
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding DP4: application to drug compounds and automation†
Kristaps
Ermanis
a,
Kevin E. B.
Parkes
b,
Tatiana
Agback
b and
Jonathan M.
Goodman
*c
aCentre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: ke291@cam.ac.uk
bMedivir AB, PO Box 1086, SE-141 22 Huddinge, Sweden
cCentre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: jmg11@cam.ac.uk; Tel: +44 (0)1223 336434
First published on 23rd March 2016
Abstract
The DP4 parameter, which provides a confidence level for NMR assignment, has been widely used to help assign the structures of many stereochemically-rich molecules. We present an improved version of the procedure, which can be downloaded as Python script instead of running within a web-browser, and which analyses output from open-source molecular modelling programs (TINKER and NWChem) in addition to being able to use output from commercial packages (Schrodinger's Macromodel and Jaguar; Gaussian). The new open-source workflow incorporates a method for the automatic generation of diastereomers using InChI strings and has been tested on a range of new structures. This improved workflow permits the rapid and convenient computational elucidation of structure and relative stereochemistry.
Introduction
Computational methods for the prediction of NMR spectra are well established and have become an invaluable tool in the structure elucidation.1 When DFT calculations are used to predict the NMR spectra of candidate structures, the next step is to decide which of the predictions match the experimental NMR data best. Many different ways to quantitatively evaluate goodness of fit have been developed, including mean absolute error, corrected mean absolute error and correlation coefficient. In addition to these, we have developed CP3 and DP4 statistical parameters which help choosing the correct structure when several sets or just one set of experimental NMR data are available, respectively.2,3 Both of these parameters generally appear to give higher confidence in the correct structure than other parameters. DP4 in particular has seen wide use in structure elucidation of many complex natural products4 and also synthetic compounds.5A typical NMR prediction process starts with a conformational search on all of the isomers. DFT GIAO calculations are then run on all low-energy conformers, the predicted shifts are combined using Boltzmann weighting and finally, the DP4 analysis is employed to decide which structure is the most likely to be correct. DP4 analysis achieves this by first empirically scaling all the predicted chemical shifts to remove any systematic errors. Errors are then calculated for each of the signals. Then, assuming that each error follows a normal or t distribution with known parameters, the probability of encountering each error in a correct structure is calculated. Next, all of the probabilities for signals in a candidate structure are multiplied to give the overall absolute probability, that the candidate structure is the correct one. Finally, these resulting probabilities are converted to a set of relative probabilities that each candidate structure is the correct one using Bayes’ theorem.
DP4 was originally developed for the elucidation of the relative stereochemistry of natural products. Drug compounds is another other class of compounds that also exhibit diverse stereochemistry. They inhabit a distinct chemical space characterized by higher number of heteroatoms and aromatic systems. While DP4 should in principle be applicable to any organic compound, its performance in drug compounds is all but untested.
So far in our investigations we have been using MacroModel6 for conformational searches and Jaguar7 or Gaussian8 for DFT and GIAO calculations. However, there exist open-source alternatives for both conformational search and for DFT calculations. Incorporation and validation of this software in the DP4 process would significantly increase its accessibility.
Results and discussion
Application to drug molecules
DP4 was originally developed with a focus on natural products and their fragments.3 We wanted to explore the application of the DP4 workflow in a less familiar chemical space. Drug compounds occupy one such region – in general, they contain less stereocentres, but more heteroatoms and aromatic systems when compared to natural products. A large portion of drug compounds also contain basic nitrogen atoms or systems with several possible tautomeric states, which can make the prediction of NMR spectra challenging. For this study a selection of stereochemically rich drug compounds from the Top 100 drugs was chosen (Fig. 1).9 The selected compounds all had 2–64 plausible diastereomers and had a well defined tautomeric state. Nucleoside analogs in particular were not included in this study because of the potential tautomeric and protonated states. | ||
| Fig. 1 Stereochemically rich drug molecules selected for DP4 study. Diastereomer count shown. Diastereomers labelled with letters a–bl, a being the correct diastereomer. Configuration was varied at chiral centres marked with ‘*’. | ||
The compounds and their diastereomers were submitted to MacroModel conformational search, all conformers with energy less than 10 kJ mol−1 relative to the global minimum were then submitted to DFT single point calculation and NMR GIAO calculation10 using Gaussian quantum chemistry software. Our earlier studies demonstrated that this protocol provides an effective balance of precision and speed.3 Finally, the predicted NMR shifts were submitted to DP4 analysis. For mometasone, 4, and testosterone, 3, only the structures diastereomeric at the peripheral stereocentres were considered. In the case of tiotropium, 7, the epoxide chiral centres were varied in concert, as the trans isomers would be too strained to exist. For the rest of the compounds all possible diastereomers were considered. The results of this study are shown in Fig. 2.
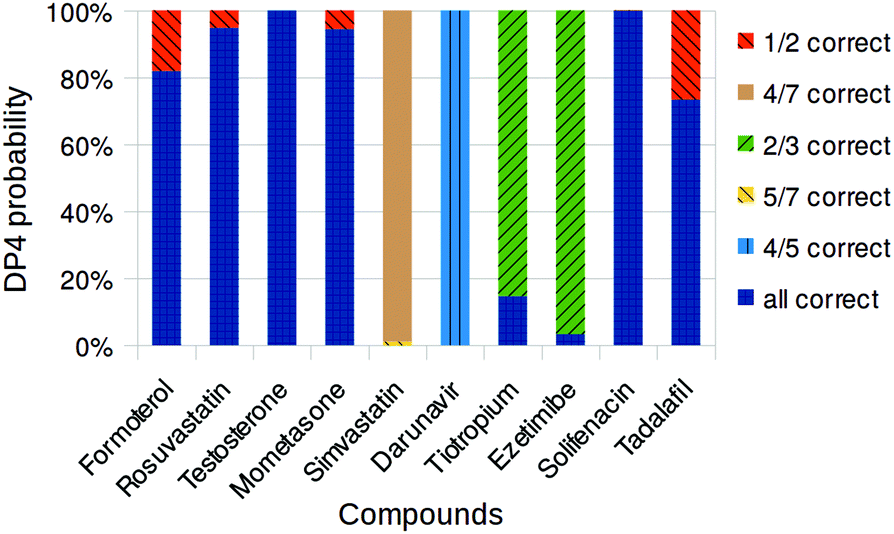 | ||
| Fig. 2 Overall DP4 probabilities assigned to drug compound diastereomers using MacroModel and Gaussian. Only diastereomers with probabilities greater than 0.1% shown. | ||
Ideally, the DP4 parameter will give 100% confidence for the correct diastereoisomer, but all certainty beyond a random selection is useful, and certainty on some stereocentres is helpful. In the figure, the colours correspond to the utility of the assignment. Dark blue is the probability of the completely correct structure. The other colours relate to the utility – six out of seven stereocentres correct is a better result than three out of four. We note that all of the structures considered are C1 symmetry, and so the number of diastereoisomers that need to be considered is always 2N−1.
Early in our investigations it was found that inclusion of the solvent in the DFT calculations reduced the errors of the chemical shift prediction and improved the identification of the correct diastereomer. Therefore the PCM solvent model11 was used for all NMR GIAO calculations. In most cases DP4 is able to correctly determine the relative stereochemistry of these compounds with good confidence and shows the generality and robustness of the DP4 approach. Even highly flexible structures like rosuvastatin, 2, and formoterol, 1, were correctly identified. In the case of ezetimibe, 8, however DP4 favoured the structure epimeric at the remote OH centre as the most likely with 98% confidence, with the correct structure as a remote second most likely candidate. Similarly in the case of darunavir, 6, the most likely structure was identified as the one with epimeric OH centre. However, the relative stereochemistry of the remaining four stereocentres was identified correctly, despite the flexible nature of the molecule. The case of simvastatin, 5, was particularly challenging as this compound is quite flexible and has seven stereocentres and 64 possible diastereomers. Unfortunately this particular problem appears to be too complex for the current DP4 methodology and the diastereomers identified as likely candidates were completely different from the correct structure. However, the DP4 conclusion that four diastereomers have significant probability may suggest the challenge of this highly flexible structure to the user and thus would likely not lead to incorrect conclusions.
Several of our previous studies have shown that optimization of the geometries at the DFT level provides only limited improvement at high computational cost. We decided to test if this still holds true on the compounds in the current dataset and we chose two compounds with the most potential for improvement – tiotropium, 7, and ezetimibe, 8. The geometries were optimized at the same DFT level as used for shift calculation and the DP4 analysis was repeated at a cost of approximately 30-fold increase in the computational time. For tiotropium, DFT optimization reduced the average corrected NMR shift error by 0.27 ppm for carbon and by 0.02 ppm for proton, and left the separate DP4 confidences based on carbon and proton NMR essentially unchanged. However, the overall confidence was increased from 14.7% to 68.2% because the carbon and proton were now in conflict as to which is the most likely structure and this let the correct structure emerge as the overall top result. For ezetimibe, DFT optimization reduced the average corrected NMR shift error by 0.77 ppm for carbon, but increased the error by 0.01 ppm for proton. The overall confidence improved from 2.8% to 72.0%. Therefore improvement can be gained by optimizing the geometries at the DFT level for these anomalous examples. However, the computational cost makes this approach impractical in many cases: to repeat the DP4 analysis with DFT optimization on darunavir and simvastatin would require about 12 and 36 CPU months on our desktop setup, respectively.
Overall the results from this study are very encouraging. In majority of cases DP4 is capable of elucidating the relative stereochemistry of the drug compounds despite the fact that the statistical model was developed for natural products and their fragments.
Integration of open-source software
Computing power nowadays is more accessible than ever and this often means that costly software licences are a greater barrier to the use of computational chemistry than the availability of hardware. This is particularly true for the occasional users. To alleviate this problem in the context of structure elucidation, we decided to test open-source software as alternatives to MacroModel, Gaussian and Jaguar.One well-known open-source DFT package with NMR GIAO implementation is NWChem.12 We have previously shown that the differences between Gaussian and Jaguar DFT packages do not significantly affect the outcome of DP4 calculations, and, therefore, we hoped that the same would be true for NWChem. Probably the most significant difference between Gaussian and NWChem are the available solvent models. While Gaussian has several versions of PCM models implemented, the only solvent model available in NWChem is COSMO.13 It was hoped that the different solvent models would not cause large differences to the chemical shift calculations. To test this, NWChem was used for DP4 workflow and the process was repeated for six drug molecules (Fig. 3). The results were very similar to those achieved with Gaussian, with the mean absolute difference in the predicted shifts being 0.34 ppm for carbon and 0.08 ppm for proton NMR. This shows that NWChem can be used in place of Jaguar or Gaussian with little effect on DP4 performance, using the standard workflow, which uses molecular geometries generated by force-fields.
 | ||
| Fig. 3 Overall DP4 probabilities assigned to drug compound diastereomers using MacroModel and NWChem. Only diastereomers with probabilities greater than 0.1% shown. | ||
One significant difference was in the case of formoterol, 1. While the MacroModel/Gaussian combination gives 81.9% overall DP4 probability for the correct diastereomer, MacroModel/NWChem combination gives only 0.5% for the correct structure. This arises from conflicting conclusions from carbon and proton spectra in both cases. The proton data gives high (>95%) confidence in the correct structure, while carbon data gives high (>95%) confidence in the incorrect one. The confidence in the correct proton assignment is slightly lower in the NWChem case, probably because of the different solvent model. This causes the confidence in the carbon NMR based assignment to rise and also causes a switch in the overall assignment. In all other cases the overall DP4 probabilities are very similar with the Gaussian version of the workflow.
TINKER is an open-source molecular mechanics package14 which we tested as an alternative for MacroModel in perfoming conformational searches. The performance of TINKER/Gaussian workflow was tested by repeating the calculations on the drug molecules (Fig. 4). In our experience TINKER conformational searches were slower than the same searches run using MacroModel and generated many more conformations. As a result, we were not able to complete the calculations for darunavir, 6, and simvastatin, 5. For the rest of the molecules, however, the DP4 results are quite similar to the MacroModel workflow and in general seem equally good at identifying the correct diastereomer. For tiotropium, 7, the DP4 performance improved the outcome and the confidence in the correct structure increased from 14.7% when using MacroModel to 60.9% with TINKER. In contrast, the confidences for the correct diastereomers of tadalafil, 10, and solifenacin, 9, were reduced. Both structures can only have two diastereomers and in the case of tadalafil the confidence in the correct one is only 50.5%, essentially having no predictive value. In the case of solifenacin, the confidence was reduced even further to 42.8%. The cause for these differences is most likely the differences in the search algorithm implementations. On average, however, the DP4 results appear to be of similar quality regardless of the molecular mechanics software used.
 | ||
| Fig. 4 Overall DP4 probabilities assigned to drug compound diastereomers using Tinker and Gaussian. Only diastereomers with probabilities greater than 0.1% shown. | ||
Finally, the fully open-source version of the process was tested using TINKER and NWChem. Calculations were run on the same set of molecules as in the MacroModel/NWChem case (Fig. 5). Overall the results appear to be very similar to the previous results. As in the case of TINKER/Gaussian workflow, results for solifenacin, 9, and tadalafil, 10, were indecisive. In all other cases, however, the correct diastereomer was identified with high confidence. The average performance of this workflow once again appears to be similar to the other three investigated. This demonstrates a fully open-source version of the DP4 workflow and should significantly improve its accessibility to chemists in all areas of organic chemistry.
 | ||
| Fig. 5 Overall DP4 probabilities assigned to drug compound diastereomers using Tinker and NWChem. Only diastereomers with probabilities greater than 0.1% shown. | ||
Automatic generation of diastereomers
The most common use of DP4 is for the determination of the relative stereochemistry of complex molecules. The number of possible diastereomers quickly increases with the number of stereocentres and the manual input of the candidate structures becomes cumbersome and error-prone. User-friendliness would be significantly improved if all candidate diastereomers could be generated from a single base structure. There has been little previous work on this problem and the solutions developed so far are not general.15 We reasoned that the most straightforward way of generating diastereomers would be through InChI strings.16 InChIs are a compact and consistent way of representing chemical structures and because of the layered structure, they are easy to manipulate by computer programs (Fig. 6). | ||
| Fig. 6 Key features of InChI strings and their use in automatic diastereomer generation. | ||
To generate diastereomers, the base candidate 3D structure is first converted into an InChI string. This can be done using one of several utilities and frameworks freely available online, including IUPAC utilities,16 OpenBabel17 and Marvin MolConverter.18 From this, InChI strings for diastereomeric structures are generated by modifying the relative stereochemistry layer to represent all possible combinations of the configurations. Finally, InChIs are converted back into 3D structures. Several utilities offer this service, and we found that only Marvin MolConverter, which is available as free software from ChemAxon,18 produced good 3D structures. After this, structures could be immediately used in the DP4 process without any further modification. Both the generation of all diastereomers and diastereomers at particular chiral centres have been implemented as a Python script with Marvin Molconverter as the backend for the generation of InChI strings from structures and the generation of structures from InChI strings. This process was successfully used in the drug study to generate all the candidate structures. The only exception was tiotropium, 7, because some of the automatically generated structures would be too strained to be viable and manually drawn structures were analyzed in this case.
Full automation of the workflow
Taking the idea of DP4 automation further, a Python application PyDP4 was developed that integrates and automates the whole process (Fig. 7). The application takes the 3D geometry of the candidate structures as an SDF file and prepares the input files for the conformational search done by either MacroModel or TINKER. After the completion of the conformational searches, the low-energy conformations are selected and Gaussian, Jaguar or NWChem input files are prepared and run. Finally, it extracts the predicted NMR shifts, performs the Boltzmann weighting and combines the computational NMR data with the experimental NMR description and passes it on to DP4 analysis. It is important to note that there is no user interaction required after the submission of the initial 3D structures and NMR spectra description. | ||
| Fig. 7 Integrated and automated PyDP4 workflow for relative stereochemistry elucidation. | ||
The script also alleviates the shortcomings in TINKER conformation pruning. TINKER only removes duplicate conformations based on energy; therefore the number of conformations generated is much larger than in the case of MacroModel. To keep the number of conformations submitted to the DFT calculations down, we implemented RMSD pruning of conformations. RMSD pruning works by calculating the differences in atom positions in different conformers and if the RMSD value exceeds a chosen cut-off value, the conformations are considered similar and one of the conformations is discarded. This procedure requires the molecules to be aligned and to achieve this, QTRFIT alignment algorithm was also implemented in Python.19
All of these improvements in combination mean that DP4 structure elucidation and verification can now be done quickly and easily. In our experience, a full elucidation of the relative stereochemistry could be done with as little as one hour of active user effort. In principle, new developments in this area, such as Sarotti's recent modification of DP4,22 could be incorporated into a similar workflow.
Computational methods
All molecular mechanics calculations were performed using either MacroModel6 (Version 9.9) or TINKER.14 All conformational searches were done using MMFF force field20 and Low Mode following search algorithms.21 The conformational searches were done in the gas phase and the step count for MacroModel was adjusted so that all low-energy conformers were found at least 5 times.Quantum mechanical calculations were carried out using Gaussian '09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and NWChem 6.5,12 B3LYP functional23 and 6-31G(d,p) basis set.24 PCM and COSMO solvent models were used in the case of Gaussian and NWChem, respectively.11,13 NMR shielding constant calculations used the GIAO method.10 Calculation setup, data extraction and DP4 analysis were done using the PyDP4 script written in Python 2.7. The script is available on the group website (http://www-jmg.ch.cam.ac.uk/tools/nmr), as well as on GitHub (https://github.com/KristapsE/PyDP4) and in the ESI† under the MIT license. TINKER, NWChem and Marvin Molconverter can be obtained directly from their authors.
8 and NWChem 6.5,12 B3LYP functional23 and 6-31G(d,p) basis set.24 PCM and COSMO solvent models were used in the case of Gaussian and NWChem, respectively.11,13 NMR shielding constant calculations used the GIAO method.10 Calculation setup, data extraction and DP4 analysis were done using the PyDP4 script written in Python 2.7. The script is available on the group website (http://www-jmg.ch.cam.ac.uk/tools/nmr), as well as on GitHub (https://github.com/KristapsE/PyDP4) and in the ESI† under the MIT license. TINKER, NWChem and Marvin Molconverter can be obtained directly from their authors.
Conclusions
DP4 analysis has been applied to a novel chemical space and was successfully used for assigning stereochemistry of drug molecules. Open-source alternatives to MacroModel and Gaussian/Jaguar were tested. TINKER molecular mechanics package and NWChem ab initio software have been integrated in a fully open-source DP4 workflow and this should significantly improve accessibility of DP4 analysis.A novel and general approach to automatic generation of diastereomers was developed and relies on the use of InChI strings. Finally, a full automation of the DP4 workflow has reduced user interaction to a minimum – verification or elucidation of the stereochemical structure can now be done in as little as one hour of active effort.
Acknowledgements
The authors wish to thank Medivir for the generous financial support.Notes and references
- For reviews in the area see: M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839 CrossRef CAS PubMed; D. J. Tantillo, Nat. Prod. Rep., 2013, 30, 1079 RSC.
- S. G. Smith and J. M. Goodman, J. Org. Chem., 2009, 74, 4597 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946 CrossRef CAS PubMed.
- (a) A. Gutiérrez-Cepeda, A. H. Daranas, J. J. Fernández, M. Norte and M. L. Souto, Mar. Drugs, 2014, 12, 4031 CrossRef PubMed; (b) N. Tanaka, M. Takekata, S. Kurimoto, K. Kawazoe, K. Murakami, D. Damdinjav, E. Dorjibal and Y. Kashiwada, Tetrahedron Lett., 2015, 56, 817 CrossRef CAS; (c) T. Iwai, T. Kubota and J. Kobayashi, J. Nat. Prod., 2014, 77, 1541 CrossRef CAS PubMed; (d) H. J. Domínguez, J. G. Napolitano, M. T. Fernández-Sánchez, D. Cabrera-García, A. Novelli, M. Norte, J. J. Fernández and A. H. Daranas, Org. Lett., 2014, 16, 4546 CrossRef PubMed; (e) L.-B. Dong, Y.-N. Wu, S.-Z. Jiang, X.-D. Wu, J. He, Y.-R. Yang and Q.-S. Zhao, Org. Lett., 2014, 16, 2700 CrossRef CAS PubMed; (f) M. Menna, A. Aiello, F. D'Aniello, C. Imperatore, P. Luciano, R. Vitalone, C. Irace and R. Santamaria, Eur. J. Org. Chem., 2013, 3241 CrossRef CAS; (g) S. G. Brown, M. J. Jansma and T. R. Hoye, J. Nat. Prod., 2012, 75, 1326 CrossRef CAS PubMed; (h) T. P. Wyche, J. S. Piotrowski, Y. Hou, D. Braun, R. Deshpande, S. McIlwain, I. M. Ong, C. L. Myers, I. A. Guzei, W. M. Wrestler, D. R. Andes and T. S. Bugni, Angew. Chem., Int. Ed., 2014, 126, 11767 CrossRef; (i) I. Rodríguez, G. Genta-Jouve, C. Alfonso, K. Calabro, E. Alonso, J. A. Sánchez, A. Alfonso, O. P. Thomas and L. M. Botana, Org. Lett., 2015, 17, 2392 CrossRef PubMed; (j) E. D. Gussem, W. Herrebout, S. Specklin, C. Meyer, J. Cossy and P. Bultinck, Chem. – Eur. J., 2014, 20, 17385 CrossRef PubMed; (k) D. J. Shepherd, P. A. Broadwidth, B. S. Dyson, R. S. Paton and J. W. Burton, Chem. – Eur. J., 2013, 19, 12644 CrossRef CAS PubMed; (l) I. Paterson, S. M. Dalby, J. C. Roberts, G. I. Naylor, E. A. Guzmán, R. Isbrucker, T. P. Pitts, P. Linley, D. Divlianska, J. K. Reed and A. E. Wright, Angew. Chem., Int. Ed., 2011, 50, 3219 CrossRef CAS PubMed; (m) I. Paterson and G. W. Haslett, Org. Lett., 2013, 15, 1338 CrossRef CAS PubMed; (n) N. Tanaka, M. Asai, T. Kusama, J. Fromont and J. Kobayashi, Tetrahedron Lett., 2015, 56, 1388 CrossRef CAS; (o) I. H. Hwang, J. Oh, W. Zhou, S. Park, J.-H. Kim, A. G. Chittiboyina, D. Ferreira, G. Y. Song, S. Oh, M. Na and M. T. Hamann, J. Nat. Prod., 2015, 78, 453 CrossRef CAS PubMed; (p) F. Cen-Pacheco, J. Rodríguez, M. Norte, J. J. Fernández and A. H. Daranas, Chem. – Eur. J., 2013, 19, 8525 CrossRef CAS PubMed; (q) M. W. Lodewyk and D. J. Tantillo, J. Nat. Prod., 2011, 74, 1339 CrossRef CAS PubMed; (r) H. J. Domínguez, G. D. Crespín, A. J. Santiago-Benítez, J. A. Gavín, J. J. Fernández and A. H. Daranas, Mar. Drugs, 2014, 12, 176 CrossRef PubMed; (s) F. Cen-Pacheco, A. J. Santiago-Benítez, C. García, S. J. Álvarez-Méndez, A. J. Martín-Rodríguez, M. Norte, V. S. Martín, J. A. Gavín, J. J. Fernández and A. H. Daranas, J. Nat. Prod., 2015, 78, 712 CrossRef CAS PubMed; (t) S. D. Micco, A. Zampella, M. V. D'Auria, C. Festa, S. D. Marino, R. Riccio, C. P. Butts and G. Bifulco, Beilstein J. Org. Chem., 2013, 9, 2940 CrossRef PubMed; (u) T. D. Tran, N. B. Pham and R. J. Quinn, Eur. J. Org. Chem., 2014, 4805 CrossRef CAS; (v) M. Omar, Y. Matsuo, H. Maeda, Y. Saito and T. Tanaka, Org. Lett., 2014, 16, 1378 CrossRef CAS PubMed; (w) W. Zhou, J. Oh, W. Lee, S. Kwak, W. Li, A. G. Chittiboyina, D. Ferreira, M. T. Hamann, S. H. Lee, J.-S. Bae and M. Na, Biochim. Biophys. Acta, 2014, 1840, 2042 CrossRef CAS PubMed; (x) S. K. Amini, Magn. Reson. Chem., 2013, 51, 328 CrossRef CAS PubMed; (y) J. Rodríguez, R. M. Nieto, M. Blanco, F. A. Valeriote, C. Jiménez and P. Crews, Org. Lett., 2014, 16, 464 CrossRef PubMed; (z) T. P. Wyche, Y. Hou, D. Braun, H. C. Cohen, M. P. Xiong and T. S. Bugni, J. Org. Chem., 2011, 76, 6542 CrossRef CAS PubMed; (a a) Q. Li, Y.-S. Xu, G. A. Ellis, T. S. Bugni, Y. Tang and R. P. Hsung, Tetrahedron Lett., 2013, 54, 5567 CrossRef CAS PubMed; (a b) S. Khokhar, Y. Feng, M. R. Campitelli, R. J. Quinn, J. N. A. Hooper, M. G. Ekins and R. A. Davis, J. Nat. Prod., 2013, 76, 2100 CrossRef CAS PubMed; (a c) A. E. Nugroho, M. Okuda, Y. Yamamoto, Y. Hirasawa, C.-P. Wong, T. Kaneda, O. Shirota, A. H. A. Hadi and H. Morita, Tetrahedron, 2013, 69, 4139 CrossRef CAS; (a d) F. Cen-Pacheco, M. Norte, J. J. Fernández and A. H. Daranas, Org. Lett., 2014, 16, 2880 CrossRef CAS PubMed.
- (a) J. Cho, S. Lee, S. Hwang, S. H. Kim, J. S. Kim and S. Kim, Eur. J. Org. Chem., 2013, 4614 CrossRef CAS; M. J. Bartlett, P. T. Northcote, M. Lein and J. E. Harvey, J. Org. Chem., 2014, 79, 5521 Search PubMed; (b) I. Paterson, M. Xuan and S. M. Dalby, Angew. Chem., Int. Ed., 2014, 53, 7286 CrossRef CAS PubMed.
- MacroModel, version 9.9, Schrödinger, LLC, New York, NY, 2009 Search PubMed.
- Jaguar, version 9.9, Schrödinger, LLC, New York, NY, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- 2012 data used, general reference: N. A. McGrath, M. Brichacek and J. T. Njardson, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS.
- (a) F. London, J. Phys. Radium, 1937, 8, 397 CrossRef CAS; (b) R. Ditchfield, J. Chem. Phys., 1972, 56, 5688 CrossRef CAS; (c) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS.
- (a) M. T. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef; (b) M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS; (c) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 2009, 113, 6378 CrossRef CAS PubMed; (b) A. Klamt and G. Schüürman, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC; (c) T. N. Truong and E. V. Stefanovich, Chem. Phys. Lett., 1995, 240, 253 CrossRef CAS.
- J. W. Ponder and F. M. Richards, J. Comput. Chem., 1987, 8, 1016–1024 CrossRef CAS.
- (a) M. L. Contreras, J. Alvarez, D. Guajardo and R. Rozas, J. Chem. Inf. Model., 2006, 46, 2288–2298 CrossRef CAS PubMed; (b) J. G. Nourse, R. E. Carhart, D. H. Smith and C. Djerassi, J. Am. Chem. Soc., 1979, 101, 1216 CrossRef CAS.
- S. Heller, A. McNaught, S. Stein, D. Tchekhovskoi and I. Pletnev, J. Cheminf., 2013, 5, 7 CAS.
- (a) N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchinson, J. Cheminf., 2011, 3, 33 CrossRef PubMed; (b) The Open Babel Package, version 2.3.1 http://openbabel.org.
- Marvin MolConverter, version 15.5.18.0, ChemAxon Kft., Budapest, Hungary Search PubMed.
- D. J. Heisterberg, QTRFIT, Ohio Supercomputer Center, Columbus, OH, USA Search PubMed.
- (a) T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS; (b) T. A. Halgren, J. Comput. Chem., 1996, 17, 520 CrossRef CAS; (c) T. A. Halgren, J. Comput. Chem., 1996, 17, 553 CrossRef CAS; (d) T. A. Halgren and R. B. Nachbar, J. Comput. Chem., 1996, 17, 587 CAS; (e) T. A. Halgren, J. Comput. Chem., 1996, 17, 616 CrossRef CAS; (f) T. A. Halgren, J. Comput. Chem., 1999, 20, 720 CrossRef CAS; (g) T. A. Halgren, J. Comput. Chem., 1999, 20, 730 CrossRef CAS.
- (a) I. Kolossováry and W. C. Guida, J. Am. Chem. Soc., 1996, 118, 5011 CrossRef; (b) I. Kolossováry and W. C. Guida, J. Comput. Chem., 1999, 20, 1671 CrossRef.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PyDP4 Python script. See DOI: 10.1039/c6ob00015k |
| This journal is © The Royal Society of Chemistry 2016 |