Azulenium chemistry: towards new derivatives of photochromic dihydroazulenes†
Received
9th December 2015
, Accepted 13th January 2016
First published on 13th January 2016
Abstract
Here we present the preparation of a selection of azulenium ions by hydride abstraction from photochromic 1,8a-dihydroazulenes (1,8a-DHAs) incorporating two cyano groups at C-1. The reactivity of the electrophilic tropylium ring of these molecules towards lithium triisopropylsilylacetylide was investigated. The position of attack by the nucleophile depended on the substitution pattern of the azulenium cation but was in general found to occur preferentially at positions C-4, C-5, and C-6, and to a minor extent at positions C-7 and C-8. The outcome was a mixture of non-photochromic, regioisomeric DHAs. One of these compounds containing the ethynyl substituent at position C-4 was partly tautomerized to the photochromic 1,8a-dihydroazulene, which was isolated and its switching properties were investigated by UV-Vis absorption spectroscopy. Upon irradiation, it undergoes a ring-opening reaction to form a vinylheptafulvene (VHF), which in turn returns to the original DHA. The half-life of this reaction was significantly smaller than for a derivative with the alkynyl substituent placed at C-7. This fast switching behavior was according to the calculations explained by an enhancement in the stability of the reactive s-cis conformer of the VHF relative to the, still more stable, s-trans conformer, and by a smaller activation energy for this s-cis conformer to undergo ring-closure.
Introduction
Dihydroazulene 1 (DHA) is a photoswitch that upon exposure to light undergoes a ring-opening to vinylheptafulvalene 2 (VHF).1 VHF returns, in turn, to DHA (Scheme 1) with a rate of ring-closure that is solvent dependent and is believed to proceed via a zwitterionic transition state. We have recently focused on exploiting these reversible switching properties in molecular electronics devices2 and further efforts are currently underway to employ the DHA/VHF for the development of solar thermal energy storage systems and other advanced materials such as photoresponsive liquid crystals.3
 |
| | Scheme 1 DHA-VHF photo/thermoswitch and numbering of the DHA core. | |
In order to tune the DHA–VHF properties, in particular in regard to the forward and backward switching events, the DHA and VHF absorption maxima, the energy storage capacity of the metastable VHF, and the possibility for liquid crystallinity, it is desirable to achieve reliable synthetic protocols for regioselectively placing substituents in all positions of the DHA core. So far, it has been possible to selectively brominate the 3- and the 7-positions,4 the latter giving an appropriate functional handle for further transformations using palladium cross-coupling methodologies.5 In fact, it has been found that the electronic character of substituent groups at position 7 strongly influences the rate of the thermal VHF to DHA back-reaction.5a,b,6
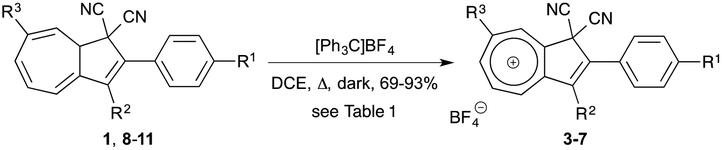
Rather than subjecting DHA 1 to further functionalization, an alternate approach employed phenyltropylium as an electrophilic starting material for constructing a substituted DHA core.7 This method gave, however, a mixture of regioisomers with the phenyl substituent at different positions in the seven-membered ring. From this reaction, small amounts of the 5-Ph substituted isomer could be isolated pure, but the lack of selectivity made this strategy rather inconvenient. Recently, it has been shown that DHA 1 could be oxidized by means of hydride abstraction using the tritylium cation to affect the formation of the azulenium salt 3 in good yield (Fig. 1).8 The same method has been used to form azulenium cations from other dihydroazulenes.9 We became interested in preparing a large selection of such azulenium cations with aryl substituents at various positions (4–7) from DHAs 8–11 and in investigating the scope of nucleophilic additions to such species. This approach was stimulated by the fact that some regioselectivity was previously observed for the reaction of aryltropylium species with nucleophiles,10 depending on the bulkiness of the nucleophile and the electronic character of the aryl group. The parent 2H-azulenium cation has previously been shown to have a clear vinyltropylium ion character and with the charge predominantly localized on C-3a, C-5, C-7, and C-8a in the seven-membered ring and on C2 and C3 in the five-membered ring.11 Synthetic protocols for donor- (OMe) and acceptor- (CN) substituted DHAs 8 and 9 have previously been reported,5d as the protocols for the 2,3-diphenyl DHA 10.12 Synthesis of the new 2,3,7-triphenyl substituted DHA 11 will be presented below.
 |
| | Fig. 1 Retrosynthetic route to azulenium salts 3–7 by hydride abstraction from DHA precursors 1, 8–11. | |
Results and discussion
Synthesis
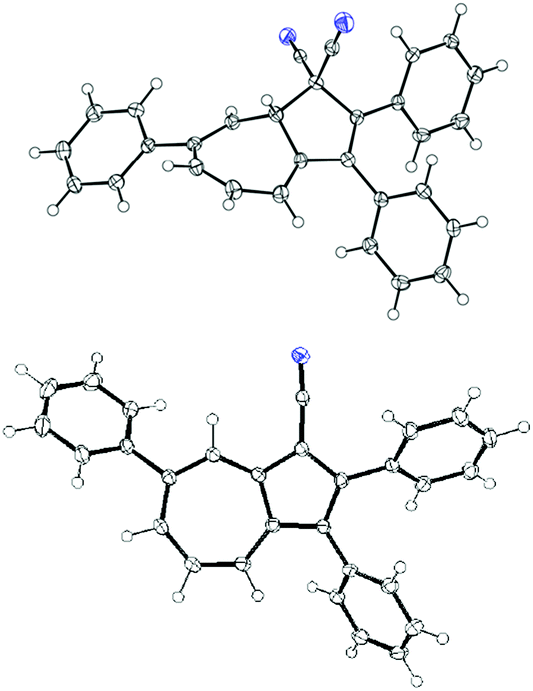
Synthesis of DHA 11 was performed according to Scheme 2. First, DHA 10 was subjected to the bromination–elimination protocol providing the intermediate 12, which was then subjected to a Suzuki reaction with phenylboronic acid yielding DHA 11 in reasonable yield over three steps. In addition to 11, azulenes 13 and 14 were isolated from the reaction mixture, a result of loss of hydrogen cyanide. Single crystals of both 11 and 14 were subjected to X-ray crystallographic analysis, which confirmed the identities of both structures (Fig. 2).
 |
| | Scheme 2 Synthesis of 2,3,7-triphenyl-substituted DHA 11. RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl. | |
 |
| | Fig. 2 Top: molecular structure of 11 (CCDC 1441529). The crystals were grown from dichloromethane/ethanol. Bottom: molecular structure of 14 (CCDC 1441530). The crystals were grown from THF/heptane. The blue spheres represent nitrogen atoms. | |
Subjecting DHA 11 to light produced the corresponding VHF as evidenced by a redshift in the absorption maximum in the UV-Vis absorption spectrum (Fig. 3). The VHF underwent a very fast back-reaction with a half-life of ca. 11 s at 25 °C in acetonitrile. The half-life is thus halved relative to that of the VHF corresponding to DHA 10, for which a half-life of 20 s at 25 °C has been reported.12
 |
| | Fig. 3 UV-Vis absorption spectra of pure DHA 11 and the corresponding VHF in MeCN at 25 °C. On account of the fast VHF to DHA conversion, the spectrum of VHF contains overlapping DHA absorptions. | |
Our next objective was to study the possibility of converting DHAs 1 and 8–11 to the corresponding azulenium ions. Hydride abstraction of these DHAs was accomplished by treatment with [Ph3C]BF4 in refluxing 1,2-dichloroethane (DCE) with reaction times varying from one night to two days depending upon the substitution pattern of the DHA (Scheme 3 and Table 1). Noteworthily, generation of the azulenium ion required a long reaction time in the presence of the electron-withdrawing cyano substituent (R1). The salts 3–7 were conveniently precipitated by the addition of diethyl ether to the cooled reaction mixtures. All these salts were generally poorly soluble in most organic solvents although it was possible to obtain full NMR spectroscopic analyses using CD3CN as the solvent, despite a limited stability in this solvent. The yield of the salt 3 was slightly improved in comparison with our previous report8 (from 86 to 93%) by performing the reaction under an inert atmosphere.
 |
| | Scheme 3 Synthesis of azulenium tetrafluoroborate salts 3–7. DCE = 1,2-dichloroethane. | |
Table 1 Synthesis of azulenium salts – reaction times and yields
| DHA – Starting material |
R1 |
R2 |
R3 |
Reaction time (h) |
Azulenium salt – product |
Yield (%) |
|
1
|
H |
H |
H |
17 |
3
|
93 |
|
8
|
OMe |
H |
H |
15 |
4
|
85 |
|
9
|
CN |
H |
H |
38 |
5
|
71 |
|
10
|
H |
Ph |
H |
24 |
6
|
69 |
|
11
|
H |
Ph |
Ph |
39 |
7
|
78 |
The absorption profiles for the pure azulenium tetrafluoroborates 3–7 were measured in both CH3CN and CH2Cl2 (see the ESI†). Table 2 lists the longest-wavelength absorption maxima. When going from CH3CN to CH2Cl2 the salts experience a redshift in the absorption maximum of ca. 35 nm.
Table 2 Longest-wavelength absorption maxima of azulenium salts
| Azulenium salt |
λ
max (CH3CN) (nm) |
λ
max (CH2Cl2) (nm) |
|
3
|
435 |
468 |
|
4
|
509 |
545 |
|
5
|
438 |
475 |
|
6
|
438 |
473 |
|
7
|
463 |
501 |
Azulenium salts 3–7 were then subjected to nucleophilic attack with lithium triisopropylsilylacetylide. This nucleophile was chosen as we found that other nucleophiles such as alcohols tended to give labile products. In addition, molecules with potential for subsequent acetylenic coupling reactions (after desilylation) are desirable. The product outcomes are summarized in Scheme 4 and Table 3. The general reaction conditions involved the addition of a THF solution of the nucleophile at −78 °C to a suspension of the azulenium salt also held at −78 °C in THF. In these cases, low temperature was used to avoid the formation of azulene products through an elimination of hydrogen cyanide, a side reaction which typically can occur under basic conditions.4a The addition product ratios were determined after the reaction mixture was passed through a silica gel column to remove any unwanted impurities and all these mixtures were determined to be analytically pure. The regioisomers could be separated from their respective mixtures using subsequent flash column chromatography, which allowed for the assignment of some of the products.
 |
| | Scheme 4 Addition of triisopropylsilylacetylide anion to azulenium ions 3–7. | |
Table 3 Product distributions from nucleophilic attack of lithium triisopropylsilylacetylide on cations 3–7
| Entry |
Starting material |
Product |
Overall yield (%) |
% of isomersa |
|
a
|
b
|
c
|
d
|
e
|
|
Determined by 1H NMR spectroscopic analysis of the pure regioisomeric mixture (obtained after chromatographic purification).
Compound 20 (resulting from conversion of 19b).
|
| 1 |
3
|
15
|
73 |
29 |
47 |
9 |
10 |
5 |
| 2 |
4
|
16
|
57 |
28 |
28 |
27 |
12 |
5 |
| 3 |
5
|
17
|
40 |
19 |
30 |
39 |
9 |
3 |
| 4 |
6
|
18
|
44 |
6 |
51 |
20 |
16 |
7 |
| 5 |
7
|
19
|
25 |
25 |
21b |
54 |
— |
— |
It was found that the reaction of the acetylide with 3 (Entry 1) gave a mixture of isomers, with the addition pattern predominating in the 5- (15b) and 4-positions (15a). Using flash column chromatography, it was possible to isolate and characterize these two main isomers. All addition products have proton connectivity spanning the 7-membered ring from positions 4 to 7, where the point of attack is sp3 hybridized. Coupling constants could be used to distinguish the nature of the protons on the adjacent carbons, as cis-alkene couplings (ca. 10 Hz) were distinguishable from couplings between the protons on the adjacent “alkene units” (ca. 7 Hz), which assisted in elucidating the structural framework. The orientation relative to the 5-membered ring could be verified by a NOESY correlation from the proton at the 3-position. The reactions with the azulenium 4 bearing an electron-donating OMe group and the azulenium 5 bearing an electron-withdrawing CN group showed minor changes to the addition pattern (entries 2 and 3), but unfortunately these reactions appeared to be less regioselective. It was envisaged that the phenyl substituent in position 3 of azulenium 6 could potentially block attack at position 4 (18a). Indeed, it was found that attack on position 4 was reduced in the treatment of 6 with the acetylide anion (entry 4), and the substitution at the 5-position (18b) was the predominant product. The addition of triisopropylsilylacetylide to the highly substituted system gave 19a and 19c in addition to an unexpected product 20. In this case addition to the 5-position had occurred but with the formation of a norcaradiene product. This type of product13 has previously been observed in the reaction of 1,3-diphenyltropylium with nucleophiles.14
None of the products formed are photoactive in regard to a ring-opening reaction. They all require a double bond migration to move a hydrogen to the 8a position in order to generate the photoactive DHA. A 1,5-sigmatropic shift has been reported to occur in alkynyl substituted cycloheptatriene in warm DMF.15 As the photoactive DHA has the longest conjugation pathway, one could imagine sigmatropic shifts to terminate at the presumably thermodynamically most stable, photoactive DHA. Due to the tedious purification procedure in separating the isomers from the addition reaction, the double bond isomerization reaction was examined on only two derivatives, 15a and 15b featured in Scheme 5. It was found that heating the 4-isomer 15a was very destructive but resulted in both formation of the photoactive compound 21 (in 11%) and the corresponding azulene 22 (in 16%), arising from the loss of hydrogen cyanide. Purification of the DHA was made possible by preparative TLC, where the plate was continuously irradiated using several TLC lamps (355 nm), as the VHF form of 21, formed under the illumination, is more polar allowing for an easier purification. On the other hand, thermolysis of the 5-substituted isomer 15b in DMF did not yield any photoswitching products, instead significant conversion to the azulene 23 was obtained, the structure of which was confirmed by X-ray crystallography (Fig. 4). Heating of both 15a and 15b in other solvents was investigated, but they did not facilitate the formation of neither photoactive species nor azulene by-products and due to such low yields of both the addition products and the isomerization, this was not further pursued.
 |
| | Scheme 5 Tautomerization of dihydroazulene products. | |
 |
| | Fig. 4 Top: molecular structure of 23 (CCDC 1441531). The crystals were grown from evaporating ethyl acetate. The blue sphere represents nitrogen and the yellow sphere silicon. | |
Switching studies of 4-alkynyl substituted DHA 21
With the first, pure 4-substituted 1,8a-DHA 21 in hand, its photoswitching properties were studied using UV-Vis absorption spectroscopy. Upon irradiation of 21 with light (ca. 375 nm), the corresponding VHF 24 was formed with a characteristic absorption maximum at 473 nm (Fig. 5); the VHF returned in the dark to 21 with an identical absorption spectrum as that measured before photolysis and with spectral evolution with clean isosbestic points, suggesting no conversion to the congested 8a-isomer 25 (with a Z-VHF as open form, Scheme 6). In contrast, it was previously found that a ring-opening/closing cycle of a 7-substituted DHA results in the formation of a mixture of 6- and 7-substituted DHAs accompanied by a red-shift in the characteristic DHA absorption, arising from the contributions from the 6-isomer.4a This isomerism has also been observed in experiments upon 5-phenyl substituted DHA (5-/8-substituted isomers), and this study was complimented by NMR spectroscopy. The lack of isomerization of the 4-substituted isomer is in agreement with the actual purification procedure that was performed where the compound was constantly irradiated. DFT calculations also show that DHA 25 is significantly higher in Gibbs free energy than the isomer 21, by 30.6–38.8 kJ mol−1, depending on the DFT functional utilized (see the ESI†). This clearly indicates that there should be no conversion to 25 in an opening–closure cycle since it is thermochemically disfavored.
 |
| | Fig. 5 Top: absorption spectra of DHA 21 and its corresponding VHF 24 in MeCN at 25 °C. Bottom: Arrhenius plot for the thermal VHF to DHA ring-closure. Ea = 95.8 ± 1.9 kJ mol−1 and A = 7.10 × 1012 s−1. | |
 |
| | Scheme 6 Ring-opening/closure of DHA 21. | |
The VHF 24 to DHA 21 conversion is readily followed by the first-order decay of the VHF absorption at 473 nm. The back-reaction was followed at five different temperatures in order to make an Arrhenius plot for the kinetics of the ring-closure. This plot provided an activation energy for the back-reaction of Ea = 95.8 ± 1.9 kJ mol−1 and a pre-exponential factor of A = 7.10 × 1012 s−1. The rate constant at 25 °C was determined to be 1.165 × 10−4 s−1, corresponding to a half-life of 99.2 min. In comparison, the VHF 26 (Fig. 6) of the isomeric 7-triisopropylsilylethynyl substituted DHA was previously found to undergo ring-closure with a half-life of 670 min at 25 °C.4a Thus, VHF 24 undergoes a significantly faster ring-closure. It is known from previous studies of the ring-closure of substituted phenyl VHFs that electron-withdrawing or donating groups can affect the rate of the thermal back reaction (Hammett correlations),6 yet in the present case the position of the substituent on the heptafulvene core seems of importance. The VHF has two conformers, s-cis and s-trans, which exist in equilibrium through a single bond rotation (Fig. 6). It is only s-cis that can close back to the DHA, yet the s-trans is usually more stable. If the energy difference between the two conformers is lower, then the VHF equilibrium lies closer to the reactive s-cis form, and ring-closure should then occur faster. To shed further light on this, we subjected the VHF isomers to DFT calculations (at different levels, see the ESI†). In all cases the s-trans conformation is found more stable than the corresponding s-cis conformation. The s-trans conformation of Z-26 is found to be 10–11 kJ mol−1 lower in Gibbs free energy than the corresponding s-cis conformation. For E-24, the Gibbs free energy difference is found somewhat lower, with the s-trans conformation now being only 2.9 to 4.1 kJ mol−1 lower in Gibbs free energy than the s-cis conformation. This supports that VHF E-24 closes faster due to a lower energy difference between the s-cis and s-trans conformations, shifting the thermal equilibrium more towards the reactive s-cis conformation. The reactivity of the s-cis conformers of 24 and 26 may also depend on the position of the alkyne unit. For example, conjugation effects could play a role. Therefore, we calculated the transition state (TS) energies (in MeCN) of the related s-cis VHFs where the i-Pr groups were substituted with Me groups (for calculation reasons). The corresponding TS structures are shown in the ESI,† and we obtain differences in Gibbs free energies of activation at 25 °C for “24-SiMe3” and “26-SiMe3”, which vary between 1.0 and 3.4 kJ mol−1 depending on the functional used. At the CAM-B3LYP/6-311 + G(d) level, free energies of activation of 91.5 (“24-SiMe3”) and 94.9 kJ mol−1 (“26-SiMe3”) were obtained, which correspond to a factor of 4 in rate constants (using the Eyring equation), while that observed experimentally is ca. 6.8. The influence was, however, smaller at the M06-2X level (difference in TS energies of +1.0 kJ mol−1). In all, it seems that not only does the larger content of the s-cis conformer of 24 relative to that of 26 play a role for its faster back-reaction, but the s-cis conformer of 24 also seems to be more reactive.
 |
| | Fig. 6 Conformations of VHFs E-24 and Z-26. | |
Conclusions
It is possible to synthesize a host of azulenium salts from their respective DHAs. Addition of lithium triisopropylsilylacetylide to these salts has shown that the positions 4, 5, and 6 are the most predominant points of attack in anotherwise reaction of low selectivity. The electronic influence of either donating or withdrawing groups upon the para-position of a phenyl ring at C-2 did little to enhance the selectivity of this addition, yet the placement of a phenyl blocking group in the 3-position of the azulenium prevented to some extent attack in the 4-position. One 4-substituted derivative was found to undergo a 1,5-sigmatropic shift to form a photoactive DHA (with a hydrogen on position 8a), which allowed switching studies of this 4-substituted DHA/VHF system. An opening–closure cycle gave none of the isomeric 8a-substituted isomer, which according to DFT calculations is of significantly higher energy. Interestingly, the VHF to DHA ring-closure rate was significantly enhanced compared to the previously studied 7-substituted analogue. For the ring-opened VHF a lower energy difference between s-cis and s-trans conformations was calculated (but still in favor of s-trans) than for the VHF obtained from the 7-substituted DHA. More molecules are thus in the reactive s-cis conformation on account of the steric constraints imposed by the bulky triisopropylsilyl group. The calculations also indicate that the s-cis conformer is more reactive by itself when the alkyne substituent is next to the exocyclic double bond.
While previous experiments have shown that fast VHF to DHA ring-closure can be promoted by (i) suitable donor–acceptor substitution,6 (ii) by having a phenyl substituent at position 3,1b,12 or (iii) by locking the VHF in its s-cis conformation via a covalent bridge (fused ring system),16 the current study has shown that a bulky substituent at position 4 also provides a means of achieving fast switching.
Experimental
General methods
NMR spectra were acquired with a Bruker Avance UltraShield 500 Plus instrument equipped with a cryoprobe or a Bruker 500 MHz Avance III system with a broadband probe (frequencies of 500 and 125 MHz for 1H and 13C NMR, respectively). All chemical-shift values in 1H and 13C NMR spectra were referenced to the residual solvent peak (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; CD3CN: δH = 1.96 ppm, δC = 1.94 ppm; C6D6: δH = 7.16 ppm, δC = 128.06 ppm). For compounds 19a, 19c, and 20, two 13C signals were observed for the TIPS CH3 carbon nuclei at 300 K in CDCl3, which we ascribe to slow conversion between rotamers. Thus, recording a sample of 19a at 323 K caused coalescence of these two signals into one signal. All starting materials were either purchased from Sigma–Aldrich, Combiblocks or synthesized; compounds 1,178,5d9,5d and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 were prepared according to literature procedures. Palladium catalysed cross couplings and all reactions involving the formation and use of azulenium salts were performed under an argon atmosphere. HPLC grade solvents were used as received with the exception of anhydrous THF and diethyl ether which were freshly distilled from a sodium/benzophenone couple. Flash column chromatography was performed on 40–63 μm SiO2, and TLC was performed on commercially available precoated silica plates and examined under UV light. All manipulations of DHA in solution were done so in the dark and the vessels were covered with aluminium foil. Melting points were measured with a Büchi apparatus and are uncorrected. Microanalyses were performed either at the University of Copenhagen, Department of Chemistry, or at London Metropolitan University. Electrospray (ESP) ionization mass spectra were acquired with a Bruker MicroTOF-Q II-system, whereas matrix-assisted laser desorption ionization (MALDI) mass spectra were acquired with a Bruker FT-ICR instrument equipped with a 7 T magnet. Prior to the experiments, the instrument was calibrated by using Na TFA cluster ions. UV-Vis absorption spectroscopy measurements were performed using a 1 cm-path-length cuvette. UV-Vis absorption spectra were obtained by scanning the wavelength from 800 to 200 nm in a 1 cm-path-length cuvette using a Cary 50 Bio UV-visible spectrophotometer. Photoswitching experiments were performed using a 150-W Xenon arc lamp equipped with a monochromator. The thermal back-reaction was performed by heating the sample (cuvette) by a Peltier unit in the UV-Vis spectrophotometer.
12 were prepared according to literature procedures. Palladium catalysed cross couplings and all reactions involving the formation and use of azulenium salts were performed under an argon atmosphere. HPLC grade solvents were used as received with the exception of anhydrous THF and diethyl ether which were freshly distilled from a sodium/benzophenone couple. Flash column chromatography was performed on 40–63 μm SiO2, and TLC was performed on commercially available precoated silica plates and examined under UV light. All manipulations of DHA in solution were done so in the dark and the vessels were covered with aluminium foil. Melting points were measured with a Büchi apparatus and are uncorrected. Microanalyses were performed either at the University of Copenhagen, Department of Chemistry, or at London Metropolitan University. Electrospray (ESP) ionization mass spectra were acquired with a Bruker MicroTOF-Q II-system, whereas matrix-assisted laser desorption ionization (MALDI) mass spectra were acquired with a Bruker FT-ICR instrument equipped with a 7 T magnet. Prior to the experiments, the instrument was calibrated by using Na TFA cluster ions. UV-Vis absorption spectroscopy measurements were performed using a 1 cm-path-length cuvette. UV-Vis absorption spectra were obtained by scanning the wavelength from 800 to 200 nm in a 1 cm-path-length cuvette using a Cary 50 Bio UV-visible spectrophotometer. Photoswitching experiments were performed using a 150-W Xenon arc lamp equipped with a monochromator. The thermal back-reaction was performed by heating the sample (cuvette) by a Peltier unit in the UV-Vis spectrophotometer.
2,3-Diphenylazulene-1,1(8aH)-dicarbonitrile (10).
Compound 10 was synthesized according to the literature procedure.121H NMR (500 MHz, CDCl3) δ = 7.39–7.31 (m, 5H), 7.30–7.26 (m, 3H), 7.20–7.12 (m, 2H), 6.56 (dd, J = 11.1, 6.0 Hz, 1H), 6.51 (dd, J = 11.1, 5.7 Hz, 1H), 6.34 (ddd, J = 10.0, 5.7, 2.0 Hz, 1H), 6.07 (br d, J = 6.0 Hz, 1H), 5.82 (dd, J = 10.0, 4.0 Hz, 1H), 3.73 (ddd, J = 4.0, 2.0, 2.0 Hz, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 145.7, 140.1, 136.2, 132.6, 131.7, 131.2, 130.9, 129.4, 129.4, 129.0, 129.0, 129.0, 128.8, 127.7, 121.1, 119.7, 115.8, 113.1, 50.2, 47.4 ppm.
2,3,7-Triphenylazulene-1,1(8aH)-dicarbonitrile (11).
To a stirring solution of 10 (1.07 g, 3.22 mmol) in CH2Cl2 (30 mL) at −78 °C was added dropwise a solution of Br2 (4.15 mL, 0.78M in CH2Cl2, 3.24 mmol) and stirring was continued for 1 h. The solvent was removed under reduced pressure to give the dibromide as an off-orange solid. The dibromide was dissolved in dry THF (50 mL), under an argon atmosphere and cooled to 0 °C, after which time a solution of LiHMDS (3.5 mL, 1.0M in toluene, 3.5 mmol) was slowly added and the contents were stirred for 2 h, where the temperature was allowed to slowly reach rt. The reaction was quenched with saturated aqueous NH4Cl (20 mL) and the contents were diluted with water (80 mL). The mixture was extracted with a THF/Et2O mixture (1:9; 2 × 100 mL). The combined organics were dried over MgSO4, filtered and the solvent was removed in vacuo. The residue was taken up in toluene (100 mL) and water (20 mL). To this degassed biphasic mixture containing 12 were sequentially added potassium phosphate (1.99 g, 9.38 mmol), phenylboronic acid (516 mg, 4.23 mmol), RuPhos (156 mg, 0.334 mmol) and Pd(OAc)2 (39 mg, 0.174 mmol), and the resulting mixture was heated to 55 °C for 16 h. Since the reaction was not done, the reaction mixture was degassed again and additional potassium phosphate (1.07 g, 5.04 mmol), phenylboronic acid (264 mg, 2.17 mmol), RuPhos (88 mg, 0.189 mmol) and Pd(OAc)2 (25 mg, 0.111 mmol) were added, and the reaction mixture was heated to 65 °C for 8 h. The contents of the vessel were diluted with water (100 mL) and the phases separated. The aqueous phase was extracted with CH2Cl2 (2 × 100 mL) and the combined organics dried over MgSO4. Filtration and removal of the solvent in vacuo gave a crude residue which was subjected to flash column chromatography (CH2Cl2/heptane 1:1) to afford pure 11 (579 mg, 44%) as a yellow solid. The reaction also yielded 13 (78 mg, 8%) as a purple solid and 14 (102 mg, 8%) as a green solid.
(11): M.p. = 172–175 °C. Rf = 0.38 (1:1 CH2Cl2/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.47–7.27 (m, 13H), 7.22–7.16 (m, 2H), 6.84–6.79 (m, 2H), 6.12 (ddd, J = 4.9, 1.5, 1.5 Hz, 1H), 6.03 (br d, J = 4.9 Hz, 1H), 3.83 (dd, J = 4.9, 1.5 Hz, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 145.1, 142.0, 139.9, 139.8, 136.8, 132.7, 132.4, 131.9, 131.7, 129.5, 129.4, 129.1, 129.0, 129.0, 128.9, 128.7, 128.2, 127.9, 120.4, 116.3, 115.9, 113.2, 50.2, 47.3 ppm. HRMS (MALDI +ve) calcd for C30H20N2 [M+]: m/z = 408.1616; found m/z = 408.1630. Anal. calcd (%) for C30H20N2 (408.50): C 88.21, H 4.94, N 6.86; found C 88.23, H 4.85, N 6.90.
2,3-Diphenylazulene-1-carbonitrile (13).
M.p. = 201–216 °C (decomposes). Rf = 0.26 (CH2Cl2/heptane 1:1). 1H NMR (500 MHz, CDCl3) δ = 8.47 (d, J = 9.7 Hz, 1H), 8.04 (d, J = 9.8 Hz, 1H), 7.60 (d, J = 7.7 Hz, 2H), 7.14–7.04 (m, 8H), 7.03–6.87 (m, 1H), 6.71 (t, J = 9.7 Hz, 1H), 6.65 (t, J = 9.8 Hz, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 151.3, 144.3, 140.1, 139.1, 137.2, 136.4, 135.5, 134.6, 131.6, 130.9, 129.9, 128.8, 128.8, 128.4, 127.5, 127.4, 127.3, 117.4, 97.8 ppm. HRMS (MALDI +ve) calcd for C23H15N [M+]: m/z = 305.1199; found m/z = 305.1203.
2,3,7-Triphenylazulene-1-carbonitrile (14).
M.p. >230 °C. Rf = 0.31 (CH2Cl2/heptane 1:1). 1H NMR (500 MHz, C6D6) δ = 9.02 (d, J = 1.8 Hz, 1H), 8.04 (d, J = 9.8 Hz, 1H), 7.71–7.56 (m, 2H), 7.44 (dt, J = 10.4, 0.9 Hz, 1H), 7.39–7.29 (m, 2H), 7.29–7.07 (m, 10H), 7.06–6.95 (m, 1H), 6.74 (t, J = 10.1 Hz, 1H) ppm. 13C NMR (125 MHz, C6D6) δ = 151.8, 143.7, 143.6, 141.1, 140.0, 139.5, 136.9, 136.1, 135.5, 134.6, 131.6, 130.9, 129.6, 129.3, 128.9, 128.8, 128.6, 128.4, 127.4, 127.1, 117.5, 98.6 ppm, 1C masked. HRMS (MALDI +ve) calcd for C29H19N [M+]: m/z = 381.1512; found m/z = 381.1522. Anal. calcd (%) for C29H19N (381.48): C 91.31, H 5.02, N 3.67; found C 91.22, H 5.14, N 3.59.
1,1-Dicyano-2-phenyl-1H-azulenium tetrafluoroborate (3).
Prepared from a modified literature procedure.8 A solution of 1 (1.56 g, 6.08 mmol) and [Ph3C]BF4 (3.01 g, 9.13 mmol) in CH2ClCH2Cl (60 mL) was heated to a reflux point for 17 h under an argon atmosphere, after which time, the reaction mixture was allowed to cool to rt and precipitation was induced by the addition of dry Et2O (40 mL). The precipitate was isolated by filtration and washed with dry Et2O (3 × 20 mL) yielding 3 (1.94 g, 93%) as a yellow solid. M.p. = 178 °C (decomposes). 1H NMR (300 MHz, CD3CN) δ = 9.44 (dd, J = 8.9, 1.5 Hz, 1H), 9.18–9.09 (m, 2H), 9.08–8.97 (m, 2H), 8.29 (s, 1H), 8.23–8.16 (m, 2H), 7.86–7.73 (m, 3H) ppm. 13C NMR (75 MHz, CD3CN) δ = 170.9, 157.6, 157.5, 155.5, 153.6, 153.1, 148.0, 147.7, 136.8, 135.7, 131.4, 129.8, 128.6, 110.4, 46.2 ppm. MS (FAB +ve); m/z = 255 [M+]. Anal. calcd (%) for C18H11N2BF4 (342.10): C 63.04, H 3.00, N 8.14; found C 63.18, H 3.24, N 8.19.
1,1-Dicyano-2-(4-methoxyphenyl)-1H-azulenium tetrafluoroborate (4).
To a solution of 8 (1.05 g, 3.67 mmol) in CH2ClCH2Cl (60 mL) was added [Ph3C]BF4 (1.45 g, 4.40 mmol), and the resulting solution was heated to a reflux point for 15 h under an argon atmosphere. To the cooled reaction mixture was added dry Et2O (40 mL) and the resulting precipitate was collected by suction filtration and washed with dry Et2O (3 × 80 mL) giving 4 (1.16 g, 85%) as golden brown microcrystals. M.p. = 209–220 °C (decomposes). 1H NMR (500 MHz, CD3CN) δ = 9.20 (dd, J = 8.7, 1.4 Hz, 1H), 8.94–8.83 (m, 2H), 8.82–8.68 (m, 2H), 8.17–8.11 (m, 2H), 8.04 (s, 1H), 7.31–7.23 (m, 2H), 3.99 (s, 3H) ppm. 13C NMR (125 MHz, CD3CN) δ = 171.8, 166.8, 156.8, 156.7, 156.3, 151.9, 151.4, 146.7, 146.1, 133.9, 132.8, 121.3, 117.3, 110.6, 57.1, 45.6 ppm. MS (ESP+): m/z = 258 [M+]. Anal. calcd (%) for C19H13N2OBF4 (372.13): C 61.32, H 3.52, N 7.53; found C 61.65, H 3.07, N 7.42.
1,1-Dicyano-2-(4-cyanophenyl)-1H-azulenium tetrafluoroborate (5).
A solution of 9 (1.00 g, 3.58 mmol) and [Ph3C]BF4 (1.52 g, 4.60 mmol) in CH2ClCH2Cl (60 mL) was heated to a reflux point for 14 h under an argon atmosphere. [Ph3C]BF4 (792 mg, 2.40 mmol) was added and the mixture was refluxed for a further 8 h, and extra [Ph3C]BF4 (892 mg, 2.70 mmol) was added, and the mixture was refluxed for a further 16 h. The contents of the reaction vessel were cooled to rt and the product was precipitated by addition of dry Et2O (40 mL). The precipitate was collected by filtration and washed with dry Et2O (3 × 20 mL) and a dry Et2O/CH2Cl2 mixture (1:1, 3 × 15 mL) giving 5 (931 mg, 71%) as a yellow powder. M.p. = 208–217 °C (decomposes). 1H NMR (500 MHz, CD3CN) δ = 9.53 (d, J = 9.2 Hz, 1H), 9.37–8.79 (m, 4H), 8.39 (s, 1H), 8.26 (d, J = 8.5 Hz, 2H), 8.07 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (125 MHz, CD3CN) δ = 169.7, 158.1, 158.1, 154.7, 154.3, 152.3, 148.8, 148.8, 139.7, 134.9, 132.3, 130.0, 118.6, 117.6, 109.9, 46.4 ppm. HRMS (ESP +ve) calcd for C19H10N3: m/z = 280.0867; found m/z = 280.0869. Anal. calcd (%) for C19H10N3BF4 (367.11): C 62.16, H 2.75, N 11.45; found C 62.42, H 2.51, N 10.92.
1,1-Dicyano-2,3-diphenyl-1H-azulenium tetrafluoroborate (6).
A solution of 10 (1.56 g, 4.70 mmol) and [Ph3C]BF4 (2.33 g, 7.05 mmol) in CH2ClCH2Cl (60 mL) was heated to a reflux point for 15 h under an argon atmosphere, after which extra [Ph3C]BF4 (1.23 g, 3.73 mmol) was added, and the mixture was refluxed for a further 9 h. Then the reaction mixture was allowed to cool to rt and precipitation was induced by the addition of dry Et2O (40 mL). The precipitate was isolated by filtration and washed with dry Et2O (3 × 50 mL) affording 6 (1.36 g, 69%) as a yellow powder. M.p. = 208–217 °C (decomposes). 1H NMR (500 MHz, CD3CN) δ = 9.55 (dd, J = 9.3, 1.0 Hz, 1H), 9.21–8.98 (m, 3H), 8.79–8.65 (m, 1H), 7.70–7.63 (m, 5H), 7.62–7.58 (m, 1H), 7.53–7.46 (m, 4H) ppm. 13C NMR (125 MHz, CD3CN) δ = 171.2, 157.7, 157.3, 154.2, 154.0, 150.2, 149.9, 148.1, 147.0, 133.9, 131.8, 131.2, 130.7, 130.7, 130.6, 130.0, 129.9, 110.4, 48.1 ppm. HRMS (MALDI +ve) calcd for C24H15N2m/z = 331.1230; exp = 331.1228. Anal. calcd (%) for C24H15N2BF4 (418.20): C 68.93, H 3.62, N 6.70; found C 69.02, H 3.59, N 6.79.
1,1-Dicyano-2,3,7-triphenyl-1H-azulenium tetrafluoroborate (7).
A solution of 11 (1.06 g, 2.59 mmol) and [Ph3C]BF4 (1.28 mg, 3.89 mmol) in CH2ClCH2Cl (60 mL) was heated to a reflux point for 16 h under an argon atmosphere, after which extra [Ph3C]BF4 (669 mg, 2.03 mmol) was added, and the reaction mixture was refluxed for a further 23 h. Then the reaction mixture was allowed to cool to rt and precipitation was induced by the addition of dry Et2O (50 mL). The precipitate was isolated by filtration and washed with a dry Et2O/CH2Cl2 mixture (1:1; 2 × 20 mL). The title compound 7 was isolated as an orange powder (1.00 g, 78%). M.p. >230 °C. 1H NMR (500 MHz, CD3CN) δ = 9.68 (d, J = 2.1 Hz, 1H), 9.19 (ddd, J = 10.2, 2.1, 0.8 Hz, 1H), 8.97 (t, J = 10.2 Hz, 1H), 8.58 (dd, J = 10.2, 0.8 Hz, 1H), 8.07–8.05 (m, 2H), 7.95–7.74 (m, 3H), 7.72–7.56 (m, 6H), 7.56–7.45 (m, 4H) ppm. 13C NMR (125 MHz, CD3CN) δ = 168.0, 167.1, 156.1, 155.4, 152.2, 149.7, 149.1, 146.5, 144.9, 139.4, 134.5, 133.6, 131.8, 131.7, 131.2, 131.0, 130.7, 130.7, 130.6, 130.1, 110.7, 48.6 ppm, 1C masked. HRMS (MALDI +ve) calcd for C30H19N2 [M+]: m/z = 407.1543; found m/z = 407.1543. Anal. calcd (%) for C30H19N2BF4 (494.30): C 72.90, H 3.87, N 5.76; found C 72.24, H 3.65, N 5.49.
Reaction of 3 with lithium triisopropylsilylacetylide.
To a degassed solution of triisopropylsilylacetylene (1.2 mL, 5.35 mmol) at −40 °C in Et2O (25 mL) was added LiHMDS (3.2 mL, 1.0 M in toluene, 3.2 mmol) and the vessel was allowed to warm to −20 °C. The reaction contents were cooled to −78 °C and added via a cannula to a vessel containing a degassed suspension of 3 (978 mg, 2.86 mmol) in THF (50 mL) at −78 °C. The reaction mixture was stirred for 1 h at −78 °C, quenched with saturated aqueous NH4Cl (50 mL) and allowed to warm to ambient temperature. The reaction mixture was diluted with Et2O (100 mL) and water (50 mL) and the phases were separated. The aqueous component was extracted with Et2O (50 mL) and the combined organic extracts were dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude residue was purified by flash column chromatography (toluene/heptane 2:3) to give a regioisomeric mixture of 15 (914 mg, 73%) as a yellow oil. Anal. calcd (%) for C29H32N2Si (436.67): C 79.77, H 7.39, N 6.42; found C 79.73, H 7.47, N 6.49. Mixture 15 could be subject to further flash column chromatography (2:23 EtOAc/heptane) to give pure samples of the following products.
2-Phenyl-4-[(triisopropylsilyl)ethynyl]-1,4-dihydroazulene-1,1-dicarbonitrile (15a).
A yellow solid. M.p. = 98–100 °C. Rf = 0.49 (2:23 EtOAc/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.86–7.65 (m, 2H), 7.53–7.45 (m, 2H), 7.43–7.37 (m, 1H), 7.30 (s, 1H), 6.95 (d, J = 11.2 Hz, 1H), 6.84 (dd, J = 11.2, 6.1 Hz, 1H), 6.29 (ddd, J = 9.7, 6.1, 1.2 Hz, 1H), 5.46 (dd, J = 9.7, 5.0 Hz, 1H), 3.46 (dd, J = 5.0, 1.2 Hz, 1H), 1.16–1.14 (m, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 142.3, 139.3, 133.5, 132.7, 132.6, 130.2, 129.8, 129.6, 127.2, 125.5, 125.0, 122.4, 112.1, 112.0, 105.2, 85.2, 43.4, 32.2, 18.8, 11.4 ppm. HRMS (MALDI +ve) calcd for C29H31N2Si [(M − H)+]: m/z = 435.2251; found m/z = 435.2258. Anal. calcd (%) for C29H32N2Si (436.67): C 79.77, H 7.39, N 6.42; found C 79.35, H 7.40, N 6.34.
2-Phenyl-5-[(triisopropylsilyl)ethynyl]-1,5-dihydroazulene-1,1-dicarbonitrile (15b).
A yellow solid. M.p. = 110–112 °C. Rf = 0.45 (2:23 EtOAc/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.73–7.68 (m, 2H), 7.50–7.46 (m, 2H), 7.45–7.40 (m, 1H), 7.18 (d, J = 6.5 Hz, 1H), 7.01 (s, 1H), 6.35 (ddd, J = 9.4, 6.5, 1.4 Hz, 1H), 5.56–5.53 (m, 2H), 3.22 (ddd, J = 5.4, 5.4, 1.4 Hz, 1H), 1.11–1.08 (m, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 139.3, 138.4, 136.7, 132.0, 130.3, 130.0, 129.5, 126.1, 125.8, 123.6, 117.9, 114.1, 113.4, 107.7, 82.2, 43.9, 32.4, 18.8, 11.3 ppm, 1C masked. HRMS (MALDI +ve) calcd for C29H31N2Si [(M − H)+]: m/z = 435.2251; found m/z = 435.2262. Anal. calcd (%) for C29H32N2Si (436.67): C 79.77, H 7.39, N 6.42; found C 79.54, H 7.41, N 6.34.
2-Phenyl-6-[(triisopropylsilyl)ethynyl]-1,5-dihydroazulene-1,1-dicarbonitrile (15c).
A yellowish oil. Rf = 0.40 (2:23 EtOAc/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.75–7.72 (m, 2H), 7.50–7.47 (m, 2H), 7.44–7.41 (m, 1H), 7.14 (s, 1H), 6.75 (dd, J = 9.3, 1.4 Hz, 1H), 6.51 (dd, J = 9.3, 1.4 Hz, 1H), 5.60–5.54 (m, 2H), 2.92 (dddd, J = 5.6, 5.6, 1.4, 1.4 Hz, 1H), 1.10–1.09 (m, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 146.2, 139.6, 137.3, 133.5, 129.9, 129.9, 129.6, 126.9, 125.9, 125.0, 121.6, 119.7, 112.4, 112.3, 107.8, 82.2, 44.3, 33.3, 18.8, 11.3 ppm. HRMS (MALDI +ve) calcd for C29H31N2Si [(M − H)+]: m/z = 435.2251; found m/z = 435.2249.
2-Phenyl-7-[(triisopropylsilyl)ethynyl]-1,5-dihydroazulene-1,1-dicarbonitrile (15d).
A yellowish oil. Rf = 0.31 (2:23 EtOAc/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.71–7.69 (m, 2H), 7.50–7.47 (m, 2H), 7.45–7.41 (m, 1H), 7.14 (s, 1H), 6.70 (d, J = 6.4 Hz, 1H), 6.34 (ddd, J = 9.9, 6.4, 1.2 Hz, 1H), 6.20 (d, J = 5.4 Hz, 1H), 5.44 (dd, J = 9.9, 5.4 Hz, 1H), 3.40 (ddd, J = 5.4, 5.4, 1.2 Hz, 1H), 1.09–1.08 (m, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 141.1, 137.2, 136.0, 134.9, 130.3, 130.0, 129.5, 126.7, 126.4, 126.3, 126.2, 124.1, 113.9, 113.4, 106.9, 82.6, 42.4, 32.2, 18.8, 11.3 ppm. HRMS (MALDI +ve) calcd for C29H31N2Si [(M − H)+]: m/z = 435.2251; found m/z = 435.2256.
2-Phenyl-8-[(triisopropylsilyl)ethynyl]-1,5-dihydroazulene-1,1-dicarbonitrile (15e).
A yellowish oil. Rf = 0.26 (2:23 EtOAc/heptane). 1H NMR (500 MHz, CDCl3) δ = 7.70–7.68 (m, 2H), 7.47–7.44 (m, 2H), 7.40–7.37 (m, 1H), 7.03 (s, 1H), 6.78 (dd, J = 10.6, 6.2 Hz, 1H), 6.71 (d, J = 10.6 Hz, 1H), 6.22 (br dd, J = 9.2, 6.2 Hz, 1H), 5.38 (dd, J = 9.2, 6.2 Hz, 1H), 3.73 (dd, J = 6.2, 1.0 Hz, 1H), 1.13 (br s, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 141.1, 138.8, 133.6, 132.6, 130.1, 129.5, 129.5, 129.4, 125.9, 124.1, 112.1, 112.0, 87.0, 43.7, 29.0, 18.8, 11.4 ppm, 3C masked. HRMS (MALDI +ve) calcd for C29H31N2Si [(M − H)+]: m/z = 435.2251; found m/z = 435.2257.
Reactions of 4–7 with lithium triisopropylsilylacetylide were performed in an analogous manner; for details, see the ESI.†
Tautomerization of 15a.
A solution of 15a (106 mg, 0.243 mmol) in freshly distilled degassed DMF (7 mL) was heated to 85 °C for 3.5 h, after which time the volatiles were removed under reduced pressure. The crude material was subjected to flash column chromatography (toluene/heptane 1:1), which gave 22 (16.3 mg, 16%), as a dark green glassy solid, in addition to an impure component, which contained 21. This solution was concentrated and irradiated at 353 nm for 10 h. The then formed VHF was purified by preparative TLC (toluene/heptane 3:1) whilst the plate was subjected to continuous irradiation at 353 nm. To remove the final small impurities the residue was subjected to flash column chromatography (toluene/heptane 1:1), which furnished 21 (12 mg, 11%) as a yellow solid.
1,1-Dicyano-2-phenyl-4-triisopropylsilylacetyl-1,8a-dihydroazulene (21).
M.p. = 115–118 °C. Rf = 0.41 (toluene/heptane 1:1). 1H NMR (500 MHz, CDCl3) δ = 7.81–7.67 (m, 2H), 7.61–7.40 (m, 3H), 7.24 (s, 1H), 6.64 (d, J = 11.4 Hz, 1H), 6.47 (dd, J = 11.4, 5.9 Hz, 1H), 6.28 (ddd, J = 10.1, 5.9, 2.1 Hz, 1H), 5.89 (dd, J = 10.1, 4.0 Hz, 1H), 3.82 (dd, J = 4.0, 2.1 Hz, 1H), 1.13–1.15 (m, 21H) ppm. 13C NMR (125 MHz, CDCl3) δ = 143.7, 141.6, 133.5, 131.7, 130.7, 130.6, 130.5, 129.5, 127.6, 126.5, 121.1, 115.4, 115.0, 112.7, 104.0, 96.6, 51.0, 45.3, 18.9, 11.4 ppm. HRMS (ESP +ve) calcd for C29H33N2Si: m/z = 437.2424; found m/z = 437.2407.
2-Phenyl-4-[(triisopropylsilyl)ethynyl]azulene-1-carbonitrile (22).
R
f = 0.16 (toluene/heptane 1:1). 1H NMR (500 MHz, C6D6) δ = 8.33 (d, J = 9.5 Hz, 1H), 8.24 (dd, J = 8.3, 1.0 Hz, 2H), 8.08 (s, 1H), 7.27 (d, J = 10.2 Hz, 1H), 7.24–7.21 (m, 2H), 7.11–7.07 (m, 1H), 6.82 (dd, J = 10.2, 10.0 Hz, 1H), 6.63 (ddd, J = 10.0, 9.5, 1.5 Hz, 1H), 1.22–1.19 (m, 21H) ppm. 13C NMR (125 MHz, C6D6) δ = 152.5, 145.1, 142.1, 136.5, 135.8, 134.9, 131.7, 131.3, 129.8, 129.5, 129.0, 127.6, 117.6, 117.5, 108.0, 101.5, 96.5, 18.9, 11.7 ppm. HRMS (ESP +ve) calcd for C28H32NSi [(M + H)+]: m/z = 410.2300; found m/z = 410.2299.
Tautomerization of 15b.
A solution of 15b (102 mg, 0.234 mmol) in distilled degassed DMF (10 mL) was heated to 90 °C for 5.5 h. The solution was allowed to cool to rt, and the DMF was removed under reduced pressure. The crude mixture was purified by flash column chromatography (toluene/heptane 1:1) to give 23 (57 mg, 60%) as a blue solid.
2-Phenyl-5-[(triisopropylsilyl)ethynyl]azulene-1-carbonitrile (23).
M.p. = 125–128 °C. Rf = 0.21 (toluene/heptane 1:1). 1H NMR (500 MHz, C6D6) δ = 8.26 (ddd, J = 1.6 Hz, 1H), 8.18 (ddd, J = 9.5, 0.9, 0.9 Hz, 1H), 8.05–8.03 (m, 2H), 7.48 (ddd, J = 10.5, 1.6, 0.9 Hz, 1H), 7.23–7.19 (m, 2H), 7.13–7.10 (m, 1H), 6.91 (s, 1H), 6.50 (dd, J = 10.5, 9.6 Hz, 1H), 1.25–1.24 (m, 21H) ppm. 13C NMR (125 MHz, C6D6) δ = 152.8, 145.2, 141.3, 140.8, 140.8, 135.6, 134.4, 129.8, 129.4, 129.0, 126.4, 122.7, 118.3, 117.4, 110.5, 96.9, 92.1, 18.9, 11.7 ppm. HRMS (ESP +ve) calcd for C28H31NNa [(M + Na)+]: m/z = 432.2118, found m/z = 432.2118. Anal. calcd (%) for C28H31NSi (409.65): C 82.10, H 7.63, N 3.42; found C 81.58, H 7.34, N 3.74.
Acknowledgements
The University of Copenhagen and the Danish Council for Independent Research|Technology and Production Sciences (#12-126668) are acknowledged for financial support. We thank Dr Søren L. Broman for the generous donation of starting materials.
Notes and references
-
(a) J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., Int. Ed. Engl., 1984, 23, 960 CrossRef;
(b)
T. Mrozek, A. Ajayaghosh and J. Daub, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2001, pp. 63–106 Search PubMed;
(c) S. L. Broman and M. B. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 21172 RSC.
-
(a) S. Lara-Avila, A. V. Danilov, S. E. Kubatkin, S. L. Broman, C. R. Parker and M. B. Nielsen, J. Phys. Chem. C, 2011, 115, 18372 CrossRef CAS;
(b) S. L. Broman, S. Lara-Avila, C. L. Thisted, A. D. Bond, S. Kubatkin, A. Danilov and M. B. Nielsen, Adv. Funct. Mater., 2012, 22, 4249 CrossRef CAS;
(c) T. Li, M. Jevric, J. R. Hauptmann, R. Hviid, Z. Wei, R. Wang, N. E. A. Reeler, E. Thyrhaug, S. Petersen, J. A. S. Meyer, N. Bovet, T. Vosch, J. Nygård, X. Qui, W. Hu, Y. Liu, G. C. Solomon, H. G. Kjaergaard, T. Bjørnholm, M. B. Nielsen, B. W. Laursen and K. Nørgaard, Adv. Mater., 2013, 25, 4164 CrossRef CAS PubMed.
-
(a) S. T. Olsen, J. Elm, F. E. Storm, A. N. Gejl, A. S. Hansen, M. H. Hansen, J. R. Nikolajsen, M. B. Nielsen, H. G. Kjaergaard and K. V. Mikkelsen, J. Phys. Chem. A, 2015, 119, 896 CrossRef CAS PubMed;
(b) M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. B. Nielsen, Chem. – Eur. J., 2015, 21, 7454 CrossRef CAS PubMed;
(c) A. U. Petersen, M. Jevric, R. J. Mandle, E. J. Davis, S. J. Cowling, J. W. Goodby and M. B. Nielsen, RSC Adv., 2015, 5, 89731 RSC.
-
(a) M. Å. Petersen, S. L. Broman, A. Kadziola, K. Kilså and M. B. Nielsen, Eur. J. Org. Chem., 2009, 2733 CrossRef CAS;
(b) V. Mazzanti, M. Cacciarini, S. L. Broman, C. R. Parker, M. Schau-Magnussen, A. D. Bond and M. B. Nielsen, Beilstein J. Org. Chem., 2012, 8, 958 CrossRef CAS PubMed;
(c) M. Cacciarini, S. L. Broman and M. B. Nielsen, ARKIVOC, 2014, i, 249 Search PubMed.
-
(a) S. L. Broman, M. Å. Petersen, C. G. Tortzen, A. Kadziola, K. Kilså and M. B. Nielsen, J. Am. Chem. Soc., 2010, 132, 9165 CrossRef CAS PubMed;
(b) M. Å. Petersen, S. L. Broman, K. Kilså, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2011, 1033 CrossRef CAS;
(c) M. Jevric, S. L. Broman and M. B. Nielsen, J. Org. Chem., 2013, 78, 4348 CrossRef CAS PubMed;
(d) S. L. Broman, M. Jevric, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2014, 79, 41 CrossRef CAS PubMed.
- S. L. Broman, M. Jevric and M. B. Nielsen, Chem. – Eur. J., 2013, 19, 9542 CrossRef CAS PubMed.
- L. Skov, M. Å. Petersen, S. L. Broman, A. D. Bond and M. B. Nielsen, Org. Biomol. Chem., 2011, 9, 6498 CAS.
- S. L. Broman, A. U. Petersen, C. G. Tortzen, J. Vibenholt, A. D. Bond and M. B. Nielsen, Org. Lett., 2012, 14, 318 CrossRef CAS PubMed.
-
(a) S. Itoh, N. Morita, R. Miyatake and S. Kuroda, Chem. Lett., 1997, 1011 Search PubMed;
(b) M. Oda, A. Sakamoto, T. Uchiyama, T. Kajioka, R. Miyatake and S. Kuroda, Tetrahedron Lett., 1999, 40, 3595 CrossRef CAS;
(c) M. Oda, H. Kainuma, T. Uchiyama, R. Miyatake and S. Kuroda, Tetrahedron, 2003, 59, 2831 CrossRef CAS;
(d) M. Oda, N. Nakajima, N. C. Thanh, T. Kajioka and S. Kuroda, Tetrahedron, 2006, 62, 8177 CrossRef CAS;
(e) M. Oda, N. Nakajima, N. C. Thanh, K. Kitahara, R. Miyatake and S. Kuroda, Eur. J. Org. Chem., 2008, 5301 CrossRef CAS.
-
(a) W. Abraham, K. Buck, S. Neubert and D. Kreysig, Z. Anorg. Allg. Chem., 1979, 19, 97 CAS;
(b) M. Balci, Chim. Acta Turc., 1983, 11, 31 CAS.
- T. Okazaki and K. K. Laali, Org. Biomol. Chem., 2003, 1, 3078 CAS.
- T. Mrozek, H. Görner and J. Daub, Chem. – Eur. J., 2001, 7, 1028 CrossRef CAS.
- For a review on the cycloheptatriene-norcaradiene conversion, see: O. C. McNamara and A. R. Maguire, Tetrahedron, 2011, 67, 9 CrossRef CAS.
-
(a) T. Mukai, H. Kubota and T. Toda, Tetrahedron Lett., 1967, 37, 3581 CrossRef;
(b) S. W. Staley, M. A. Fox and A. Cairncross, J. Am. Chem. Soc., 1977, 99, 4524 CrossRef CAS.
- M. Tamm, T. Bannenberg, K. Baum, R. Frolich, T. Steiner, T. Meyer-Friederichsen and J. Heck, Eur. J. Inorg. Chem., 2000, 6, 1161 Search PubMed.
- S. L. Broman, O. Kushnir, M. Rosenberg, A. Kadziola, J. Daub and M. B. Nielsen, Eur. J. Org. Chem., 2015, 4119 CrossRef.
- S. L. Broman, S. L. Brand, C. R. Parker, M. Å. Petersen, C. G. Tortzen, A. Kadziola, K. Kilså and M. B. Nielsen, ARKIVOC, 2011, ix, 51 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for reactions of 4–7 with triisopropylsilyl acetylide, NMR and UV-Vis spectra and calculated data. CCDC 1441529–1441531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02523k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a