
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of inositol phosphate-based competitive antagonists of inositol 1,4,5-trisphosphate receptors†
Vera
Konieczny
a,
John. G.
Stefanakis
b,
Efstratios D.
Sitsanidis
b,
Natalia-Anastasia T.
Ioannidou
b,
Nikolaos V.
Papadopoulos
b,
Konstantina C.
Fylaktakidou
c,
Colin W.
Taylor
*a and
Alexandros E.
Koumbis
*b
aDepartment of Pharmacology, Tennis Court Road, Cambridge, CB2 1PD, UK. E-mail: cwt1000@cam.ac.uk
bLaboratory of Organic Chemistry, Chemistry Department, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: akoumbis@chem.auth.gr
cLaboratory of Organic, Bioorganic and Natural Product Chemistry, Molecular Biology and Genetics Department, Democritus University of Thrace, 68100 Alexandroupolis, Greece
First published on 20th January 2016
Abstract
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are intracellular Ca2+ channels that are widely expressed in animal cells, where they mediate the release of Ca2+ from intracellular stores evoked by extracellular stimuli. A diverse array of synthetic agonists of IP3Rs has defined structure–activity relationships, but existing antagonists have severe limitations. We combined analyses of Ca2+ release with equilibrium competition binding to IP3R to show that (1,3,4,6)IP4 is a full agonist of IP3R1 with lower affinity than (1,4,5)IP3. Systematic manipulation of this meso-compound via a versatile synthetic scheme provided a family of dimeric analogs of 2-O-butyryl-(1,3,4,6)IP4 and (1,3,4,5,6)IP5 that compete with (1,4,5)IP3 for binding to IP3R without evoking Ca2+ release. These novel analogs are the first inositol phosphate-based competitive antagonists of IP3Rs with affinities comparable to that of the only commonly used competitive antagonist, heparin, the utility of which is limited by off-target effects.
Introduction
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are intracellular Ca2+ channels that are almost ubiquitously expressed in animal cells.1,2 IP3Rs are essential links between receptors in the plasma membrane that stimulate phospholipase C and release of Ca2+ from the endoplasmic reticulum (ER). The resulting cytosolic Ca2+ signals regulate many diverse cellular processes.3 The three subtypes of IP3Rs expressed in vertebrates (IP3R1-3) are closely related proteins and they are each regulated by both (1,4,5)IP3 (1, Fig. 1) and Ca2+, but they differ in their sensitivity to other forms of regulation and in their subcellular and tissue distributions.1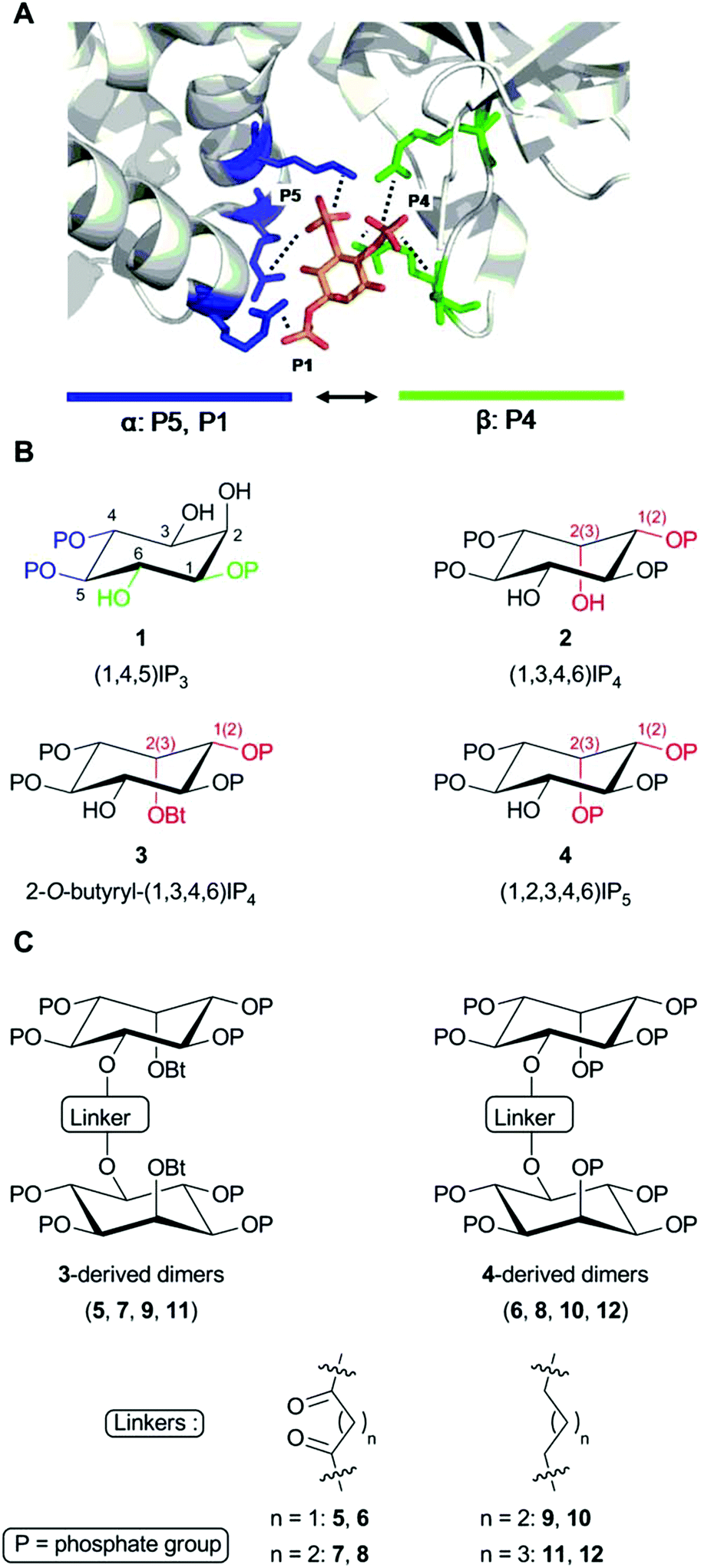 | ||
| Fig. 1 Structures of the ligands used. (A) Key contacts between (1,4,5)IP3 and residues within the IP3-binding core (IBC) of IP3R1 (Protein Data Bank 1N4K).9 P4 and P5 form the most extensive interactions (not all are shown) with residues in the β- (in green) and α-domains (in blue) of the IBC, respectively, pulling the two domains towards each other.23 P1 and the 6-hydroxyl enhance affinity through hydrogen bonding with one residue each within the α-domain. (B) (1,4,5)IP3 (1) showing the essential vicinal (4,5)-bisphosphate (blue) and 6-hydroxyl and 1-phosphate required for high-affinity binding (green). Analogs (2–4) are shown in configurations most likely to reflect their interaction with IP3R to allow comparison with (1,4,5)IP3. Numbers in brackets refer to equivalent groups of (1,4,5)IP3. Differences from (1,4,5)IP3 are highlighted in red. (C) Structures of the dimeric analogs and the acyl or alkyl linkers used. | ||
Extensive structure–activity studies,4–8 reinforced by a high-resolution structure of (1,4,5)IP3 bound to the IP3-binding core of IP3R1 (Fig. 1A),9 established that the vicinal 4,5-bisphosphate moiety is essential for (1,4,5)IP3 binding and the equatorial 6-hydroxyl and 1-phosphate confer high affinity (Fig. 1B). All high-affinity agonists of IP3R have structures equivalent to these substituents. The only endogenous inositol phosphate likely to bind to IP3Rs under physiological conditions is (1,4,5)IP3, the immediate water-soluble product of phospholipase C-catalyzed hydrolysis of the membrane lipid phosphatidylinositol 4,5-bisphosphate. However, synthetic ligands of IP3Rs, including many inositol phosphates7 and derivatives of adenophostins,10–12 have provided insight into the structural determinants of IP3R activation. These ligands include analogs of (1,4,5)IP3 that are resistant to degradation,13 fluorescent analogs,14 partial agonists,6 and synthetic derivatives of adenophostins.10 There are, however, no ligands of IP3R that distinguish effectively between IP3R subtypes,5,15,16 and the only available antagonists have severe limitations.17 The commonly used antagonists are heparin, 2-aminoethoxydiphenyl borate (2-APB), xestospongins and high concentrations of caffeine. The limitations of these antagonists include off-target effects, notably interactions with other Ca2+ channels, Ca2+ pumps, G proteins and other signalling pathways; membrane-impermeability (heparin) and, for xestospongins, an inconsistent history of effectiveness as discussed recently.17 This study was undertaken with the aim of developing more effective antagonists of IP3R.
(1,3,4,6)IP4, which retains the essential pharmacophore of an IP3R agonist (Fig. 1B), stimulates Ca2+ release via IP3R, but its affinity is between 10 and 100-fold lower than that of (1,4,5)IP3.18–21 Some,19,22 though not all,21 studies have suggested that (1,3,4,6)IP4 may be a partial agonist, namely that it less effectively activates IP3R than full agonists like (1,4,5)IP3. It seems likely that the inverted position of the 2-OH in (1,3,4,6)IP4 (equivalent to the 3-OH of (1,4,5)IP3 when the structures are compared in orientations likely to reflect their interactions with IP3R, Fig. 1B) is a major determinant of the reduced affinity.5,20 Although (1,3,4,6)IP4 is produced endogenously from (1,3,4)IP3, it is unlikely to attain concentrations that regulate IP3Rs.24 Nevertheless, we chose (1,3,4,6)IP4 to attempt development of novel antagonists of IP3R because it and analogs in which its free hydroxyls are modified (3, 4) are meso compounds that make synthesis more straightforward, and we had initially supposed that (1,3,4,6)IP4 might have reduced efficacy.19,22 We previously reported that dimers of inositol phosphates are high-affinity partial agonists of IP3R.6 We have now developed a family of antagonists of IP3Rs (5–12 in Fig. 1C) from the (1,3,4,6)IP4 backbone by modification of its free hydroxyls and dimerization of the modified structures. Diesteric or dietheric linkages of various sizes (n = 1–3) were chosen for these 5-O-homodimers, which were synthesized by means of a diverse and versatile approach. The most useful of these ligands (8, 10 and 12) bind to IP3R1 with an affinity comparable to that of the best available competitive antagonist of IP3R, heparin, the utility of which is limited by its off-target effects.
Results and discussion
Chemistry
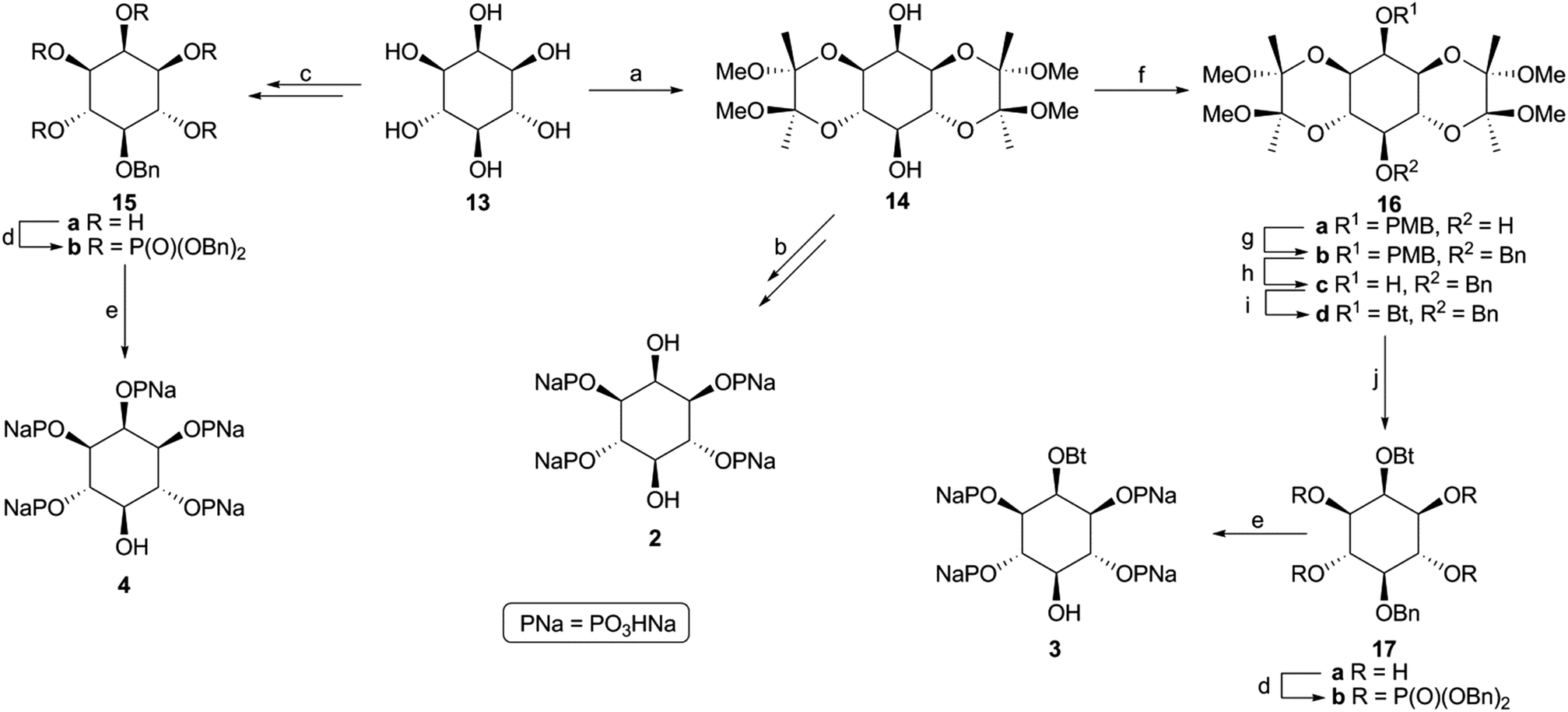 | ||
Scheme 1 Synthesis of phosphates 2–4. Reagents and conditions: (a) ref. 25; (b) ref. 26; (c) ref. 27; (d) i. (BnO)2PN(iPr)2, 1H-tetrazole, CH3CN, 25 °C, 48 h; ii. m-CPBA, CH2Cl2, −50 to 0 °C, 5 h, for 15b 73%, for 17b 63%; (e) Pd/C, H2 (1 atm), NaHCO3, EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 25 °C, 48–96 h, for 3 and 4 100%; (f) i. NaH, DMF, 0 °C, 1 h; ii. PMBCl, 0 to 25 °C, 12 h, 67%; (g) i. NaH, DMF, 0 °C, 1 h; ii. BnBr, 0 to 25 °C, 12 h, 90%; (h) DDQ, CH2Cl2/H2O (10:1), 25 °C, 24 h, 71%; (i) Bt2O, Et3N, DMAP, CH2Cl2, 25 °C, 12 h, 91%; (j) 90% aq. TFA, CH2Cl2, 25 °C, 2 h, 100%. :1), 25 °C, 48–96 h, for 3 and 4 100%; (f) i. NaH, DMF, 0 °C, 1 h; ii. PMBCl, 0 to 25 °C, 12 h, 67%; (g) i. NaH, DMF, 0 °C, 1 h; ii. BnBr, 0 to 25 °C, 12 h, 90%; (h) DDQ, CH2Cl2/H2O (10:1), 25 °C, 24 h, 71%; (i) Bt2O, Et3N, DMAP, CH2Cl2, 25 °C, 12 h, 91%; (j) 90% aq. TFA, CH2Cl2, 25 °C, 2 h, 100%. | ||
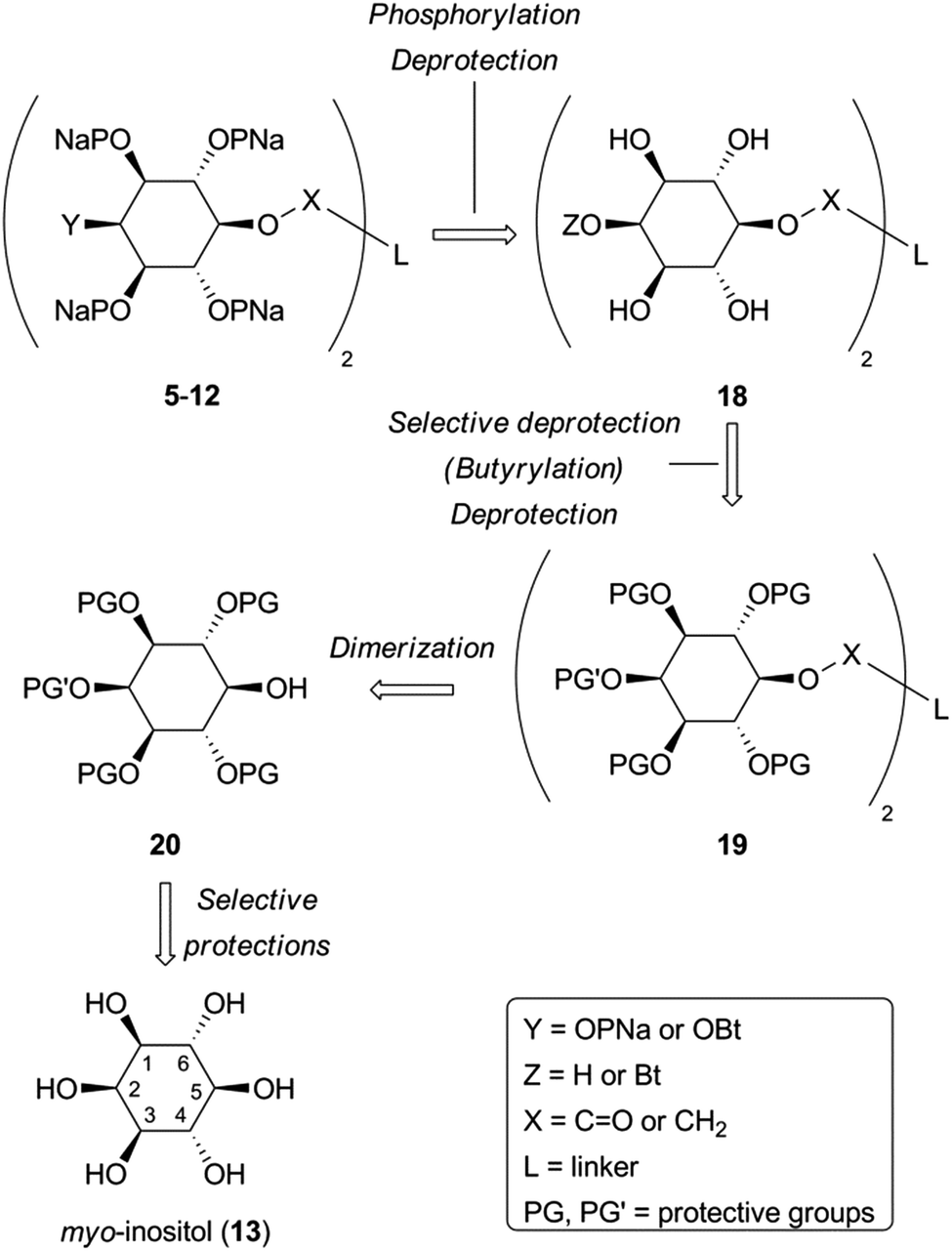 | ||
| Scheme 2 Retrosynthetic analysis for target dimers 5–12. | ||
Monobenzyl ether 21 (Scheme 3) was recognized as a suitable derivative, appropriately functionalized to play the role of 20. Moreover 21 is easily accessible25,29,30 from myo-inositol through butanedione bisacetal 14. Direct dimerization of this compound was initially investigated using the Steglich esterification approach31 and employing malonic (n = 1) and succinic acid (n = 2) as linkers (Scheme 3 and Table S1 in ESI†). However, these apparently simple couplings were found to be complicated, under various reaction conditions tested, by the formation of acetate 22 (in the first case) and N-acylureas 24a–c (in both cases). Thus, for malonic acid reactions, the presence of DMAP seemed to solely favor the decarboxylation process, regardless of the carbodiimide (DCC or DIC) and the solvent used.32 We could not securely determine whether this decarboxylation occurred prior to or after the first esterification. However, in other runs we isolated the N-acylureas 24a and 24b, suggesting that acetate 22 is formed from malonic monoester. Although replacing DMAP with DIPEA eliminated this problem, the only product isolated was N-acylurea 24b, in very low yield, whereas starting material was quantitatively recovered when EDC was used. On the other hand, the reactions performed in the absence of base33 were productive, yielding the desired dimer (23a) along with the corresponding N-acylurea (24a or 24b). The best results were obtained in the case of the DCC-promoted coupling.34 Surprisingly, applying the same conditions (DCC in Et2O) for the coupling of 21 with succinic acid was unsuccessful. In order to reach dimer 23b the presence of DMAP was a crucial factor using either DCC or EDC in CH2Cl2.34 Again, the reaction with DCC furnished an inseparable mixture of dimer 23b and N-acylurea 24c, which was subsequently resolved upon hydrogenolysis. In contrast to esters 23a,b, the synthesis of dimeric ethers 25a,b was accomplished in a more facile way. Williamson etherifications, through the in situ formed (NaH) sodium alkoxide of 21, were initially attempted in DMF using the required diiodo- or dibromo-alkanes, but with poor results. Replacing halo-electrophiles with the more reactive ditosylates 2635 and 2736 and applying a protocol37 which involved KOH as base and a more polar solvent (DMSO) furnished the desired dimers (25a,b) in a clean way and in good yields.38
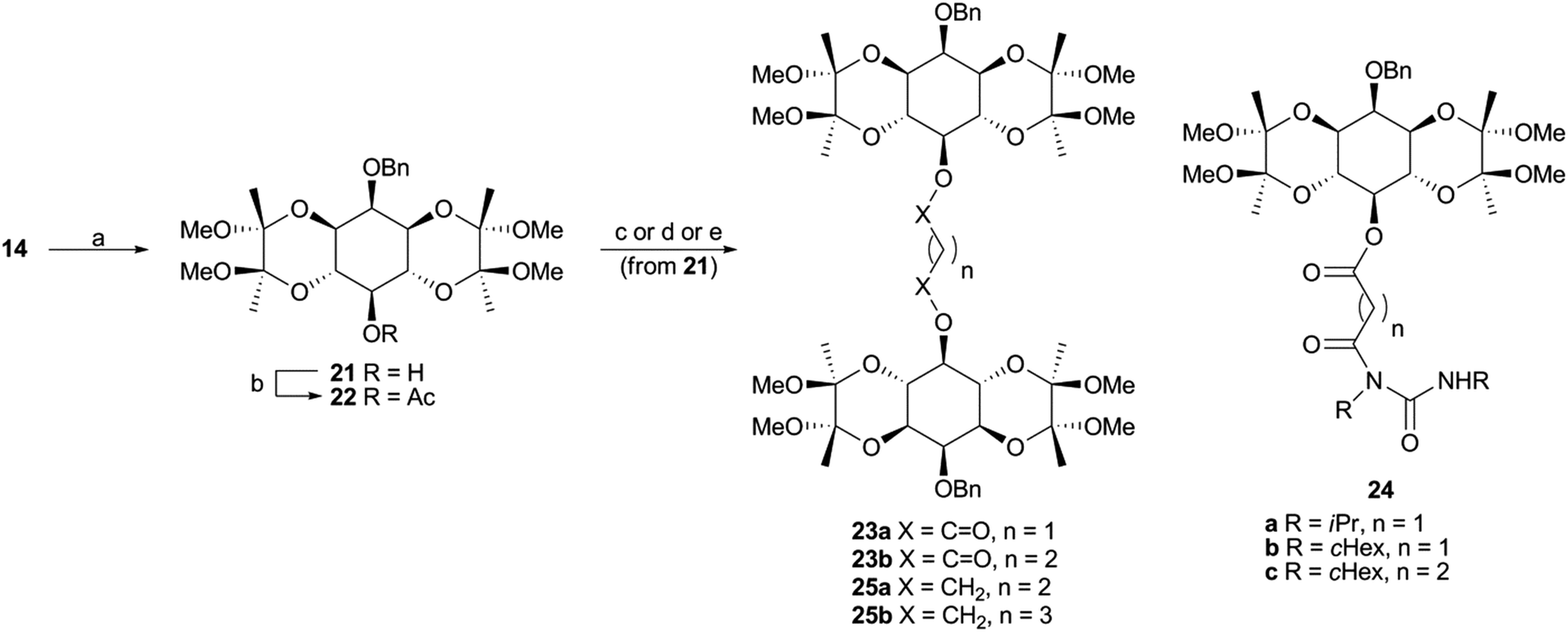 | ||
| Scheme 3 Synthesis of dimers 23 and 25. Reagents and conditions: (a) ref. 29, 30; (b) CH2(COOH)2, DCC or DIC, DMAP, CH2Cl2, 25 °C, 24 h, 37–41%; (c) CH2(COOH)2, DCC, Et2O, 25 °C, 24 h, 62% of 23a and 12% of 24b; (d) HOCO(CH2)2COOH, DCC, DMAP, CH2Cl2, 25 °C, 96 h, 56% of 23b and 12% of 24c; (e) TsOCH2(CH2)nCH2OTs (26 or 27), KOH, BnH/DMSO (4:1), 55 °C, 120 h, for 25a 45%, for 25b 62%. | ||
With the key intermediate dimers in our hands, we proceeded to the next steps, which involved installation of the butyryl and phosphate groups. Pd-catalyzed hydrogenolysis of 23a,b and 25a,b led to the corresponding diols 28, which were esterified upon exposure to butyric anhydride to give 29 in very good yields (Scheme 4).
 | ||
| Scheme 4 Synthesis of dimeric phosphates 5–12. Reagents and conditions: (a) Pd/C, H2 (1 atm), MeOH, 25 °C, 24 h, 93–100%; (b) Bt2O, Et3N, DMAP, CH2Cl2, 25 °C, 24 h, 79–97%; (c) 90% aq. TFA, CH2Cl2, 25 °C, 2 h, 98–100%; (d) i. (BnO)2PN(iPr)2, 1H-tetrazole, CH3CN, 25 °C, 48 h; ii. m-CPBA, CH2Cl2, −50 to 0 °C, 5 h, 60–78%; (e) Pd/C, H2 (1 atm), NaHCO3, EtOH/H2O (1:1), 25 °C, 48–72 h, 95–100%. | ||
Both 28 and 29 were then used to reach the final targets. Thus, careful treatment of these dimers (especially in the case of 29) with aqueous TFA furnished octaols and decaols 30, in nearly quantitative yields (Scheme 4). Perphosphorylation of these crude polyols was accomplished as described for 17b (Scheme 1) to obtain the protected polyphosphates 31. The latter were debenzylated upon hydrogenolysis in the presence of sodium bicarbonate to yield the octakis and decakis phosphate salts 5–12.39
Biology
 | ||
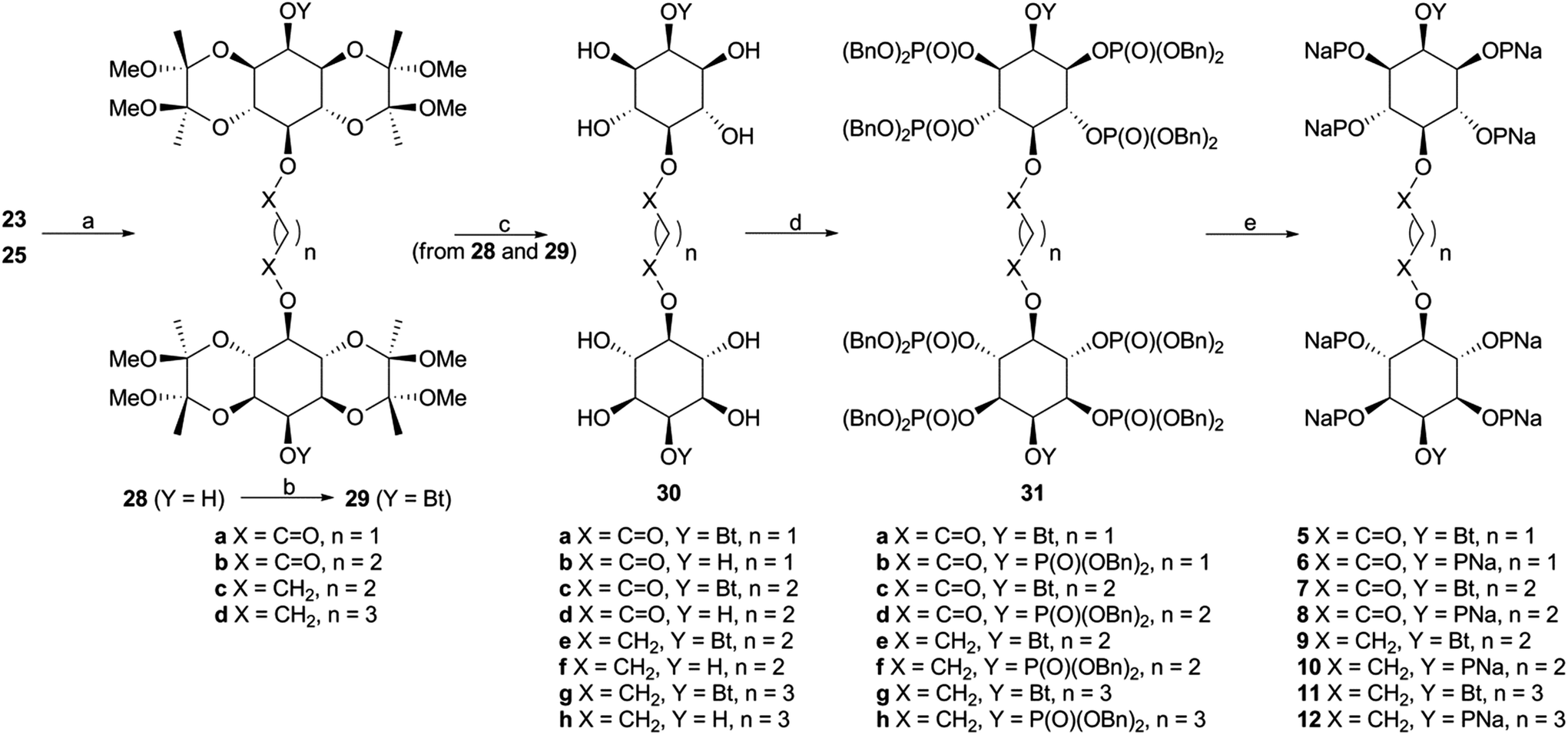
| Fig. 2 2-O-Butyryl-(1,3,4,6)IP4 (3) is a partial agonist and (1,2,3,4,6)IP5 (4) is an antagonist of IP3R1. (A) Typical experiment showing Ca2+ uptake into the ER of permeabilized DT40-IP3R1 cells after addition of MgATP (1.5 mM), recorded with a luminal Ca2+ indicator (mag-fluo4). Addition of (1,4,5)IP3 (concentrations in nM), with cyclopiazonic acid (CPA, 10 μM) to inhibit the Ca2+ pump, reveals the concentration-dependent effect of (1,4,5)IP3 on Ca2+ release. Results show fluorescence (relative fluorescence units, RFU) as means from triplicate determinations in a single experiment. (B) Summary results show effects of the indicated analogs on Ca2+ release (% of Ca2+ content of intracellular stores). (C) Equilibrium competition binding with 3H-(1,4,5)IP3 and the indicated analogs using membranes from Sf9-IP3R1 cells in CLM containing 1.5 mM MgATP. Results in B and C are means ± s.e.m., n = 3. The inset shows a representative Western blot (n = 2) demonstrating expression of IP3R1 in membranes from rat cerebellum (5 μg protein) and Sf9-IP3R1 cells (0.3 μg). Data summarized in ESI in Table S2.† | ||
Because both agonists (1 and 2) released the same amount of Ca2+ at maximally effective concentrations, a comparison of EC50 and Kd values allows the effectiveness with which each promotes opening of the IP3R Ca2+ channel to be determined. A partial agonist needs to occupy more receptors to elicit the same response, which is then reflected in a higher EC50/Kd ratio (and a lower value for pEC50–pKd, where p denotes the negative log).6 (1,3,4,6)IP4 and (1,4,5)IP3 did not differ significantly in their pEC50–pKd values (Table S2 in ESI†) suggesting that (1,4,5)IP3 and (1,3,4,6)IP4 have similar efficacies. We conclude that (1,3,4,6)IP4 is a full agonist with lower affinity than (1,4,5)IP3, in agreement with a previous report,21 but inconsistent with suggestions that it is a partial agonist.19,22
A maximally effective concentration of 2-O-butyryl-(1,3,4,6)IP4 released a smaller fraction of the intracellular Ca2+ stores than did (1,4,5)IP3 (Fig. 2B) and it bound to the IP3R1 with 50 ± 22-fold lower affinity than (1,4,5)IP3 (Fig. 2C). The lesser maximal Ca2+ release evoked by 2-O-butyryl-(1,3,4,6)IP4, suggests that it is less efficacious than (1,4,5)IP3. Although 2-O-butyryl-(1,3,4,6)IP4 and (1,3,4,6)IP4 differed in their ability to evoke Ca2+ release, they bound to IP3R with similar affinities (Fig. 2C and Table S2 in ESI†). Hence, addition of a butyryl moiety to the 2-position of (1,3,4,6)IP4 decreased efficacy without affecting affinity. 2-O-Butyryl-(1,3,4,6)IP4 (3) thus replaced (1,3,4,6)IP4 as a lead compound from which we attempted to develop ligands that bind to IP3R without activating it (i.e. competitive antagonists).
Even a very high concentration (100 μM) of (1,2,3,4,6)IP5 (4) failed to release Ca2+ (Fig. 2B), but it bound to IP3R1 with a Kd of 22.9 μM (Fig. 2C and Table S2 in ESI†). Hence, (1,2,3,4,6)IP5 has 230 ± 100-fold lower affinity than (1,4,5)IP3 for IP3R1 and significantly lower affinity than (1,3,4,6)IP4 or 2-O-butyryl-(1,3,4,6)IP4 (5.2 ± 0.5 and 4.8 ± 0.3-fold lower, respectively) (Fig. 2C, and Table S2, ESI†). (1,2,3,4,6)IP5 retains the essential pharmacophore and moieties known to be crucial for high-affinity binding (Fig. 1B), but it has an axial phosphate at the 2-position [equivalent to the 3-position of (1,4,5)IP3]. Others have reported that an axial phosphate at the 3-position of (1,4,5)IP3 reduced affinity.40 The important observation is that addition of an axial 2-phosphate to (1,3,4,6)IP4, to give (1,2,3,4,6)IP5, abolishes residual efficacy, albeit with some (5.2 ± 0.5-fold) loss of affinity.
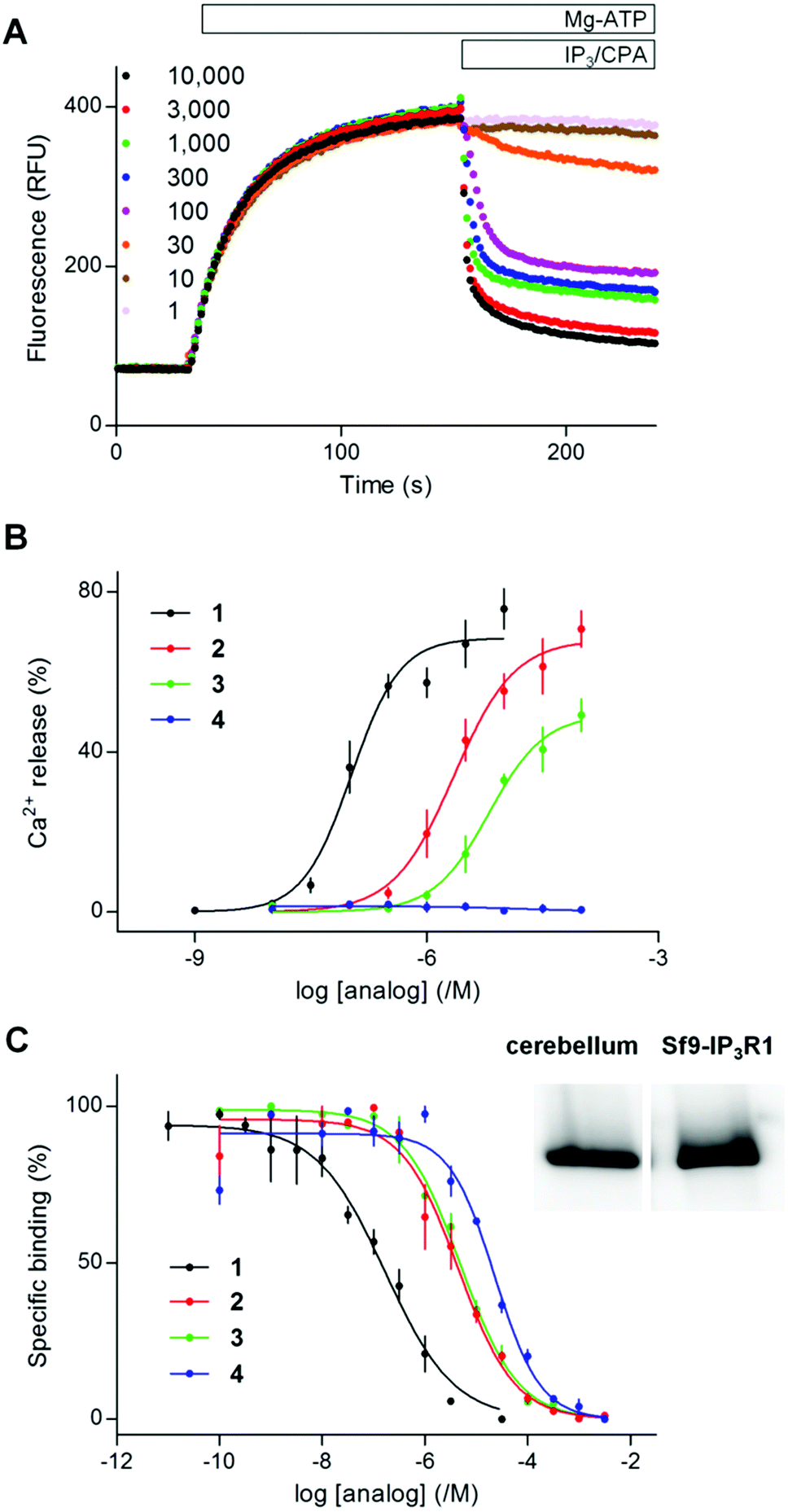
Pre-equilibration of permeabilized DT40-IP3R1 cells with (1,2,3,4,6)IP5 (100 μM, 2 min), shifted the sensitivity of the Ca2+ release evoked by (1,4,5)IP3 by 2.4 ± 0.2-fold, without affecting either the maximal Ca2+ release or Hill coefficient (Fig. 3 and Table S3, ESI†). From the dose ratios (see Experimental section), this functional analysis suggests that (1,2,3,4,6)IP5 binds to the (1,4,5)IP3-binding site of IP3R1 with a Kd of ∼70 μM. Given the non-equilibrium conditions and the different temperatures used for functional (20 °C) and radioligand binding (4 °C) experiments, this measurement is in reasonable agreement with the affinity determined from equilibrium binding to IP3R1 (Kd ∼ 23 μM) (Fig. 2C, 3B and Tables S2, S3 in ESI†). These results demonstrate that (1,2,3,4,6)IP5 is a competitive antagonist of IP3R with an affinity of ∼20–70 μM.
 | ||
| Fig. 3 (1,2,3,4,6)IP5 (4) and a dimeric analog (6) are competitive antagonists of IP3R1. (A) Typical experiment showing the Ca2+ content of the ER after addition of Mg-ATP to permeabilized DT40-IP3R1 cells, followed by addition of 4 or 6 (100 μM, antagonist) and then cyclopiazonic acid with (1,4,5)IP3 (100 nM the three upper lighter lines, or 100 μM darker lines). Results show fluorescence as means from 4 repeats within one experiment. (B) Summary shows the concentration-dependent effects of (1,4,5)IP3 on Ca2+ release alone or after preincubation with 4 or 6 (100 μM). Results are means ± s.e.m., n = 3. Summary results in ESI in Table S3.† | ||
The activities of (1,2,3,4,6)IP5 (4) and the dimer 6 are directly compared in Fig. 3. Neither 4 nor 6 (100 μM) evoked Ca2+ release, but they reduced the sensitivity of the Ca2+ release evoked by (1,4,5)IP3 by 2.4 ± 0.2 and 20.9 ± 0.7-fold, respectively, without affecting the maximal response or Hill coefficient. Hence the dimer 6, like the monomer 4, is a competitive antagonist, but 6 has an apparent affinity that is 8.8 ± 1.0-fold greater than 4 (Table S3 in ESI†).
The results with 6, suggesting that a dimer of (1,3,4,5,6)IP5 retained the lack of efficacy of (1,3,4,5,6)IP5 while displaying improved affinity, prompted analysis of seven additional dimeric analogs of 2-O-butyryl-(1,3,4,6)IP4 and (1,2,3,4,6)IP5 (Fig. 1C). None of the dimers (5–12, 100 μM) evoked Ca2+ release, and they all significantly decreased the sensitivity to (1,4,5)IP3 without affecting the maximal Ca2+ release or Hill coefficient (Fig. 4 and Table 1). All of the dimers (5–12) are therefore competitive antagonists.
 | ||
| Fig. 4 Dimers of 2-O-butyryl-(1,3,4,6)IP4 (5, 7, 9, 11) or (1,2,3,4,6)IP5 (6, 8, 10, 12) are competitive antagonists of IP3R1. (A, B) Experiments similar to those shown in Fig. 3 were used to assess the effects of the indicated concentrations of (1,4,5)IP3 on Ca2+ release from permeabilized DT40-IP3R1 cells after preincubation (2 min) with the indicated dimers (5–12, 100 μM). Results are means ± s.e.m., n = 3. Summary results in Table 1. | ||
| Ca2+ release | Binding | |||||||
|---|---|---|---|---|---|---|---|---|
| pEC50 (/M) | ΔpEC50 (/M) | EC50 (nM) | Maximal release (%) | n H | K d (μM) | pKd (/M) | K d (μM) | |
| a Summary results from Fig. 4 show the effects of (1,4,5)IP3 alone or in the presence of 100 μM of each analog. Results show pEC50, ΔpEC50 (pECcontrol50–pECantagonist50) and Hill coefficients (nH) (means ± s.e.m.) and EC50 for (1,4,5)IP3-evoked Ca2+ release (n = 3). Kd is shown calculated from functional assays and from equilibrium competition binding experiments (n = 3). Statistical differences were determined by one-way ANOVA and Tukey's post hoc test, and refer to the results with (1,4,5)IP3 alone, *P < 0.05. | ||||||||
| (1,4,5)IP3 | 7.03 ± 0.02 | — | 94 | 70 ± 1 | 1.31 ± 0.18 | — | 6.90 ± 0.19 | 0.13 |
| +5 | 6.51 ± 0.06* | 0.51 ± 0.03 | 306 | 65 ± 6 | 0.86 ± 0.17 | 44 | ||
| +6 | 6.03 ± 0.06* | 1.00 ± 0.07 | 931 | 58 ± 4 | 1.28 ± 0.08 | 11 | 4.79 ± 0.05 | 16.4 |
| +7 | 6.14 ± 0.08* | 0.89 ± 0.08 | 719 | 59 ± 4 | 1.67 ± 0.57 | 15 | ||
| +8 | 5.89 ± 0.11* | 1.14 ± 0.09 | 1300 | 60 ± 5 | 1.49 ± 0.39 | 8 | ||
| +9 | 6.24 ± 0.07* | 0.79 ± 0.09 | 571 | 64 ± 4 | 1.63 ± 0.42 | 20 | ||
| +10 | 5.86 ± 0.06* | 1.17 ± 0.05 | 1393 | 64 ± 3 | 1.58 ± 0.17 | 7 | ||
| +11 | 6.27 ± 0.13* | 0.76 ± 0.15 | 537 | 59 ± 4 | 1.08 ± 0.17 | 21 | ||
| +12 | 5.84 ± 0.16* | 1.19 ± 0.18 | 1449 | 60 ± 5 | 2.13 ± 0.55 | 7 | 5.11 ± 0.08 | 7.7 |
Although 2-O-butyryl-(1,3,4,6)IP4 is a partial agonist with a Kd of 4.8 μM (Fig. 2B and Table S2 in ESI†), its dimeric analogs are competitive antagonists with slightly reduced apparent affinities (Kd = 15–44 μM) (Table 1). The decreased affinity is consistent with evidence from analogs of (1,4,5)IP3, where substitution of the 6-hydroxyl (equivalent to the 5-hydroxyl of 3, through which the dimers are linked) reduced affinity.41,42 The 6-hydroxyl of (1,4,5)IP3 is thought to stabilize interactions of the IP3-binding core.7 However, the reduction in affinity between 3 and its dimers is modest by comparison with the 70 to 100-fold decrease for 6-deoxy-(1,4,5)IP3 and 6-methoxy-(1,4,5)IP3 relative to (1,4,5)IP3.41,42 Hence, dimerization of 2-O-butyryl-(1,3,4,6)IP4, to give 5, 7, 9 and 11, successfully reduced efficacy, but without improving affinity (Table 1).
The antagonist 12 is one of three dimers of (1,2,3,4,6)IP5 (4) with equally high affinity, and it shifted the EC50 for (1,4,5)IP3 by 19.4 ± 6.5-fold, suggesting an apparent Kd of ∼7 μM (Fig. 4B). Given the similar affinities of the dimers 8, 10 and 12 (Kd 7–8 μM) in functional assays (Table 1), we examined only 12 in equilibrium competition binding experiments. The Kd value for 12 determined in these experiments (7.7 μM) concurs with the results from functional analyses (Table 1).
These results establish that 8, 10 and 12 are competitive antagonists of IP3R with low-micromolar affinity. Although modifications of the 6-hydroxyl of (1,4,5)IP3 reduced affinity,41,42 dimerization through the analogous 5-hydroxyls of 3 and 4 caused more modest decreases or increases in affinity, respectively (Table 1). That pattern is similar across the four different linkers used (Fig. 1C). For each linker, dimers of 4 had 2 to 3-fold greater affinity than dimers of 3, even though monomeric 4 has significantly lower affinity than monomeric 3 (Fig. 4, Tables 1 and S2 in ESI†). For dimers of both 3 and 4, the shortest linker (n = 1, Fig. 1C) less effectively increased affinity than did the longer linkers (n = 2–3) (Table 1).
Conclusions
There is a need for selective antagonists of IP3Rs.17 Aiming to discover new lead-compounds of this type, a series of novel (1,3,4,6)IP4 and (1,2,3,4,6)IP5 homodimers were synthesized following a practical synthetic strategy. Among these homodimers, ligands 8, 10 and 12 were the antagonists with greatest affinity for IP3R (Kd 7–8 μM). 5-Carboxymethyl-(1,4)IP2 was reported to partially inhibit IP3-evoked Ca2+ release, but only at an extremely high concentration (5 mM).43 (1,3,4,5,6)IP5 is the only other inositol phosphate previously shown to be a competitive antagonist, but it bound to IP3R1 with lower affinity (Kd ∼40 μM)44 than (1,2,3,4,6)IP5 (4, Kd ∼ 23 μM) and with substantially lower affinity than the dimers of 4. These comparisons are consistent with our observation that 10 μM (1,2,4,5,6)IP5 had no detectable effect on (1,4,5)IP3-evoked Ca2+ release,6 whereas the same concentration of 12 caused a 2.8-fold decrease in (1,4,5)IP3-sensitivity (not shown). A dimeric benzene with six attached phosphate groups (biphenyl 2,2′,4,4′,5,5′-hexakisphosphate) was recently reported to be a rather high-affinity (Kd ∼ 200 nM) antagonist of IP3R, but it inhibited IP3 5-phosphatase with very similar potency.45 Compounds 8, 10 and 12 are the most potent inositol phosphate-based antagonists of IP3R so far reported. The affinity of these antagonists for IP3R1 (Kd 7–8 μM) is comparable to that of heparin (Kd ∼ 4 μM),17 but the new dimeric antagonists are smaller than heparin (Mr ∼1200 and ∼5000, respectively), and less likely to interact with as many additional intracellular targets. None of these antagonists is membrane-permeant, but based on the versatility of our synthetic approach, it may be feasible to esterify the phosphate groups of the dimeric antagonists to allow them to cross the plasma membrane and then be de-esterified by endogenous intracellular esterases.46Experimental
Chemistry
:1 to 2:1) to give 1.78 g (62%) of diester 23a and 510 mg (12%) of ureido derivative 24b.
:1 to 1:1) to give 820 mg (56%) of diester 23b and 260 mg (12%) of ureido derivative 24c.
:1 mixture of toluene and DMSO (2.5 mL), powdered KOH (140 mg, 2.5 mmol) was added and the mixture was warmed up to 55 °C. Then, 2635 or 2736 (0.5 mmol) was added in one portion and the resulting slurry was heated at the same temperature for 120 h, while the progress of the reaction was monitored by TLC. Upon completion, the mixture was neutralized with the addition of a saturated aqueous NH4Cl solution. Then, water was added to dissolve all solids and the clear solution was extracted with toluene (50 mL) and CH2Cl2 (2 × 50 mL). The combined organic phases were dried over Na2SO4, and concentrated in vacuo. The residue was purified with flash column chromatography (hexanes/EtOAc 5:1 to 1:1) to give ethers 25a,b.
:1 to 1:1) to give butyrates 29.
:1 to 1:2 and then 2–5% CH3OH in EtOAc).
:1 mixture of EtOH and deionized H2O (3 × 30 mL). The combined filtrates were evaporated under reduced pressure (55 °C), and the resulting residue was dried under high vacuum for 24 h to yield the desired phosphate salt.
Biology
000g, 5 min, 4 °C). The pellet was washed with 700 μL of CLM, resuspended in 200 μL of CLM, and radioactivity was determined by liquid scintillation counting. Culture of Sf9 cells, infection with baculovirus encoding rat IP3R1, and preparation of membranes were as described previously.49 Quantification of IP3R1 expression by Western blotting, using an anti-peptide antiserum to IP3R149 was performed as described.50
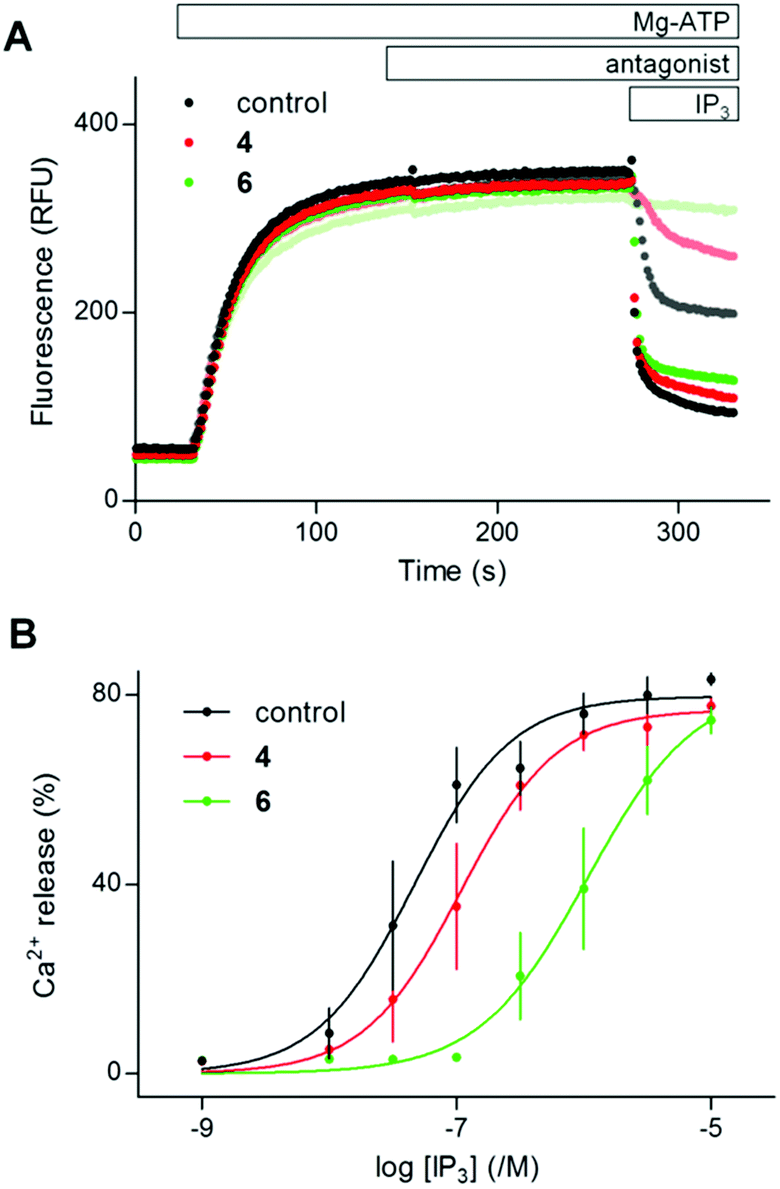
The dose ratio (DR = EC′50/EC50, where EC′50 and EC50 are the EC50 values for (1,4,5)IP3-evoked Ca2+ release determined in the presence and absence of antagonist, respectively) was used to calculate the apparent affinity (Kd) of IP3R1 for antagonists from functional assays:
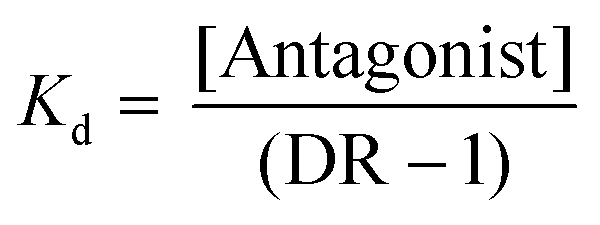
From equilibrium competition binding experiments, the Kd of competing ligands was calculated from the concentration (IC50) required to cause 50% displacement of the specifically bound 3H-(1,4,5)IP3:

The [3H-(1,4,5)IP3] was 1.5 nM, and Kd(1,4,5)IP3 (127 nM) (ESI Table S2†). pKd values were then used for statistical analyses.51
For comparisons of differences between pEC50 and pKd values (pEC50–pKd), the standard deviation of the difference (σpEC50–Kd) was calculated from the individual variances (σpEC50 and σpKd):52
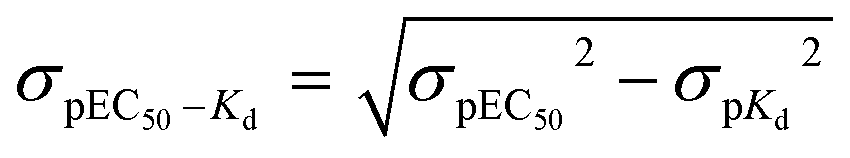
Author contributions
V. K. and J. G. S. contributed equally. A. E. K. initiated the study and supervised the chemistry. C. W. T. designed, interpreted and supervised the biological analyses. V. K. designed, performed and analyzed the biological experiments. J. G. S. and E. D. S. designed the chemical part, performed the synthesis of dimers and interpreted all spectral data. N.-A. T. I. and N. V. P. performed the synthesis of monomers. K. C. F. contributed to formulating the initial rationale. All authors have contributed to the manuscript and approved the final version.Abbreviations
| Bt | Butyryl |
| CLM | Cytosol-like medium |
| m-CPBA | m-Chloro-perbenzoic acid |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DIC | N,N′-Diisopropylcarbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| EC50 (IC50) | Half-maximal effective (inhibitory) concentration |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ER | Endoplasmic reticulum |
| IP3 | Inositol trisphosphate |
| IP3R | IP3 receptor |
| IP4 | Inositol tetrakisphosphate |
| IP5 | Inositol pentakisphosphate (structures of the analogs and their codes are shown in Fig. 1) |
| K d | Equilibrium dissociation constant |
| pEC50 (pKd) | −logEC50 (Kd) |
| PMB | p-Methoxybenzyl |
| TFA | Trifluoroacetic acid |
Acknowledgements
Supported by a Senior Investigator Award from the Wellcome Trust 101844 (to C. W. T.), Biotechnology and Biological Sciences Research Council UK and the German Academic Exchange Service (to V. K.). A. E. K. thanks the Research Committee of AUTh for financial support. C. W. T. and V. K. thank Dr S. B. Shears (N. I. E. H. S, U. S. A.) for his helpful advice.Notes and references
- J. K. Foskett, C. White, K. H. Cheung and D. O. Mak, Physiol. Rev., 2007, 87, 593 CrossRef CAS PubMed.
- C. W. Taylor and S. C. Tovey, Cold Spring Harbor Perspect. Biol., 2010, 2, a004010 CAS.
- M. J. Berridge, M. D. Bootman and H. L. Roderick, Nat. Rev. Mol. Cell Biol., 2003, 4, 517 CrossRef CAS PubMed.
- R. A. Wilcox, W. U. Primrose, S. R. Nahorski and R. A. J. Challiss, Trends Pharmacol. Sci., 1998, 19, 467 CrossRef CAS PubMed.
- E. P. Nerou, A. M. Riley, B. V. L. Potter and C. W. Taylor, Biochem. J., 2001, 355, 59 CrossRef CAS PubMed.
- A. M. Rossi, A. M. Riley, S. C. Tovey, T. Rahman, O. Dellis, E. J. A. Taylor, V. G. Veresov, B. V. L. Potter and C. W. Taylor, Nat. Chem. Biol., 2009, 5, 631 CrossRef CAS PubMed.
- B. V. L. Potter and D. Lampe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1933 CrossRef CAS.
- A. P. Kozikowski, V. I. Ognyanov, A. H. Fauq, S. R. Nahorski and R. A. Wilcox, J. Am. Chem. Soc., 1993, 115, 4429 CrossRef CAS.
- I. Bosanac, J.-R. Alattia, T. K. Mal, J. Chan, S. Talarico, F. K. Tong, K. I. Tong, F. Yoshikawa, T. Furuichi, M. Iwai, T. Michikawa, K. Mikoshiba and M. Ikura, Nature, 2002, 420, 696 CrossRef CAS PubMed.
- A. M. Rossi, A. M. Riley, B. V. L. Potter and C. W. Taylor, Curr. Top. Membr., 2010, 66, 209 CAS.
- A. M. Vibhute, V. Konieczny, C. W. Taylor and K. M. Sureshan, Org. Biomol. Chem., 2015, 13, 6698 CAS.
- K. M. Sureshan, A. M. Riley, M. P. Thomas, S. C. Tovey, C. W. Taylor and B. V. Potter, J. Med. Chem., 2012, 55, 1706 CrossRef CAS PubMed.
- C. W. Taylor, M. J. Berridge, A. M. Cooke and B. V. L. Potter, Biochem. J., 1989, 259, 645 CrossRef CAS PubMed.
- Z. Ding, A. M. Rossi, A. M. Riley, T. Rahman, B. V. L. Potter and C. W. Taylor, Mol. Pharmacol., 2010, 77, 995 CrossRef CAS PubMed.
- H. Saleem, S. C. Tovey, T. Rahman, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2012, 8, e54877 Search PubMed.
- H. Saleem, S. C. Tovey, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2013, 8, e58027 CAS.
- H. Saleem, S. C. Tovey, T. F. Molinski and C. W. Taylor, Br. J. Pharmacol., 2014, 171, 3298 CrossRef CAS PubMed.
- I. Ivorra, R. Gigg, R. F. Irvine and I. Parker, Biochem. J., 1991, 273, 317 CrossRef CAS PubMed.
- R. A. Wilcox, S. T. Safrany, D. Lampe, S. J. Mills, S. R. Nahorski and B. V. L. Potter, Eur. J. Biochem., 1994, 223, 115 CrossRef CAS PubMed.
- N. T. Burford, S. R. Nahorski, S.-K. Chung, Y.-T. Chang and R. A. Wilcox, Cell Calcium, 1997, 21, 301 CrossRef CAS PubMed.
- P.-J. Lu, D.-M. Gou, W.-R. Shieh and C.-S. Chen, Biochemistry, 1994, 33, 11586 CrossRef CAS PubMed.
- D. J. Gawler, B. V. L. Potter, R. Gigg and S. R. Nahorski, Biochem. J., 1991, 276, 163 CrossRef CAS PubMed.
- M.-D. Seo, S. Velamakanni, N. Ishiyama, P. B. Stathopulos, A. M. Rossi, S. A. Khan, P. Dale, C. Li, J. B. Ames, M. Ikura and C. W. Taylor, Nature, 2012, 483, 108 CrossRef CAS PubMed.
- F. S. Menniti, K. G. Oliver, K. Nogimori, J. F. Obie, S. B. Shears and J. W. Putney Jr., J. Biol. Chem., 1990, 265, 11167 CAS.
- S. J. Mills, A. M. Riley, C. Liu, M. F. Mahon and B. V. L. Potter, Chem. – Eur. J., 2003, 9, 6207 CrossRef CAS PubMed.
- A. E. Koumbis, C. D. Duarte, C. Nicolau and J. M. Lehn, ChemMedChem, 2011, 6, 169 CrossRef CAS PubMed.
- M. T. Rudolf, T. Kaiser, A. H. Guse, G. W. Mayr and C. Schultz, Liebigs Ann./Recl., 1997, 1861 CrossRef CAS.
- The reaction was closely monitored in order to avoid the oxidative removal of the benzyl group.
- A. M. Riley, H. Wang, J. D. Weaver, S. B. Shears and B. V. L. Potter, Chem. Commun., 2012, 48, 11292 RSC.
- H. Wang, H. Y. Godage, A. M. Riley, J. D. Weaver, S. B. Shears and B. V. L. Potter, Chem. Biol., 2014, 21, 689 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef.
- We have practically recovered the rest of unreacted starting material (21).
- V. V. Mozhaev, C. L. Budde, J. O. Rich, A. Y. Usyatinsky, P. C. Miehels, Y. L. Khmelnitsky, D. S. Clark and J. S. Dordiek, Tetrahedron, 1998, 54, 3971 CrossRef CAS.
- For large scale runs we have found it more convenient to subject the mixture of 23 and 24 to hydrogenolysis (see Scheme 4), since the debenzylated derivatives were more easily separable.
- A. E. Martin, T. M. Ford and J. E. Bulkowski, J. Org. Chem., 1982, 47, 412 CrossRef CAS.
- D. H. Bus, D. J. Olszanski, J. C. Stevens, W. P. Schammel, M. Kojima, N. Herron, L. L. Zimmer, K. A. Holter and J. Mocak, J. Am. Chem. Soc., 1981, 103, 1472 CrossRef.
- P. Goueth, A. Ramiz, G. Ronco, G. Mackenzie and P. Villa, Carbohydr. Res., 1995, 266, 171 CrossRef CAS.
- Only traces of the corresponding 5-O-but-3-enyl- and 5-O-pent-4-enyl-derivatives (elimination products) were detected in the reaction mixture.
- For compounds 5 and 6 a rapid H–D exchange of malonic protons in the NMR solvent (D2O) occurs. Therefore, these protons disappear in the 1H NMR spectrum, whereas a quintet is observed in the 13C NMR spectrum for the CD2 group (around 40 ppm). The complete insolubility of these compounds in non-protic solvents did not allow us to run other NMR experiments. The provided data (NMR and HRMS) are consisted with the given structures.
- A. P. Kozikowski, V. I. Ognyanov, A. H. Fauq, R. A. Wilcox and S. R. Nahorski, J. Chem. Soc., Chem. Commun., 1994, 599 RSC.
- M. A. Polokoff, G. H. Bencen, J. P. Vacca, S. J. DeSolms, S. D. Young and J. R. Huff, J. Biol. Chem., 1988, 63, 11922 Search PubMed.
- S. T. Safrany, R. J. H. Wojcikiewicz, J. Strupish, S. R. Nahorski, D. Dubreuil, J. Cleophax, S. D. Gero and B. V. L. Potter, FEBS Lett., 1991, 278, 252 CrossRef CAS PubMed.
- N. S. Keddie, Y. Ye, T. Aslam, T. Luyten, D. Bello, C. Garnham, G. Bultynck, A. Galione and S. J. Conway, Chem. Commun., 2011, 47, 242 RSC.
- P. J. Lu, W. R. Shieh and C. S. Chen, Biochem. Biophys. Res. Commun., 1996, 220, 637 CrossRef CAS PubMed.
- S. J. Mills, T. Luyten, C. Erneux, J. B. Parys and B. V. L. Potter, Messenger, 2012, 1, 167 CrossRef PubMed.
- S. J. Conway and G. J. Miller, Nat. Prod. Rep., 2007, 24, 687 RSC.
- E. Pantazaka and C. W. Taylor, J. Biol. Chem., 2011, 286, 23378 CrossRef CAS PubMed.
- S. C. Tovey, Y. Sun and C. W. Taylor, Nat. Protocols, 2006, 1, 259 CAS.
- F. Wolfram, E. Morris and C. W. Taylor, Biochem. J., 2010, 428, 483 CrossRef CAS PubMed.
- S. C. Tovey, S. G. Dedos, E. J. A. Taylor, J. E. Church and C. W. Taylor, J. Cell Biol., 2008, 183, 297 CrossRef CAS PubMed.
- T. P. Kenakin, A pharmacology primer. Theory, application, and methods, Elsevier, San Diego, 2004 Search PubMed.
- D. Colquhoun, Lectures in biostatistics, Clarendon Press, Oxford, 1971 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, compound characterization and copies of NMR spectra. See DOI: 10.1039/c5ob02623g |
| This journal is © The Royal Society of Chemistry 2016 |