
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 1,2-biphenylethane based single-molecule diodes†
Elena
Galán
a,
Mickael L.
Perrin
b,
Martin
Lutz
c,
Herre S. J.
van der Zant
b,
Ferdinand C.
Grozema
a and
Rienk
Eelkema
*a
aDepartment of Chemical Engineering, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: r.eelkema@tudelft.nl
bKavli Institute of Nanoscience, Delft University of Technology, Lorentzweg 1, 2628 CJ Delft, The Netherlands
cBijvoet Center for Biomolecular Research, Crystal and Structural Chemistry, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
First published on 19th January 2016
Abstract
We have described the synthesis of novel biphenylethane-based wires for molecular electronics. Exceptional single-molecule diode behavior was predicted for unsymmetrically substituted biphenylethane derivatives, synthesized here using the so far unexplored unsymmetrically substituted 1,2-bis(4-bromophenyl)ethanes as key intermediates, which were obtained from the corresponding tolane precursor by selective hydrogenation.
Since the proposal of the molecular diode by Aviram and Ratner,1 fabrication of molecular junctions has been a promising approach to build future nanoscale electronic devices. Recently, symmetric molecules consisting of two conjugated arms connected by a non-conjugated segment have shown an intrinsic and pronounced negative differential conductance effect in the current–voltage characteristics of a single molecule in a break junction.2 This phenomenon is widely used to amplify electric signals and for switching applications. Theoretically, it has been predicted that by introducing an asymmetry into these molecules, their properties can be significantly changed, turning them into single-molecule diodes. Diodes are among the basic functional units in electronics and have attracted much attention experimentally at the single-molecule level.3 Based on these principles, we recently proposed a new theoretical model showing the realization of single-molecule diodes with very high rectification ratios (RR).4 RR is defined as the ratio of the forward to backward current measured at the same absolute bias. The proposed diodes are molecular wires with an unsymmetrically substituted biphenylethane-based backbone (see molecule T in Scheme 1 for a specific example). In this design, an ethane bridge breaks the conjugation between the two phenylethynylbenzene halves of the molecule. The molecule is end capped with thiols as the anchoring unit for coupling to metallic electrodes. The required asymmetry comes from the substitution on one half of the molecule with electron-withdrawing groups (EWG). By changing the EWG, the RR can be optimized. The model predicts RR as high as 1500, far beyond typically reported experimental values (RR ≤ 10).3b–f,5 Only very recently, single-molecule diodes with high RR (in excess of 200) have been reported, however the rectification obtained there does not arise from the molecule, but it is generated from an asymmetry in the metallic electrodes.6 When introducing two fluorine substituents into one phenyl, the predicted rectification ratios depend heavily on the location of these fluorine atoms on the phenyl ring. The highest rectification ratio (RR = 751) was predicted for the isomer with two fluorine atoms ortho with respect to the ethane bridge. Here, we report the challenging synthesis of these novel wires as well as several control compounds to enable their exploration as intrinsic molecular diodes.
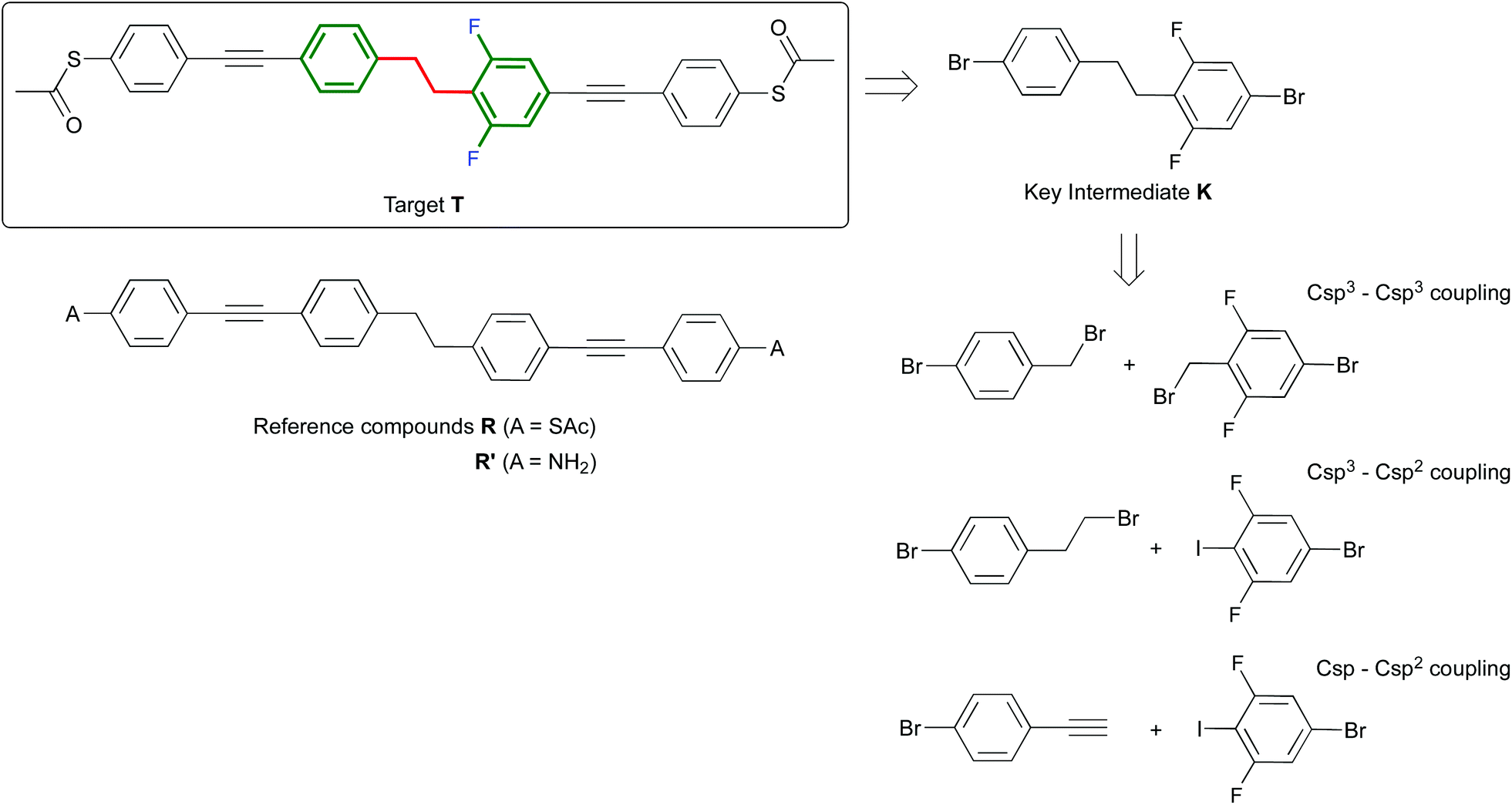 | ||
| Scheme 1 Structure of the target compound T and reference compounds R and R′ and retrosynthetic pathways to T through intermediate K. | ||
The symmetric biphenylethane derivatives R and R′ (Scheme 1) without an EWG were obtained successfully as reference compounds starting from 1,2-bis(4-bromophenyl)ethane (1),7 see Scheme 2. Amine-functionalized R′ is directly obtained through Sonogashira coupling between 1 and commercially available ethynylaniline. In the case of thioacetyl-derived R, the coupling reaction between 1 and either trimethylsilylacetylene (TMSA) or 4-ethynyl-1-thioacetylbenzene8 did not take place. Thus, the bromines in 1 were first exchanged for iodines. This was easily accomplished by the lithium–halogen exchange using nBuLi at −84 °C followed by the addition of I2 into THF, to produce 2,9 in high yield. 2 was then coupled with TMSA under Sonogashira conditions using Pd(PPh3)2Cl2 as a catalyst to afford 3. After cleavage of the trimethylsilyl protecting groups in the presence of tetra-n-butylammoniumfluoride (TBAF), the terminal alkyne generated was finally cross-coupled with 4-iodo-1-thioacetylbenzene,8 yielding reference compound R.
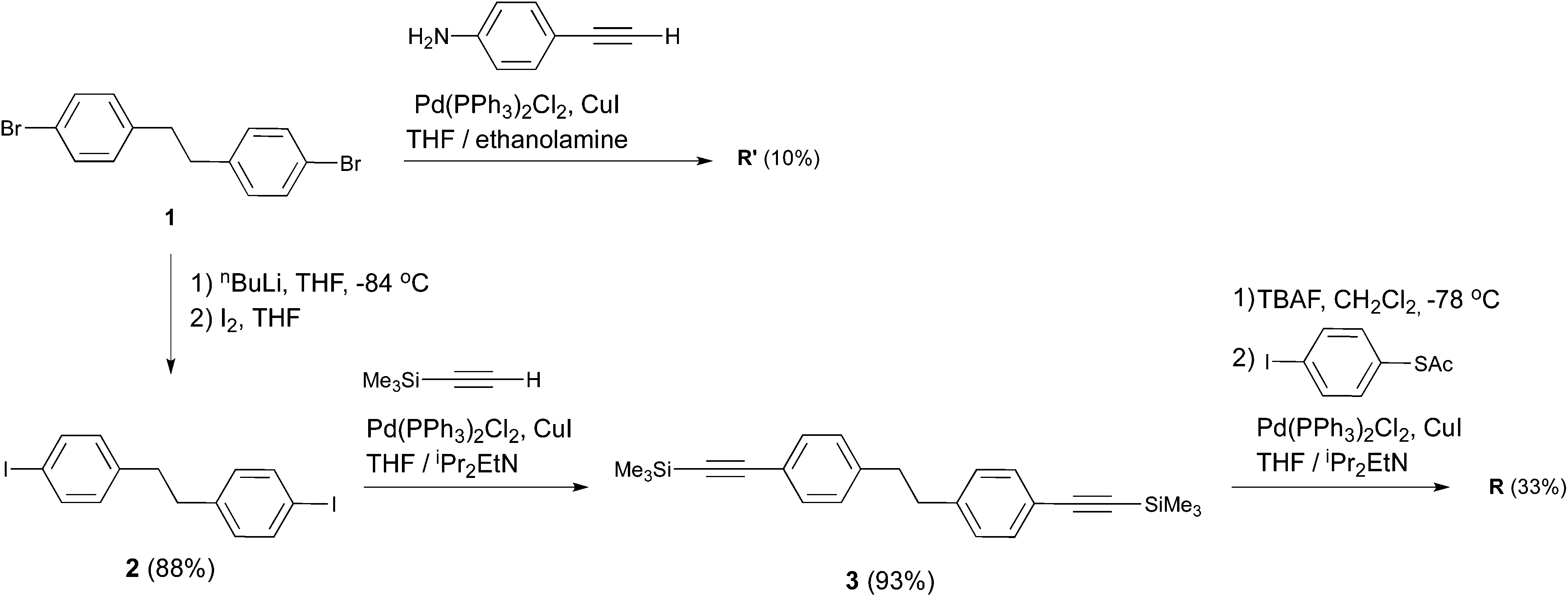 | ||
| Scheme 2 Synthesis of R and R′. | ||
For the synthesis of the target molecule T, a variety of starting materials is commercially available, which allows us to explore a range of synthetic routes. The molecule contains acetyl-protected thiol end groups, and as EWG two fluorine atoms are present on one half of the molecule, both in ortho position with respect to the ethane bridge. With the purpose of maintaining some flexibility regarding the late stage introduction of the anchoring groups through Sonogashira coupling, our synthetic approach to T has bromide-derived biphenylethane K as the key intermediate. In contrast to the synthesis of the symmetric analogue R, the route to T presents some significant challenges, which are mainly associated with the obtainment of its key intermediate K. Whereas the synthesis of 1,2-bis(4-bromophenyl)ethane (1) (key intermediate to obtain R) is well known in the literature, no unsymmetrically substituted analogues (such as K) have been described so far.
To develop a new synthetic pathway towards K, we first considered the Csp3–Csp3 bond formation approach (Scheme 1). For instance, we attempted to synthesize K by mixing the two corresponding benzylic halides in the presence of Fe/CuCl in water.7b When exploiting this methodology using two different reactants, a statistical mixture of homo- and hetero-coupled products is expected.10 However, in our particular case, only one spot was observed by TLC and the main product we obtained after precipitation was 1,2-bis(4-bromophenyl)ethane. Alternatively, lithiation of 4-bromobenzylbromide and then treatment with the corresponding electrophile (4-bromo-2,6-difluorobenzyl bromide) also did not lead to the desired compound. In this case, 4-bromobenzylbromide undergoes Wurtz coupling, in which the lithiated species initially formed react competitively with the starting material to produce again homo-coupled products. Unsymmetrically substituted biphenylethane derivatives can be obtained by exploiting alkyl–aryl cross-coupling reactions.11 Based on these results, we also explored the Csp3–Csp2 bond formation approach (Scheme 1) towards K. In the first step towards alkyl–aryl cross-coupling reactions, selective formation of the metal–alkyl complex is required (frequently a Grignard reagent). In our particular case, we were not able to obtain the magnesium–alkyl complex selectively starting from 4-bromophenethylbromide. This is understandable taking into account that our alkyl derivative has two bromine functionalities, and the fact that the reaction of organic bromides with magnesium is considered to be among the least selective of organic reactions.12 For instance, we observed that when a solution of (2-bromoethyl)benzene and pinacolborane is treated with magnesium, the desired boronic ester is formed. However, when using the same conditions, but starting with 4-bromophenethylbromide, a complex mixture of compounds is formed, according to 1H NMR. Attempts to either purify this complex mixture or to further cross-couple it with 1-bromo-3,5-difluoro-4-iodobenzene were unsuccessful.
As it stands, the described strategies did not lead to a successful synthesis of the key intermediate K. Therefore, we present here an alternative strategy that exploits the formation of K through a Csp2–Csp bond formation approach (see Scheme 1). In particular, K is obtained through selective hydrogenation of the corresponding alkyne precursor 6 (see Scheme 3). Molecule 6 can be easily obtained through two different synthetic routes in 25% or 67% yield starting from 4 or 7, respectively. Reaction of 4 with TMSA under Sonogashira conditions was studied first. Here, the addition of an excess of TMSA (1.3 eq. with respect to 4) had a negative effect. In this case, both mono-TMSA (5) and di-TMSA adducts were obtained, and this mixture cannot be separated by regular column chromatography. Adding an equimolar amount of TMSA solved this issue. In this case, starting material 4 and product 5 were present in the crude reaction but the compounds can be separated by column chromatography. After purification, 5 together with a small amount of 1,4-bis(trimethylsilyl)butadiyne was obtained and used directly in the next reaction. In order to cleave off TMS, we used TBAF because when using KOH/MeOH some unknown side-products were generated during the reaction. After cleavage of TMS, the terminal alkyne obtained was immediately used because it was not very stable. Thus, by reaction with 1-bromo-4-iodobenzene (2 eq.) under Sonogashira conditions, diphenylacetylene 6 was obtained pure, but in overall low yield (25%). On the other hand, using 7 as the starting material, its TMS cleavage in the presence of KOH/MeOH is quantitative. Furthermore, the terminal alkyne generated was stable under ambient conditions. Next, the reaction of the terminal alkyne with 4 (1.1 eq. with respect to terminal alkyne) under Sonogashira conditions provided a mixture of 4 and product 6. After column chromatography, pure 6 was obtained in overall good yield (67%).
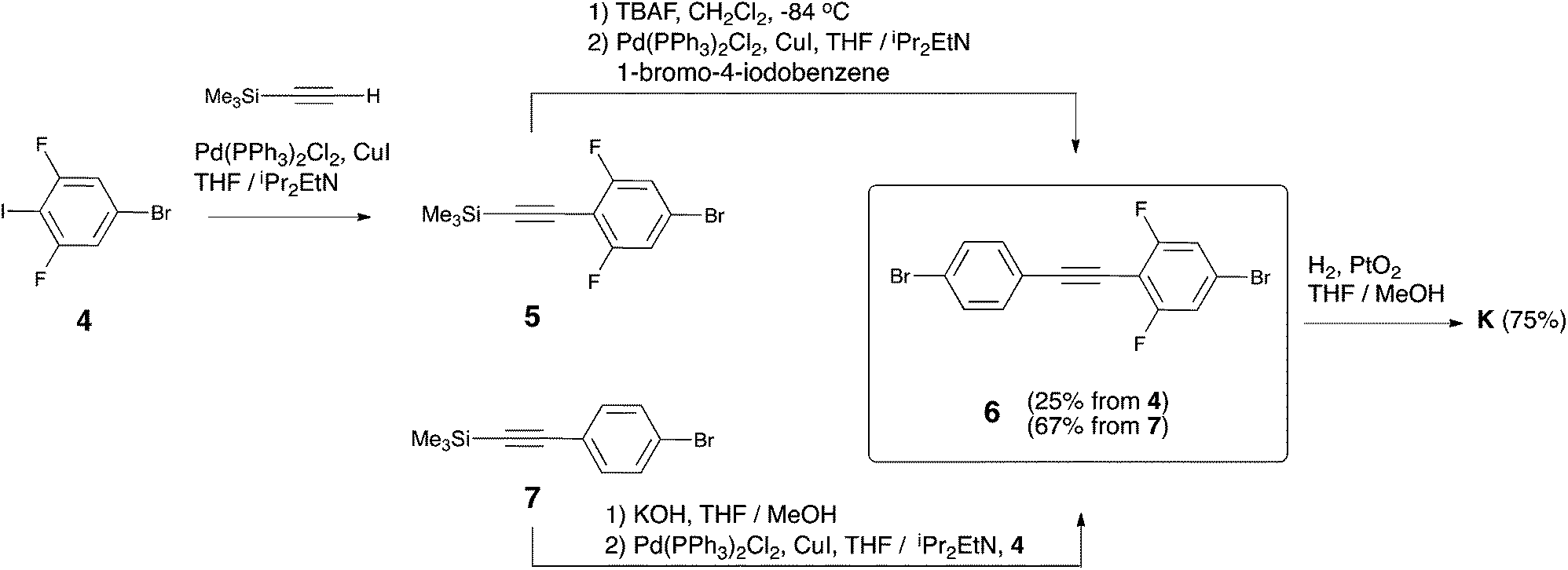 | ||
| Scheme 3 Synthesis of intermediate acetylene 6 and alkane K. | ||
In the final and most crucial step, selective hydrogenation of 6 in the presence of a catalytic amount of PtO2 at room temperature and atmospheric hydrogen pressure13 provides access to the key intermediate K. The selectivity of this hydrogenation reaction is very sensitive to the reaction conditions (see section 2 of the ESI†). Thus, to obtain reproducible results it is very important to stir the reaction thoroughly because the generation of side-products is minimized when the reaction is completed in the shortest time possible (typically 2–3 hours). If the reaction is left overnight, some of the phenyl–bromine bonds are cleaved. On the other hand, if the reaction is stopped before reaching full conversion, the desired product together with the intermediate containing an ethene bridge is obtained. In both cases, these mixtures of compounds cannot be easily separated by column chromatography. It was also noticed that the reaction cannot be pushed to completion when it is carried out in neat THF (in this case, a mixture between an intermediate containing the ethene bridge and K is always obtained even for overnight reactions). A mixture of THF and MeOH as the solvent for the reaction was found to afford the desired reactivity and selectivity, providing K in 75% yield after purification. Only two 6-type molecules are known in the literature.14 Thus, in order to explore the synthetic scope of the approach, the synthesis of two additional unsymmetric diphenylethanes (see Scheme 4: dichloro (11) and trifluoromethyl (13) derived diphenylethanes) was carried out from the corresponding new dibromotolane precursors.
 | ||
| Scheme 4 Synthesis of intermediate acetylenes 10 and 12 and alkanes 11 and 13. | ||
After cleavage of TMS from 7, the terminal alkyne was reacted with 8 under Sonogashira conditions to provide 10. Subsequent hydrogenation of 10 in the presence of a catalytic amount of PtO2 at room temperature and atmospheric hydrogen pressure provides access to chloro-derived diphenylethane 11. In this case, the reaction was carried out in THF overnight. After column chromatography, a small fraction containing the intermediate alkene was also collected. Alternatively, after cleavage of TMS from 7, the terminal alkyne was reacted with 9 under Sonogashira conditions to afford 12. Subsequent hydrogenation of 12 in THF/MeOH and in the presence of a catalytic amount of PtO2 at room temperature and atmospheric hydrogen pressure provides access to the trifluoromethyl-derived diphenylethane 13. In order to completely purify the compound, recrystallization in ethanol was carried out, affording 13 in moderate yield (39%).
It is noteworthy that K-type molecules have not been explored so far in the literature. For this reason, single crystals of K were subjected to an X-ray crystal structure determination. Compound K crystallizes as extremely thin plates in the centrosymmetric space group P21/c (no. 14). The two phenyl rings of the molecule are approximately coplanar with an interplanar angle of 1.7(3)°. The torsion angles of the phenyl rings to the central C–C bond are −99.8(8) and −79.4(9)°, respectively. The ethane bridge shows an anti-staggered conformation (Fig. 1).
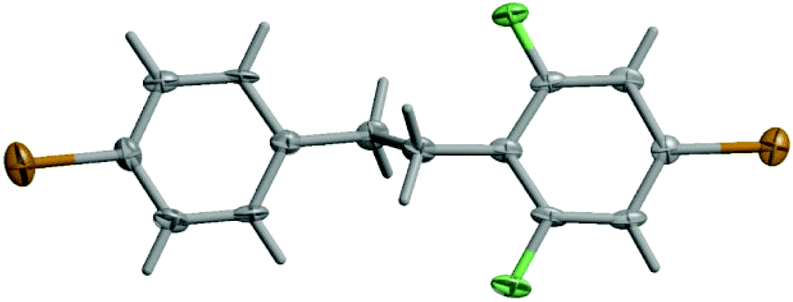 | ||
| Fig. 1 Molecular structure of K in the crystal. Displacement parameters are drawn at the 50% probability level. | ||
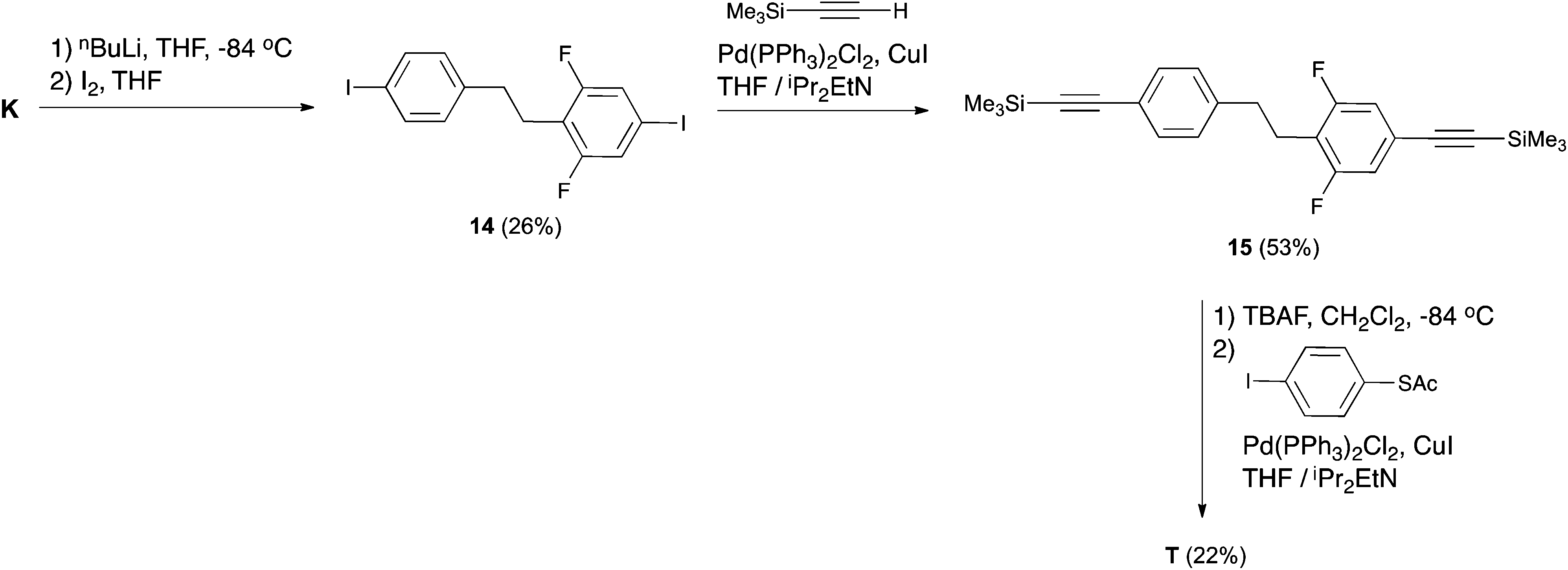
As pointed out in the Introduction, our aim was to use intermediate K to obtain T to enable experimental verification of its theoretically predicted exceptional rectification properties. Henceforth, compound T was obtained starting from the key intermediate K in three steps, according to Scheme 5. We initially attempted to react K with TMSA under Sonogashira conditions, but K proved unreactive. We then exchanged the bromine groups on K for iodines. This was accomplished by the lithium–halogen exchange using nBuLi at −84 °C followed by the addition of I2 to produce 14. The low yield of this reaction can be related to the fact that the fluorines in the meta position with respect to the bromine activate hydrogens in the ortho position, in such a way that deprotonation of these activated hydrogens may compete with the lithium–halogen exchange. Attempts to increase the yield of this reaction by modifying either the solvent or the organometallic reagent were unsuccessful. Compound 14 proved reactive to TMSA under Sonogashira conditions using Pd(PPh3)2Cl2 as a catalyst. Compound 15 was desilylated in the presence of TBAF and the terminal alkyne generated was coupled with 4-iodo-1-thioacetylbenzene8 in the presence of Pd(PPh3)2Cl2 to afford the target compound T.
 | ||
| Scheme 5 Synthesis of the molecular wire T. | ||
In summary, we have presented here a synthetic approach that gives access to biphenylethane-based molecules for application in single-molecule electronics. We have shown that whereas the synthesis of the symmetrically substituted biphenylethane-containing molecular wire R is straightforward, synthesis of the unsymmetrically substituted analogue T presents some additional challenges, mainly concerning the synthesis of intermediate K. Obvious strategies involving alkyl–alkyl or alkyl–aryl couplings afforded either no desired product or complex reaction mixtures. To overcome these problems, we have introduced a strategy to obtain the fluorine-containing 1,2-bis(4-bromophenyl)ethane K by selective hydrogenation of the corresponding alkyne precursor 6. In order to check the scope of this synthetic pathway towards unsymmetrically substituted 1,2-bis(4-bromophenyl)ethane derivatives, the synthesis of two additional biphenylethanes (9 and 11) was also demonstrated. K was subsequently used to synthesize a fully functionalized molecular diode. The outstanding performance of these molecules as single-molecule diodes was recently confirmed using the mechanically controllable break junction technique, and these results will be published in due course.15
We thank the Dutch Foundation for Fundamental Research on Matter (FOM), the Dutch Organisation for Scientific Research (NWO), the Ministry of Education, Culture and Science (OCW), and the EU (ERC advanced grant Mols@Mols). We thank T. D. Tiemersma-Wegman (University of Groningen) and Kersti Karu (UCL Chemistry Mass Spectrometry Facility, London) for carrying out high-resolution mass spectroscopy measurements.
Notes and references
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- M. L. Perrin, R. Frisenda, M. Koole, J. S. Seldenthuis, J. Celis Gil, H. Valkenier, J. C. Hummelen, N. Renaud, F. C. Grozema, J. M. Thijssen, D. Dulić and H. S. J. van der Zant, Nat. Nanotechnol., 2014, 9, 830–834 CrossRef CAS PubMed.
- (a) M. Elbing, R. Ochs, M. Koentopp, M. Fischer, C. von Hanisch, F. Weigend, F. Evers, H. B. Weber and M. A. Mayor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8815–8820 CrossRef CAS PubMed; (b) I. Diez-Perez, J. Hihath, Y. Lee, L. Yu, L. Adamska, M. A. Kozhushner, I. I. Oleynik and N. Tao, Nat. Chem., 2009, 1, 635–641 CrossRef CAS PubMed; (c) J. Hihath, C. Bruot, H. Nakamura, Y. Asai, I. Diez-Perez, Y. Lee, L. Yu and N. Tao, ACS Nano, 2011, 5, 8331–8339 CrossRef CAS PubMed; (d) E. Loertscher, B. Gotsmann, Y. Lee, L. Yu, C. Rettner and H. Riel, ACS Nano, 2012, 6, 4931–4939 CrossRef PubMed; (e) A. Batra, P. Darancet, Q. Chen, J. S. Meisner, J. R. Widawsky, J. B. Neaton, C. Nuckolls and L. Venkataraman, Nano Lett., 2013, 13, 6233–6237 CrossRef CAS PubMed; (f) A. Batra, J. S. Meisner, P. Darancet, Q. Chen, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Faraday Discuss., 2014, 174, 79–89 CAS.
- M. L. Perrin, E. Galán, R. Eelkema, F. C. Grozema, J. M. Thijssen and H. S. J. van der Zant, J. Phys. Chem. C, 2015, 119, 5697–5702 CAS.
- T. Kim, Z. F. Liu, C. Lee, J. Neaton and L. Venkataraman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10928–10932 CrossRef CAS PubMed.
- B. Capozzi, J. L. Xia, O. Adak, E. J. Dell, Z. F. Liu, J. C. Taylor, J. B. Neaton, L. M. Campos and L. Venkataraman, Nat. Nanotechnol., 2015, 10, 522 CrossRef CAS PubMed.
- See for instance: (a) C. D. Mboyi, S. Gaillard, M. D. Mabaye, N. Pannetier and J.-L. Renaud, Tetrahedron, 2013, 69, 4875–4882 CrossRef CAS; (b) J. Liu and B. Li, Synth. Commun., 2007, 37, 3273–3278 CrossRef CAS.
- Z. F. Shi, L.-J. Wang, H. Wang, X.-P. Cao and H.-L. Zhang, Org. Lett., 2007, 9, 595–598 CrossRef CAS PubMed.
- M. Giedyk, S. N. Fedosov and D. Gryko, Chem. Commun., 2014, 50, 4674–4676 RSC.
- R. Filler, G. L. Cantrell and E. W. Choe, J. Org. Chem., 1987, 52, 511–515 CrossRef CAS.
- (a) E. Negishi, H. Matsushita, M. Kobayashi and C. L. Rand, Tetrahedron Lett., 1983, 24, 3823–3824 CrossRef CAS; (b) J. D. St. Denis, C. C. G. Scully, C. F. Lee and A. K. Yudin, Org. Lett., 2014, 16, 1338–1341 CrossRef CAS PubMed.
- (a) H. R. Rogers, C. L. Hill, Y. Fujiwara, R. J. Rogers, H. L. Mitchell and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 217–226 CrossRef CAS; (b) H. R. Rogers, J. Deutch and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 226–231 CrossRef CAS; (c) J. J. Barber and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 239–243 CrossRef CAS.
- H. Cho, Y. Iwama, K. Okano and H. Tokuyama, Synlett, 2013, 0813–0816 CrossRef CAS.
- (a) G. Li, X. Wang and F. Wang, Tetrahedron Lett., 2005, 46, 8971–8973 CrossRef CAS; (b) X. Yang, S. Kajiyama, J.-K. Fang, F. Xu, Y. Uemura, N. Koumura, K. Hara, A. Orita and J. Otera, Bull. Chem. Soc. Jpn., 2012, 85, 687–697 CrossRef CAS.
- M. L. Perrin, E. Galán, R. Eelkema, J. M. Thijssen, F. C. Grozema and H. S. J. van der Zant, 2015, submitted for publication.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, characterization and X-ray data (CIF) for the products. CCDC 1445199. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob00008h |
| This journal is © The Royal Society of Chemistry 2016 |