
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple and rapid quantification of phospholipids for supramolecular membrane transport assays†
Robert
Hein
,
Can B.
Uzundal‡
and
Andreas
Hennig
*
Department of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: a.hennig@jacobs-university.de
First published on 15th January 2016
Abstract
We introduce a simple 1H NMR method for quantification of the phospholipid content of liposomes. The method is validated by comparison with the established Stewart assay, which revealed significant uncertainties in phospholipid quantification of established liposome preparations used in supramolecular membrane transport assays.
Liposomes are spherical vesicles composed of phospholipids, which self-assemble in the form of lipid bilayers and enclose an aqueous interior. They have been most intensively investigated as carriers for drug delivery,1 and as model systems for biological membranes.2 More recently, supramolecular chemists used liposomes to study the efficiency and mechanism of membrane transport systems as well as lipid phase organization.3,4 Liposome-based transport assays are particularly useful for supramolecular chemists, because they do not require specialized equipment, such as a planar lipid bilayer workstation, but can still be used to rapidly screen relevant characteristics and quantitatively evaluate various activities exerted by synthetic supramolecules. To set-up such an assay, large unilamellar vesicles (LUVs) of defined size and membrane composition are commonly prepared with an internally entrapped fluorescent dye. To remove the external, unencapsulated material, in particular the fluorescent dye, size exclusion chromatography (SEC) is used as the final step.5 To verify the nature and identity of the prepared LUVs, various analytical methods have been developed, which allow e.g. determination of lamellarity, size, trapping efficiency, as well as phospholipid composition and content of LUVs. Especially the latter is of high importance, because the total phospholipid concentration may largely determine the overall activity of supramolecular transport systems. For example, it may influence membrane partitioning and concentration-dependent self-assembly of transient supramolecular structures inside the lipid bilayer.6
However, a routine determination of the total phospholipid concentration is only rarely performed. Most often, the initial amount of phospholipids used to prepare the lipid film is reported, but loss of material during extrusion and SEC as well as potentially altered concentrations of phospholipid stock solutions are not accounted for (see examples in Table S1 in ESI†). At best, the classical Stewart assay or one of its alternatives is used,7 which are, however, relatively tedious and have specific drawbacks. We now report a rapid and simple 1H NMR method,§ which provides accurate total phospholipid concentrations in less than 15 min on a routine 400 MHz NMR including sample preparation.
In our 1H NMR method, we simply dilute a small amount (ca. 50–175 μL) of the prepared LUV stock solution with deuterated organic solvent (methanol/chloroform or methanol). The amounts of LUV solution and organic solvent were optimized to fully dissolve all tested liposomes regardless of their lipid composition (containing lipids with palmitoyl and oleoyl fatty acid side chains, with choline, serine, and ethanolamine headgroups, as well as the lipid mixture egg yolk phosphatidylcholine, i.e. EYPC) leading to sharp signals (Fig. 1). Concentrated liposome stock solutions could be simply dissolved with ca. 85% methanol (Method A in ESI†), whereas less concentrated liposome solutions additionally required ca. 20% chloroform to account for the presence of larger amounts of water (Method B in ESI†). For quantification, the mixture contains additionally a defined amount of 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TMSP) as standard. Fortunately, a signal at ca. 0.88 ppm, assigned to the terminal methyl groups of the phospholipid's fatty acid side chains,8 is well separated from all other peaks. Therefore, comparison of the integrated peak areas of that peak with the peak from the standard conveniently affords the total phospholipid concentration (Fig. 1). Noteworthy, we also attempted the non-destructive quantification of LUVs with a nested inner tube filled with standard and D2O to provide a lock signal, which gave, however, NMR signals too broad to be reliably integrated.
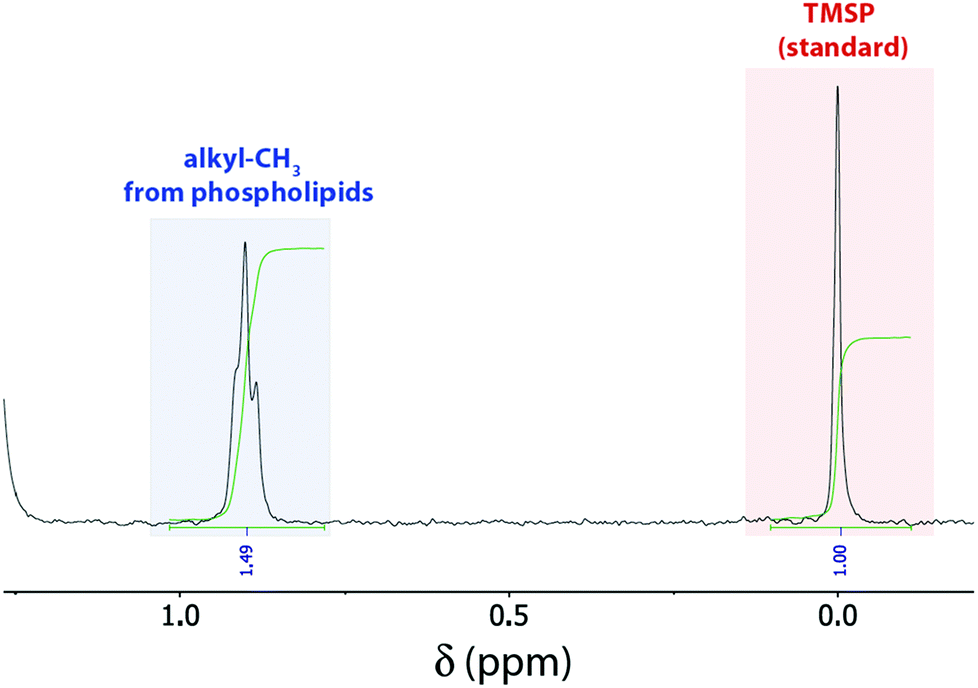 | ||
| Fig. 1 1H NMR spectrum of 100 μL POPC LUV stock solution in 10 mM phosphate buffer, pH 7.4 diluted with 430 μL CD3OD/100 μL CDCl3 and 20 μL D2O containing 5 mM TMSP as internal standard. | ||
We then explored the sensitivity, application scope, and robustness of this simple 1H NMR method and compared our results with the classical Stewart assay, and one of its alternatives, the Rouser assay (Table 1).7,9 The Stewart assay is based on the formation of a red-colored complex between the phospholipid headgroups and ammonium ferrothiocyanate, which can be quantified spectrophotometrically after extraction into chloroform.7 Consequently, the Stewart assay is relatively tedious and requires the determination of specific calibration curves for different phospholipid headgroups as well as mixtures thereof.7 The Rouser assay is based on the hydrolysis of phospholipids into orthophosphate by incubation with concentrated perchloric acid at 180 °C and subsequent spectrophotometric quantification of inorganic phosphate after reaction with ammonium molybdate and ascorbic acid in a boiling water bath.9 The additional hydrolysis step enables accurate quantification of lipid mixtures, but sacrifices the possibility to analyze liposomes prepared in phosphate buffers. Furthermore, the requirement for multiple incubation steps renders the Rouser assay even more time-demanding than the Stewart assay.
| Assay | Time requireda | Simplicityb | Lipid mixtures | Phosphate buffer | C.V.d | LOQe | LODe |
|---|---|---|---|---|---|---|---|
| a Depends on the phospholipid concentration and the desired accuracy. b Assessed based on the number of steps to be carried out. c The Stewart assay requires separate calibration curves for different phospholipid headgroups. d Coefficient of variation (n ≥ 7). e Limits of quantification and detection (see ESI for details). f Determined on a routine 400 MHz NMR (see ESI for details). | |||||||
| Stewart | >45 min | Medium | Not analyzablec | Tolerated | 6.6% | 7 nmol | 2.3 nmol |
| Rouser | >60 min | Difficult | Analyzable | Not tolerated | 6.7% | 15 nmol | 5 nmol |
| 1H NMR | ca. 15 min | Very simple | Analyzable | Tolerated | 6.3% | 65 nmolf | 21.5 nmolf |
In contrast to these established assays, our 1H NMR method allows determination of total phospholipid content of lipid mixtures with different head groups (phosphatidylcholine, -serine, and -ethanolamine) and fatty acid acyl side chains as well as in phosphate buffer. We also tested whether substances required for standard supramolecular transport assays,4,5 such as macrocycles, dyes and quenchers as well as different buffers (see Chart S1 in ESI†) interfere with the spectral region used for integration, and found that none of these substances shows NMR peaks below ca. 1.2 ppm or otherwise detrimentally interacts with the phospholipids (see Fig. S3 to S10 in ESI†). Furthermore, no undesirable precipitation of buffer ingredients or other substances was observed upon diluting the aqueous LUV stock solutions with methanol or methanol/chloroform mixtures.
To independently confirm the phospholipid content obtained by our 1H NMR method, we validated the phospholipid content of randomly selected samples by the Stewart assay (Fig. 2). This gave consistent values within error except for LUVs prepared with encapsulated carboxyfluorescein (CF) and with a supramolecular complex of lucigenin (LCG) and p-sulfonatocalix[4]arene (CX4).
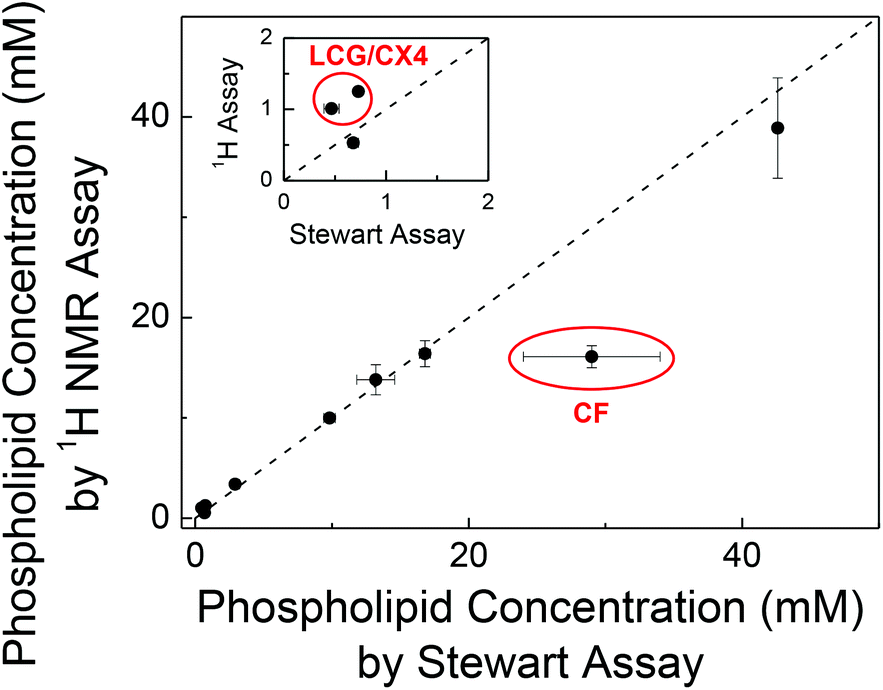 | ||
| Fig. 2 Mutual validation of the 1H NMR assay with the Stewart assay. Each data point presents an individually prepared batch of LUVs. The error bars are from triplicate measurements of the solutions. The inset is an expansion of the data at low concentrations. | ||
For LUVs with entrapped CF, we determined (16.1 ± 1.1) mM with the 1H NMR method and (29 ± 5) mM with the Stewart assay (Fig. 2). The latter value is unusually high (cf. Table S1 in ESI†) and has a comparatively large error. This is traced back to CF being partially extracted into the chloroform phase in presence of phospholipids,10 which strongly absorbs in the wavelength region of the Stewart assay, thus leading to a systematic overestimate of the phospholipid concentration. For LUVs with entrapped LCG/CX4, the concentrations obtained from the Stewart assay were lower than the values obtained from 1H NMR (inset of Fig. 2). Since LUVs with only LCG gave consistent values, we assume that CX4 interferes with the Stewart assay, e.g. by binding to the choline headgroup thereby preventing formation of the red-colored complex with ferrothiocyanate. These results were surprising and are to the best of our knowledge unreported despite the ubiquitous use of CF-loaded vesicles in membrane transport experiments.
Finally, the sensitivity and reproducibility of the Stewart assay, the Rouser assay, and our 1H NMR method were compared (Table 1). Therefore the limits of detection (LOD) and quantification (LOQ) were determined from the standard deviation of the blanks for the Stewart and Rouser assay, and from the signal-to-noise ratio for the 1H NMR method (see ESI† for details). This indicated that the Stewart and Rouser assay are similarly sensitive in accordance with the literature,7,9 whereas the 1H NMR method is less sensitive by a factor of five to ten. The sensitivity is nonetheless excellent for an NMR-based method, since the peak used for quantification originates from six protons of the terminal methyl groups per phospholipid molecule. Furthermore, the sensitivity is fully sufficient for a rapid routine analysis of LUV stock solutions prepared for supramolecular transport assays. For example, to exceed the LOQ in the 1H NMR method, only few μL need to be sacrificed for LUV stock solutions prepared from 2.5 to 25 mg ml−1 phospholipids. Moreover, the reproducibility expressed as the coefficient of variation was comparable for all three methods.
Conclusions
In conclusion, we have presented a rapid and simple method to determine the phospholipid content of liposomes, and in particular of LUVs with entrapped dyes for investigating supramolecular membrane transport. Established assays require significantly more preparation and measurement time and are either unable to quantify lipid mixtures or suffer from interference with phosphate buffers. Our 1H NMR assay only requires transferral of an aliquot of the prepared LUV stock solution into a standard NMR tube, addition of solvents and acquisition of a standard proton NMR to provide the phospholipid concentration with excellent precision and sufficient sensitivity. Certain types of phospholipids or additives may, however, require particular attention. For example, cholesterol, which is sometimes added to afford more stable LUVs, may prevent phospholipid quantification owing to overlapping signals above 0.8 ppm, although the signal around 0.67 ppm originating from the C18 methyl group of cholesterol could be used to quantify cholesterol instead.Since access to NMR, which may be a bottleneck for some biochemists and biophysical chemists, is usually unproblematic for synthetic chemists, we believe that the NMR method reported herein is highly useful for synthetic chemists interested in supramolecular chemistry of liposomes and membrane transport.
Acknowledgements
We thank Jacobs University Bremen for generous support and Dr Andrea Barba-Bon, Mr Adam Lalewicz, and Ms Yu-Chen Pan for their assistance with the assay validation.Notes and references
- S. Vemuri and C. Rhodes, Pharm. Acta Helv., 1995, 70, 95 CrossRef CAS PubMed
; M. Frohlich, V. Brecht and R. Peschka-Suss, Chem. Phys. Lipids, 2001, 109, 103 CrossRef PubMed
- J. Sloniec, M. Schnurr, C. Witte, U. Resch-Genger, L. Schröder and A. Hennig, Chem. – Eur. J., 2013, 19, 3110 CrossRef CAS PubMed
- H. Valkenier, N. López Mora, A. Kros and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2137 CrossRef CAS PubMed
- G. Ghale, A. G. Lanctôt, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762 CrossRef CAS PubMed
-
S. Matile, N. Sakai and A. Hennig, in Supramolecular Chemistry: From Molecules to Nanomaterials, 8 Volume Set, ed. J. W. Steed and P. A. Gale, Wiley, 2012, p. 473 Search PubMed
- See for example: T. Takeuchi, V. Bagnacani, F. Sansone and S. Matile, ChemBioChem, 2009, 10, 2793 CrossRef CAS PubMed
- J. C. M. Stewart, Anal. Biochem., 1980, 104, 10 CrossRef CAS PubMed
- H. C. Gaede and R. E. Stark, J. Chem. Educ., 2001, 78, 1248 CrossRef CAS
- G. Rouser, S. Fleische and A. Yamamoto, Lipids, 1970, 5, 494 CrossRef CAS PubMed
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details on liposome preparations and ESI figures. See DOI: 10.1039/c5ob02480c |
| ‡ Permanent address: Department of Chemistry, Bilkent University, 06800 Bilkent, Ankara, Turkey. |
| § Initially, we also attempted to use 31P NMR with neat LUV stock solutions in water using a nested inner tube with the standard, but the signals were too broad to be reliably integrated. Addition of organic solvents to dissolve the liposomes sacrificed the sensitivity to such an extent that the 31P NMR method became impractical for routine analysis. |
| This journal is © The Royal Society of Chemistry 2016 |