
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pushing the limits of catalytic C–H amination in polyoxygenated cyclobutanes†
Pierre-Antoine
Nocquet
a,
Raphaël
Hensienne
a,
Joanna
Wencel-Delord‡
a,
Eugénie
Laigre
a,
Khadidja
Sidelarbi
b,
Frédéric
Becq
b,
Caroline
Norez
b,
Damien
Hazelard
a and
Philippe
Compain
*a
aLaboratoire de Synthèse Organique et Molécules Bioactives (SYBIO), Université de Strasbourg/CNRS (UMR 7509), Ecole Européenne de Chimie, Polymères et Matériaux (ECPM), 25 rue Becquerel, 67087 Strasbourg, France. E-mail: philippe.compain@unistra.fr
bLaboratoire Signalisation et Transports Ioniques Membranaires (STIM), Université de Poitiers et CNRS (ERL7368), 1 rue Georges Bonnet, 86000 Poitiers, France
First published on 28th January 2016
Abstract
A synthetic route to a new class of conformationally constrained iminosugars based on a 5-azaspiro[3.4]octane skeleton has been developed by way of Rh(II)-catalyzed C(sp3)–H amination. The pivotal stereocontrolled formation of the quaternary C–N bond by insertion into the C–H bonds of the cyclobutane ring was explored with a series of polyoxygenated substrates. In addition to anticipated regioselective issues induced by the high density of activated α-ethereal C–H bonds, this systematic study showed that cyclobutane C–H bonds were, in general, poorly reactive towards catalytic C–H amination. This was demonstrated inter alia by the unexpected formation of a oxathiazonane derivative, which constitutes a very rare example of the formation of a 9-membered ring by way of catalyzed C(sp3)–H amination. A complete stereocontrol could be however achieved by activating the key insertion position as an allylic C–H bond in combination with reducing the electron density at the undesired C–H insertion sites by using electron-withdrawing protecting groups. Preliminary biological evaluations of the synthesized spiro-iminosugars were performed, which led to the identification of a new class of correctors of the defective F508del-CFTR gating involved in cystic fibrosis.
Introduction
Over the past decade, catalytic C–H amination of C(sp3)–H bonds has established itself as a powerful tool for the synthesis of relevant nitrogen-containing compounds.1 The direct and selective functionalization of unactivated C–H bonds is indeed a strategy of choice to achieve a major simplification of synthetic sequences since no prior incorporation of functional groups is needed.2 Based on pioneering studies by Breslow and Gellman,3 the group of Du Bois developed in the early 2000s a powerful process for the intramolecular amination of C(sp3)–H bonds through Rhodium nitrene intermediates using carbamate or sulfamic ester substrates (Fig. 1).1,4 In the following years, various catalytic systems and new nitrene precursors were identified, and relevant mechanistic insights were disclosed.1 The process developed by Du Bois is still the best method in terms of practicality, mild experimental conditions and functional group tolerance. The broad synthetic utility of Rh(II)-catalyzed C–H amination has been indeed superbly demonstrated by the total synthesis of complex natural molecules such as (−)-tetrodotoxin.5 In addition, this highly regioselective, stereospecific process is valuable in the retrosynthetic analysis of enantiopure target molecules since it occurs with complete retention of configuration at the insertion site. Another strategic advantage of synthetic design is that the regioselectivity of the amination reaction may be predicted. Amination reactions performed with carbamates led almost exclusively to five-membered rings whereas sulfamic esters afforded, in general, six-membered rings.1 In addition to structural parameters related to elongated S–N/S–O bonds and unfavourable compression of the N–S–O angle in 5-membered cyclic sulfamates,1,4 electronic factors are also at play. Sites adjacent to electron-donating groups as well as benzylic, allylic and tertiary C–H bonds are generally favoured. Recent examples concerning the formation of 5- to 10-membered products from sulfamate substrates6 have nevertheless complicated the matter further. In some cases, conformational control, in combination or not with stereoelectronic effects, may dominate purely electronic factors.6 The major challenge associated with C(sp3)–H amination thus remain the control of regioselectivity in C–N bond formation, especially for complex substrates displaying “non-classical” conformation or a high density of reactive C–H bonds. | ||
| Fig. 1 Intramolecular catalysed C–H amination of sulfamic esters and carbamates. | ||
In connection with our interest in biologically relevant glycomimetics,7 we recently described the synthesis of the first examples of conformationally constrained iminosugars 1–2 based on four-membered ring-containing spirocycles (Fig. 2).8
 | ||
| Fig. 2 Some examples of iminosugars and of constrained analogues thereof. | ||
These compounds were designed as analogues of 1-deoxynojirimycin (DNJ, 3), a common motif found in many bioactive iminosugars, such as the antidiabetic Glyset™ or Zavesca™ (Gaucher disease, cystic fibrosis).9,10 In addition to exploring unfrequented regions of chemical and intellectual property spaces, we were interested in performing the first catalytic C–H amination of cyclobutanes11 to generate the pivotal C–N spiranic bond. An additional challenge was to design efficient strategies to secure high regioselectivity in polyoxygenated substrates with up to four contiguous reactive C–H bonds. Herein we wish to describe the full details of this synthetic study that led to unexpected regioselectivity and to a new class of bioactive iminosugars.
Results and discussion
The consecutive retrosynthetic analyses presented hereafter were mainly focused on achieving a high level of regiocontrol in the C–H amination critical step. Our synthetic strategies were based on guidelines established in the literature and competition experiments employing bifunctional substrates. From these precedents, the order of reactivity for C–H bond insertion may be roughly formulated as follows: allylic > α-ethereal ∼ 3° ∼ benzylic > 2° ≫ 1°.1l,12–14 Sites adjacent to electron-withdrawing groups are strongly disfavoured.4bFirst strategy: C–H amination of non-allylic C–H bonds using sulfamate esters
Our first synthetic strategy takes advantage of cyclobutanol 4, an advanced intermediate synthesized recently in our group from vitamin C (Fig. 3).7a The sulfamate function was easily introduced on the carbon side chain at C4 by the two-step conversion of the ester group in 4. | ||
| Fig. 3 Retrosynthetic analysis. | ||
Sulfamic ester 7 was expected to be a promising reaction substrate in terms of regioselectivity since C–H insertion into a tertiary C–H bond to form a 6-membered cyclic sulfamidate is supposed to be highly favoured. Insertion into the α-ethereal C–H bonds at C1 or C3 to form a less favourable 7-membered ring could nevertheless not be ruled out. We first envisioned original iminosugars of type I since sulfamidates II could be direct precursors to 1-azaspiro[3.3]heptanes following a two-step protocol involving a ring-opening/ring-closing cascade.15 After the activation of the oxathiazinane ring by N-acylation, the desired azetidine ring was indeed expected to be obtained by a one-pot reaction with NaI followed by NaH. Compound 7 was obtained in 3 steps from 4 (Scheme 1). First attempts to protect the secondary alcohol in 4 with a benzyl group under classical basic conditions (NaH, BnBr) led to sluggish conversion. Better results were obtained for protection as a silyl ether and 5 could be eventually obtained in 80% yield. After the reduction of ester 5 with LAH, the corresponding alcohol 6 was reacted with sulfamoyl chloride and DMAP to afford sulfamic ester 7. Quite surprisingly, exposure of 7 to the standard C–H amination protocol using PhI(OAc)2, MgO and a catalytic amount of Rh2(OAc)4 did not lead to any expected C–H insertion products but instead afforded alcohol 8 in 32% yield. The structure of 8 was unambiguously determined by using a heteronuclear multiple-bond correlation (HMBC) between benzylic protons and C-2 (see the ESI† for the HMBC spectrum of 8). The highly regioselective deprotection of the benzyloxy group at C1 could be explained by the formation of a 9-membered cyclic sulfamidate (Scheme 1). C–H insertion into the activated methylene of the benzylic ether would generate a labile hemiaminal that could be readily hydrolysed during the work-up procedure to give 8. It is noteworthy that only one example of the formation of a 9-membered ring by way of intramolecular-catalyzed C–H amination has been reported so far in the literature.6c In the aforementioned study, the macrocyclic sulfamidate was nevertheless obtained in low yields (16%) from (Z)-hex-4-en-1-yl sulfamate, the major product being the corresponding expected oxathiazinane derivative.
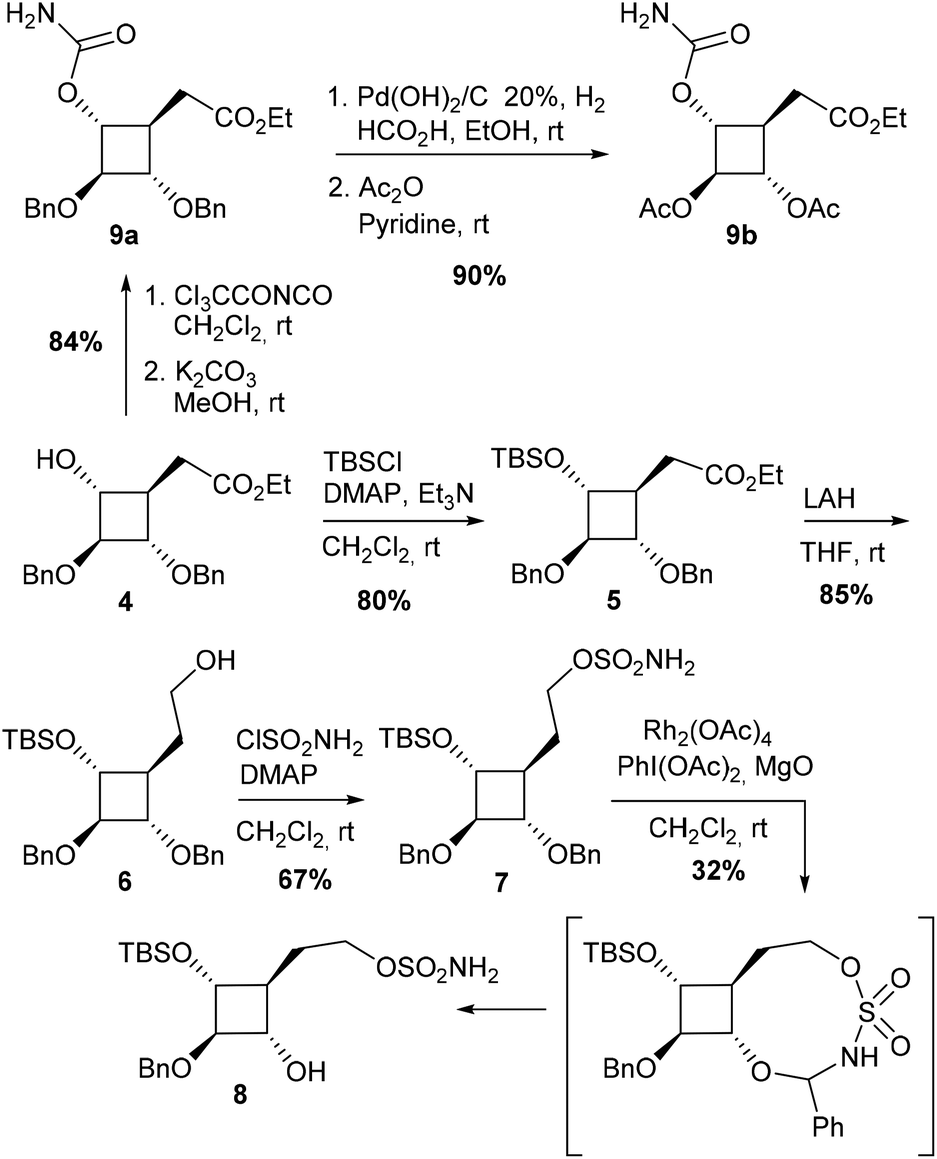 | ||
| Scheme 1 Synthesis of compounds 7 and 9. | ||
The unexpected outcome of the C–H amination of 7 prompted us to adopt a different strategy since the regioselectivity observed showed quite clearly that C–H bonds in cyclobutane were poorly reactive. No C–H insertions were indeed observed either at C4 or at C1 or C3 despite the fact that tertiary C–H bonds and tertiary α-ethereal C–H bonds are known to be favoured towards Rh(II)-catalyzed C–H amination. These results are consistent with the significantly lower s-character of the exocyclic bonds on four-membered hydrocarbon rings.11b
Second strategy: C–H amination of non-allylic C–H bonds using carbamates
To synthesize more rapidly substrates for C–H amination and to reduce the number of possible regioisomers, we turned our attention to the synthesis of carbamate 9a (Scheme 1). This compound was obtained in 84% yield from 4 by treatment with Cl3CC(O)NCO followed by K2CO3. Reaction of carbamate 9a with different Rh(II) catalysts and stoichiometric amounts of PhI(OAc)2 and MgO afforded hemiaminal 11a in 46–61% yields as the only regioisomer (Table 1, entries 1–3). The presence of the ester in the β-position is likely to have strongly deactivated the position at C4. These results were nonetheless encouraging since they demonstrated that catalytic amination of cyclobutane C–H bonds was feasible. To favour the formation of the desired regioisomer 10a, the benzyloxy group was replaced by a much more electron-withdrawing protecting group to reduce the electronic density at C2. Hydrogenolysis of 9a afforded the corresponding diol which was protected as acetates to yield compound 9b (Scheme 1). Unfortunately, the presence of four electron-withdrawing groups around the cyclobutane ring completely abolished the reactivity of the cyclobutane C–H bonds and no C–H amination product could be obtained (Table 1, entries 4 and 5).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | Reaction time (h) | R | 10 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
9c |
| a See the Experimental section for the experimental conditions. b Isolated yields. c Recovered after purification on a silica gel. d The reaction was performed at 60 °C. | |||||||
| 1 | Rh2(OAc)4 (5) | CH2Cl2 | 7 | Bn | — | 54% | 12% |
| 2 | Rh2(esp)2 (2) | CH2Cl2 | 7 | Bn | — | 61% | 18% |
| 3 | Rh2(tpa)4 (5) | CH2Cl2 | 15 | Bn | — | 46% | 31% |
| 4 | Rh2(esp)2 (2) | CH2Cl2 | 8 | Ac | — | — | 72% |
| 5 | Rh2(tpa)4 (10) | C6H6d |
8 | Ac | — | — | ∼10% |
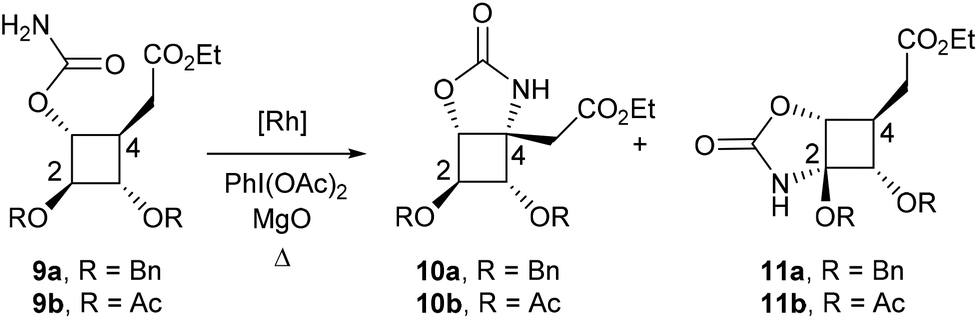
A different tactic was designed to generate the desired C–N bond at C4, taking advantage of the C–H amination product 11a. The objective was to cleave the oxazolidinone ring to afford a hemiaminal function, as a masked ketone, and a secondary alcohol at C3 (compound 13) that may be converted into the corresponding carbamate to perform a second C–H amination reaction (Scheme 2).
 | ||
| Scheme 2 Synthesis and evaluation of carbamate 14 as the substrate for C–H amination. | ||
After N-Boc protection, oxazolidinone 12 was reacted with cesium carbonate16 to provide alcohol 13 which was converted into the corresponding carbamate 14 in 51% yield for the three steps. Unfortunately, despite several attempts using typical rhodium-catalyzed conditions, no C–H amination product could be obtained from 14 and only the starting material was recovered in up to 52% yield.
Another strategy to avoid regioselectivity issues was to introduce the carbamate function on the carbon side chain at C4 (Scheme 3). Due to the strong bias of carbamates for 5-membered ring formation, C–H insertion was expected to occur exclusively at C4 from substrates 16. Three carbamates protected (Ac, TBS) or not at C3 were synthesized from alcohol 15.7a None of these compounds led to the desired azaspiranic products under typical Rh(II)-catalyzed C–H amination conditions. In each case, the starting material was the only compound that could be isolated by chromatography on a silica gel.
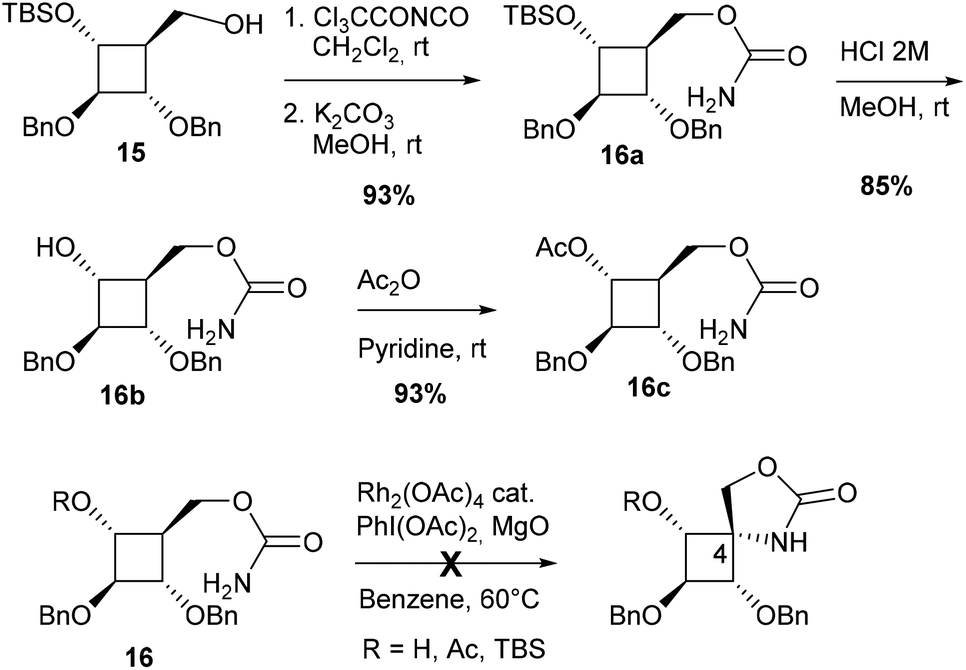 | ||
| Scheme 3 Synthesis and evaluation of carbamates 16 as the substrate for C–H amination. | ||
These results indicated that even in the absence of a strong electron-withdrawing group in the β-position, as in carbamate 9, the cyclobutane tertiary C–H bond was not reactive enough to undergo the C–H insertion process.
Third strategy: C–H amination of allylic C–H bonds using carbamates
Considering the apparent low reactivity of the tertiary C–H bond at C4 discussed above and the regioselectivity issues due to the high density of α-oxygenated C–H bonds, we decided to introduce a vinylic group onto the cyclobutane skeleton (Fig. 4). Allylic C–H bonds were indeed shown in several studies to be favoured over α-oxygenated C–H bonds.12 To further discriminate between cyclobutane C–H bonds, the carbamate function was directly introduced on the 4-membered ring to deactivate the α-oxygenated C–H bond at C3. With these substrates VII thus designed, we shifted our focus towards iminosugars of type III based on a 5-azaspiro[3.4]octane skeleton (Fig. 4). The next logical step to rapidly obtain spiranic iminosugars was indeed to perform olefin ring-closing metathesis (RCM) of the C–H amination products VI after N-allylation. | ||
| Fig. 4 Retrosynthetic analysis towards azaspiro[3.4]octane derivatives. | ||
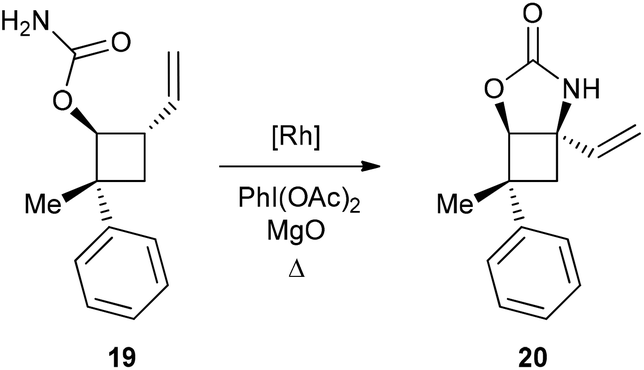
Combining C–H amination and RCM has two main advantages in terms of synthetic efficiency. First of all, the vinylic group introduced to direct the C–H insertion at C4 is directly involved in the construction of the second ring of our azaspiranic targets. Secondly, no additional steps are required to avoid the poisoning of the metathesis catalyst since the nitrogen atom is deactivated by an electron-withdrawing group.17 The feasibility of our strategy was first evaluated on a simplified cyclobutane derivative, carbamate 19 (Scheme 4). This compound was synthesized by carbamoylation of the readily available racemic alcohol 18 obtained in 2 steps from vinyl epoxide.18 Another advantage of using 19 as a model substrate is that it reproduces the same structural pattern found in VII – a trans-1-carbamoyl-2-vinylcyclobutane motif – but with minimal regioselectivity issues.
 | ||
| Scheme 4 Model study on compound 19. | ||
Encouragingly, treatment of 19 with 10 mol% of Rh(esp)2 or Rh2(oct)4 and stoichiometric amounts of PhI(OAc)2 and MgO in CH2Cl2 provided the expected C–H insertion product 20 in 41% yield (Table 2, entries 1 and 2). Increasing the amount of catalyst to 20 mol% or performing the reaction in refluxing dichloroethane (DCE) led to improved yields up to 58% (entries 3 and 4). Replacing PhI(OAc)2 with PhI(OPiv)2 did not increase the efficiency of the C–H insertion process (entry 5). The modest yields observed for the formation of cyclic carbamate 20 further confirmed the low reactivity of cyclobutane C–H bonds towards catalytic C–H amination.
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | Reaction time (h) | T |
20b |
19c |
| a See the Experimental section for the experimental conditions. b Isolated yields. c Recovered after purification on a silica gel. d PhI(OPiv)2 was used instead of PhI(OAc)2. | ||||||
| 1 | Rh2(esp)2 (10) | CH2Cl2 | 17 | Δ | 41% | 39% |
| 2 | Rh2(oct)4 (10) | CH2Cl2 | 32 | Δ | 41% | 6% |
| 3 | Rh2(esp)2 (20) | CH2Cl2 | 55 | Δ | 52% | 19% |
| 4 | Rh2(esp)2 (15) | DCE | 43 | Δ | 58% | 16% |
| 5d | Rh2(esp)2 (15) | CH2Cl2 | 32 | Δ | 47% | 22% |
The C–H allylic bonds also provide an opportunity for performing C–H amination via π-allyl species. To apply the methodology recently developed by the group of White based on electrophilic Pd(II)/sulfoxide catalysis,19 alcohol 18 was converted into the corresponding N-tosylcarbamate 21 by treatment with TsNCO (Scheme 5). Under standard allylic C–H amination conditions using phenyl bis-sulfoxide/Pd(OAc)2 and a stoichiometric amount of phenyl-benzoquinone (PhBQ), the desired N-tosylcarbamate 22 was obtained in only 9% yield (15% based on the recovered starting materials). In addition, 22 is obtained as a mixture with PhBQ since the two compounds are difficult to separate by flash chromatography. No improvement was obtained when the reaction was performed under an oxygen atmosphere or in the presence of additives including AcOH,20 Na2S2O8, PhI(OAc)2 or Ag2CO3. To the best of our knowledge, this result is a very rare example of a C–H amination of a tertiary allylic C–H bond using White's methodology.21
 | ||
| Scheme 5 Model study on compound 21. | ||
Despite the modest yields observed in the model study, carbamate 23a was synthesized by carbamoylation of alcohol 177a,8 and subjected to a Rh(II)-catalyzed C–H amination (Table 3, entry 1). Disappointingly, in contrast to previous results obtained with pyran or acyclic substrates,12 the regioselectivity of insertion was strongly in favor of the methine group adjacent to the oxygen atom over the allylic C–H bond at C4. After an extensive study, the best results in terms of yields and regioselectivity were obtained when carbamate 23a was treated with 20 mol% of Rh2(esp)2 and stoichiometric amounts of PhI(OAc)2 and MgO in refluxing CH2Cl2.8 These reaction conditions led to a complete conversion of 23a and to the desired allylic C–H insertion 24a but in only 17% yield, carbamate 25a being still the major product (56% yield). To overcome this unexpected obstacle, we designed a strategy to strongly disfavour the formation of the unwanted regioisomer 25. Our objective was to reduce the electron density of the α-oxygenated C–H bond at C2 by using electron-withdrawing protecting groups instead of benzyloxy groups. However, such a tactic may be seen as a double-edged sword since the presence of three electron-withdrawing groups around the cyclobutane ring may also reduce the reactivity of the C–H bond at C4. Acetate and benzoate esters were firstly selected as hydroxyl-protecting groups.
a
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Rh2(esp)2 (mol%) | Reaction time (h) | R | δ(H2) ppmb | δ(H4) ppmb |
24c |
25c |
23d |
| a See the Experimental section for the experimental conditions. b From the 1H NMR spectrum of compounds 23 recorded in CDCl3. c Isolated yields. d Recovered after purification on a silica gel. e DCE was used instead of CH2Cl2. | ||||||||
| 1 | 20 | 27 | Bn | 3.95 | 2.48 | 17% | 56% | — |
| 2 | 15 | 8 | Ac | 5.09 | 2.65 | 26% | — | 10% |
| 3 | 15 | 24 | Ac | 5.09 | 2.65 | 31% | — | — |
| 4 | 20 | 23 | p-MeOBz | 5.49 | 2.85 | 40% | — | 12% |
| 5 | 20 | 23 | Bz | 5.53 | 2.90 | 40% | — | 10% |
| 6e | 25 | 40 | Bz | 5.53 | 2.90 | 32% | — | 23% |
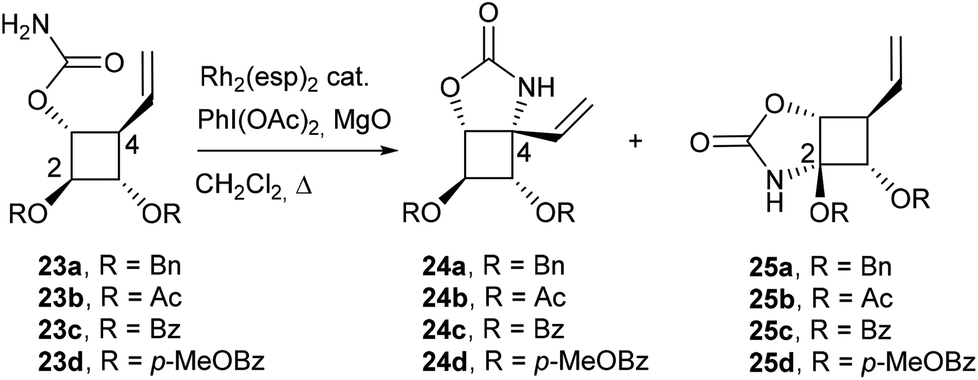
To fine tune the electronic environment around the reactive cyclobutane C–H bond, para-methoxybenzoate ester 23d was also prepared as a close analogue of substrate 23c. The esters 23b, 23c and 23d were synthesized in 2 steps from carbamate 23a by deprotection of the benzyl group followed by treatment with Ac2O, BzCl or para-methoxybenzoyl chloride (p-MeOBzCl) in the presence of pyridine (Scheme 6).
 | ||
| Scheme 6 Synthesis of compounds 23. | ||
To our delight, the electron-withdrawing group strategy reached its goal, allowing complete regioselectivity towards the desired C–H insertion products 24 (Table 3, entries 2–6). As with substrate 23a, the reaction time has to be relatively long (∼24 h) to ensure total conversion of the starting material (entries 2 and 3) and acetate-protected carbamate 24b was obtained in up to 31% yield. The use of the more electron-withdrawing benzoate group was found to increase the yield by ∼20% (entries 3 and 5). In contrast to the results obtained in the model study (Table 2), reactions performed in DCE do not increase the yield of the process (entry 6). The addition of an electron-donating group to the benzoate phenyl ring had no impact on the yield of the C–H amination reaction and carbamate 24d was obtained in 40% yield (entry 4). As shown by the 1H NMR of compounds 23 (Table 3), the signals of H2 shifted downfield upon increasing the electron-withdrawing ability of the hydroxyl protecting groups, an observation consistent with a decrease of electron density around H2. The H2 chemical shifts correlated nicely with the yields of carbamates 24 and the level of regiocontrol achieved. As suggested by the downfield shifts of H4 protons from 2.48 ppm (R = Bn) to 2.90 ppm (R = Bz), the addition of two electron-withdrawing groups impacted also the electron density at C4 and thus the efficiency of the desired C–H amination reaction. Despite the modest yields observed, the electron-withdrawing group strategy was nonetheless efficient if one considers the complete regiocontrol achieved and the yields obtained with the related carbamate 19, a substrate with minimal regioselectivity issues. In addition, the yields of the C–H amination reaction provided the azaspiranic intermediate 24c in quantities sufficient to complete the synthesis of the targeted iminosugars 1–2. To pursue our synthetic goal, we took advantage of the readily available model substrate 26 obtained by N-allylation of 20 to evaluate the key RCM step. Pleasingly, the highly constrained cyclobutane-containing tricycle 27 was obtained in 94% yield using 5 mol% Grubbs II catalyst in refluxing CH2Cl2 (Scheme 7). This result provides a further example of the powerfulness of the RCM process considering the large additional ring strain generated by the 5-membered ring closure.
 | ||
| Scheme 7 Model study (RCM reaction). | ||
The same process was applied to carbamate 24c with a similar efficiency for the RCM step (89% yield) to provide the 5-azaspiro[3.4]octane skeleton of our targets. The spiranic iminosugars 1–2 were then obtained in 2 or 3 steps from the common intermediate 28 thus generated (Scheme 8).8
 | ||
| Scheme 8 Synthesis of spiranic iminosugars 1 and 2. | ||
Biological evaluation
The potential of the three DNJ analogues 1–2 was first evaluated for the treatment of cystic fibrosis. This life-threatening inherited disease is caused by a mutation in the gene for the protein Cystic Fibrosis Transmembrane conductance Regulator (CFTR).22 Zavesca™ (N-Bu DNJ), a clinical candidate for the treatment of cystic fibrosis,22b is able to act as a CFTR corrector by inhibiting endoplasmic reticulum-resident trimming α-glucosidases and thus overcoming the processing defect of the mutant protein.23 To evaluate the impact of a constrained iminosugar conformation on F508del-CFTR defective trafficking, the rescue of the F508del-CFTR function was assessed using the iodide efflux technique on CF cells treated for 4 hours with 100 μM of iminosugars (Fig. 5).24 In these experiments, N-Bu DNJ was used as a reference compound with regard to its therapeutic potential. Results presented in Fig. 5A show the rescue of the F508del-CFTR function by N-Bu DNJ but also by N-butyl spiranic iminosugar 2b and to a less extent by 2a. The presence of a butyl chain and of more than three hydroxyl groups was found to play an important role since the best corrector of the spiranic series, iminosugar 2b, displayed an F508del-CFTR activity rescue not significantly different from the N-Bu DNJ-induced one.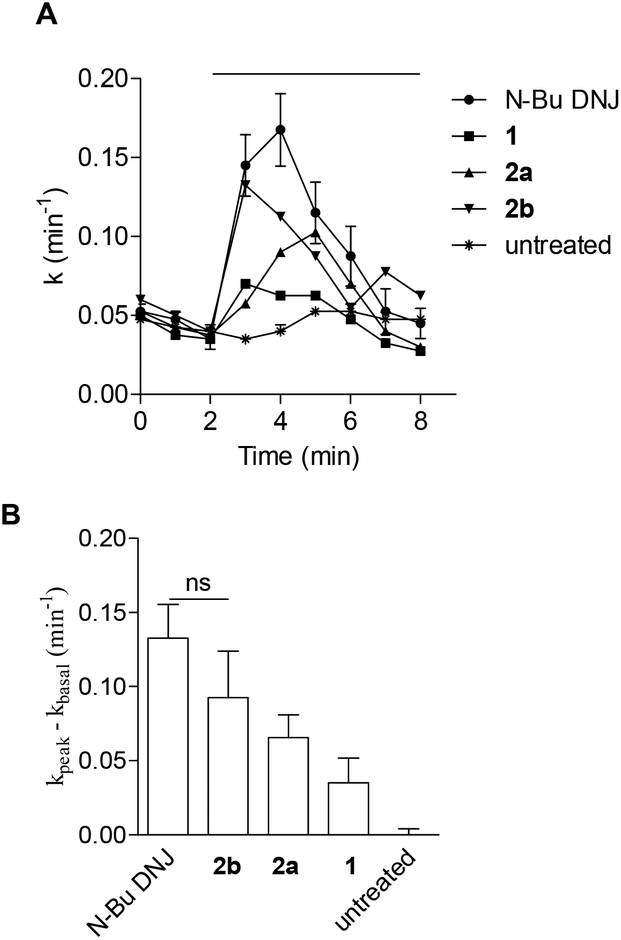 | ||
| Fig. 5 (A) Rescue of functional F508del-CFTR activity: iodide efflux curves on untreated or iminosugar-treated F508del-CFTR stably transfected HeLa cells. Iodide effluxes were stimulated by forskolin/genistein as indicated by the horizontal bar above the traces. Each value represents the mean ± SEM (n = 4). (B) Summary of the functional evaluation of F508del-CFTR: histograms represent the mean (±SEM) of four experiments obtained by the iodide efflux technique in CF cells untreated or treated for 4 h with 100 μM of iminosugar as indicated. A mixture of forskolin (10 μM) + genistein (30 μM) was used to activate CFTR. ns, not significant. | ||
In our previous study on Gaucher disease, a rare genetic disorder caused by the deficiency of β-glucocerebrosidase (GCase), we identified α-1-C-nonyl-iminoxylitol (29) as a promising lead for a pharmacological chaperone therapy (Fig. 6).25 This therapeutic strategy is based on the use of competitive inhibitors capable of enhancing GCase residual hydrolytic activity at sub-inhibitory concentrations.26,27 Reversible competitive inhibitors are indeed believed to modify and/or stabilize the three-dimensional structure of the deficient but still catalytically active glycosidase, preventing its premature degradation by the endoplasmic reticulum quality-control system before trafficking to lysosomes.27 α-1-C-Nonyl-iminoxylitol (29) was found to be a very specific nanomolar inhibitor of GCase.25 It was hypothesized that the qualitative leap in inhibitory potency between 29 and 30,25 the corresponding analogue in the D-gluco series, was due to a piperidine ring inversion from a classical 4C1(D) to a 1C4(D) conformation in which all hydroxyl groups are axial and the alkyl chain is equatorial.25,26,28
 | ||
| Fig. 6 Inhibition of GCase. | ||
In a preliminary study, we evaluate whether further improvement of the activity might be gained for structures of type III that may be considered constrained mimetics of iminosugar 29 with the hydroxyl groups in pseudo axial orientation. Considering that the presence of an alkyl chain is important for GCase inhibitory activity but cytotoxicity may be associated with long alkyl chain iminosugar derivatives, first preliminary inhibitory assays were performed with spiro-iminosugar 2b (Fig. 6). This compound was found to display weak inhibitory activity (IC50 > 100 μM) and showed no significant improvement in the inhibition of GCase relative to the corresponding analogues in the D-gluco series, α-1-C-butyl DNJ (31)29 and N-butyl DNJ (Zavesca™).
Conclusions
In conclusion we have developed a synthetic route to conformationally constrained iminosugars based on four-membered ring-containing spirocycles. The key step of our strategy was the formation of the pivotal quaternary C–N bond of the 5-azaspiro[3.4]octane skeleton by way of Rh(II)-catalyzed C(sp3)–H amination. In addition to the goal of developing a new class of bioactive iminosugars, our aim was to explore the limits of this powerful stereospecific process with a series of substrates. The main anticipated challenge was to secure a high level of regioselectivity from polyoxygenated substrates with a high density of activated α-ethereal C–H bonds. An additional and rather unexpected issue was found to be the low reactivity of cyclobutane C–H bonds towards catalytic C–H amination, which led to modest yields and unusual regioselectivity. A complete stereocontrol could be nevertheless achieved by using a combination of electron-withdrawing and activating groups. The mere introduction of a vinylic group was indeed not sufficient to reach high regioselectivity even though insertion into allylic C–H bonds has been described to be favoured over α-ethereal C–H bonds. Consequently, electron-withdrawing protecting groups were required to reduce the electron density at the undesired C–H insertion site. First preliminary biological evaluations of the potential of the synthesized spiro-iminosugars for the treatment of Gaucher disease and cystic fibrosis were performed, which led to the identification of a new class of CFTR correctors.Experimental section
Tetrahydrofuran (THF) was dried by passing through an activated alumina column under Ar or distilled over Na/benzophenone under Ar. Dichloromethane (CH2Cl2) and dichloroethane (DCE) were distilled over CaH2 under Ar. Pyridine and triethylamine were distilled over KOH under Ar and stored over KOH. Dried DMF was purchased from Sigma-Aldrich. All the reactions were performed in standard glassware under Ar unless otherwise specified. Flash chromatographies were performed on silica gel 60 (230–400 mesh, 0.040–0.063 mm) purchased from E. Merck or using an automatic flash chromatography device. Thin Layer Chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 purchased from E. Merck. IR spectra (cm−1) were recorded on a Perkin-Elmer Spectrum One spectrophotometer. NMR spectra were recorded on 300 MHz or 400 MHz spectrometers with solvent peaks as the reference. Carbon multiplicities were assigned by distortionless enhancement by polarization transfer (DEPT) experiments. The 1H and 13C signals were assigned by 2D experiments (COSY, HSQC, HMBC). For convenience, the assignment of 1H and 13C for all the molecules were based on the same numbering (see the ESI†). ESI-HRMS mass spectra were recorded on a TOF-spectrometer. Specific rotations were determined at room temperature (20 °C) on a Perkin-Elmer 241 polarimeter for sodium (λ = 589 nm). Rh2(esp)2 (CAS: 819050-89-0), Rh2(OAc)4 (CAS: 15956-28-2), Rh2(oct)4 (CAS: 73482-96-9) and Rh2(tpa)4 (CAS: 142214-04-8) were purchased from Sigma-Aldrich.Ethyl 2-((1S,2S,3R,4R)-2,3-bis(benzyloxy)-4-((tert-butyldimethylsilyl)oxy)cyclobutyl)acetate (5)
To a solution of alcohol 47a (200 mg, 0.54 mmol, 1 equiv.) in CH2Cl2 (1 mL) were added TBSCl (122 mg, 0.81 mmol, 1.5 equiv.), DMAP (33 mg, 0.27 mmol, 0.5 equiv.) and Et3N (0.15 mL, 1.08 mmol, 2 equiv.). The solution was stirred for 18.5 h. Water was added and the product was extracted with Et2O (3×). The combined organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:19) to afford the desired cyclobutane 5 (209 mg, 80%) as a pale yellow oil.
R
f
0.26 (EtOAc/petroleum ether, 1:19), [α]20D +0.5 (c 1.0, CHCl3), IR (film) 1733, 1097, 835 cm−1, 1H NMR (400 MHz, CDCl3) δ 7.41–7.28 (m, 10H, Ph), 4.66 (d, J = 11.7 Hz, 1H, CH2Ph), 4.61 (d, J = 11.7 Hz, 1H, CH2Ph), 4.56 (s, 2H, CH2Ph), 4.16 (q, J = 7.1 Hz, 2H, CH2CH3), 3.83 (m, 1H, H-2), 3.65 (m, 1H, H-1 or H-3), 3.49 (m, 1H, H-1 or H-3), 2.57 (dd, J = 15.0, 5.9 Hz, 1H, H-1′a), 2.50 (dd, J = 14.9, 6.9 Hz, 1H, H-1′b), 2.17 (m, 1H, H-4), 1.28 (t, J = 7.2 Hz, 3H, CH2CH3), 0.95 (s, 9H, C(CH3)3), 0.14 (s, 3H, SiCH3), 0.12 (s, 3H, SiCH3), 13C NMR (100 MHz, CDCl3) δ 171.7 (CO), 138.4 (Cq-Ar), 138.2 (Cq-Ar), 128.42 (2 CH-Ar), 128.40 (2 CH-Ar), 127.8 (4 CH-Ar), 127.70 (CH-Ar), 127.67 (CH-Ar), 86.0 (C-2), 77.3 (C-1 or C-3), 71.6 (2 CH2Ph), 71.1 (C-1 or C-3), 60.5 (CH2CH3), 41.2 (C-4), 36.3 (C-1′), 25.8 (C(CH3)3), 18.0 (C(CH3)3), 14.3 (CH2CH3), −4.5 (SiCH3), −4.6 (SiCH3), HRMS (ESI) m/z 507.250 ([M + Na]+, calcd for C28H40O5SiNa: 507.254).
2-((1S,2S,3R,4R)-2,3-Bis(benzyloxy)-4-((tert-butyldimethylsilyl)oxy)cyclobutyl)ethanol (6)
LiAlH4 (23 mg, 0.61 mmol, 1.5 equiv.) was added to a solution of ester 5 (199 mg, 0.41 mmol, 1 equiv.) in THF (2.3 mL), cooled to 0 °C. The solution was stirred at rt for 2.5 h. After cooling to 0 °C, water (0.02 mL), 10% aqueous NaOH (0.03 mL) and water (0.05 mL) were successively added. The solution was stirred for 40 min at rt, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:9 to 1:3) to afford the desired alcohol 6 (155 mg, 85%) as a colorless oil.
R
f
0.51 (EtOAc/petroleum ether, 1:3), [α]20D −5 (c 1.0, CHCl3), IR (film) 3449, 1061, 836 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.27 (m, 10H, Ph), 4.63 (d, J = 11.6 Hz, 1H, CH2Ph), 4.61–4.54 (m, 2H, CH2Ph), 4.52 (d, J = 11.6 Hz, 1H, CH2Ph), 3.80 (t, J = 5.7 Hz, 1H, H-2), 3.67 (m, 2H, H-2′), 3.53 (m, 1H, H-1 or H-3), 3.36 (m, 1H, H-1 or H-3), 2.50 (t, J = 6.6 Hz, 1H, OH), 1.86–1.68 (m, 3H, H-4, H-1′), 0.90 (s, 9H, C(CH3)3), 0.11 (s, 3H, SiCH3), 0.09 (s, 3H, SiCH3), 13C NMR (75 MHz, CDCl3) δ 138.1 (Cq-Ar), 137.9 (Cq-Ar), 128.6 (2 CH-Ar), 128.5 (2 CH-Ar), 128.0 (3 CH-Ar), 127.9 (2 CH-Ar), 127.8 (CH-Ar), 86.0 (C-2), 77.8 (C-1 or C-3), 71.9 (C-1 or C-3), 71.8 (CH2Ph), 71.7 (CH2Ph), 61.9 (C-2′), 42.7 (C-4), 34.9 (C-1′), 25.8 (C(CH3)3), 17.9 (C(CH3)3), −4.4 (SiCH3), −4.5 (SiCH3), HRMS (ESI) m/z 465.241 ([M + Na]+, calcd For C26H38O4SiNa: 465.243).
2-((1R,2S,3S,4R)-2,3-Bis(benzyloxy)-4-((tert-butyldimethylsilyl)oxy)cyclobutyl)ethyl sulfamate (7)
Preparation of sulfamoyl chloride: formic acid (0.78 mL, 20.7 mmol, 0.9 equiv.) was added slowly to chlorosulfonyl isocyanate (2 mL, 23.0 mmol, 1 equiv.) cooled at 0 °C. The solution was stirred at rt for 4.5 h. The solution was cooled at 0 °C and benzene (30 mL) was added. The solution was filtered and concentrated under reduced pressure to give a white to brown solid of ClSO2NH2 (1.44 g, 40%) which was used without further purification.IR (neat) 1374, 1174 cm−1.
DMAP (91 mg, 0.75 mmol, 2 equiv.) followed by ClSO2NH2 (65 mg, 0.56 mmol, 1.5 equiv.) were added to a solution of 6 (165 mg, 0.37 mmol, 1 equiv.) in CH2Cl2 (5.8 mL). After 2 h of stirring, a second portion of DMAP (91 mg, 0.75 mmol, 2 equiv.) followed by ClSO2NH2 (65 mg, 0.56 mmol, 1.5 equiv.) were added. The solution was stirred for 15 h. Water (a few drops) was added and the solution was filtered. The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:1) to afford 7 (130 mg, 67%) as a colorless oil.
R
f
0.42 (EtOAc/petroleum ether, 1:3), [α]20D +5 (c 0.9, MeOH), IR (film) 3355, 1359 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.41–7.27 (m, 10H, Ph), 4.75 (s, 2H, NH2), 4.65 (d, J = 11.7 Hz, 1H, CH2Ph), 4.60–4.52 (m, 2H, CH2Ph), 4.48 (d, J = 11.2 Hz, 1H, CH2Ph), 4.22 (m, 2H, H-2′), 3.79 (t, J = 5.6 Hz, 1H, H-2), 3.53 (m, 1H, H-1 or H-3), 3.39 (m, 1H, H-1 or H-3), 2.07 (m, 1H, H-4), 1.84 (m, 2H, H-1′), 0.91 (s, 9H, C(CH3)3), 0.11 (s, 3H, SiCH3), 0.09 (s, 3H, SiCH3), 13C NMR (75 MHz, CDCl3) δ 137.9 (Cq-Ar), 137.5 (Cq-Ar), 128.8 (2 CH-Ar), 128.6 (2 CH-Ar), 128.4 (2 CH-Ar), 128.3 (CH-Ar), 128.0 (3 CH-Ar), 86.3 (C-2), 77.6 (C-1 or C-3), 72.3 (C-1 or C-3), 71.79 (CH2Ph), 71.76 (CH2Ph), 69.6 (C-2′), 41.1 (C-4), 31.6 (C-1′), 25.9 (C(CH3)3), 18.0 (C(CH3)3), −4.2 (SiCH3), −4.5 (SiCH3), HRMS (ESI) m/z 544.212 ([M + Na]+, calcd for C26H39NO6SSiNa: 544.216).
2-((1S,2R,3R,4S)-3-(Benzyloxy)-2-((tert-butyldimethylsilyl)oxy)-4-hydroxycyclobutyl)ethyl sulfamate (8)
MgO (10 mg, 0.25 mmol, 2.3 equiv.), PhI(OAc)2 (39 mg, 0.12 mmol, 1.1 equiv.) and Rh2(OAc)2 (2.4 mg, 0.0055 mmol, 0.05 equiv.) were added to a solution of 7 (58 mg, 0.11 mmol, 1 equiv.) in degassed CH2Cl2 (0.69 mL). The solution was stirred at rt for 7 h. After filtration through Celite, the solution was concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:7 to 4:1) to afford 8 (15 mg, 32%) as a white solid.
R
f
0.25 (EtOAc/petroleum ether, 2:1), [α]20D −8 (c 1.0, MeOH), IR (neat) 3386, 1385 cm−1, 1H NMR (300 MHz, CD3OD) δ 7.40–7.22 (m, 5H, Ph), 4.69 (d, J = 11.7 Hz, 1H, CH2Ph), 4.57 (d, J = 11.7 Hz, 1H, CH2Ph), 4.20 (t, J = 6.9 Hz, 2H, H-2′), 3.60 (t, J = 6.1 Hz, 1H, H2), 3.48 (m, 1H, H-1 or H-3), 3.40 (m, 1H, H-1 or H-3), 2.12–1.87 (m, 2H, H-1′), 1.66 (m, 1H, H-4), 0.90 (s, 9H, C(CH3)3), 0.10 (s, 6H, 2 SiCH3), 13C NMR (75 MHz, CD3OD) δ 139.6 (Cq-Ar), 129.5 (2 CH-Ar), 129.0 (2 CH-Ar), 128.8 (CH-Ar), 88.7 (C-2), 72.60 (C-1 or C-3), 72.57 (C-1 or C-3), 72.3 (CH2Ph), 69.2 (C-2′), 44.0 (C-4), 32.5 (C-1′), 26.4 (C(CH3)3), 18.9 (C(CH3)3), −4.2 (SiCH3), −4.3 (SiCH3), HRMS (ESI) m/z 454.169 ([M + Na]+, calcd for C19H33NO6SSiNa: 454.169).
Ethyl 2-((1S,2S,3R,4R)-2,3-bis(benzyloxy)-4-(carbamoyloxy)cyclobutyl)acetate (9a)
Trichloroacetyl isocyanate (84 μL, 0.70 mmol, 1.2 equiv.) was added to a solution of 47a (218 mg, 0.59 mmol, 1 equiv.) in CH2Cl2 (1.6 mL) cooled to 0 °C. The solution was stirred at rt for 7 h and concentrated under reduced pressure. The residue was dissolved in MeOH (1.3 mL) and K2CO3 (8 mg, 0.059 mmol, 0.1 equiv.) was added. The solution was stirred for 14 h. Saturated aqueous NH4Cl (2 mL) was added and the product was extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:1) to afford the desired carbamate 9a (205 mg, 84%) as a white solid.
R
f
0.45 (EtOAc/petroleum ether, 1:2), [α]20D = −12 (c 1.0, MeOH), IR (neat) 3440, 1723, 1666, cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.23 (m, 10H, Ph), 4.64–4.46 (m, 7H, 2 CH2Ph, NH2, H-3), 4.12 (q, J = 7.2 Hz, 2H, CH2CH3), 3.94 (m, 1H, H-2), 3.55 (m, 1H, H-1), 2.65 (dd, J = 16.1, 7.4 Hz, 1H, H-1′a), 2.56 (dd, J = 16.1, 6.2 Hz, 1H, H-1′b), 2.27 (m, 1H, H-4), 1.24 (t, J = 7.1 Hz, 3H, CH2CH3), 13C NMR (75 MHz, MeOD) δ 173.4 (CO), 158.8 (NCO), 139.4 (Cq-Ar), 139.2 (Cq-Ar), 129.38 (2 CH-Ar), 129.35 (2 CH-Ar), 129.1 (2 CH-Ar), 128.9 (2 CH-Ar), 128.8 (2 CH-Ar), 83.8 (C-2), 77.9 (C-1), 73.2 (C-3), 72.7 (CH2Ph), 72.3 (CH2Ph), 61.7 (CH2CH3), 40.4 (C-4), 36.8 (C-1′), 14.5 (CH2CH3), HRMS (ESI) m/z 436.172 ([M + Na]+, calcd for C23H27NO6Na: 436.173).
(1S,2R,3R,4S)-3-(Carbamoyloxy)-4-(2-ethoxy-2-oxoethyl)cyclobutane-1,2-diyl diacetate (9b)
Pd(OH)2/C 20% (15 mg) and HCO2H (2 drops) were added to a solution of 9a (95 mg, 0.23 mmol, 1 equiv.) in EtOH (3 mL). The solution was placed under a H2 atmosphere and stirred until disappearance of the starting material (25 h). The solution was filtered through Celite and concentrated under reduced pressure. The residue was dissolved in pyridine (6 mL) and Ac2O (1.7 mL) was added. The solution was stirred at rt for 17 h. The solution was concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:1 to 2:1) to afford the desired ester 9b (66 mg, 90%) as a white solid.
R
f
0.31 (silica gel, EtOAc/petroleum ether, 2:1), [α]20D = +2 (c 1.0, CHCl3), IR (film) 3474, 1725, 1225 cm−1, 1H NMR (300 MHz, CDCl3) δ 5.09 (t, J = 6.3 Hz, 1H, H-2), 5.02 (br s, 2H, NH2), 4.68 (dd, J = 7.7, 6.3 Hz, 1H, H-1 or H-3), 4.61 (dd, J = 7.8, 6.3 Hz, 1H, H-1 or H-3), 4.08 (q, J = 7.2 Hz, 2H, CH2CH3), 2.72 (m, 2H, H-1′), 2.38 (m, 1H, H-4), 2.04 (s, 3H, CH3), 2.03 (s, 3H, CH3), 1.21 (t, J = 7.2 Hz, 3H, CH2CH3), 13C NMR (75 MHz, CDCl3) δ 171.4 (CO), 170.4 (CO), 170.0 (CO), 155.8 (NCO), 74.4 (C-2), 70.7 (C-1 or C-3), 70.0 (C-1 or C-3), 60.7 (CH2CH3), 38.9 (C-4), 35.4 (C-1′), 20.8 (2 CH3), 14.2 (CH2CH3), HRMS (ESI) m/z 340.099 ([M + Na]+, calcd for C13H19NO8Na: 340.100).
General procedure A for C–H amination of carbamates (Tables 1–3)
MgO (2.3 equiv.), PhI(OAc)2 (1.4 equiv.) and catalyst (2 mol%–20 mol%) were added to a solution of carbamate (1 equiv., 0.2 mmol) in degassed CH2Cl2, DCE or benzene (1.8 ml). The solution was refluxed (CH2Cl2, DCE) or heated at 60 °C (benzene). After cooling, the solution was filtered through a pad of Celite and concentrated under reduced pressure. The crude product was purified by flash chromatography.Ethyl 2-((1R,5R,6S,7S)-5,6-bis(benzyloxy)-3-oxo-2-oxa-4-azabicyclo[3.2.0]heptan-7-yl)acetate (11a) (Table 1, entries 1–3)
According to general procedure A, 11a was obtained as a white solid after flash chromatography (EtOAc/petroleum ether, 1:5 to 1:1).
R
f
0.22 (EtOAc/petroleum ether, 1:2), [α]20D −25 (c 1.0, CHCl3), IR (film) 3286, 1757, 1730 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.24 (m, 10H, Ph), 5.61 (s, 1H, NH), 4.62–4.48 (m, 3H, CH2Ph), 4.48–4.41 (m, 2H, CH2Ph, H-3), 4.14 (q, J = 7.1 Hz, 2H, CH2CH3), 4.00 (dd, J = 6.9, 1.1 Hz, 1H, H-1), 2.56 (dd, J = 16.0, 5.9 Hz, 1H, H-1′a), 2.44 (dd, J = 16.0, 7.4 Hz, 1H, H-1′b), 2.33 (m, 1H, H-4), 1.24 (t, J = 7.1 Hz, 3H, CH2CH3), 13C NMR (75 MHz, CDCl3) δ 170.9 (CO), 158.7 (NCO), 137.4 (Cq-Ar), 136.9 (Cq-Ar), 128.69 (2 CH-Ar), 128.66 (2 CH-Ar), 128.4 (CH-Ar), 128.3 (2 CH-Ar), 128.2 (CH-Ar), 127.8 (2 CH-Ar), 91.7 (C-2), 80.8 (C-1 or C-3), 75.8 (C-1 or C-3), 72.8 (CH2Ph), 65.8 (CH2Ph), 61.0 (CH2CH3), 42.3 (C-4), 34.7 (C-1′), 14.3 (CH2CH3), HRMS (ESI) m/z 434.154 ([M + Na]+, calcd for C23H25NO6Na: 434.157).
Ethyl 2-(N-tertbutyloxycarbonyl-(1R,5R,6S,7S)-5,6-bis(benzyloxy)-3-oxo-2-oxa-4-azabicyclo[3.2.0]heptan-7-yl)acetate (12)
Boc2O (83 mg, 0.38 mmol, 2 equiv.), Et3N (53 μL, 0.38 mmol, 2 equiv.) and DMAP (7 mg, 0.057 mmol, 0.3 equiv.) were added to a solution of 11a (78 mg, 0.19 mmol, 1 equiv.) in CH2Cl2 (5 mL). The solution was stirred for 3 h and concentrated under reduced pressure. The residue was dissolved in EtOAc (6 mL). The organic phase was washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:2) to afford the desired bicyclic compound 12 (82 mg, 84%) as a white solid.
R
f
0.56 (EtOAc/petroleum ether, 1:2), [α]20D −42 (c 1.0, CHCl3), IR (film) 1816, 1736 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.24 (m, 10H, Ph), 4.78 (d, J = 12.1 Hz, 1H, CH2Ph), 4.59 (d, J = 11.2 Hz, 1H, CH2Ph), 4.53 (d, J = 11.9 Hz, 1H, CH2Ph), 4.47 (d, J = 11.2 Hz, 1H, CH2Ph), 4.36 (m, 1H, H-1 or H-3), 4.10 (q, J = 7.1 Hz, 2H, CH2CH3), 4.09–4.02 (m, 1H, H-1 or H-3), 2.50 (m, 1H, H-1′a), 2.41–2.27 (m, 2H, H-1′b, H-4), 1.47 (s, 9H, C(CH3)3), 1.22 (t, J = 7.2 Hz, 3H, CH2CH3), 13C NMR (75 MHz, CDCl3) δ 170.7 (CO), 152.8 (NCO), 149.1 (NCO), 137.7 (Cq-Ar), 136.4 (Cq-Ar), 128.7 (2 CH-Ar), 128.5 (2 CH-Ar), 128.3 (CH-Ar), 128.0 (3 CH-Ar), 127.8 (2 CH-Ar), 94.3 (C-2), 84.2 (C(CH3)3), 80.2 (C-1 or C-3), 73.1 (CH2Ph), 72.8 (C-1 or C-3), 66.4 (CH2Ph), 61.1 (CH2CH3), 42.3 (C-4), 34.5 (C-1′), 28.0 (C(CH3)3), 14.3 (CH2CH3), HRMS (ESI) m/z 534.204 ([M + Na]+, calcd for C28H33NO8Na: 534.204).
Ethyl 2-((1R,2S,3S,4R)-2,3-bis(benzyloxy)-3-((tert-butoxycarbonyl)amino)-4-hydroxycyclobutyl)acetate (13)
Cs2CO3 (10 mg, 0.029 mmol, 0.2 equiv.) was added to a solution of 12 (83 mg, 0.16 mmol, 1 equiv.) in EtOH (6 mL). The solution was stirred for 21 h and concentrated under reduced pressure. Water (2 mL) was added and pH was adjusted to 7 with 0.1 N HCl. The product was extracted with CH2Cl2 (5×). The combined organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:1) to afford the desired cyclobutane 13 (57 mg, 72%) as a colorless oil.
R
f
0.49 (EtOAc/petroleum ether, 1:2), [α]20D −25 (c 1.0, CHCl3), IR (film) 3417, 1732, 1483 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.23 (m, 10H, Ph), 5.98 (br s, 1H, NH), 4.61 (d, J = 11.7 Hz, 1H, CH2Ph), 4.56 (d, J = 11.9 Hz, 1H, CH2Ph), 4.55 (d, J = 11.4 Hz, 1H, CH2Ph), 4.43 (d, J = 11.6 Hz, 1H, CH2Ph), 4.13 (q, J = 7.2 Hz, 2H, CH2CH3), 3.91 (d, J = 7.8 Hz, 1H, H-1 or H-3), 3.56 (d, J = 7.8 Hz, 1H, H-1 or H-3), 2.51 (dd, J = 6.9, 2.3 Hz, 2H, H-1′), 2.24 (m, 1H, H-4), 1.45 (s, 9H, C(CH3)3), 1.24 (t, J = 7.1 Hz, 3H, CH2CH3), 13C NMR (75 MHz, CDCl3) δ 172.3 (CO), 156.3 (NCO), 138.1 (Cq-Ar), 137.6 (Cq-Ar), 128.6 (2 CH-Ar), 128.5 (2 CH-Ar), 128.21 (CH-Ar), 128.17 (2 CH-Ar), 127.9 (2 CH-Ar), 127.7 (CH-Ar), 89.3 (C(CH3)3 or C-2), 80.9 (C(CH3)3 or C-2), 77.7 (C-1 or C-3), 73.6 (C-1 or C-3), 72.6 (CH2Ph), 65.7 (CH2Ph), 60.8 (CH2CH3), 41.8 (C-4), 36.2 (C-1′), 28.3 (C(CH3)3), 14.3 (CH2CH3), HRMS (ESI) m/z 508.223 ([M + Na]+, calcd for C27H35NO7Na: 508.231).
Ethyl 2-((1S,2S,3R,4R)-2,3-bis(benzyloxy)-3-((tert-butoxycarbonyl)amino)-4-(carbamoyloxy)cyclobutyl)acetate (14)
Trichloroacetyl isocyanate (35 μL, 0.29 mmol, 1.3 equiv.) was added to a solution of 13 (109 mg, 0.22 mmol, 1 equiv.) in CH2Cl2 (1 mL) cooled to 0 °C. The solution was stirred at rt for 16 h and concentrated under reduced pressure. The residue was dissolved in MeOH (0.7 mL) and K2CO3 (3 mg, 0.022 mmol, 0.1 equiv.) was added. The solution was stirred for 7.5 h. Saturated aqueous NH4Cl (2 mL) was added and the product was extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:2 to 1:1) to afford the desired carbamate 14 (101 mg, 85%) as a white solid.
R
f
0.13 (silica gel, EtOAc/petroleum ether, 1:2), [α]20D −33 (c 1.0, CHCl3), IR (film) 3423, 2931, 1724, 1158 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.37–7.17 (m, 10H, Ph), 5.69 (br s, 1H, NHBoc), 4.87 (br s, 2H, NH2), 4.78 (d, J = 7.6 Hz, 1H, H-1 or H-3), 4.65 (m, 1H, CH2Ph), 4.56–4.37 (m, 3H, CH2Ph), 4.05 (q, J = 7.1 Hz, 2H, CH2CH3), 3.58 (d, J = 7.5 Hz, 1H, H-1 or H-3), 2.59–2.44 (m, 2H, H-1′), 2.38 (m, 1H, H-4), 1.42 (s, 9H, C(CH3)3), 1.18 (t, J = 7.2 Hz, 3H, CH2CH3), 13C NMR (100 MHz, CDCl3) δ 171.5 (CO), 155.3 (2 NCO), 138.2 (Cq-Ar), 137.7 (Cq-Ar), 128.6 (2 CH-Ar), 128.4 (3 CH-Ar), 128.1 (3 CH-Ar), 127.9 (CH-Ar), 127.6 (CH-Ar), 89.3 (C-2), 79.8 (C(CH3)3), 78.5 (C-1 or C-3), 73.4 (C-1 or C-3), 73.0 (CH2Ph), 65.9 (CH2Ph), 60.8 (CH2CH3), 40.6 (C-4), 36.0 (C-1′), 28.3 (C(CH3)3), 14.3 (CH2CH3), HRMS (ESI) m/z 551.230 ([M + Na]+, calcd for C28H36N2O8Na: 551.236).
(1S,2S,3R,4R)-2,3-Bis(benzyloxy)-4-((tert-butyldimethylsilyl)oxy)cyclobutyl)methyl carbamate (16a)
Trichloroacetyl isocyanate (27 μL, 0.22 mmol, 1.3 equiv.) was added to a solution of 157a (74 mg, 0.17 mmol, 1 equiv.) in CH2Cl2 (0.5 mL) cooled to 0 °C. The solution was stirred for 15 h and concentrated under reduced pressure. The residue was dissolved in MeOH (0.42 mL) and K2CO3 (2 mg, 0.017 mmol, 0.1 equiv.) was added. The solution was stirred for 23.5 h. Saturated aqueous NH4Cl (1 mL) was added and the product was extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:3) to afford the desired carbamate 16a (76 mg, 93%) as a white solid.
R
f
0.41 (EtOAc/petroleum ether, 1:2), [α]20D −2 (c 1.0, CHCl3), IR (film) 3353, 1720, 1331 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.30–7.16 (m, 10H, Ph), 4.64 (br s, 2H, NH2), 4.55 (d, J = 11.7 Hz, 1H, CH2Ph), 4.49 (d, J = 11.7 Hz, 1H, CH2Ph), 4.48 (d, J = 11.9 Hz, 1H, CH2Ph), 4.43 (d, J = 11.7 Hz, 1H, CH2Ph), 4.09 (m, 2H, H-1′), 3.76 (t, J = 5.6 Hz, 1H, H-2), 3.59 (m, 1H, H-1 or H-3), 3.41 (m, 1H, H-1 or H-3), 1.93 (m, 1H, H-4), 0.82 (s, 9H, C(CH3)3), 0.02 (s, 3H, CH3), 0.00 (s, 3H, CH3), 13C NMR (75 MHz, CDCl3) δ 156.8 (NCO), 138.2 (Cq-Ar), 138.1 (Cq-Ar), 128.54 (2 CH-Ar), 128.52 (2 CH-Ar), 127.92 (2 CH-Ar), 127.86 (4 CH-Ar), 86.0 (C-2), 74.7 (C-1 or C-3), 71.74 (CH2Ph), 71.73 (CH2Ph), 68.4 (C-1 or C-3), 63.9 (C-1′), 44.3 (C-4), 25.8 (C(CH3)3), 18.0 (C(CH3)3), −4.5 (CH3), −4.7 (CH3), HRMS (ESI) m/z 494.237 ([M + Na]+, calcd for C26H37NO5SiNa: 494.233).
(1R,2S,3S,4R)-2,3-Bis(benzyloxy)-4-hydroxycyclobutyl)methyl carbamate (16b)
HCl (2 M, 0.5 mL, 1.0 mmol, 2 equiv.) was added to a solution of 16a (240 mg, 0.51 mmol, 1 equiv.) in MeOH (2 mL). The solution was stirred at rt for 2.5 h. NaHCO3 was added and the solution was stirred for 5 min. Water was added and the product was extracted with EtOAc (3×). The combined organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (MeOH/CH2Cl2, 5:95) to afford the desired alcohol 16b (155 mg, 85%) as a white solid.
R
f
0.16 (MeOH/CH2Cl2, 5:95), [α]20D −4 (c 1.0, CHCl3), IR (film) 3351, 1705, 1331 cm−1, 1H NMR (300 MHz, CD3OD) δ 7.39–7.23 (m, 10H, Ph), 4.65 (d, J = 11.7 Hz, 1H, CH2Ph), 4.57 (d, J = 11.7 Hz, 1H, CH2Ph), 4.56 (d, J = 11.7 Hz, 1H, CH2Ph), 4.51 (d, J = 11.9 Hz, 1H, CH2Ph), 4.12 (d, J = 5.1 Hz, 2H, H-1′), 3.78 (t, J = 5.8 Hz, 1H, H-2), 3.57 (m, 1H, H-1 or H-3), 3.49 (m, 1H, H-1 or H-3), 1.92 (m, 1H, H-4), 13C NMR (75 MHz, CD3OD) δ 159.8 (NCO), 139.6 (Cq-Ar), 139.4 (Cq-Ar), 129.36 (2 CH-Ar), 129.34 (2 CH-Ar), 129.0 (4 CH-Ar), 128.74 (CH-Ar), 128.70 (CH-Ar), 86.6 (C-2), 75.7 (C-1 or C-3), 72.6 (CH2Ph), 72.2 (CH2Ph), 68.9 (C-1 or C-3), 64.4 (C-1′), 45.5 (C-4), HRMS (ESI) m/z 380.145 ([M + Na]+, calcd for C20H23NO5Na: 380.147).
(1R,2R,3S,4S)-2,3-Bis(benzyloxy)-4-((carbamoyloxy)methyl)cyclobutyl acetate (16c)
Ac2O (1 mL) was added to a solution of 16b (68 mg, 0.19 mmol, 1 equiv.) in pyridine (5 mL). The solution was stirred at rt for 15 h. Water was added and the product was extracted with EtOAc (1×). The organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:1) to afford the desired ester 16c (71 mg, 93%) as a white solid.
R
f
0.17 (EtOAc/petroleum ether, 1:1), [α]20D = −5 (c 1.0, CHCl3), IR (film) 3363, 2923, 1738, 1238 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.41–7.24 (m, 10H, Ph), 4.71 (br s, 2H, NH2), 4.63–4.55 (m, 4H, H-1 or H-3, CH2Ph), 4.52 (d, J = 11.7 Hz, 1H, CH2Ph), 4.28 (dd, J = 11.4, 4.5 Hz, 1H, H-1′a), 4.18 (dd, J = 11.4, 5.5 Hz, 1H, H-1′b), 3.97 (t, J = 5.8 Hz, 1H, H-2), 3.66 (m, 1H, H-1 or H-3), 2.13 (m, 1H, H-4), 2.03 (s, 3H, CH3), 13C NMR (75 MHz, CDCl3) δ 170.3 (CO), 156.7 (NCO), 138.0 (Cq-Ar), 137.8 (Cq-Ar), 128.59 (2 CH-Ar), 128.57 (2 CH-Ar), 128.1 (2 CH-Ar), 128.01 (CH-Ar), 127.98 (CH-Ar), 127.9 (2 CH-Ar), 82.7 (C-2), 74.7 (C-1 or C-3), 72.0 (CH2Ph), 71.8 (CH2Ph), 69.2 (C-1 or C-3), 63.6 (C-1′), 42.6 (C-4), 21.0 (CH3), HRMS (ESI) m/z 422.153 ([M + Na]+, calcd for C22H25NO6Na: 422.157).
(1S*,2R*,4S*)-2-Methyl-2-phenyl-4-vinylcyclobutyl carbamate (19)
Trichloroacetyl isocyanate (0.11 mL, 0.94 mmol, 1.3 equiv.) was added to a solution of 1818 (135 mg, 0.72 mmol, 1 equiv.) in CH2Cl2 (2 mL) cooled to 0 °C. The solution was stirred at rt for 15 h and concentrated under reduced pressure. The residue was dissolved in MeOH (1.8 mL) and K2CO3 (10 mg, 0.072 mmol, 0.1 equiv.) was added. The solution was stirred for 8 h. Saturated aqueous NH4Cl (2 mL) was added and the product was extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:5 to 1:3) to afford the desired carbamate 19 (169 mg, 96%) as a white solid.
R
f
0.44 (EtOAc/toluene, 2:8), IR (neat) 3354, 1715, cm−1, 1H NMR (400 MHz, CDCl3) δ 7.37–7.27 (m, 2H, Ph), 7.24–7.14 (m, 3H, Ph), 5.99 (ddd, J = 17.2 Hz, 10.3, 6.6 Hz, 1H, H-1′), 5.15 (d, J = 8.0 Hz, 1H, H-3), 5.08 (dt, J = 17.5, 1.5 Hz, 1H, H-2′a), 5.02 (dt, J = 10.4, 1.4 Hz, 1H, H-2′b), 4.92 (br s, 2H, NH2), 3.14–3.01 (m, 1H, H-4), 2.20 (t, J = 10.4 Hz, 1H, H-1a), 1.84 (t, J = 10.3 Hz, 1H, H-1b), 1.46 (s, 3H, CH3), 13C NMR (100 MHz, CDCl3) δ 156.3 (NCO), 149.8 (Cq-Ar), 139.0 (C-1′), 128.5 (2 CH-Ar), 126.0 (CH-Ar), 125.1 (2 CH-Ar), 114.6 (C-2′), 78.0 (C-3), 45.7 (C-2), 42.7 (C-4), 32.8 (C-1), 24.5 (CH3), HRMS (ESI) m/z 254.116 ([M + Na]+, calcd for C14H17NO2Na: 254.115).
(1R*,5R*,7R*)-7-Methyl-7-phenyl-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptan-3-one (20) (Table 2)
According to general procedure A, 20 was obtained as a white solid after flash chromatography (EtOAc/petroleum ether, 1:9 to 1:3).
R
f
0.27 (EtOAc/ toluene, 2:8), IR (film) 3281, 1750 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.44–7.30 (m, 2H, Ph), 7.29–7.15 (m, 3H, Ph), 6.49 (br s, 1H, NH), 5.90 (dd, J = 17.2 Hz, 10.6 Hz, 1H, H-1′), 5.26 (d, J = 17.5 Hz, 1H, H-2′a), 5.21 (d, J = 10.7 Hz, 1H, H-2′b), 4.97 (s, 1H, H-3), 2.81 (d, J = 12.9 Hz, 1H, H-1a), 2.48 (d, J = 12.9 Hz, 1H, H-1b), 1.55 (s, 3H, CH3), 13C NMR (75 MHz, CDCl3) δ 160.8 (NCO), 148.3 (Cq-Ar), 137.8 (C-1′), 128.9 (2 CH-Ar), 126.5 (CH-Ar), 125.3 (2 CH-Ar), 116.3 (C-2′), 86.5 (C-3), 59.7 (C-4), 45.5 (C-2), 43.6 (C-1), 26.3 (CH3), HRMS (ESI) m/z 252.100 ([M + Na]+, calcd for C14H15NO2Na: 252.099).
(1S*,2R*,4S*)-2-Methyl-2-phenyl-4-vinylcyclobutyl tosylcarbamate (21)
p-Toluenesulfonyl isocyanate (184 μL, 1.2 mmol, 1 equiv.) was added dropwise to a solution of 1818 (227 mg, 1.2 mmol, 1 equiv.) in THF (3.2 mL) cooled to 0 °C. The solution was stirred at 0 °C for 10 min and then allowed to warm to rt. The reaction mixture was stirred for 1.5 h at rt and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 2:8 to 2:3). Traces of p-toluenesulfonyl isocyanate residues were still present. The residue was dissolved in 150 mL of cyclohexane and then 150 mL of H2O were added. The solution was stirred at rt overnight. The two layers were separated and the organic layer was dried over MgSO4, filtered and concentrated under reduced pressure to afford the desired N-tosylcarbamate 21 (405 mg, 87%) as a white solid.
R
f
0.25 (EtOAc/petroleum ether, 2:8), IR (film) 3241, 1740 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.95 (d, J = 8.1 Hz, 2H, Ar), 7.36 (d, J = 8.2 Hz, 2H, Ar), 7.30–7.21 (m, 2H, Ar), 7.21–7.12 (m, 1H, Ar), 7.09–7.01 (m, 2H, Ar), 5.96–5.80 (m, 1H, H-1′), 5.04 (d, J = 7.9 Hz, 1H, H-3), 5.00–4.90 (m, 2H, H-2′), 3.08–2.92 (m, 1H, H-4), 2.46 (s, 3H, CH3-Ph), 2.19 (t, J = 10.3 Hz, 1H, H-1a), 1.82 (t, J = 10.3 Hz, 1H, H-1b), 1.38 (s, 3H, CH3-Ph), 13C NMR (100 MHz, CDCl3) δ 149.7 (NCO or Cq-Ar), 148.9 (NCO or Cq-Ar), 145.2 (NCO or Cq-Ar) 138.2 (C-1′), 135.8 (Cq-Ar), 129.8 (2 CH-Ar), 128.6 (2 CH-Ar), 128.4 (2 CH-Ar), 126.2 (CH-Ar), 124.9 (2 CH-Ar), 115.2 (C-2′), 80.4 (C-3), 45.6 (C-2), 42.3 (C-4), 32.5 (C-1), 24.6 (CH3), 21.8 (CH3), HRMS (ESI) m/z 408.126 ([M + Na]+, calcd for C21H23NO4SNa: 408.124).
(1R*,5R*,7R*)-7-Methyl-7-phenyl-4-tosyl-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptan-3-one (22)
Phenyl-benzoquinone30 (48.3 mg, 0.26 mmol, 1.05 equiv.) and 1,2-bis(phenylsulfinyl)ethane palladium(II) acetate (12.6 mg, 0.025 mmol, 0.1 equiv.) were added to a solution of N-tosylcarbamate 21 (96.4 mg, 0.25 mmol, 1 equiv.) in degassed THF. The flask was sealed and the reaction mixture was stirred 66 h at 45 °C. The reaction mixture was diluted with CH2Cl2. The organic layer was washed with saturated aqueous NH4Cl and then brine. The aqueous layer was extracted (4×) with CH2Cl2. The combined organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:9 to 15:85) to afford the desired product 21 (14 mg) contaminated with phenyl-benzoquinone. The yield of 21 (9%) was estimated by 1H NMR analysis of the fraction containing 21 and PhBQ.
R
f
0.5 (EtOAc/petroleum ether, 1:9) IR (film) 1785 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H, Ar), 7.40–7.30 (m, 4H, Ar), 7.25–7.20 (m, 1H, Ar), 7.17 (d, J = 7.7 Hz, 2H, Ar), 6.04 (dd, J = 17.3, 10.6 Hz, 1H, H-1′), 5.39 (d, J = 10.2 Hz, 1H, H-2a′), 5.36 (d, J = 16.8 Hz, 1H, H-2a′), 4.87 (s, 1H, H-3), 3.00 (s, 2H, H-1), 2.45 (s, 3H, CH3-Ph), 1.47 (s, 3H, CH3-Ph), 13C NMR (100 MHz, CDCl3) δ 153.3 (NCO), 147.6 (Cq-Ar), 145.7 (Cq-Ar), 135.8 (C-1′), 135.6 (Cq-Ar), 130.3 (2 CH-Ar), 129.05 (2 CH-Ar), 129.0 (2 CH-Ar), 126.8 (CH-Ar), 125.1 (2 CH-Ar), 119.0 (C-2′), 84.1 (C-3), 65.6 (C-4), 45.3 (C-2), 40.9 (C-1), 26.3 (CH3), 21.9 (CH3), HRMS (m/z 406.109 ([M + Na]+, calcd for C21H21NO4SNa: 406.108).
(1R,2R,3S,4S)-2,3-Bis(benzyloxy)-4-vinylcyclobutyl carbamate (23a)
Trichloroacetyl isocyanate (0.20 mL, 1.72 mmol, 1.3 equiv.) was added to a solution of 177a,8 (403 mg, 1.3 mmol, 1 equiv.) in CH2Cl2 (3.7 mL) at 0 °C. The solution was stirred at rt for 16 h and concentrated under reduced pressure. The residue was dissolved in MeOH (3.2 mL) and K2CO3 (18 mg, 0.13 mmol, 0.1 equiv.) was added. The solution was stirred at rt for 8 h. Saturated aqueous NH4Cl (5 mL) was added and the product was extracted with CH2Cl2 (3×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:3 to 1:1) to afford carbamate 23a (431 mg, 94%) as a white solid.
R
f
0.46 (EtOAc/petroleum ether, 1:2), [α]20D +3 (c 1.0, CHCl3), IR (film) 3354, 1718, 1326, 1087 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.41–7.20 (m, 10H, Ph), 5.95 (ddd, J = 17.3, 10.2, 7.2 Hz, 1H, H-1′), 5.19 (d, J = 17.3 Hz, 1H, H-2′a), 5.09 (d, J = 10.2 Hz, 1H, H-2′b), 4.72–4.42 (m, 7H, 2 CH2Ph, NH2, H-1 or H-3), 3.95 (t, J = 5.9 Hz, 1H, H-2), 3.60 (m, 1H, H-1 or H-3), 2.48 (q, J = 7.5 Hz, 1H, H-4), 13C NMR (75 MHz, CDCl3) δ 156.0 (NCO), 137.84 (Cq-Ar), 137.82 (Cq-Ar), 136.3 (C-1′), 128.52 (2 CH-Ar), 128.50 (2 CH-Ar), 128.0 (2 CH-Ar), 127.9 (4 CH-Ar), 116.4 (C-2′), 82.6 (C-2), 77.1 (C-1 or C-3), 72.4 (C-1 or C-3), 71.62 (CH2Ph), 71.57 (CH2Ph), 46.3 (C-4), HRMS (ESI) m/z 376.148 ([M + Na]+, calcd for C21H23NO4Na: 376.152).
Debenzylation of 23a
BCl3 (1 M in CH2Cl2, 14 mL, 14 mmol, 12 equiv.) was added to a solution of carbamate 23a (411 mg, 1.16 mmol, 1 equiv.) in CH2Cl2 (12 mL) at −60 °C. The solution was allowed to warm slowly to rt overnight. MeOH/H2O (20:1, 50 mL) was added and the solution was concentrated under reduced pressure. The process was repeated once. The crude product was purified by flash chromatography (MeOH/CH2Cl2, 10:90 to 15:85) to afford (1R,2R,3S,4S)-2,3-dihydroxy-4-vinylcyclobutyl carbamate (169 mg, 84%) as a white solid.8
(1S,2R,3R,4S)-3-(Carbamoyloxy)-4-vinylcyclobutane-1,2-diyl diacetate (23b)
Ac2O (0.31 mL, 3.30 mmol, 6 equiv.) and DMAP (136 mg, 1.11 mmol, 2 equiv.) were added to a solution of (1R,2R,3S,4S)-2,3-dihydroxy-4-vinylcyclobutyl carbamate8 (96 mg, 0.55 mmol, 1 equiv., obtained by debenzylation of 23a) in pyridine (5.5 mL). The solution was stirred at rt for 17 h. Et2O (38 mL) was added and the organic layer was washed successively with 1% aqueous HCl (22 mL), saturated aqueous NaHCO3 (38 mL) and brine (38 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:2 to 1:1) to afford 23b (115 mg, 81%) as a colorless oil.
R
f
0.33 (EtOAc/petroleum ether, 1:1), [α]20D −9 (c 1.0, CHCl3), IR (film) 3374, 1728, 1220 cm−1, 1H NMR (400 MHz, CDCl3) δ 5.95 (ddd, J = 17.2, 10.4, 6.7 Hz, 1H, H-1′), 5.22 (d, J = 17.2 Hz, 1H, H-2′a), 5.15 (d, J = 10.4 Hz, 1H, H-2′b), 5.09 (t, J = 6.3 Hz, 1H, H-2), 4.99–4.86 (br s, 2H, NH2), 4.73 (m, 1H, H-1 or H-3), 4.68 (m, 1H, H-1 or H-3), 2.65 (q, J = 7.5 Hz, 1H, H-4), 2.06 (s, 6H, CH3), 13C NMR (100 MHz, CDCl3) δ 170.13 (CO), 170.10 (CO), 155.6 (NCO), 134.2 (C-1′), 117.3 (C-2′), 75.2 (C-2), 70.7 (C-1 or C-3), 69.9 (C-1 or C-3), 45.1 (C-4), 20.84 (CH3), 20.80 (CH3), HRMS (ESI) m/z 280.078 ([M + Na]+, calcd for C11H15NO6Na: 280.079).
(1S,2R,3R,4S)-3-(Carbamoyloxy)-4-vinylcyclobutane-1,2-diyl dibenzoate (23c)
BzCl (0.19 mL, 1.64 mmol, 4.6 equiv.) was added to a solution of (1R,2R,3S,4S)-2,3-dihydroxy-4-vinylcyclobutyl carbamate8 (62 mg, 0.36 mmol, 1 equiv., obtained by debenzylation of 23a) in pyridine (3.6 mL) at 0 °C. The solution was stirred at 0 °C for 1 h. MeOH was added and the solution was diluted in EtOAc. The organic layer was washed with water and 1 N HCl, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:2 to 1:1) to afford cyclobutane 23c (118 mg, 87%) as a white solid.
R
f 0.25 (silica gel, EtOAc/petroleum ether, 1:2), [α]20D +48 (c 1.0, CHCl3), IR (film) 3374, 1721, 1275 cm−1, 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 8.1 Hz, 4H, Ph), 7.62–7.53 (m, 2H, Ph), 7.49–7.40 (m, 4H, Ph), 6.12 (ddd, J = 17.2, 10.5, 6.5 Hz, 1H, H-1′), 5.53 (t, J = 6.3 Hz, 1H, H-2), 5.34 (dt, J = 17.2, 1.7 Hz, 1H, H-2′a), 5.23 (dt, J = 10.4, 1.2 Hz, 1H, H-2′b), 5.16 (dd, J = 7.9, 6.3 Hz, 1H, H-1 or H-3), 4.96 (dd, J = 8.1, 6.3 Hz, 1H, H-1 or H-3), 4.76 (br s, 2H, NH2), 2.90 (q, J = 7.6 Hz, 1H, H-4), 13C NMR (75 MHz, CDCl3) δ 165.72 (CO), 165.67 (CO), 155.4 (NCO), 134.3 (C-1′), 133.5 (2 CH-Ar), 130.1 (3 CH-Ar), 130.0 (3 CH-Ar), 129.5 (Cq-Ar), 129.4 (Cq-Ar), 128.55 (CH-Ar), 128.54 (CH-Ar), 117.5 (C-2′), 75.6 (C-2), 71.0 (C-1 or C-3), 70.6 (C-1 or C-3), 45.8 (C-4), HRMS (ESI) m/z 404.109 ([M + Na]+, calcd for C21H19NO6Na: 404.110).
(1S,2R,3R,4S)-3-(Carbamoyloxy)-4-vinylcyclobutane-1,2-diyl bis(4-methoxybenzoate) (23d)
p-Anisoyl chloride (0.22 mL, 1.59 mmol, 4.6 equiv.) was added to a solution of (1R,2R,3S,4S)-2,3-dihydroxy-4-vinylcyclobutyl carbamate8 (60 mg, 0.35 mmol, 1 equiv., obtained by debenzylation of 23a) in pyridine (3.5 mL) cooled at 0 °C. The solution was stirred at 0 °C for 2 h. The reaction mixture was quenched with ice and diluted with Et2O. The product was extracted with Et2O (4×). The combined organic layer was washed with 1 N HCl until pH 5, water, saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 2:3) to afford the desired cyclobutane 23d (110 mg, 72%) as a white solid.
R
f
0.19 (EtOAc/petroleum ether, 2:3), [α]20D +85 (c 1.0, CHCl3), IR (film) 1712, 1604, 1251, 1167, 1095 cm−1, 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.3 Hz, 4H, Ph), 6.95–6.87 (m, 4H, Ph), 6.11 (ddd, J = 17.1, 10.5, 6.5 Hz, 1H, H-1′), 5.49 (t, J = 6.3 Hz, 1H, H-2), 5.33 (d, J = 17.2 Hz, 1H, H-2′a), 5.22 (d, J = 10.4 Hz, 1H, H-2′b), 5.12 (m, 1H, H-1 or H-3), 4.93 (m, 1H, H-1 or H-3), 4.80 (br s, 2H, NH2), 3.86 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 2.85 (m, 1H, H-4), 13C NMR (100 MHz, CDCl3) δ 165.42 (Cq-OCH3 or CO), 165.36 (Cq-OCH3 or CO), 163.8 (2 Cq-OCH3 or CO), 155.5 (NCO), 134.4 (C-1′), 132.2 (2 CH-Ar), 132.1 (2 CH-Ar), 121.83 (Cq-Ar), 121.77 (Cq-Ar), 117.3 (C-2′), 113.80 (2 CH-Ar), 113.78 (2 CH-Ar), 75.3 (C-1 or C-2 or C-3), 71.1 (C-1 or C-2 or C-3), 70.3 (C-1 or C-2 or C-3), 55.59 (OCH3), 55.58 (OCH3), 45.9 (C-4), HRMS (ESI) m/z 464.130 ([M + Na]+, calcd for C23H23NO8Na: 464.132).
(1R,5R,6S,7R)-6,7-Bis(benzyloxy)-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptan-3-one (24a) and (1R,5R,6S,7S)-5,6-bis(benzyloxy)-7-vinyl-2-oxa-4-azabicyclo[3.2.0]heptan-3-one (25a) (Table 3, entry 1)
According to general procedure A, compounds 25a and 24a were obtained as pale yellow oils after flash chromatography (EtOAc/petroleum ether, 1:5 to 1:2).
:3), [α]20D −7 (c 0.6, CHCl3), IR (film) 3272, 1760 cm−1, 1H NMR (400 MHz, CDCl3) δ 7.41–7.26 (m, 10H, Ph), 5.86 (m, 1H, H-1′), 5.57 (br s, 1H, NH), 5.17 (d, J = 1.0 Hz, 1H, H-2′a), 5.14 (d, J = 6.6 Hz, 1H, H-2′b), 4.63 (d, J = 11.9 Hz, 1H, CH2Ph), 4.57 (d, J = 11.5 Hz, 1H, CH2Ph), 4.50 (d, J = 11.9 Hz, 1H, CH2Ph), 4.44 (d, J = 11.5 Hz, 1H, CH2Ph), 4.30 (d, J = 4.7 Hz, 1H, H-1 or H-3), 4.02 (dd, J = 7.2, 1.0 Hz, 1H, H-1 or H-3), 2.68 (m, 1H, H-4), 13C NMR (100 MHz, CDCl3) δ 158.2 (NCO), 137.2 (Cq-Ar), 136.8 (Cq-Ar), 134.3 (C-1′), 128.76 (2 CH-Ar), 128.72 (2 CH-Ar), 128.4 (CH-Ar), 128.3 (CH-Ar), 128.2 (2 CH-Ar), 127.9 (2 CH-Ar), 117.4 (C-2′), 91.1 (C-2), 81.0 (C-1 or C-3), 76.4 (C-1 or C-3), 72.5 (CH2Ph), 65.9 (CH2Ph), 49.0 (C-4), HRMS (ESI) m/z 374.136 ([M + Na]+, calcd for C21H21NO4Na: 374.136).
:2), [α]20D +54 (c 1.0, CHCl3), IR (film) 3279, 1755 cm−1, 1H NMR (400 MHz, CDCl3) δ 7.40–7.24 (m, 10H, Ph), 5.81 (dd, J = 17.3, 10.6 Hz, 1H, H-1′), 5.71 (br s, 1H, NH), 5.24 (d, J = 17.6 Hz, 1H, H-2′a), 5.22 (d, J = 10.6 Hz, 1H, H-2′b), 4.57 (d, J = 11.5 Hz, 1H, CH2Ph), 4.56 (d, J = 11.9 Hz, 1H, CH2Ph), 4.51 (d, J = 11.8 Hz, 1H, CH2Ph), 4.47 (d, J = 12.0 Hz, 1H, CH2Ph), 4.32 (d, J = 2.1 Hz, 1H, H-1 or H-3), 4.11 (dd, J = 5.3, 3.5 Hz, 1H, H-2), 3.96 (d, J = 5.5 Hz, 1H, H-1 or H-3), 13C NMR (100 MHz, CDCl3) δ 159.2 (NCO), 137.1 (Cq-Ar), 136.9 (Cq-Ar), 136.2 (C-1′), 128.74 (2 CH-Ar), 128.73 (2 CH-Ar), 128.4 (CH-Ar), 128.3 (CH-Ar), 128.24 (2 CH-Ar), 128.21 (2 CH-Ar), 116.7 (C-2′), 84.2 (C-2), 81.3 (C-1 or C-3), 78.9 (C-1 or C-3), 72.22 (CH2Ph), 72.21 (CH2Ph), 62.1 (C-4), HRMS (ESI) m/z 374.148 ([M + Na]+, calcd for C21H21NO4Na: 374.136).
(1R,5R,6S,7R)-3-Oxo-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptane-6,7-diyl diacetate (24b) (Table 3, entry 3)
According to general procedure A, compound 24b was obtained as a colorless oil after purification by flash chromatography (EtOAc/petroleum ether, 1:5 to 2:1).
R
f
0.46 (Et2O/CH2Cl2, 1:1), [α]20D +57 (c 1.0, CHCl3), IR (film) 3293, 1747, 1220 cm−1, 1H NMR (300 MHz, CDCl3) δ 6.03 (dd, J = 17.3, 10.6 Hz, 1H, H-1′), 5.99 (s, 1H, NH), 5.43 (d, J = 14.4 Hz, 1H, H-2′a), 5.38 (d, J = 7.6 Hz, 1H, H-2′b), 5.15 (dd, J = 5.8, 3.5 Hz, 1H, H-2), 5.08 (dd, J = 5.8, 1.7 Hz, 1H, H-1), 4.62 (dd, J = 3.5, 1.7 Hz, 1H, H-3), 2.14 (s, 3H, CH3), 2.12 (s, 3H, CH3), 13C NMR (75 MHz, CDCl3) δ 170.0 (CO), 169.4 (CO), 158.8 (NCO), 134.9 (C-1′), 118.1 (C-2′), 78.4 (C-3), 76.9 (C-2), 74.4 (C-1), 62.6 (C-4), 20.7 (CH3), 20.6 (CH3), HRMS (ESI) m/z 278.063 ([M + Na]+, calcd for C11H13NO6Na: 278.064).
(1R,5R,6S,7R)-3-Oxo-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptane-6,7-diyl dibenzoate (24c) (Table 3, entries 5 and 6)
According to general procedure A, compound 24c was obtained as a colorless oil after purification by flash chromatography (EtOAc/toluene, 1:7 to 1:6).
R
f
0.48 (EtOAc/toluene, 1:3), [α]20D +97 (c 1.0, CHCl3), IR (film) 3321, 1762, 1722, 1248 cm−1, 1H NMR (300 MHz, CDCl3) δ 8.12–8.04 (m, 4H, Ph), 7.65–7.56 (m, 2H, Ph), 7.51–7.43 (m, 4H, Ph), 6.20 (dd, J = 17.3, 10.7 Hz, 1H, H-1′), 5.96–5.90 (br s, 1H, NH), 5.54 (d, J = 17.3 Hz, 1H, H-2′a), 5.50–5.43 (m, 3H, H-1, H-2, H-2′b), 4.85 (dd, J = 2.8, 2.0 Hz, 1H, H-3), 13C NMR (75 MHz, CDCl3) δ 165.6 (CO), 165.2 (CO), 158.8 (NCO), 135.0 (C-1′), 134.0 (CH-Ar), 133.9 (CH-Ar), 130.2 (2 CH-Ar), 130.1 (2 CH-Ar), 128.82 (Cq-Ar), 128.78 (2 CH-Ar), 128.7 (2 CH-Ar), 128.6 (Cq-Ar), 118.2 (C-2′), 78.7 (C-3), 77.6 (C-1 or C-2), 75.1 (C-1 or C-2), 63.0 (C-4), HRMS (ESI) m/z 402.094 ([M + Na]+, calcd for C21H17NO7Na: 402.095).
(1R,5R,6S,7R)-3-Oxo-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptane-6,7-diyl bis(4-methoxybenzoate) (24d) (Table 1, entry 4)
According to general procedure A, compound 24d was obtained as a colorless oil after purification by flash chromatography (EtOAc/toluene, 1:4).
R
f
0.27 (EtOAc/toluene, 1:4), [α]20D = +144 (c 1.0, CHCl3), IR (film) 3326, 1761, 1716, 1605, 1250, 1168, 1100 cm−1, 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 6.7 Hz, 2H, Ph), 8.01 (d, J = 6.6 Hz, 2H, Ph), 6.93 (d, J = 6.6 Hz, 4H, Ph), 6.19 (dd, J = 17.3, 10.7 Hz, 1H, H-1′), 5.88 (br s, 1H, NH), 5.53 (d, J = 17.2 Hz, 1H, H-2′a), 5.47–5.38 (m, 3H, H-1 or H-3, H-2, H-2′b), 4.82 (dd, J = 3.1, 1.5 Hz, 1H, H-1 or H-3), 3.87 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 13C NMR (100 MHz, CDCl3) δ 165.3 (Cq-OCH3 or CO), 164.9 (Cq-OCH3 or CO), 164.2 (Cq-OCH3 or CO), 164.1 (Cq-OCH3 or CO), 158.8 (NCO), 135.2 (C-1′), 132.3 (2 CH-Ar), 132.2 (2 CH-Ar), 121.1 (Cq-Ar), 120.8 (Cq-Ar), 118.0 (C-2′), 114.04 (2 CH-Ar), 113.96 (2 CH-Ar), 78.8 (C-1 or C-2 or C-3), 77.4 (C-1 or C-2 or C-3), 75.0 (C-1 or C-2 or C-3), 63.0 (C-4), 55.65 (OCH3), 55.64 (OCH3), HRMS (ESI) m/z 462.110 ([M + Na]+, calcd for C23H21NO8Na: 462.116).
(1R*,5R*,7R*)-4-Allyl-7-methyl-7-phenyl-5-vinyl-2-oxa-4-azabicyclo[3.2.0]heptan-3-one (26)
NaH (60% in oil, 11.5 mg, 0.29 mmol, 1.2 equiv.) was added to a solution of carbamate 20 (55 mg, 0.24 mmol, 1 equiv.) in DMF (0.9 mL) cooled at 0 °C. The solution was stirred for 30 min at 0 °C and then 1 h at rt. An additional portion of DMF (0.4 mL) was added and allyl bromide (42 μL, 0.48 mmol, 2 equiv.) was added and the solution was stirred at rt for 2.5 h. Saturated aqueous NH4Cl was added, the solution was diluted with Et2O and the product was extracted with Et2O (4×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 8:92 to 10:90) to afford diene 26 (60 mg, 93%) as a colorless oil.
R
f
0.65 (EtOAc/petroleum ether, 3:7), IR (film) 1748 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.40–7.30 (m, 2H, Ph), 7.35–7.15 (m, 3H, Ph), 6.01–5.85 (m, 1H, H-2′), 5.83 (dd, J = 17.3, 10.6 Hz, 1H, H-1′′), 5.34 (d, J = 10.6 Hz, 1H, H-2′′a), 5.31–5.15 (m, 3H, H-2′′b, H-3′), 4.84 (s, 1H, H-3), 4.07 (ddt, J = 15.5, 5.6, 1.4 Hz, 1H, H-1′a), 3.70 (dd, J = 15.4, 7.6, 1H, H-1′b), 2.82 (d, J = 12.9 Hz, 1H, H-1a) 2.47 (d, J = 13.0 Hz, 1H, H-1b), 1.49 (s, 3H, CH3), 13C NMR (100 MHz, CDCl3) δ 157.8 (NCO), 147.4 (Cq-Ar), 135.7 (C-1′′), 132.7 (C-2′), 127.9 (2 CH-Ar), 125.5 (CH-Ar), 124.3 (2 CH-Ar), 118.0 (C-3′ or C-2′′), 117.7 (C-3′ or C-2′′), 82.7 (C-3), 62.2 (C-4), 44.1 (C-2), 43.5 (C-1′), 39.1 (C-1), 25.3 (CH3), HRMSm/z 292.129 ([M + Na]+, calcd for C17H19NO2Na: 292.131).
(2R*,2aR*,8aR*)-2-Methyl-2-phenyl-2,2a-dihydro-1H-cyclobuta[d]pyrrolo[1,2-c]oxazol-4(6H)-one (27)
A solution of Grubbs II catalyst (7.9 mg, 93 μmol, 0.05 equiv.) in degassed CH2Cl2 (0.5 mL) was added to a solution of diene 26 (50 mg, 0.186 mmol, 1 equiv.) in degassed CH2Cl2 (2.5 mL). The solution was refluxed for 7 h. After cooling, the solution was concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:4) to afford compound 27 (42 mg, 94%) as a light brown cream solid.
R
f
0.32 (EtOAc/petroleum ether, 1:4),IR (film), 1756, 1343, 1044, 703 cm−1, 1H NMR (300 MHz, CDCl3) δ 7.43–7.34 (m, 2H, Ph), 7.29–7.21 (m, 3H, Ph), 5.95 (dt, J = 6.1, 1.7 Hz, 1H, H-3′), 5.80 (dt, J = 6.0, 2.3 Hz, 1H, H-2′), 5.26 (s, 1H, H-3), 4.41 (dt, J = 16.0, 2.1 Hz, 1H, H-1′a), 3.99–3.88 (m, 1H, H-1′b), 2.64 (d, J = 13.0 Hz, 1H, H-1a) 2.45 (d, J = 13.0 Hz, 1H, H-1b), 1.50 (s, 3H, CH3), 13C NMR (100 MHz, CDCl3) δ 164.9 (NCO), 148.5 (Cq-Ar), 129.9 (C-3′), 129.7 (C-2′), 128.9 (2 CH-Ar), 126.5 (CH-Ar), 125.4 (2 CH-Ar), 86.5 (C-3), 74.3 (C-4), 54.6 (C-1′), 45.6 (C-2), 42.9 (C-1), 25.5 (CH3), HRMSm/z 264.096 ([M + Na]+, calcd for C15H15NO2Na: 264.099).
(1S,2R,2aR,8aR)-4-oxo-2,2a,4,6-tetrahydro-1H-cyclobuta[d]pyrrolo[1,2-c]oxazole-1,2-diyl dibenzoate (28)
NaH (60%, 17 mg, 0.43 mmol, 1.05 equiv.) was added to a solution of compound 24c (156 mg, 0.41 mmol 1 equiv.) in DMF (1.6 mL) at 0 °C. The solution was stirred for 30 min at 0 °C and then 1 h at rt. Allyl bromide (72 μL, 0.82 mmol, 2 equiv.) was added and the solution was stirred at rt for 2 h. Saturated aqueous NH4Cl was added and the product was extracted with Et2O (7×). The combined organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 1:4) to afford the corresponding diene (96 mg, 55%) as a colorless oil.8 A solution of Grubbs II catalyst (5 mg, 57 μmol, 0.05 equiv.) in degassed CH2Cl2 (1 mL) was added to a solution of the diene (48 mg, 0.11 mmol, 1 equiv.) in degassed CH2Cl2 (3.6 mL). The solution was refluxed for 5 h. After cooling, the solution was concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/petroleum ether, 3:7 to 2:3) to afford compound 28 (40 mg, 89%) as a cream solid.8
R
f
0.17 (EtOAc/petroleum ether, 1:4), [α]20D +127 (c 1.0, CHCl3), IR (film) 1765, 1721, 1247, 1066 cm−1, 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 7.2 Hz, 2H, Ph), 8.04 (d, J = 7.2 Hz, 2H, Ph), 7.64–7.56 (m, 2H, Ph), 7.51–7.42 (m, 4H, Ph), 6.17–6.09 (m, 2H, H-2′, H-3′), 5.61(dd, J = 6.2, 1.4 Hz, 1H, CH–O), 5.47 (dd, J = 6.1, 3.4 Hz, 1H, CH–O), 5.15 (dd, J = 3.2, 1.5 Hz, 1H, CH–O), 4.47 (d, J = 15.9 Hz, 1H, H-1′a), 3.85 (d, J = 16.2 Hz, 1H, H-1′b), 13C NMR (100 MHz, CDCl3) δ 165.5 (CO), 165.3 (CO), 163.4 (NCO), 133.9 (2 CH-Ar), 132.8 (C-2′ or C-3′), 130.11 (2 CH-Ar), 130.10 (2 CH-Ar), 128.9 (2 Cq-Ar), 128.74 (2 CH-Ar), 128.71 (2 CH-Ar), 126.8 (C-2′ or C-3′), 78.5 (CH–O), 77.9 (CH–O), 77.1 (C-4), 74.4 (CH–O), 55.7 (C-1), HRMS (ESI) m/z 414.093 ([M + Na]+, calcd for C22H17NO6Na: 414.095).
Iodide efflux
The F508del-CFTR Cl− channel activity was assayed by measuring the rate of iodide (125I) efflux from HeLa cells stably transfected with F508del-CFTR as previously described.31 Time-dependent rates of 125I efflux were calculated from the following: ln(125I t1/125It2)/(t1 – t2), where 125It is the intracellular 125I at time t, and t1 and t2 successive time points. Curves were constructed by plotting the rates of 125I versus time. All comparisons were based on maximal values for the time-dependent rates (k = peak rates, min−1), excluding the points used to establish the baseline (k peak–k basal, min−1) (for more details see ref. 24).Enzymatic inhibition assay
The enzyme used was Cerezyme™, the recombinant enzyme of the β-glucocerebrosidase commercialized by Genzyme. The enzyme activity was measured in a 96-well plate: 13 μL of BTT buffer (pH 5.2), 2 μL of inhibitor solution in DMSO and 25 μL of enzyme solution in BTT buffer (pH 5.2, 0.1 mg mL−1) containing 0.2% sodium taurocholate and 0.1% TX-100 were added. The plate was incubated at 37 °C for 30 min. Then 60 μL of substrate solution of 4-methylumbelliferyl-β-D-glucoside in McIlvaine buffer (pH 5.2, 4 mM) were added and the plate was incubated at 37 °C for 10 min. The reaction was stopped by the addition of glycine/NaOH buffer (pH 10.6, 100 mM, 150 μL), and the fluorescence of the released 4-methylumbelliferone was measured by the use of an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Inhibition constants were generated for Cerezyme™ using 2.4 mM substrate concentration for IC50 determinations. The percentage of inhibition was plotted as a function of the logarithm of the inhibitor concentration. By means of a linear regression, the IC50 value for each compound was calculated from the value of the log of the inhibitor concentration corresponding to an inhibition of 50% of the enzyme activity.Acknowledgements
The authors are grateful for the financial support from the Institut Universitaire de France (IUF), the CNRS (UMR 7509), the University of Strasbourg, the association Vaincre La Mucoviscidose and the International Centre for Frontier Research in Chemistry (icFRC). P.-A. N. and R. H. thank the French Department of Research for their doctoral fellowships. We also thank Prof. A. Delgado (RUBAM, Institut de Química Avançada de Catalunya, Spain) for enzymatic inhibition assays and Eric Wimmer for assistance with synthetic work.Notes and references
- For recent reviews on catalytic C–H amination, see: (a) C. G. Espino and J. Du Bois, in Modern Rhodium-catalyzed Organic Reaction, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005, pp. 379–416 Search PubMed; (b) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed; (c) P. Dauban and R. Dodd, in Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, Weinheim, 2007, pp. 55–92 Search PubMed; (d) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS; (e) Z. Li and C. He, Eur. J. Org. Chem., 2006, 4313–4322 CrossRef CAS; (f) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC; (g) P. Compain and S. Toumieux, in Targets in Heterocyclic systems, Chemistry and Properties, ed. O. A. Attanasi and D. Spinelli, SCI, Rome, 2007, vol. 11, pp. 338–364 Search PubMed; (h) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed; (i) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC; (j) J.-P. Wan and Y. Jing, Belstein J. Org. Chem., 2015, 11, 2209–2222 CrossRef PubMed; (k) J. Buendlia, G. Grelier and P. Dauban, Adv. Organomet. Chem., 2015, 64, 77–118 CrossRef; (l) J. Du Bois, Chemtracts, 2005, 18, 1–13 CAS.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS.
- (a) C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935–6936 CrossRef CAS; (b) C. G. Espino and J. Du Bois, Angew. Chem., Int. Ed., 2001, 40, 598–600 CrossRef CAS.
- A. Hinman and J. Du Bois, J. Am. Chem. Soc., 2003, 125, 11510–11511 CrossRef CAS PubMed.
- (a) S. Toumieux, P. Compain and O. R. Martin, J. Org. Chem., 2008, 73, 2155–2162 CrossRef CAS PubMed; (b) S. Toumieux, P. Compain, O. R. Martin and M. Selkti, Org. Lett., 2006, 8, 4493–4496 CrossRef CAS PubMed; (c) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825–6828 CrossRef CAS PubMed; (d) B. M. Trost, B. M. O'Boyle, W. Torres and M. K. Ameriks, Chem. – Eur. J., 2011, 17, 7890–7903 CrossRef CAS PubMed; (e) S. Toumieux, P. Compain and O. R. Martin, Tetrahedron Lett., 2005, 46, 4731–4735 CrossRef CAS; (f) M. S. T. Morin, S. Toumieux, P. Compain, S. Peyrat and J. Kalinowska-Tluscik, Tetrahedron Lett., 2007, 48, 8531–8535 CrossRef CAS.
- For recent examples, see: (a) P.-A. Nocquet, D. Hazelard, G. Gruntz and P. Compain, J. Org. Chem., 2013, 78, 6751–6757 CrossRef CAS PubMed; (b) V. Chagnault, P. Compain, K. Lewinski, K. Ikeda, N. Asano and O. R. Martin, J. Org. Chem., 2009, 74, 3179–3182 CrossRef CAS PubMed; (c) P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. Mena Barragán, C. Ortiz Mellet and J.-F. Nierengarten, Angew. Chem., Int. Ed., 2010, 49, 5753–5756 CrossRef CAS PubMed; (d) C. Bonduelle, J. Huang, T. Mena-Barragán, C. Ortiz Mellet, C. Decroocq, E. Etamé, A. Heise, P. Compain and S. Lecommandoux, Chem. Commun., 2014, 50, 3350–3352 RSC.
- P.-A. Nocquet, R. Hensienne, J. Wencel-Delord, E. Wimmer, D. Hazelard and P. Compain, Org. Biomol. Chem., 2015, 13, 9176–9180 CAS.
- Iminosugars: from Synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, Wiley & Sons, Chichester, 2007 Search PubMed.
- For reviews, see: (a) B. G. Winchester, Tetrahedron: Asymmetry, 2009, 20, 645–651 CrossRef CAS; (b) R. J. Nash, A. Kato, C.-Y. Yu and G. W. J. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed; (c) G. Horne and F. X. Wilson, Prog. Med. Chem., 2011, 50, 135–176 CAS.
- (a) In the course of our study, Schomaker et al. reported the silver-catalyzed C–H amination of a sulfamoyloxyethyl cyclobutane derivative in yields less than 31%, see: J. M. Alderson, A. M. Phelps, R. J. Scamp, N. S. Dolan and J. M. Shomaker, J. Am. Chem. Soc., 2014, 136, 16720–16723 CrossRef CAS PubMed; (b) K. B. Wiberg in The Chemistry of Cyclobutanes, ed. Z. Rappoport and J. F. Liebman, Wiley & Sons, Chichester, 2005, pp. 1–16 Search PubMed.
- (a) K. A. Parker and W. Chang, Org. Lett., 2005, 7, 1785–1788 CrossRef CAS PubMed; (b) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed.
- K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS.
- J. J. Fleming, M. D. McReynolds and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 9964–9975 CrossRef CAS PubMed.
- Such a process has been recently described by Blakey et al. for the synthesis of pyrrolidine derivatives, see: A. R. Thornton, V. I. Martin and S. B. Blakey, J. Am. Chem. Soc., 2009, 131, 2434–2435 CrossRef CAS PubMed.
- T. Ishizuka and T. Kunieda, Tetrahedron Lett., 1987, 28, 4185–4188 CrossRef CAS.
- For reviews on olefin metathesis of amine-containing systems, see: (a) P. Compain, Adv. Synth. Catal., 2007, 349, 1829–1846 CrossRef CAS; (b) P. Compain and D. Hazelard in Synthesis of heterocycles by Metathesis reactions, Topics in Heterocyclic Chemistry, ed. J. Prunet, Springer, 2015, DOI:10.1007/7081_2014_139, in press.
- M. Kimura, R. Mukai, T. Tamaki, Y. Horino and Y. Tamaru, J. Am. Chem. Soc., 2007, 129, 4122–4123 CrossRef CAS PubMed.
- (a) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274–7276 CrossRef CAS PubMed; (b) For a review, see: F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863–5883 CrossRef CAS.
- F. Narah, F. Liron, G. Prestat, C. Mealli, A. Messaoudi and G. Poli, Chem. – Eur. J., 2009, 15, 11078–11082 CrossRef PubMed.
- Very recently, Nishikawa et al. reported some examples of Pd(II)-catalyzed C–H amination of tertiary allylic C–H bonds: Y. Nishikawa, S. Kimura, Y. Kato, N. Yamazaki and O. Hara, Org. Lett., 2015, 17, 888–891 CrossRef CAS PubMed.
- (a) M. E. Hodson, Respiration, 2000, 67, 595–607 CrossRef CAS PubMed; (b) A. Leonard, P. Lebecque, J. Dingemanse and T. Leal, J. Cystic Fibrosis, 2012, 11, 231–236 CrossRef CAS PubMed.
- (a) C. Norez, S. Noel, M. Wilke, M. Bijvelds, H. Jorna, P. Melin, H. DeJonge and F. Becq, FEBS Lett., 2006, 580, 2081–2086 CrossRef CAS PubMed; (b) For examples of DNJ cluster-based CFTR correctors, see: P. Compain, C. Decroocq, A. Joosten, J. de Sousa, D. Rodriguez-Lucena, T. D. Butters, J. Bertrand, R. Clément, C. Boinot, F. Becq and C. Norez, ChemBioChem, 2013, 14, 2050–2058 CrossRef CAS PubMed.
- C. Norez, G. D. Heda, T. Jensen, I. Kogan, L. K. Hughes, C. Auzanneau, R. Dérand, L. Bulteau-Pignoux, C. Li, M. Ramjeesingh, H. Li, D. N. Sheppard, C. E. Bear, J. R. Riordan and F. Becq, J. Cystic Fibrosis, 2004, 3, 119–121 CrossRef CAS PubMed.
- P. Compain, O. R. Martin, C. Boucheron, G. Godin, L. Yu, K. Ikeda and N. Asano, ChemBioChem, 2006, 7, 1356–1359 CrossRef CAS PubMed.
- P. Compain, Synlett, 2014, 1215–1240 CrossRef.
- For selected reviews on pharmacological chaperone therapy see: (a) J.-Q. Fan, in Iminosugars: from Synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, Wiley-VCH, Weinheim, 2007, pp. 225–247 Search PubMed; (b) J.-Q. Fan, Trends Pharmacol. Sci., 2003, 24, 355–360 CrossRef CAS PubMed; (c) Z. Yu, A. R. Sawkar and J. W. Kelly, FEBS J., 2007, 274, 4944–4950 CrossRef CAS PubMed; (d) G. Parenti, EMBO Mol. Med., 2009, 1, 268–279 CrossRef CAS PubMed; (e) T. M. Wrodnigg and A. E. Stütz, Curr. Enzyme Inhib., 2012, 8, 47–99 CrossRef CAS.
- (a) F. Oulaïdi, S. Front-Deschamps, E. Gallienne, E. Lesellier, K. Ikeda, N. Asano, P. Compain and O. R. Martin, ChemMedChem, 2011, 6, 353–361 CrossRef PubMed; (b) W. Schönemann, E. Gallienne, P. Compain, K. Ikeda, N. Asano and O. R. Martin, Bioorg. Med. Chem., 2010, 18, 2645–2650 CrossRef PubMed.
- L. Yu, K. Ikeda, A. Kato, I. Adachi, G. Godin, P. Compain, O. R. Martin and N. Asano, Bioorg. Med. Chem., 2006, 14, 7736–7744 CrossRef CAS PubMed.
- Phenyl-benzoquinone is commercially available but can be prepared according to: Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed.
- W. Kammouni, B. Moreau, F. Becq, A. Saleh, A. Pavirani, C. Figarella and M. D. Merten, Am. J. Respir. Cell Mol. Biol., 1999, 20, 684–691 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02602d |
| ‡ Present address: SynCat, Université de Strasbourg/CNRS (UMR 7509), Ecole Européenne de Chimie, Polymères et Matériaux (ECPM), 25 rue Becquerel, 67087 Strasbourg, France. |
| This journal is © The Royal Society of Chemistry 2016 |