PubMed; (c) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (d) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 105, 2875 CrossRef PubMed; (e) D. Crich and A. Banerjee, Acc. Chem. Res., 2007, 40, 151 CrossRef CAS PubMed.
- (a) B. Z. Lu, H.-X. Wei, Y. Zhang, W. Zhao, M. Dufour, G. Li, V. Farina and C. H. Senanayake, J. Org. Chem., 2013, 78, 4558 CrossRef CAS PubMed; (b) Y. Wang, L. Ye and L. Zhang, Chem. Commun., 2011, 47, 7815 RSC; (c) A. Zanardi, J. A. Mata and E. Peris, Chem. – Eur. J., 2010, 16, 13109 CrossRef CAS PubMed; (d) N. T. Patil, V. Singh, A. Konala and A. K. Mutyala, Tetrahedron Lett., 2010, 51, 1493 CrossRef CAS; (e) W. Kong, J. Cui, Y. Yu, G. Chen, C. Fu and S. Ma, Org. Lett., 2009, 11, 1213 CrossRef CAS PubMed; (f) R. Sanz, V. Guilarte and M. P. Catroviejo, Synlett, 2008, 3006 CrossRef CAS; (g) S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526 CrossRef CAS PubMed; (h) C. Ferrer, C. H. M. Amijs and A. M. Echavarren, Chem. – Eur. J., 2007, 13, 1358 CrossRef CAS PubMed; (i) A. L. Rodriguez, C. Koradin, W. Dohle and P. Knochel, Angew. Chem., Int. Ed., 2000, 39, 2488 CrossRef CAS; (j) H.-C. Zhang, H. Ye, A. F. Moretto, K. K. Brumfield and B. E. Maryanoff, Org. Lett., 2000, 2, 89 CrossRef CAS PubMed; (k) T. D. Lee, M. V. Pickering and G. D. J. Daves, J. Org. Chem., 1974, 39, 1106 CrossRef CAS.
- For recent selected publications concerning transition-metal-catalyzed coupling of cyclic ether/cycloalkanes with heterocycles, see: (a) R. Xia, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2012, 14, 5546 CrossRef CAS PubMed; (b) P. Ren, I. Salihu, R. Scopelliti and X. Hu, Org. Lett., 2012, 14, 1748 CrossRef CAS PubMed; (c) Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971 CrossRef CAS; (d) Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600 CrossRef CAS PubMed; (e) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573 CrossRef CAS PubMed; (f) A. Correa, B. Fiser and E. Gómez-Bengoa, Chem. Commun., 2015, 51, 13365 RSC; (g) X. Wu, C. Lei, G. Yue and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 9601 CrossRef CAS PubMed; (h) C. Wang, X. Mi, Q. Li, Y. Li, M. Huang, J. Zhang, Y. Wu and Y. Wu, Tetrahedron, 2015, 6689 CrossRef CAS; (i) B. Niu, W. Zhao, Y. Ding, Z. Bian, C. U. Pittman Jr., A. Zhou and H. Ge, J. Org. Chem., 2015, 80, 7251 CrossRef CAS PubMed; (j) S. Rajamanickam, G. Majji, S. K. Santra and B. K. Patel, Org. Lett., 2015, 17, 5586 CrossRef CAS PubMed; (k) E. Kianmehr, N. Faghih, S. Karaji, Y. A. Lomedasht and K. M. Khan, J. Organomet. Chem., 2016, 10 CrossRef CAS.
- (a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 6968 CrossRef CAS PubMed; (b) X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848 CrossRef CAS PubMed; (c) M. Ghobrial, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2011, 76, 8781 CrossRef CAS PubMed.
- L.-K. Jin, L. Wan, J. Feng and C. Cai, Org. Lett., 2015, 17, 4726 CrossRef CAS PubMed.
- (a) G. J. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem. – Eur. J., 2009, 15, 333 CrossRef CAS PubMed; (b) T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016 CrossRef CAS PubMed; (c) X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704 RSC; (d) J. Zhao, H. Fang, J. Han, Y. Pan and G. Li, Adv. Synth. Catal., 2014, 356, 2719 CrossRef CAS PubMed; (e) S. K. Rout, S. Guin, W. Ali, A. Gogoi and B. K. Patel, Org. Lett., 2014, 16, 3086 CrossRef CAS PubMed; (f) G. Majji, S. Guin, S. K. Rout, A. Behera and B. K. Patel, Chem. Commun., 2014, 50, 12193 RSC; (g) A. Banerjee, S. K. Santra, N. Khatun, W. Ali and B. K. Patel, Chem. Commun., 2015, 51, 15422 RSC; (h) J. Zhao, H. Fang, R. Song, J. Zhou, J. Han and Y. Pan, Chem. Commun., 2015, 51, 599 RSC; (i) L. Fang, L. Chen, J. Yu and L. Wang, Eur. J. Org. Chem., 2015, 1910 CrossRef CAS; (j) W. Sun, Z. Xie, J. Liu and L. Wang, Org. Biomol. Chem., 2015, 13, 4596 RSC; (k) S. Ambala, T. Thatikonda, S. Sharma, G. Munagala, K. R. Yempalla, R. A. Vishwakarma and P. P. Singh, Org. Biomol. Chem., 2015, 13, 11341 RSC.
- L. Jin, J. Feng, G. Lu and C. Cai, Adv. Synth. Catal., 2015, 357, 2105 CrossRef CAS.
- F. Li, I. E. H. Elhussin, S. Li, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2015, 80, 10605 CrossRef CAS PubMed.
- (a) W. Liu, H. Cao, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS PubMed; (b) Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306 CrossRef CAS PubMed; (c) T. L. Chan, Y. Wu, P. Y. Choy and F. Y. Kwong, Chem. – Eur. J., 2013, 19, 15802 CrossRef CAS PubMed; (d) Y. Wu, P. Y. Choy and F. Y. Kwong, Org. Biomol. Chem., 2014, 12, 6820 RSC.
- (a) C. M. So, C. P. Lau and F. Y. Kwong, Chem. – Eur. J., 2011, 17, 761 CrossRef CAS PubMed; (b) P. Y. Choy, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2011, 76, 80 CrossRef CAS PubMed; (c) O. Y. Yuen, P. Y. Choy, W. K. Chow, W. T. Wong and F. Y. Kwong, J. Org. Chem., 2013, 78, 3374 CrossRef CAS PubMed; (d) P. Y. Choy, K. C. Luk, Y. Wu, C. M. So, L.-L. Wang and F. Y. Kwong, J. Org. Chem., 2015, 80, 1457 CrossRef CAS PubMed; (e) Q. Yang, P. Y. Choy, W. C. Fu, B. Fan and F. Y. Kwong, J. Org. Chem., 2015, 80, 11193 CrossRef CAS PubMed.
- (a) Y. Wu, B. Li, F. Mao, X. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed; (b) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (c) Y. Wu, L.-J. Feng, X. Lu, F. Y. Kwong and H.-B. Luo, Chem. Commun., 2014, 50, 15352 RSC; (d) See ref. 3c.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and copies of the NMR spectra. See DOI: 10.1039/c6ob00076b |
| This journal is © The Royal Society of Chemistry 2016 |