
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct conjugate alkylation of α,β-unsaturated carbonyls by TiIII-catalysed reductive umpolung of simple activated alkenes†
Plamen
Bichovski
,
Thomas M.
Haas
,
Manfred
Keller
and
Jan
Streuff
*
Institut für Organische Chemie, Albert-Ludwigs-Universität Freiburg, Albertsraße 21, 79104 Freiburg, Germany. E-mail: jan.streuff@ocbc.uni-freiburg.de; Fax: +49 761 203 8715; Tel: +49 761 203 97717
First published on 25th January 2016
Abstract
The titanium(III)-catalysed cross-selective reductive umpolung of Michael-acceptors represents a unique direct conjugate β-alkylation reaction. It allows the cross-selective preparation of 1,6- and 1,4-difunctionalised building blocks without the requirement of stoichiometric organometallic reagents. In this full paper, the development and scope of the titanium(III)-catalysed cross-selective reductive umpolung of Michael-acceptors is described. Based on the observed selectivities and additional mechanistic experiments a refined mechanistic proposal is presented.
Introduction
The metal-catalysed conjugate addition reaction to enones and related Michael-acceptors has been a thriving research field over the past two decades. Nowadays, it is possible to perform this transformation in high yield and enantioselectivity using copper-, rhodium-, or palladium-catalysis for example,1 and even the asymmetric construction of quaternary carbon centres can be achieved with high selectivity.2 Still, one drawback of the classic protocols has been the requirement of organometallic coupling precursors that need to be prepared in advance (Scheme 1a). Only a few exceptions, most being Pd- or Ni-catalysed reductive Heck reactions, have been reported.3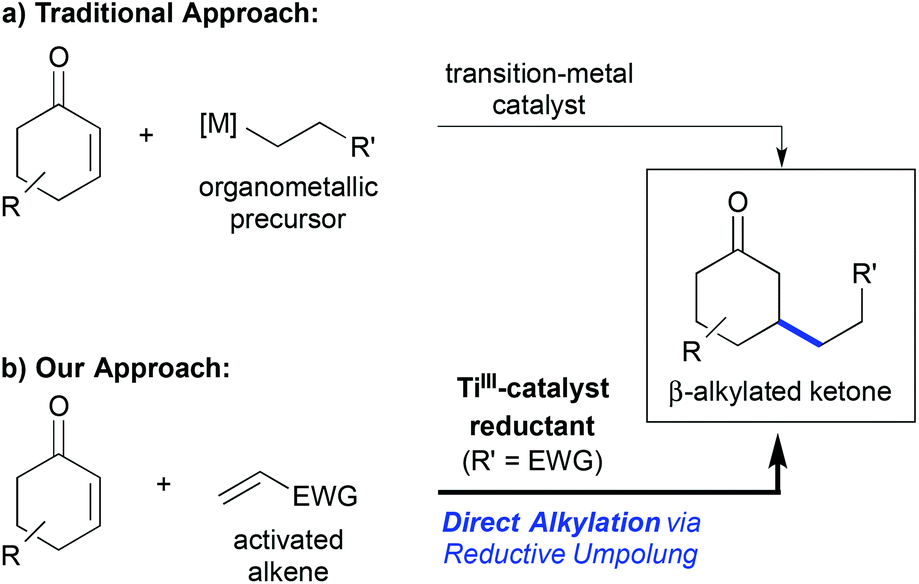 | ||
| Scheme 1 (a) Traditional β-alkylation of enones using premetallated reagents. (b) Direct titanium(III)-catalysed reductive umpolung enables the use of simple alkene precursors. | ||
Radical addition reactions to Michael-acceptors are complementary to traditional conjugate additions. They can be used to overcome this drawback and to address in particular conjugate β-alkylation reactions,4 which have remained challenging using conventional catalytic conjugate addition approaches.5 Hence, it has been shown that free radical additions using stoichiometric and catalytic conditions,6 as well as radical additions after titanium-catalysed reductive epoxide opening,7 can lead to the desired β-alkylated products in a very efficient manner. The advantage of the titanium-catalysed process was the superior catalyst control of the reaction selectivity, leading to high regio-, stereo- and even enantioselectivity.4
In 2011, we communicated a direct reductive β-alkylation of enones that enabled the use of readily available activated alkenes such as acrylonitrile as cross-coupling partners (Scheme 1b).8 Thus, the requirement of pre-metallated reagents or free radical conditions was overcome, which should be kept in mind with regard to more recent contributions in the field of reductive conjugate cross-couplings.5a–c,9 The reaction was a titanium(III)-catalysed overall umpolung reaction that led to 1,6-ketonitriles and related products. Related reductive homocoupling reactions were known before and had been applied even on industrial scale,10 but cross-selective tail-to-tail coupling of two Michael-acceptors had no precedence at that time. It should be noted that a redox-neutral NHC-catalysed cross-selective Michael umpolung was published shortly afterwards,11,12 which led to α,β-unsaturated 1,6-difunctionalized motifs.
In this full account, we wish to disclose the initial development of the titanium-catalysed cross-coupling of Michael-acceptors and the further advancement towards substrate classes such as quinolones, chromones and coumarins.13 The results lead to valuable implications for the future development of related transformations and the application of such direct β-alkylation reactions.
Results and discussion
Initial reaction optimisation
In a typical experiment, cyclohexenone (1) and 5 equiv. of inexpensive acrylonitrile (2) as coupling partner were reacted in the presence of titanocene dichloride [Cp2TiCl2] (10 mol%), zinc dust (2 equiv.), triethylamine hydrochloride (1.3 equiv.), and chlorotrimethylsilane (1.5 equiv.) in THF at 35 °C, to give alkylated ketone 3 in 87% yield after workup with aqueous HCl (Scheme 2). Manganese, a stronger reductant that has been frequently applied in catalytic reductive coupling reactions with titanocene catalysts and other metals,7,14 gave significantly reduced yields. The reaction outcome was explained by a preliminary mechanistic proposal started with a single-electron-transfer from the in situ generated titanium(III)-catalyst to the enone, generating a nucleophilic allylic radical. This radical would then add to the component with the lowest LUMO (acrylonitrile), forming the new carbon–carbon bond. The resulting electron-poor carbon radical next to the nitrile was then quickly reduced and protonated under the reaction conditions. Alternatively, a hydrogen radical abstraction (for example from THF) could take place, which still remained to be investigated. The addition of chlorotrimethylsilane was then vital for achieving turnover through silylation of the titanium(IV)-enolate that is generated in the process. This resulted in 4 as crude product. It was found that 1–2 turnovers could be achieved as well by addition of small amounts of water. The amount of Et3N·HCl was carefully balanced, since higher amounts led to the competing conjugate reduction of the enone, which was reported earlier by others.15 A five-fold excess of acrylonitrile further suppressed this conjugate reduction as well as the homo-dimerisation of the enone or its premature silylation.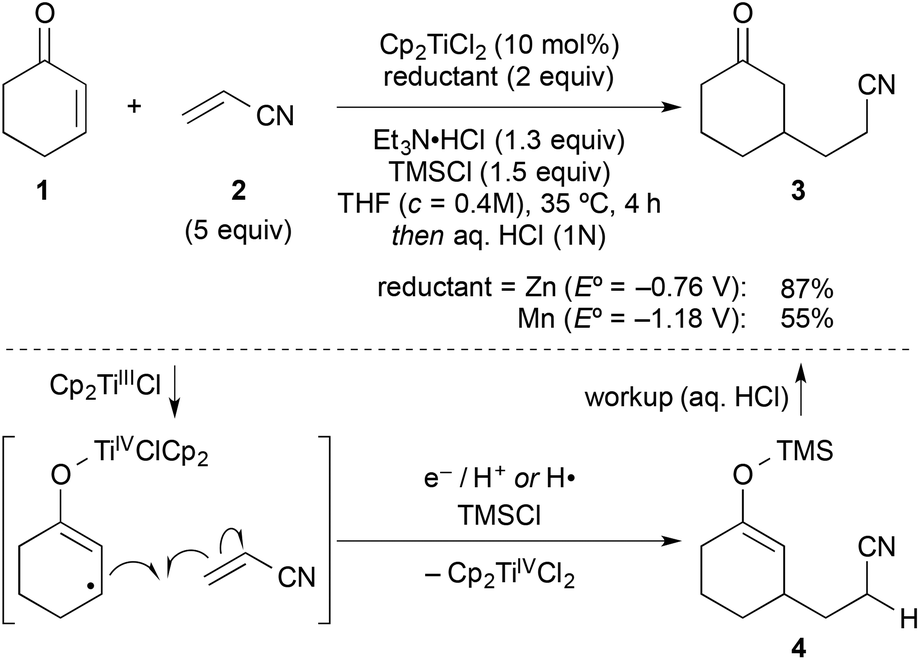 | ||
| Scheme 2 Typical coupling under the previously optimised reactions conditions und key steps of the originally proposed mechanism. Manganese gave inferior results. | ||
The reaction conditions were the result of a careful optimisation process. For example, tetrahydrofuran, which was often employed in catalyses involving single-electron-transfer reactions, was the most suitable solvent. Interestingly, a number of other solvents with a largely different dielectricity constant or Gutmann-donor number such as hexane, 1,4-dioxane, diethyl ether or dichloromethane gave reasonable yields as well. Other very similar solvents (toluene, chloroform, 1,2-dimethoxyethane) gave essentially no conversion to the product (Table 1). This illustrates that titanium(III)-chemistry is sensitive to a number of effects and reaction outcomes cannot be estimated easily. In fact, THF, which is only a moderate donor, was displaced from the TiIII-centre by acrylonitrile forming a deep-purple complex. Chelating solvents (1,2-DME) and strong donors such as acetonitrile or DMF, on the other hand, inhibited the catalyst through irreversible coordination.19 Thus we concluded, the major role of THF was to ensure a balanced solvation of the reaction partners (Et3N·HCl, is only moderately soluble, for example) and to promote an efficient reduction of TiIV to TiIII by the metallic reductant.
| Entry | Solvent |
ε
ρ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
DNb | Yieldc (%) |
|---|---|---|---|---|
| a Relative permittivity, see ref. 16. b Gutmann donor number, see ref. 17. c Determined by GC-analysis with 1,3-dimethoxybenzene as internal standard. | ||||
| 1 | n-Hexane | 1.89 (20 °C) | 0 | 60 |
| 2 | 1,4-Dioxane | 2.22 (20 °C) | 14.8 | 66 |
| 3 | CCl4 | 2.24 (20 °C) | 0 | 2 |
| 4 | Toluene | 2.39 (20 °C) | 0.1 | 3 |
| 5 | Et2O | 4.27 (20 °C) | 19.2 | 79 |
| 6 | CHCl3 | 4.81 (25 °C) | 4 | 1 |
| 7 | 1,2-DME | 7.3 (23.5 °C) | 20.0 | 0 |
| 8 | THF | 7.52 (22 °C) | 20.0 | 90 |
| 9 | CH2Cl2 | 9.14 (20 °C) | 1 | 61 |
| 10 | 1,2-DCE | 10.42 (20 °C) | 0 | 61 |
| 11 | t-BuOH | 12.5 (20 °C) | — | 0 |
| 12 | MeCN | 36.64 (20 °C) | 14.1 | 16 |
| 13 | DMF | 38.25 (20 °C) | 26.6 | 5 |
The choice of triethylamine hydrochloride as additive emerged from a screening of various ammonium salts. Without such an ammonium salt additive only poor conversion to the desired product was observed (Table 2, entry 1). Hydrochlorides within a pKa range of pKaH2O = 10–11 gave the most satisfying results. Quinuclidinium and diisopropylethylammonium salts that were within the pKa range of triethylamine gave slightly lower yields (78% and 64%, respectively). The more acidic hydrochlorides of 2,4,6-collidine and pyridine as well as hydrochlorides of secondary amines had a negative impact on the reaction (entries 3, 4, 8, and 9). Interestingly, the addition of unprotonated triethylamine was beneficial too, but also lead to the formation of larger amounts of the trimethylsilylenol ether of cyclohexenone (entry 10). The superiority of triethylamine hydrochloride, however, cannot be explained by its acidity alone and might stem from the tendency of Et3N·HCl to form a TiIII-Et3N·HCl adduct 6 (Scheme 3) with the active titanium(III) monomer 5, which was proposed to stabilize the catalyst.20
 | ||
| Scheme 3 Stabilising effect of added Et3N·HCl on [Cp2TiIIICl]. | ||
| Entry | Additive | pKa (H2O)a | Yieldb (%) |
|---|---|---|---|
| a Literature values, see ref. 18. b Determined by GC-analysis with 1,3-dimethoxybenzene as internal standard. c Significant amounts of the trimethylsilyl enol ether of cyclohexenone were observed. | |||
| 1 | None | — | 10 |
| 2 | TFA | 0.23 | 0 |
| 3 | Pyridine·HCl | 5.25 | 28 |
| 4 | Collidine·HCl | 7.48 | 55 |
| 5 | Et 3 N·HCl | 10.75 | 90 |
| 6 | Quinuclidine·HCl | 11.0 | 78 |
| 7 | iPr2NEt·TFA | ca. 11 | 64 |
| 8 | iPr2NH·TFA | 11.05 | 0 |
| 9 | Piperidine·HCl | 11.22 | 26 |
| 10 | Et3N | >20 | 48c |
Lowering the catalyst amount to 5 mol% or 3 mol% still gave 70% and 55% yield, respectively (Table 3). However, the above mentioned competing reactions (silyl enol ether formation of 1 and homo-coupling of 1) became more prominent. Without the titanocene catalyst, no product was formed.
Scope of the enone
Using the optimised conditions, a series of substrates was coupled with acrylonitrile in a similar manner to give the corresponding 1,6-ketonitriles in moderate to high yields after workup with aqueous HCl (Scheme 4). Different enone ring sizes (7–9) and substitution patterns were tolerated that enabled the construction of quaternaty carbon centres at the β-position (9, 10). The coupling proceeded in excellent diastereoselectivity regarding the newly formed C–C bond, which allowed the selective conjugate alkylation of moderately complex substrates such as (S)-carvone and (S)-verbenone (13, 14). In addition, a dihydrothiopyranone (4-thiacyclohexenone) could be employed as well giving ketonitrile 15 in a moderate 46% yield. Here, a slow addition of the dihydrothiopyranone via a syringe-pump was required to prevent the undesired reductive dimerization of the substrate.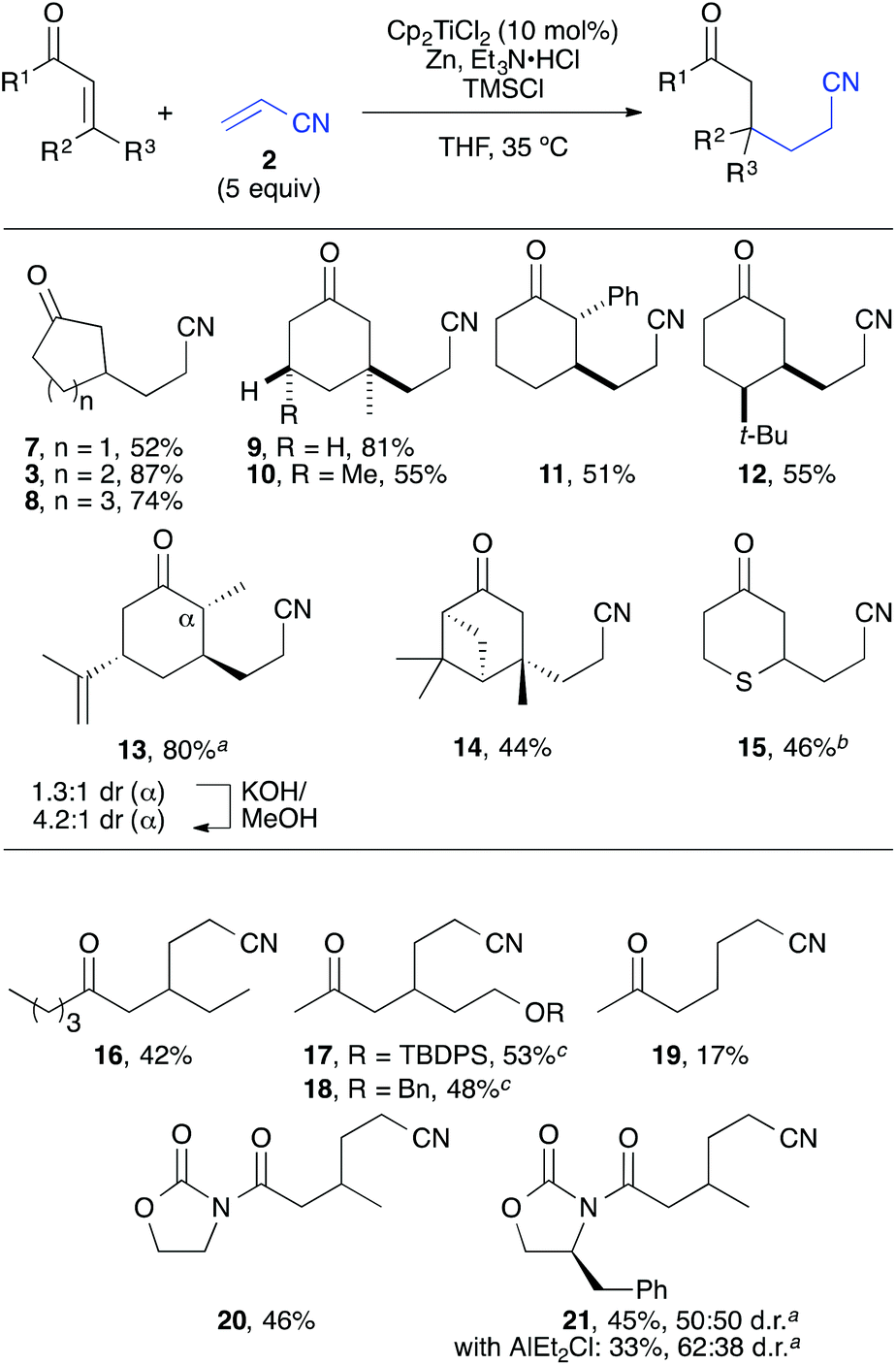 | ||
| Scheme 4 Reductive Coupling of Cyclic Enones with Acrylonitriles. Yield of isolated material. aCombined yield. bSyringe pump addition of the dihydrothiopyranone precursor. cReaction at 0 °C. | ||
The scope could be further extended towards linear enone substrates that were transformed into the corresponding 1,6-ketonitriles 16–18 in reasonable yields (42–53%). Methyl vinyl ketone, however, led to uncontrolled polymerisation under the reaction conditions and, thus, only 17% of compound 19 were isolated. In addition, α,β-unsaturated amides containing achiral and chiral oxazolidinone units could be employed as well with moderate success. However, no diastereoselectivity was observed, even if precoordination of the substrate by AlEt2Cl was attempted.
The titanium-catalysed reductive umpolung/β-alkylation could be applied to a number of quinones, chromones, and coumarines as described in the following.13 A series of substituted quinolones was treated under the same conditions with acrylonitrile as coupling partner and good yields were obtained for N-methylated and N-benzylated substrates having no further substitution (Table 4). The reaction worked also with substitution at position 7 and 8, although the yields were slightly diminished. For example, 7-methoxy, -methyl, -phenyl, -thiophen-3-yl, and -phenylethynyl groups worked well (entries 3–8). In some cases (e.g. R1 = Ph), however, significant differences in yield were observed for the N-methylated and N-benzylated precursors (entries 5 and 6). Double substitution was tolerated as well (entry 9) and importantly, halogenation of the aromatic backbone was tolerated to some extend (entries 10–12).21 This underlined the mildness of the title reaction.
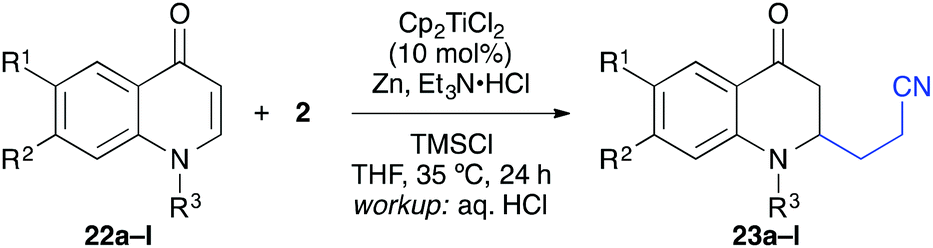

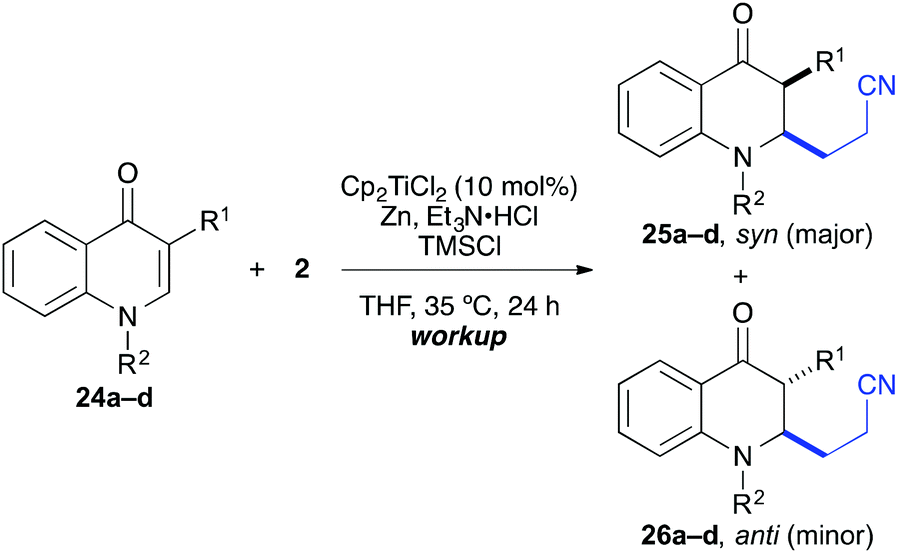
The coupling worked significantly better with 3-substituted quinolones. Here, yields between 69% and 91% were obtained for 3-methyl and 3-phenyl derivatives (Table 5). Importantly, aqueous workup under protic conditions gave exclusively the syn-diastereomer, which was a result of a pseudo-axial orientation of the cyanoethyl chain due to steric repulsion with the N-alkyl group. Quenching the silyl enol ether under controlled conditions instead produced significant amounts of the anti-diastereomer (in a 2.4:1 syn/anti ratio), which could be separated and structurally confirmed by X-ray analysis (Fig. 1).22,23 The workup had to be carried out with care and removal of the excess in acrylonitrile under reduced pressure was required. Otherwise, overalkylation in form of a subsequent Michael-addition of the enolate to acrylonitrile took place (Scheme 5). For example, if a reaction of 24a (R = Me) or 24b (R = Bn) with acrylonitrile was quenched by addition with TBAF at 0 °C, the desired products 25a and 25b were received in 30% and 42% yield, respectively, together with the corresponding double addition products 27a and 27b (42% and 39%, respectively).
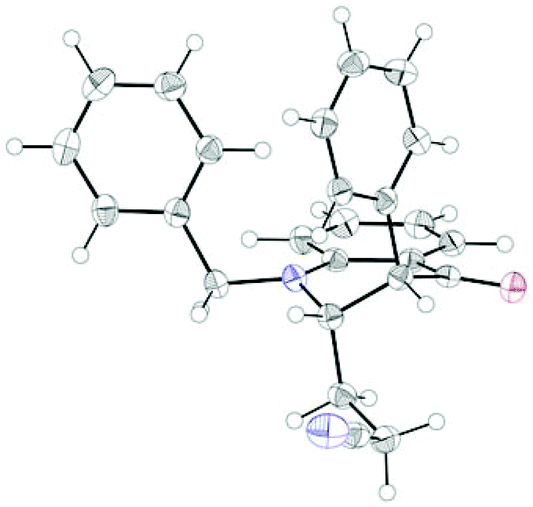 | ||
| Fig. 1 X-ray structure of 26d. Thermal ellipsoids drawn at 50% probability level. | ||
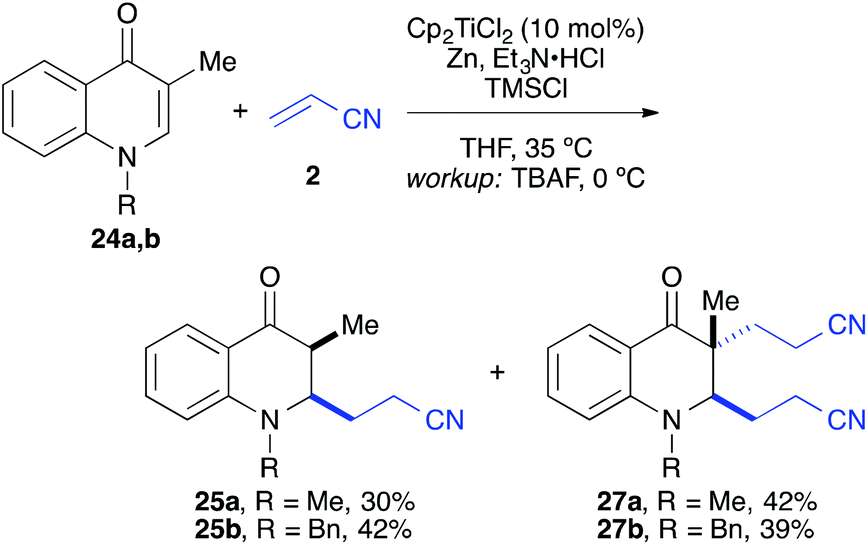 | ||
| Scheme 5 Workup with TBAF at 0 °C in presence of an excess of acrylonitrile led in part to double cyanoalkylation products. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Products | Workupa | syn/anti | Yieldb [%] |
| a HCl workup: aq. 1 N HCl, 0 °C, 3 h. TBAF workup: TBAF (1 M in THF), −78 °C, 3 h. b Yield of isolated product. | ||||||
| 1a | Me | Me | 25a, 26a | HCl | >95:5 |
86 |
| 1b | TBAF | >95:5 |
74 | |||
| 2a | Me | Bn | 25b, 26b | HCl | >95:5 |
91 |
| 2b | TBAF | 71:29 |
69 | |||
| 3a | Ph | Me | 25c, 26c | HCl | >95:5 |
72 |
| 3b | TBAF | 71:29 |
89 | |||
| 4a | Ph | Bn | 25d, 26d | HCl | >95:5 |
85 |
| 4b | TBAF | 70:30 |
69 | |||
In analogy to the quinolone substrates, C3-unsubstituted chromones were moderately successful substrates for the titanium-catalysed reductive umpolung (Table 6). Manganese powder as reductant gave slightly better reaction yields than zinc dust. Electron-donating substituents were tolerated (37–50%), but no product could be isolated with 6-bromochromone (28d). A 2-methyl substituted chromone gave only 17% product and flavone itself was transformed into the desired chromanone in 31% yield, which corresponded to two catalyst turnovers. As observed before, the yields were significantly improved when C3-substituents were present (Table 7). Interestingly, not only alkyl and aryl groups could be installed at this position, but also halides such as chloride and bromide (entries 4 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Products | Workup | syn/anti | Yielda [%] |
| a Yield of isolated product. b Isolated as diastereomeric mixture. | ||||||
| 1a | Me | H | 31a, 32a | HCl |
21:79 |
69 |
| 1b | TBAF |
78:22 |
78 | |||
| 2a | Ph | H | 31b, 32b | HCl |
21:79 |
73 |
| 2b | TBAF |
75:25 |
62 | |||
| 3a | Ph | i-PrO | 31c, 32c | HCl |
37:63 |
82 |
| 3b | TBAF |
75:25 |
81 | |||
| 4a | Cl | H | 31d, 32d | HCl |
38:62 |
49b |
| 4b | TBAF |
64:36 |
54b | |||
| 5a | Br | H | 31e, 32e | HCl |
22:78 |
42b |
| 5b | TBAF |
83:17 |
42b | |||
The relative configuration was opposite to the quinolin-4-one products and the anti-diastereomer was isolated as major component after workup with aqueous HCl.
The workup procedure drastically influenced the product distribution. The diastereoselectivity could be even switched from the favoured anti-products to the syn-products in moderate to good diastereoselectivity when workup was carried out under kinetically controlled conditions (TBAF, −78 °C).
3-Iodochromone 30f, however, was too reactive and suffered from dehalogenation under the reaction conditions and cross-coupling product 29a was isolated (Scheme 6).
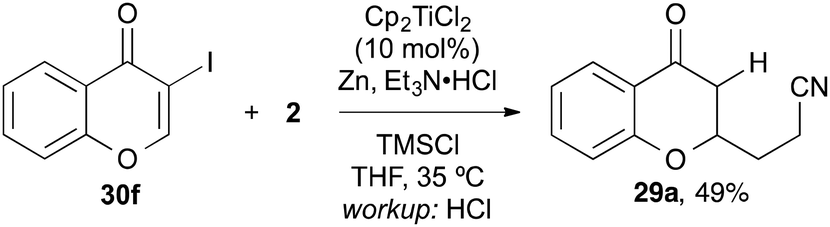 | ||
| Scheme 6 The reaction with 3-iodochromone afforded deiodinated chromanone 29a. | ||
Finally, the cross-coupling with acrylonitrile was applied to the reductive β-cyanoalkylation of coumarins. Using precursors with a diverse substitution pattern, moderate yields were achieved for the cross-coupling reaction (Table 8). Attempts to further optimize the reaction outcome were unsuccessful.24 The best yield (65%) was obtained with 6-methylcoumarin (entry 3). A quaternary stereocentre could be installed in 36% yield (entry 8) and α,β-disubstituted 2-chromanones were formed in similar quantities by the reductive cyanoethylation reaction. The diasteroselectivity was again very high and the syn-diastereomers were isolated as sole products. The relative syn-configuration was unambiguously confirmed by X-ray analysis of product 34i (Fig. 2).22
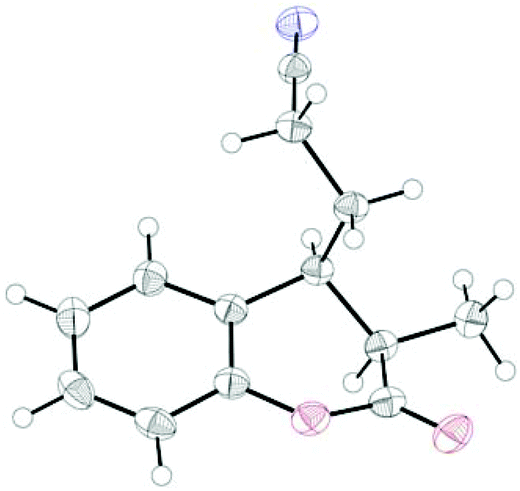 | ||
| Fig. 2 X-ray structure of 34i. Thermal ellipsoids drawn at 50% probability level. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | R5 | Product | Yielda [%] |
|
a Yield of isolated product.
b Calculated yield from an inseparable mixture with the substrate (∼1:1 ratio).
c Only the syn-isomer was formed.
d A single isomer was formed, which was assigned in analogy to 34i.
|
|||||||
| 1 | H | H | H | H | H | 34a | 42 |
| 2 | Br | H | H | H | H | 34b | 36 |
| 3 | Me | H | H | H | H | 34c | 65 |
| 4 | H | Me | H | H | H | 34d | 46 |
| 5 | H | MeO | H | H | H | 34e | 45 |
| 6 | H | Me2N | H | H | H | 34f | 26b |
| 7 | H | H | Me | H | H | 34g | 33 |
| 8 | H | H | H | Me | H | 34h | 36 |
| 9 | H | H | H | H | Me | 34i | 44c |
| 10 | H | Me2N | H | H | Me | 34j | 38b,d |
| 11 | H | Me2N | H | H | Ph | 34k | 24b,d |
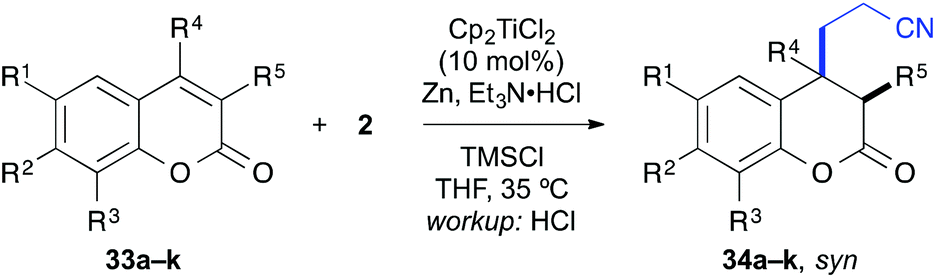
Scope of the coupling partner
Importantly, the reaction was not limited to acrylonitrile as coupling partner. Substituted acrylonitrile derivatives and a number of other activated alkenes including acrylamides and acrylates could be employed as coupling partners as well (Table 9). With cyclohexenone, we first observed that meth-acrylonitrile worked almost as well as acrylonitrile itself (entry 1) and even the quaternary carbon could be formed smoothly (entry 2). The reaction with crotononitrile was hampered (entry 3), probably due to increased sterics leading to a reduction in yield to 27%. In both cases, a 1:1 mixture of diastereomers was received. The coupling with N,N-dimethylacrylamide was unsuccessful, since this compound appeared to inhibit the catalyst (entry 4). This could be successfully addressed by the installation of a tosyl group at the amide nitrogen, which prevented the amide resonance and lowered the coordination tendency (entry 5). The coupling proceeded smoothly with 73% yield in the presence of added cinnamonitrile, which increased the yield by about 20%. Cinnamonitrile itself was an inferior coupling partner (<5%), but it was empirically found to be beneficial for this reaction. One possible rationale for this effect could be a coordination and stabilisation of the catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Product | dr | Yielda [%] | |
| a Yield of isolated product. b Combined yield. c Cinnamonitrile (20 mol%) was added to the reaction mixture. d Workup with TBAF instead of aq. HCl. | ||||
| 1 |
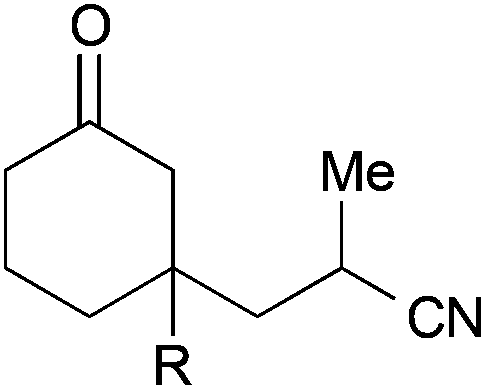
|
35, R = H | — | 71 |
| 2 | 36, R = Me | 50:50 |
70b | |
| 3 |
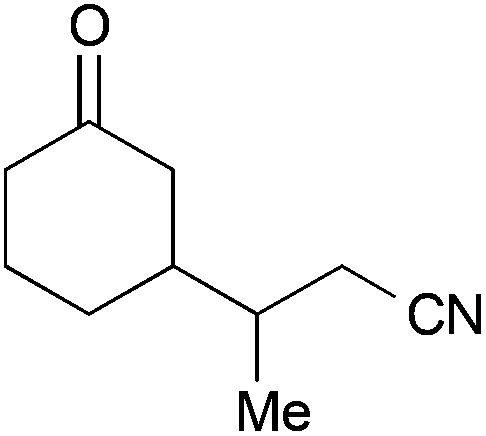
|
37 | 50:50 |
27b |
| 4 |
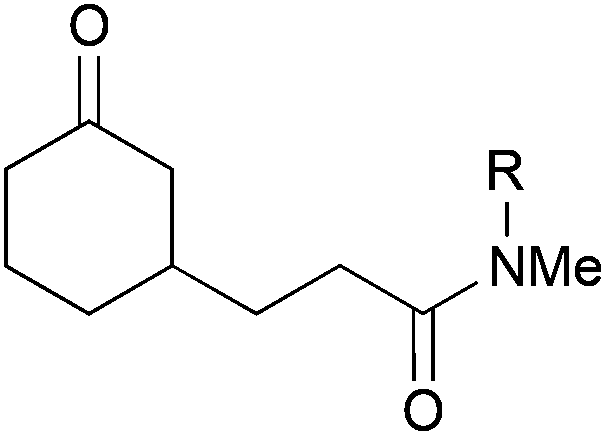
|
38, R = Me | — | 0 |
| 5 | 39, R = Ts | — | 73c | |
| 6 |
|
40, R = H | 57:43 |
36b |
| 7 | 41, R = Me | 58:42 |
90b | |
| 8 |
|
42, R = H | 55:45 |
28b |
| 9 | 43, R = Me | 62:38 |
29b | |
| 10 |
|
44 | 62:38 |
18 |
| 11 |
|
45, R = Me | — | 35d |
| 12 | 46, R = Et | — | 47d | |
| 13 | 47, R = t-Bu | — | 37d | |
| 14 | 48, R = Ph | — | 52 | |
| 15 | 49, R = Mes | — | 81 | |
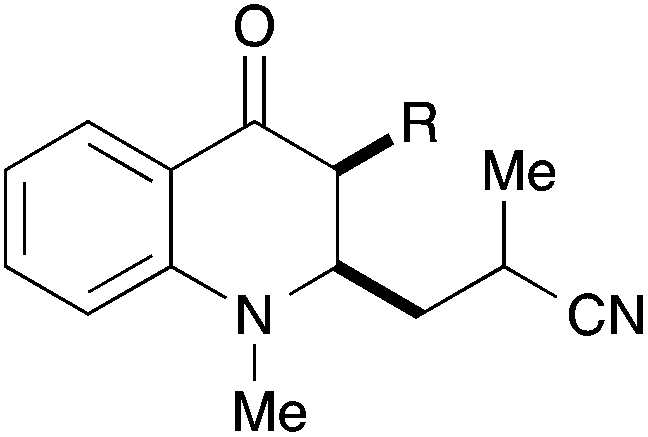
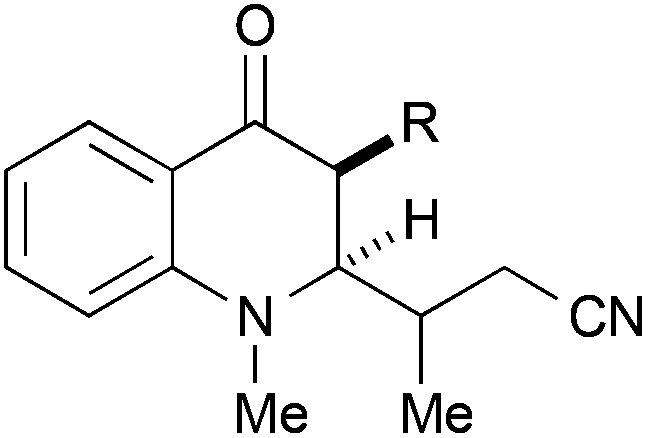
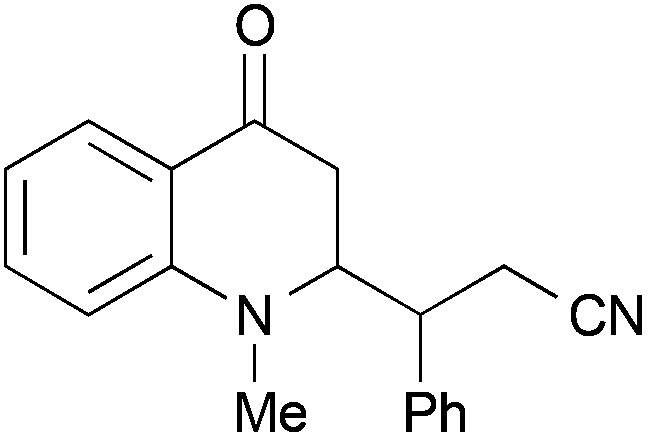
In a second series of experiments with N-methyl-4-quinolones 22a and 24a, good results were obtained for the couplings with methacrylonitrile as well. 3-Methylquinolone 24a gave again exclusively the syn-product with respect to the ring substitution in excellent 90% yield. The product was obtained as an inseparable ∼1:1-mixture of diastereomers with respect to the additional stereocentre at the nitrile α-carbon (entry 7). With crotononitrile, the yields were again reduced to ca. 30% (cf. entry 3) but a moderate diastereoselectivity of 1.6:1 dr was observed by NMR for the reaction with 24a (entry 9). Cinnamonitrile, which was employed for entry 5 as a beneficial additive, could be coupled in 18% yield to product 44 (entry 10).
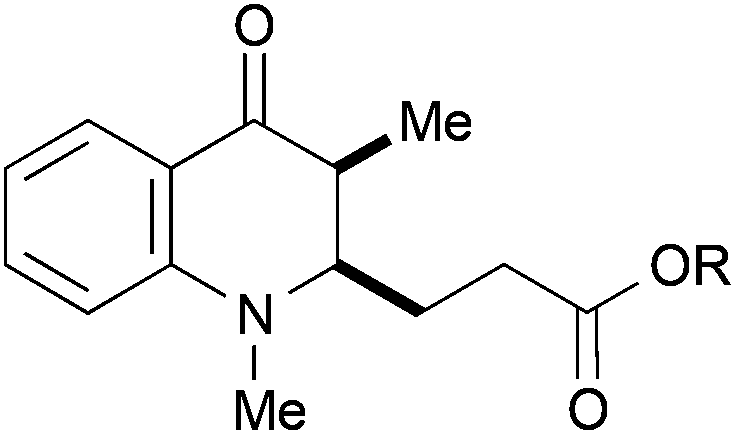
With the 3-methylated quinolone 24a as substrate, acrylates could be employed efficiently in the reductive catalytic umpolung as well (entries 11–15). Here, reasonable results were obtained with methyl, ethyl, and tert-butyl acrylate. The yield was slightly improved with the less electron-rich phenyl acrylate and with the sterically hindered mesityl acrylate,25 the coupling proceeded smoothly in 81% yield. In all cases, no cross-coupling was observed in absence of the titanocene catalyst.
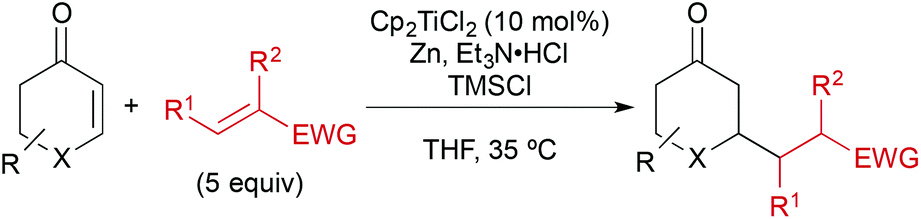
In addition, a number of other electron-deficient alkenes were tested as potential coupling partners with less success (Fig. 3). Other common nitrile-based Michael-acceptors such as 2-chloroacrylonitrile or Knoevenagel products of malononitrile or ethyl cyanoacetate did not undergo the desired reaction. This was also true for 2-nitropropene and β-nitrostyrene as well as vinyl sulfones. A saccharine-derived acrylamide, vinyl diethyl phosphonate or a propargylic ester were not suitable as well. In several cases, the reduction of the activated alkene was observed instead of the desired cross-coupling reaction. With N-acryloylsaccharine, for example, formation of the corresponding propionic amide took place.
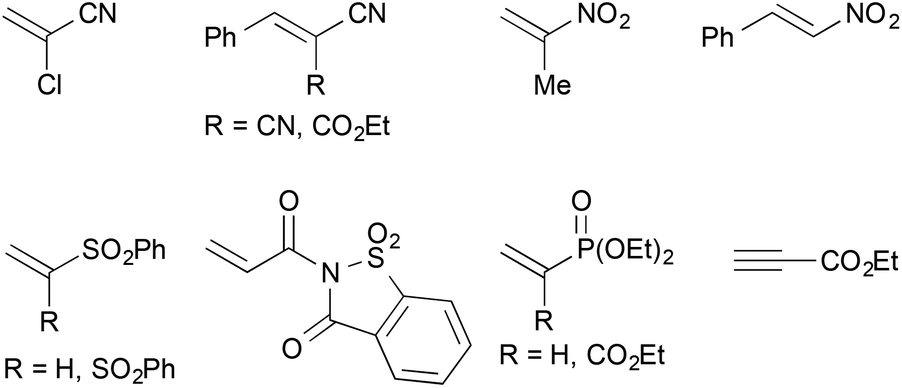 | ||
| Fig. 3 Unsuitable cross-coupling partners. | ||
Mechanistic discussion
From the observations that were made during our studies, several conclusions could be drawn regarding the underlying reaction mechanism, which allowed us to refine the initially proposed mechanism.As shown in Scheme 7, coordination of the in situ formed titanium(III)-catalyst to the enone substrate could also be interpreted as the formation of an allylic ketyl radical anion that remained coordinated to a titanium(IV)-centre. In fact, the unpaired electron was in part located at the titanium centre, at the β-carbon and at the carbonyl carbon as illustrated by the three resonance structures shown in Scheme 7. This was supported by the calculated spin density distribution at the Cp2TiIIICl–cyclohexenone complex. It was majorly located at the titanium centre and in part located at the carbonyl and β-carbon.24 A similar situation was found for an acrylonitrile–titanium(III) complex. This situation explained our experimental results: reductive coupling at the β-position leading to conjugate addition products (e.g. ketonitrile 3) was the usually preferred pathway. However, substrates with increased sterical bulk at the β-carbon led to a change in the regiochemistry and the corresponding 1,2-addition products were formed.26 For example, the reductive cross-coupling of the Wieland–Miescher ketone gave the corresponding cyanoethylated allylic alcohol 50 in 55% yield and moderate diastereoselectivity. A similar experiment with progesterone afforded the corresponding product 51 in excellent 91:9 dr and 65% yield.
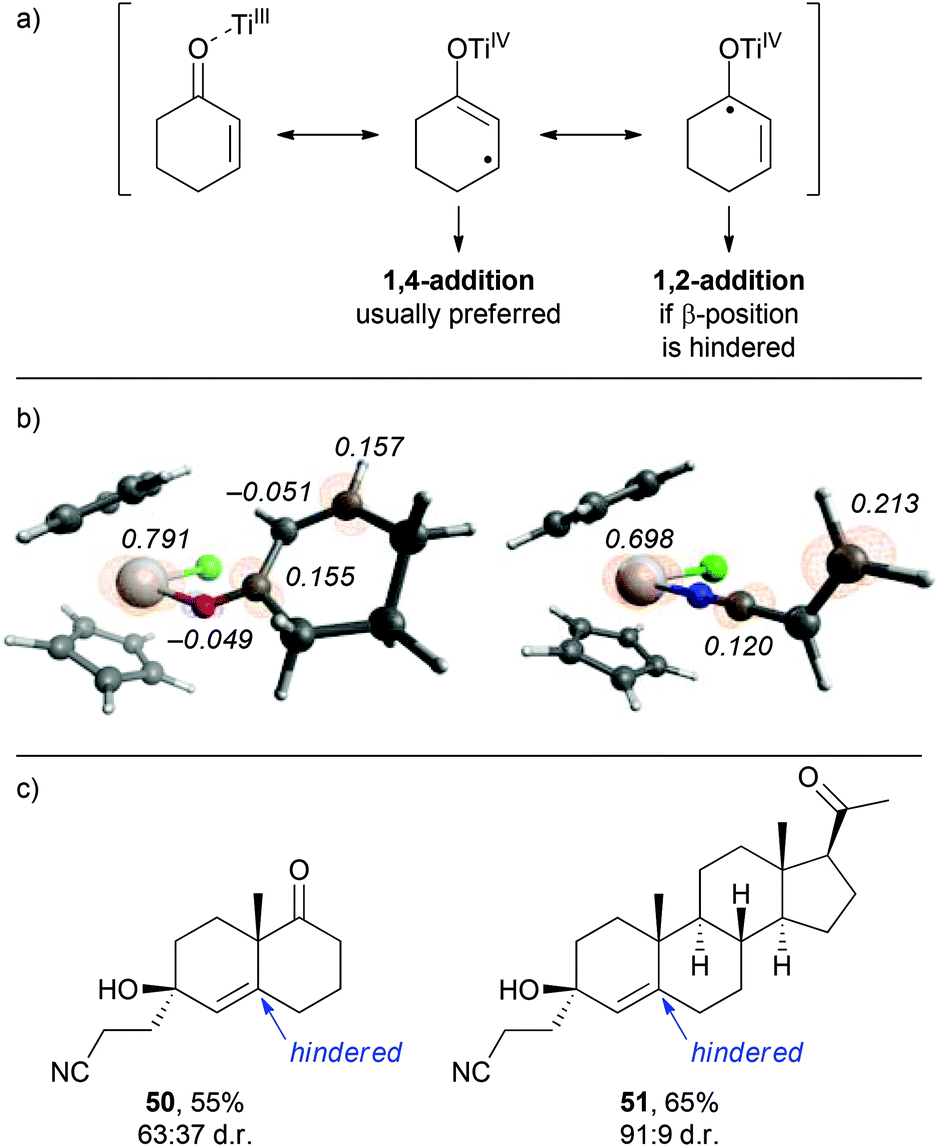 | ||
| Scheme 7 Substrate-dependent divergent regioselectivity. (a) Mesomeric forms of a TiIII–cyclohexenone complex. (b) Calculated spin density distribution for TiIII–cyclohexen-one and TiIII–acrylonitrile complexes (iso value = 0.01). (c) Observed 1,2-addition products from sterically hindered enone substrates. | ||
The origin of the hydrogen atom that was transferred to the nitrile α-carbon in course of the standard coupling between cyclohexenone and acrylonitrile was probed as well. A reaction run in THF-d8 did not lead to any deuterium incorporation into the product (Scheme 8). If a carbon-centred radical was present at this position a deuterium radical abstraction from the solvent would have been likely to occur. On the contrary, a reaction with triethylamine deuterochloride resulted in about 70% deuteration of the product at this position, which was evidence for a protonation step under the usual reaction conditions. This protonation at the nitrile α-carbon was unselective due to the absence of stereoelements in its proximity, which explains the formation of 1:1 diastereomeric mixtures in the reactions with methacrylonitrile (see Table 9, entries 2, 6, and 7).
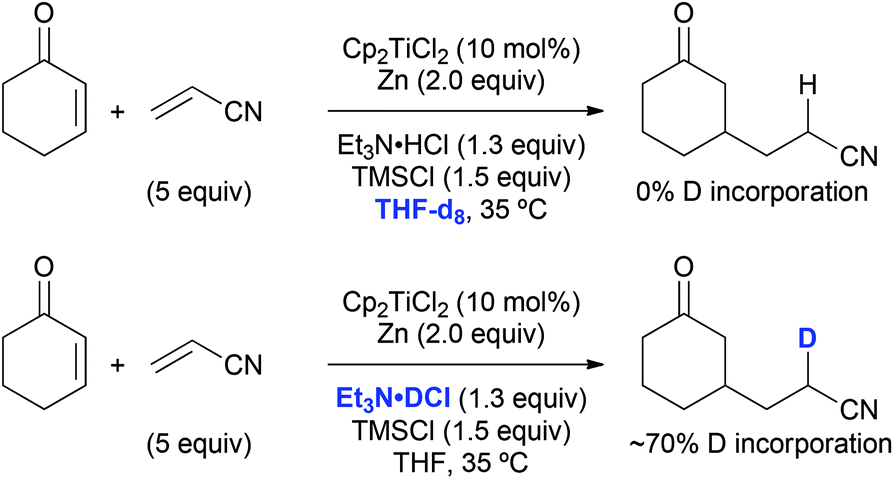 | ||
| Scheme 8 Deuterium experiments point towards a nitrile α-protonation event. | ||
Together with the results from our previous study on the mechanism of the titanium(III)-catalysed cross-acyloin type coupling,27 these observations led to a refined mechanistic proposal for the standard reaction (Scheme 9).
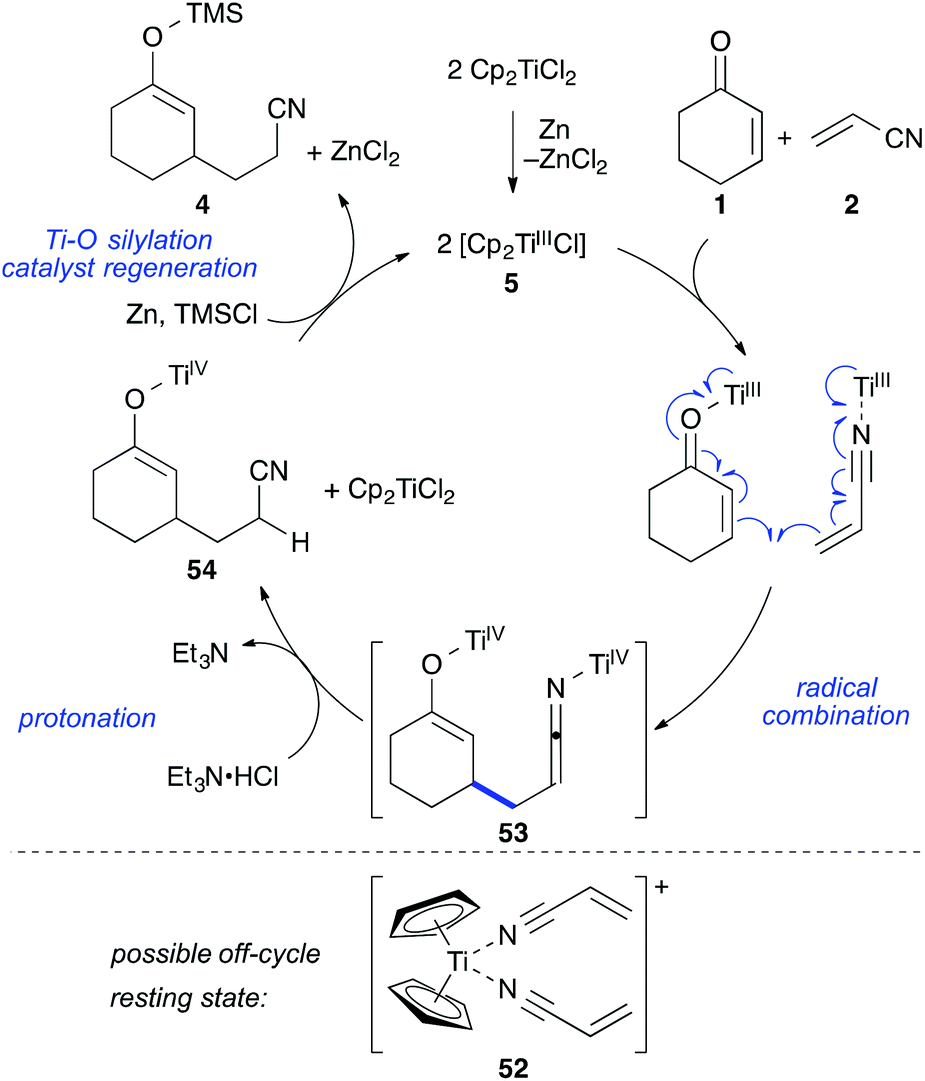 | ||
| Scheme 9 Mechanistic proposal for the reductive umpolung of Michael-acceptors. | ||
The reaction formally begins with the formation of two equivalents of 5 from [Cp2TiCl2] zinc followed by reaction with enone 1 and nitrile 2 to form coordination complexes. These complexes are in equilibrium through ligand-exchange processes. It is likely that a cationic resting state 52 is formed by solvation of the remaining chloride and coordination of a second acrylonitrile molecule (acrylonitrile was employed in a 50 fold excess with respect to the catalyst). This species could be the reason for the observed colour change to deep purple after addition of acrylonitrile and before addition of TMSCl during the reaction setup. A similar cationic resting state was previously established for the related ketone–nitrile coupling by X-ray analysis.27 The C–C bond formation would then take place in form of a catalyst-controlled radical combination, avoiding the presence of free radicals and leading to bistitanated ketenimine-enolate 53. The metallated ketenimine was quickly protonated by the hydrochloride (which was supported by the deuterium experiment) forming enolate 54. The titanium enolate was then cleaved by chlorotrimethylsilane releasing the crude product in form of silyl enol ether 4 and enabling catalyst turnover. Zinc then regenerated the titanium(III) catalyst 5. If desired, the silyl enol ether 4 could be isolated as one regioisomer in 87% yield (workup with water and filtration over florisil)8 or quenched with HCl or TBAF to afford the corresponding ketonitrile as done for the tables in this work.
Conclusions
In conclusion, we have established the titanium(III)-catalysed reductive umpolung of Michael-acceptors as an efficient cross-coupling tool for the synthesis of building blocks with functional groups in 1,6-distances. This was demonstrated on 70 examples in total including couplings with acylonitriles, acylamides and acrylates. Precursors with increased sterical hindrance could be employed for the selective synthesis of 1,4-difunctionalised products. A refined mechanistic picture was proposed based on the observed product distributions, the regio- and stereoselectivity, as well as the deuterium experiments. In the future, the development of related reductive cross-couplings will be accelerated due to the selectivity trends and mechanistic insight gained in this study. The method itself will be useful for the preparation of synthetic building blocks with functionalities in unnatural bond distances. Currently, efforts are undertaken to develop an enantioselective variant of this direct reductive β-alkylation reaction.Experimental section
Standard procedure for the TiIII-catalysed reductive umpolung
A flame-dried 50 mL-Schlenk tube containing a magnetic stirbar was charged under argon atmosphere with Cp2TiCl2 (12.4 mg, 0.05 mmol, 10 mol%), Zn (65.0 mg, 1.00 mmol, 2.0 equiv.) und Et3N·HCl (89.5 mg, 0.650 mmol, 1.3 equiv.). Stirring was started. The vessel was evacuated and backfilled with argon after a few minutes. Absolute THF (1.25 ml) was added and after 1 min the mixture had turned from red to lime-green. The substrate (e.g.1, 0.5 mmol, 1.0 equiv.) was added followed by the cross-coupling partner (e.g.2, 2.5 mmol, 5 equiv.) and TMSCl (95.2 μl, 1.5 equiv.). The reaction vessel was sealed with a greased glass-stopper and the reaction stirred for the given time at 35 °C in an oil bath or at the given temperature after which the reaction was brought back to room temperature. Unless noted otherwise, workup was carried out by addition of 1 N aqueous HCl (4 ml) and CH2Cl2 and stirring was continued for 30 minutes at room temperature (23 °C). The mixture was transferred into a separation funnel containing H2O (20 ml) and CH2Cl2 (20 ml). The biphasic mixture was shaken, the organic layer separated and the aqueous layer extracted with additional CH2Cl2 (3 × 10 ml). The combined organic extracts were dried (Na2SO4), concentrated and purified by flash chromatography as described.Workup with TBAF under kinetically controlled conditions (see Tables 6 and 7)
The reaction was setup as described in the standard procedure. After the given reaction time, all volatile components were removed under reduced pressure and heating was discontinued. The residue was treated with CH2Cl2 (5 ml) and cooled to −78 °C. At that temperature, TBAF (1 M in THF, 2.50 ml, 2.5 equiv.) was added dropwise. The mixture was stirred for another 3 h at −78 °C and then allowed to warm to room temperature (23 °C). The mixture was transferred into a separation funnel containing H2O (20 ml) and CH2Cl2 (20 ml). The biphasic mixture was shaken, the organic layer separated and the aqueous layer extracted with additional CH2Cl2 (3 × 10 ml). The combined organic extracts were dried (Na2SO4), concentrated and purified by flash chromatography as described.For a full list of materials and methods, detailed experimental data, compound characterizations and computational details, see the ESI.†
Acknowledgements
The Fonds der Chemischen Industrie (Liebig-Fellowship and Dozentenpreis to J. S.) and the Deutsche Forschungs-gemeinschaft DFG (STR 1150/3-1, STR 1150/7-1, and Heisenberg-fellowship to J. S.) are gratefully acknowledged for their support. We thank Sven Tauscher for initial contributions.References
- (a) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC; (b) S. R. Harutyuanyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef PubMed; (c) A. Gutnov, Eur. J. Org. Chem., 2008, 4547–4554 CrossRef CAS; (d) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed; (e) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279–1300 CrossRef CAS; (f) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed.
- Selected references: (a) K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902–6905 CrossRef CAS PubMed; (b) S. Lin and X. Lu, Org. Lett., 2010, 12, 2536–2539 CrossRef CAS PubMed; (c) R. Shintani, Y. Tsutsumi, M. Nagaosa, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 13588–13589 CrossRef CAS PubMed; (d) R. Shintani, W.-L. Duan and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 5628–5629 CrossRef CAS PubMed; (e) P. Mauleón and J. C. Carretero, Chem. Commun., 2005, 4961–4963 RSC.
- (a) I. R. Márquez, D. Miguel, A. Millán, M. L. Marcos, L. Álvarez de Cienfuegos, A. G. Campaña and J. M. Cuerva, J. Org. Chem., 2014, 79, 1529–1541 CrossRef PubMed; (b) P. Gao and S. P. Cook, Org. Lett., 2012, 14, 3340–3343 CrossRef CAS PubMed; (c) A. L. Gottumukkala, J. G. de Vries and A. J. Minnaard, Chem. – Eur. J., 2011, 17, 3091–3095 CrossRef CAS PubMed; (d) W. Li, A. Herath and J. Montgomery, J. Am. Chem. Soc., 2009, 131, 17024–17029 CrossRef CAS PubMed; (e) A. Minatti, X. Zheng and S. L. Buchwald, J. Org. Chem., 2007, 72, 9253–9258 CrossRef CAS PubMed; (f) M. Amatore, C. Gosmini and J. Périchon, J. Org. Chem., 2006, 71, 6130–6134 CrossRef CAS PubMed; (g) P. Shukla, Y.-C. Hsu and C.-H. Cheng, J. Org. Chem., 2006, 71, 655–658 CrossRef CAS PubMed; (h) S. Condon and J.-Y. Nédélec, Synthesis, 2004, 3070–3078 CrossRef CAS; (i) T. Tobrman and D. Dvořák, Tetrahedron Lett., 2004, 45, 273–276 CrossRef CAS; (j) K. Subburaj and J. Montgomery, J. Am. Chem. Soc., 2003, 125, 11210–11211 CrossRef CAS PubMed; (k) D. L. Comins, S. P. Joseph and Y.-m. Zhang, Tetrahedron Lett., 1996, 37, 793–796 CrossRef CAS; (l) G. K. Friestad and B. P. Branchaud, Tetrahedron Lett., 1995, 36, 7047–7050 CrossRef CAS; (m) R. Sustmann, P. Hopp and P. Holl, Tetrahedron Lett., 1989, 30, 689–692 CrossRef CAS; (n) S. A. Lebedev, V. S. Lopatina, E. S. Petrov and I. P. Beletskaya, J. Organomet. Chem., 1988, 344, 253–259 CrossRef CAS; (o) G. E. Stokker, Tetrahedron Lett., 1987, 28, 3179–3182 CrossRef CAS; (p) A. Arcadi, F. Marinelli and S. Cacchi, J. Organomet. Chem., 1986, 312, C27–C32 CrossRef CAS; (q) G. P. Boldrini, D. Savoia, E. Tagliavini, C. Trombini and A. U. Ronchi, J. Organomet. Chem., 1986, 301, C62–C64 CrossRef CAS; (r) S. Cacchi and G. Palmieri, Synthesis, 1984, 575–577 CrossRef CAS; (s) S. Cacchi and A. Arcadi, J. Org. Chem., 1983, 48, 4236–4240 CrossRef CAS.
- J. Streuff and A. Gansäuer, Angew. Chem., Int. Ed., 2015, 54, 14232–14242 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Sidera, P. M. C. Roth, R. M. Maksymowicz and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 7995–7999 CrossRef CAS PubMed; (b) R. M. Maksymowicz, P. M. C. Roth and S. P. Fletcher, Nat. Chem., 2012, 4, 649–654 CrossRef CAS PubMed; (c) R. Shrestha and D. J. Weix, Org. Lett., 2011, 13, 2766–2769 CrossRef CAS PubMed; (d) D. Martin, S. Kehrli, M. d'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416–8417 CrossRef CAS PubMed; (e) K.-s. Lee, M. K. Brown, A. W. Hird and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 7182–7184 CrossRef CAS PubMed; (f) M. d'Augustin, L. Palais and A. Alexakis, Angew. Chem., Int. Ed., 2005, 44, 1376–1378 CrossRef PubMed.
- (a) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377–10441 CrossRef CAS; (b) M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3295 CrossRef CAS PubMed.
- (a) A. Gansäuer, J. Justicia, C.-A. Fan, D. Worgull and F. Piestert, Top. Curr. Chem., 2007, 279, 25–52 CrossRef. Selected works: (b) A. Gansäuer, H. Bluhm, B. Rinker, S. Narayan, M. Schick, T. Lauterbach and M. Pierobon, Chem. – Eur. J., 2003, 9, 531–542 CrossRef PubMed; (c) A. Gansäuer, T. Lauterbach, H. Bluhm and M. Noltemeyer, Angew. Chem., Int. Ed., 1999, 38, 2909–2910 CrossRef; (d) A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120, 12849–12859 CrossRef; (e) A. Gansäuer, M. Pierobon and H. Bluhm, Angew. Chem., Int. Ed., 1998, 37, 101–103 CrossRef.
- J. Streuff, Chem. – Eur. J., 2011, 17, 5507–5510 CrossRef CAS PubMed.
- (a) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed; (b) R. Shrestha, S. C. M. Dorn and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 751–762 CrossRef CAS PubMed.
- (a) C.-C. Wang, P.-S. Lin and C.-H. Cheng, Tetrahedron Lett., 2004, 45, 6203–6206 CrossRef CAS; (b) M. M. Baizer, Tetrahedron, 1984, 40, 935–969 CrossRef CAS; (c) M. M. Baizer, U.S. Patent, 3193480, 1965 Search PubMed; (d) G. Wiemann, FR Patent, 1177602, 1959 Search PubMed; (e) J. Wiemann, Compt. Rend., 1957, 245, 172 CAS.
- A. T. Biju, M. Padmnanaban, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 8412–8415 CrossRef CAS PubMed.
- (a) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (b) J. Read de Alaniz and T. Rovis, Synlett, 2009, 1189–1207 CAS; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- P. Bichovski, T. M. Haas, D. Kratzert and J. Streuff, Chem. – Eur. J., 2015, 21, 2339–2342 CrossRef CAS PubMed.
- Selected recent works: (a) S. P. Morcillo, D. Miguel, S. Resa, A. Martín-Lasanta, A. Millán, D. Choquesillo-Lazarte, J. M. García-Ruiz, A. J. Mota, J. Justicia and J. M. Cuerva, J. Am. Chem. Soc., 2014, 136, 6943–6951 CrossRef CAS PubMed; (b) A. Rosales, J. Muñoz-Bascón, E. Roldan-Molina, M. A. Castañeda, N. M. Padial, A. Gansäuer, I. Rodríguez-García and J. E. Oltra, J. Org. Chem., 2014, 79, 7672–7676 CrossRef CAS PubMed; (c) J. Muñoz-Bascón, I. Sancho-Sanz, E. Álvarez-Manzaneda, A. Rosales and J. E. Oltra, Chem. – Eur. J., 2012, 18, 14479–14486 CrossRef PubMed.
- A. D. Kosal and B. L. Ashfeld, Org. Lett., 2010, 12, 44–47 CrossRef CAS PubMed.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, Inc., New York, 15th edn, 1999 Search PubMed.
- (a) Y. Marcus, J. Solution Chem., 1984, 13, 599–624 CrossRef CAS; (b) M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistrym, CRC/Taylor & Francis Inc., London, 3rd edn, 2006 Search PubMed.
- The pKa values from tables that were compiled by R. Williams and D. A. Evans from various literature sources: (a) http://www.chem.wisc.edu/areas/reich/pkatable ; (b) Evans http://evans.harvard.edu.
- For early studies on related nitrile complexes, see: (a) P. A. Seewald, G. S. White and D. W. Stephan, Can. J. Chem., 1988, 66, 1147–1152 CrossRef CAS; (b) P. N. Billinger, P. P. K. Claire, H. Collins and G. R. Willey, Inorg. Chim. Acta, 1988, 149, 63–67 CrossRef CAS; (c) E. J. M. de Boer and J. H. Teuben, J. Organomet. Chem., 1978, 153, 53–57 CrossRef CAS; (d) E. J. M. de Boer and J. H. Teuben, J. Organomet. Chem., 1977, 140, 41–45 CrossRef CAS.
- (a) A. Gansäuer, C. Kube, K. Daasbjerg, R. Sure, S. Grimme, G. D. Fianu, D. V. Sadasivam and R. A. Flowers II, J. Am. Chem. Soc., 2014, 136, 1663–1671 CrossRef PubMed; (b) A. Gansäuer, M. Behlendorf, D. von Laufenberg, A. Fleckhaus, C. Kube, D. V. Sadasivam and R. A. Flowers II, Angew. Chem., Int. Ed., 2012, 51, 4739–4742 CrossRef PubMed.
- The remaining material consisted of a complex product mixture and the dehalogenated substrate was not observed. The electron-poor products could potentially decompose either via a retro-Michael reaction or, alternatively, via a radical fragmentation under the reaction-/workup conditions. For an example, see: R. Matsushima and K. Sakai, J. Chem. Soc., Perkin Trans. 2, 1986, 1217–1222 RSC.
- CIF files have been deposited with the Cambridge Crystallographic Data Centre (CCDC). 26d: CCDC 1440299; 34i: CCDC 1440298.
- For the X-ray analysis of the syn-product 25b, see ref. 13.
- See the ESI.†.
- For a previous application of mesityl acrylate in reductive couplings, see: E. J. Corey and G. Z. Zheng, Tetrahedron Lett., 1997, 38, 2045–2048 CrossRef CAS.
- See also: R. E. Estévez, J. L. Oller-López, R. Robles, C. R. Melgarejo, A. Gansäuer, J. M. Cuerva and J. E. Oltra, Org. Lett., 2006, 8, 5433–5436 CrossRef PubMed.
- (a) J. Streuff, M. Feurer, G. Frey, A. Steffani, S. Kacprzak, J. Weweler, L. H. Leijendekker, D. Kratzert and D. A. Plattner, J. Am. Chem. Soc., 0000, 137, 14396–14405 CrossRef PubMed; (b) M. Feurer, G. Frey, H.-T. Luu, D. Kratzert and J. Streuff, Chem. Commun., 2014, 50, 5370–5372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional screening tables, experimental and computational details, characterisation data and NMR spectra of new compounds. CCDC 1440298 and 1440299. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02631h |
| This journal is © The Royal Society of Chemistry 2016 |