
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of dihydroisoquinolones for fragment-based drug discovery (FBDD)†
Nick
Palmer
,
Torren M.
Peakman
,
David
Norton
and
David C.
Rees
*
Astex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge CB4 0QA, UK. E-mail: david.rees@astx.com
First published on 24th December 2015
Abstract
This study describes general synthesis aspects of fragments for FBDD, as illustrated by the dihydroisoquinolones 1–3. Previous Rh(III) methodology is extended to incorporate amines, heteroatoms (N and S), and substituents (halogen, ester) as potential binding groups and/or synthetic growth points for fragment-to-lead elaboration.
Introduction
Fragment-based drug discovery
FBDD is well-established within many Pharma, biotech and academic institutions as a technology for generating new chemical matter for drug discovery.1 Over 20 compounds derived from fragment-based drug discovery (FBDD) are being evaluated in clinical trials2 and the BRAF kinase inhibitor, vemurafenib (Zelboraf) is a launched drug for late stage melanoma.3Organic chemistry is one of the enabling sciences required for FBDD and has been critical for the successful development of this field during the last decade. However, as FBDD has developed it has unearthed a pressing need to design and synthesise more fragments and to develop new synthetic methodology to convert fragments into leads. The successful prosecution of FBDD projects often requires new organic synthesis methodology which, not surprisingly, is frequently time-critical. This is increasingly an issue as more difficult protein drug discovery targets are screened. There are two areas in particular that need synthetic methodology research: (1) the design and synthesis of new fragments and (2) the elaboration of low affinity (μM or mM) fragments into high affinity (nM) leads using precisely controlled synthetic growth points (vectors).
Compared to drugs or high throughput screening (HTS) compounds, fragments are small, simple molecules. A key advantage of screening low MW fragments (in comparison with higher MW, HTS-like compounds) is the improved hit rate resulting from screening such small compounds. Fragments are designed to form efficient binding interactions with protein targets but, due to their small size, the typical binding affinity of a fragment is only mM or μM. Table 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 summarises guidelines indicating some of the chemical properties that we use today as part of the selection process for adding new fragments into the Astex screening library. These guidelines have evolved significantly compared to earlier descriptions of fragments as a result of 10 years of screening fragments by X-ray crystallography and other biophysical techniques.5 There are many other publications describing fragment library design6 and there are fragments with properties outside those listed in Table 1 that have been used to generate useful leads, so this is still an emerging area.
4 summarises guidelines indicating some of the chemical properties that we use today as part of the selection process for adding new fragments into the Astex screening library. These guidelines have evolved significantly compared to earlier descriptions of fragments as a result of 10 years of screening fragments by X-ray crystallography and other biophysical techniques.5 There are many other publications describing fragment library design6 and there are fragments with properties outside those listed in Table 1 that have been used to generate useful leads, so this is still an emerging area.
| Property | Guideline |
|---|---|
| Molecular recognition | Diverse, usually polar groups for binding to a protein (a single pharmacophore). An aspiration is to express any given binding pharmacophore in a variety of chemotypes |
| Synthetic vectors | Multiple synthetically accessible positions for fragment growth in 3 dimensions to form new binding interactions |
| Physico-chemical properties | Molecular weight: ∼140–230 |
| Non-hydrogen atoms: 10–16 | |
| Lipophilicity (clogP): ∼0.0 to 2.0 |
|
| Properties commensurate with biophysical screening at high concentrations, e.g., aqueous solubility (preferably ≥5 mM in 5% DMSO, or other screening co-solvents); stability (>24 h in solution); avoid compounds/functional groups known to be associated with high reactivity, aggregation in solution, or false positives.7 NB: aromatic C–H bonds may assist NMR screening | |
| Synthetic tractability | Typically, ∼50–100 mg and ≤4 steps from commercially available reagents |
| Shape | Variety of 2- and 3-dimensional shapes for each scaffold and pharmacophore |
| Number of freely rotatable bonds: 0–3 | |
| Number of chiral centres: 0–1, sometimes 2 |
To grow fragments into leads requires the incorporation of additional binding groups to interact with the protein target. For this, it is desirable to rapidly elaborate the fragment in any direction with either polar or non-polar groups, ideally with regio- and stereo-control utilising synthetic methodology that has been worked out in advance. These requirements place big demands on synthetic methodology and represent an opportunity to significantly improve FBDD.
Selection of dihydroisoquinolones as fragments
Dihydroisoquinolone (1, Fig. 1) is a simple, small molecule (MW = 147, clogP = 1.0) and contains a strong hydrogen-bonding motif in the form of the cis-amide. Low complexity8 plus a polar binding motif are important factors to increase the probability of a fragment being a screening hit. The related fragments 2 and 3 are examples of compounds that could be made after 1 is identified as a fragment hit from a screening campaign. Furthermore, compounds 2 and 3 are attractive as fragments in their own right due to:
• low MW (177, 204 respectively) and low clogP (0.32, 1.3 respectively)
 | ||
| Fig. 1 Dihydroisoquinolones with attractive fragment-like properties.‡ | ||
• sufficient aqueous solubility (>5 mM) for screening
• polar H-bonding groups (amide + alcohol/amine)
• numerous positions available for synthetic elaboration (fragment-to-lead stage)
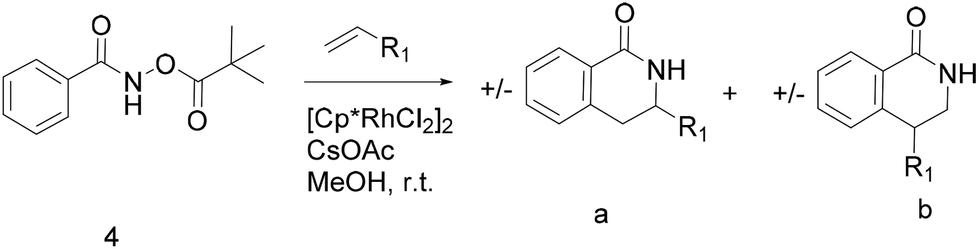
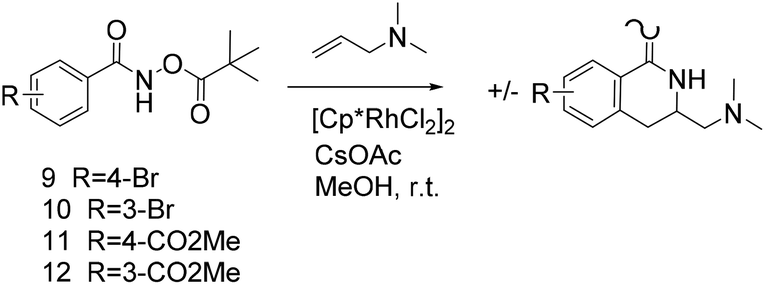
Thus, we selected the dihydroisoquinolones 2 and 3 as prototype examples to develop an FBDD-friendly synthetic methodology. The synthesis of fragment 2 has been reported previously using Rh(III) catalysed C–H bond activation methodology (Fig. 2).9 This synthesis uses readily available starting materials; proceeds at room temperature with no requirement to exclude air; is amenable to >100 mg scale and frequently the two regioisomers (a and b) can be separated by chromatography. Whilst both of these regioisomers meet many of the guidelines in Table 1 we prefer fragments not to have distally separated polar binding groups hence we prefer isomer (a) for FBDD. The methodology in Fig. 2 utilises catalytic rhodium C–H bond activation and has been developed in several subsequent publications.10 The mechanism is thought to involve metal coordination to the nitrogen moiety which directs cyclometalation to the ortho C–H bond. Alkene insertion occurs via coordination to the metal and the acyl protecting group acts as an internal oxidant. This methodology has also been reported with alkynes leading to isoquinolones and extended to substituted alkenes (for dihydroisoquinolones).9–11 The versatility of the methodology is further illustrated with other publications that describe, for related systems, chiral induction at the asymmetric carbon utilising an engineered metalloenzyme10d,e,12 or controlling the ratio of the two regioisomers by modulating the steric properties of the Rh(III) catalyst.10b
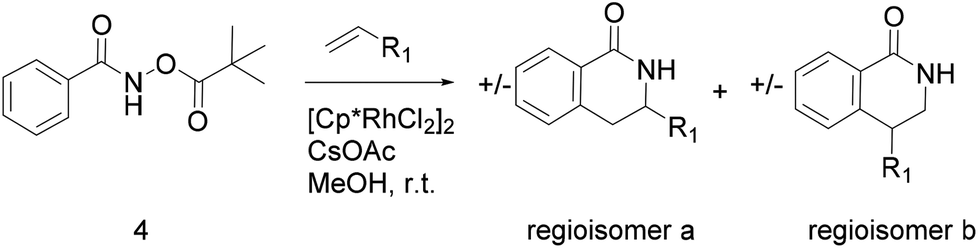 | ||
| Fig. 2 Previously published synthesis of dihydroisoquinolones utilising Rh(III) catalytic C–H activation.9,10c | ||
Extending the dihydroisoquinolone methodology for FBDD
Despite the modern synthetic methodology of Fig. 2 and the attractive fragment properties, we perceived some additional requirements from an FBDD perspective. Firstly, many drugs contain amino groups or heterocyclic aromatic rings. Fortunately, some methodology for incorporating heteroatoms into compounds related to 2 is known10a,b and we sought to apply this methodology to the functionalised dihydroisoquinolone fragments. Secondly, our FBDD projects are frequently challenged by having to work out methodology to access synthetic growth points or vectors in 3D space. One fragment may bind to different protein targets, requiring different synthetic vectors in each case, as deduced by examining the protein-fragment X-ray crystal structure. An attraction of the dihydroisoquinolone fragment template is the numerous positions available for synthetic elaboration during the fragment-to-lead stage; these are shown in Fig. 3 as blue arrows. The Rh(III) methodology has already been shown to permit several of these growth points into fragment 1.9,10,10c–e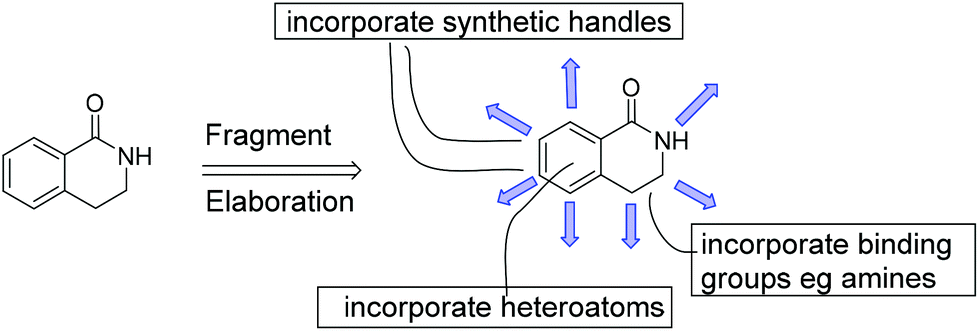 | ||
| Fig. 3 Schematic representation of synthetic elaboration of fragment 1. Growth positions are shown as blue arrows and the incorporation of aromatic heteroatoms and polar binding groups are indicated. | ||
Results and discussion
Use of substituted alkenes to give polar functionality for protein binding and/or further synthetic elaboration (Table 2)
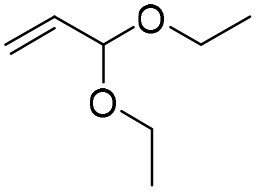
Compound 4 is readily available following literature procedures10c and was used to explore reactions with mono substituted alkenes. The alkenes selected were chosen to expand those previously described in the literature and allow introduction of new (protected) functional groups useful for subsequent transformations or protein binding interactions.|
|
||||
|---|---|---|---|---|
| Alkene | Product a | Product b | Isolated yield % | Regio-selectivity (ratio a:b estimated by NMR) |
|

|
59 | >20:1 |
|
|

|
|
a + b: 93 | 1:1 |
|
|
|
6a:42 | 1:1 |
| 6b:42 | ||||

|
|
63 | >20:1 |
|
|
|
74 | >20:1 |
|
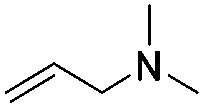
Reaction with N,N-dimethyl allylamine gives a single regioisomeric product, 3 (59% after 24 h, note long time required possibly due to the basic amine coordinating to the catalyst). This is the first example that we are aware of where an amino substituted alkene is used in this procedure. Compound 3 is interesting as a fragment itself, HAC = 15, MW = 204, clogP = 1.3, measured solubility ≥5 mM, (Fig. 1). This led us to explore this alkene in more functionalised systems (see Tables 3 and 4).
|
|
||
|---|---|---|
| Hydroxamate | Product | Isolated yield % |
|

|
54 |
|
|
53 |
|
|
84 |
|
|
56 |


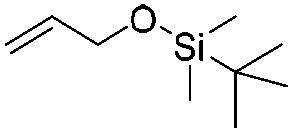
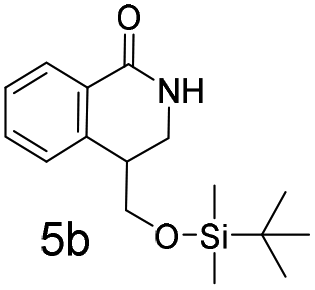
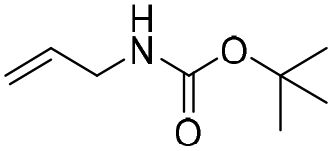
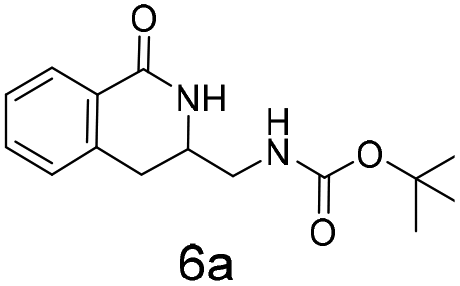
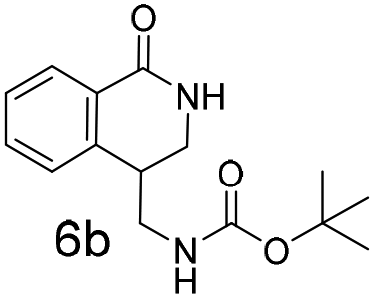
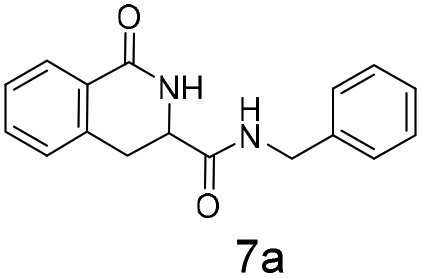
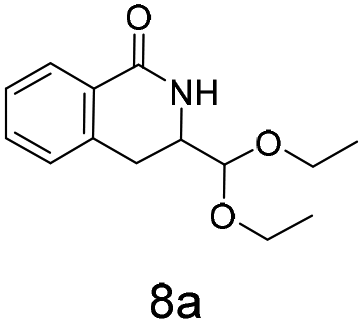
The silyl protected allyl alcohol and Boc protected allyl amine gave the cyclised products 5a,b and 6a,b respectively as 1:1 mixtures of regioisomers in good yields. These protected hydroxyl and amino substituted analogues can be deprotected to provide groups capable of forming new polar binding interactions and/or synthetic growth vectors. Benzyl substituted acrylamide gives a single regioisomer, 7a (63%). This is the first example that we are aware of using an amide in this transformation (but note that 7a has been synthesised previously by a different route13 and is commercially available in milligram quantities). If this growth vector is required, the formation of a single regioisomer is an advantage. Acrolein diethylacetal gives a single regioisomer, 8a (74%) and again the protected aldehyde can provide a handle for a range of subsequent transformations.
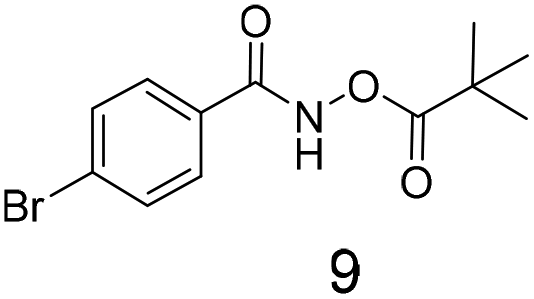
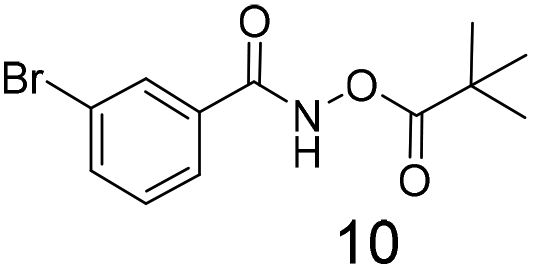
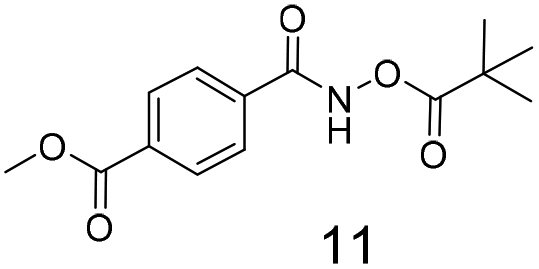
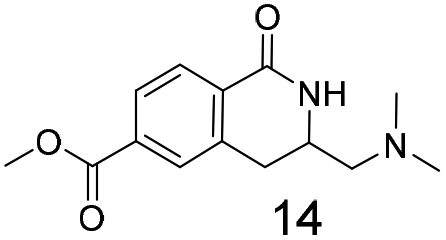
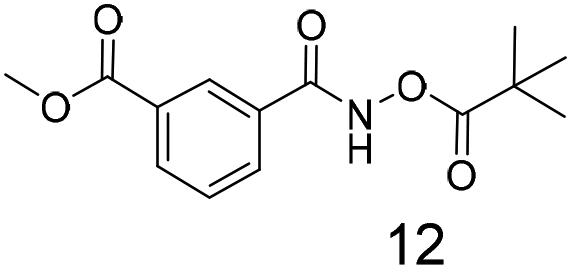
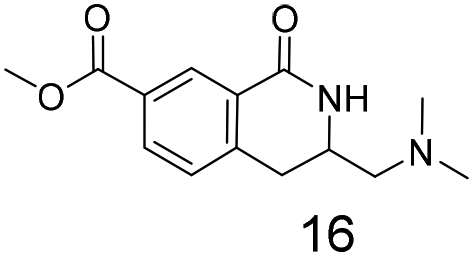
We selected aromatic bromides and esters for incorporation into the hydroxamates to probe the methodology for fragment-to-lead elaboration (Table 3). Note that compounds 13–16 are not designed to be fragments themselves. Rather, their synthesis builds confidence that if fragment 3 binds to a protein with an interaction that shows opportunity for elaboration via the meta or para positions, synthetic methodology is already established to follow-up rapidly.
Modest to good yields and a single regioisomer are observed for 13, 14, 16. The meta-bromo substituted analogue 10 gave a small amount of product corresponding to cyclisation onto the position ortho to the bromine. This product was not isolated in a pure form, but 1H NMR of a mixed fraction was consistent with this isomer (see ESI†).
There is some precedent for using the para-methyl ester substituted hydroxamate, 1110c,14 but the meta-methyl ester substituted hydroxamate, 12 has not been reported previously.
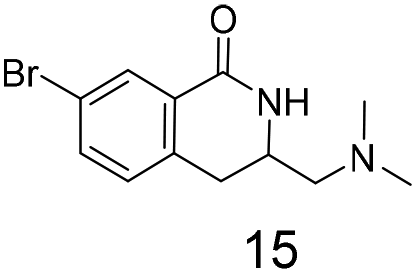
In our hands preliminary attempts to cyclise with ortho Br or OMe substituted hydroxamates were unsuccessful perhaps due to unfavourable steric interactions between the ortho substituent and the hydroxamate carbonyl (data not shown). An ortho iodide substituent on a closely related system has been reported15 but this remains an area requiring additional research.
Application of methodology to heteroaromatic analogues (Table 4 and Scheme 1)
Incorporating heteroatoms into aromatic rings of fragments is highly desirable in order to: modulate physicochemical properties (clogP, solubility etc.); probe fragment binding interactions; increase opportunities for fragment-to-lead synthetic elaboration, and to subtly alter the growth vectors. Hence, we investigated three heterocycles commonly found in biologically active leads and drugs: thiophenes, thiazoles and pyridines.
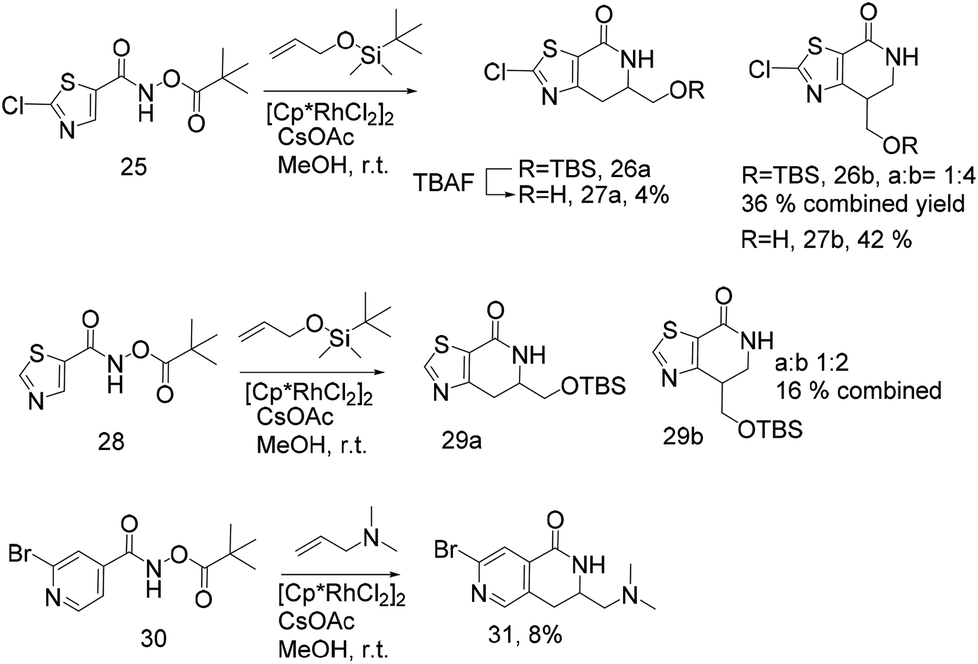 | ||
| Scheme 1 | ||
There are a few reports of the Rh(III) methodology being applied to thiophenes11 and pyridines16 (compound 17 has been used with a vinyl ester11) but from a fragment perspective the scope of the methodology is limited and we are not aware of thiazoles being used previously.
The thiophenes in Table 4 are derivatised with the same (protected) polar groups used above: amines, alcohols and amides. The aromatic bromo substituents provide potential synthetic growth points for the fragment-to-lead stage.
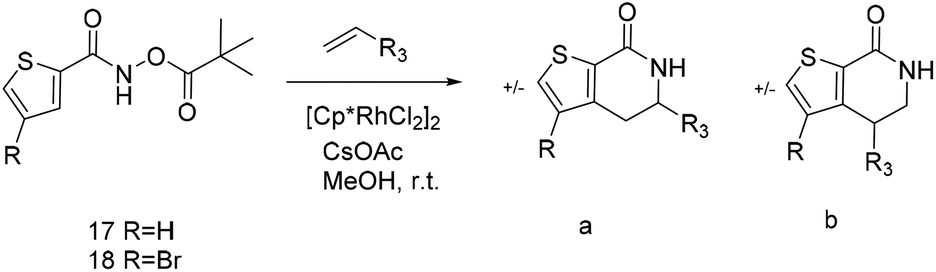
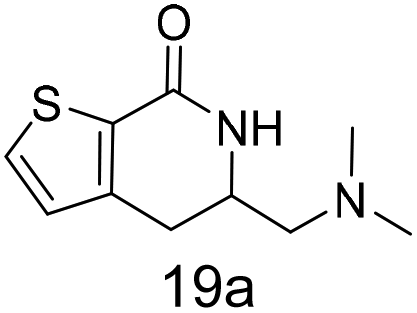
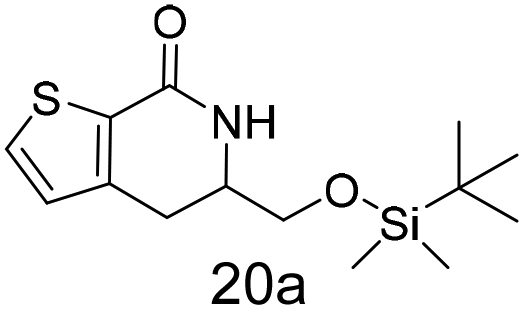
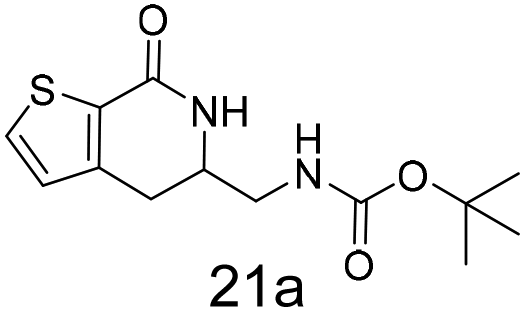
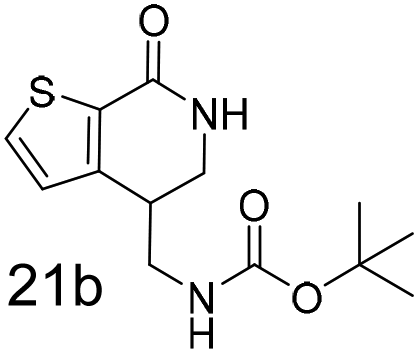
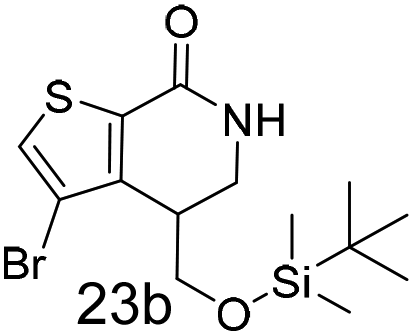
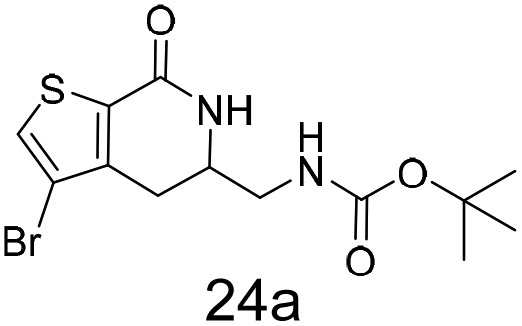
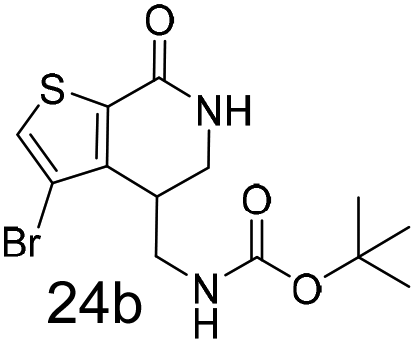
N,N-Dimethyl allyl amine reacted with both the substituted and unsubstituted thiophenes to give 19a and 22a (31, 30% yields unoptimised). Compound 19a can be considered as a fragment itself (HAC = 14, MW = 210, clogP = 1.1) and since aromatic sulphurs are known to form binding interactions with carbonyls they provide an additional protein binding possibility. Reaction of the thiophene hydroxamates 17 and 18 with the TBDMS protected allyl alcohol gives 20a,b and 23a,b respectively and Boc protected allyl amine gives 21a,b and 24a,b in good yields and with a similar isomer ratio.
Scheme 1 extends the method to thiazoles. The chloro thiazole hydroxamate, 25 was prepared using the standard conditions and treatment with TBDMS protected allyl alcohol generated the two expected isomers 26a and 26b in a 1:4 ratio. Removal of the silyl group produced a mixture of regioisomers that could be separated into 27a and 27b, albeit in low yield (4% and 42% respectively) (Scheme 1). Unoptimised attempts to use N,N-dimethyl allyl amine failed in this reaction. The TBDMS-protected alkene also reacted with the unsubstituted thiazole 28, although the unoptimised yield of 29a,b was low (16%) and regioisomer b is the major product.
Based upon literature precedent this methodology was known to be less suitable for pyridyl analogues due to the lower reactivity of these electron deficient systems and formation of regioisomer mixtures. This has been overcome to some extent by the use of a pyridine N-oxide to increase the reactivity of the pyridine ring.10a Compound 30 (Scheme 1), synthesised following standard procedures, was treated with N,N-dimethyl allylamine to give a mixture of isomers and 31 was isolated by preparative HPLC but the (unoptimised) yield is only 8% and as previously noted, higher catalyst loading was used. Our results indicate that this methodology is not yet ideal for heterocycles and given their importance in drug discovery this is another area that would benefit from additional research.
Conclusions
The emergence of FBDD has created new demands for organic synthesis. Despite the apparent simplicity of fragment-like compounds they present significant and growing challenges, e.g., methodology to design and synthesise new fragments and methodology to grow fragments into high affinity chemical leads via synthetic growth points (vectors) according to the particular fragment–protein binding interactions. These two aspects are frequently rate limiting in FBDD.This study identified the dihydroisoquinolones, 1–3 as having attractive fragment-like chemical properties (see Table 1). A published method to synthesise compound 2 utilising Rh(III) catalysed C–H bond activation9,10c has been extended to increase the utility of this template for fragment-based drug discovery. We have focused on:
• Adding polar side chains capable of exploring new binding interactions with proteins (tertiary amines and protected amines, alcohols and amides).
• Incorporating S and N heteroatoms into the aromatic ring to modulate logP, solubility, synthetic growth vectors etc.
• Adding functional groups that can be used as synthetic points for fragment growth during fragment-to-lead optimisation (aromatic bromides and esters).
These results illustrate frequently encountered synthetic organic chemistry challenges that we face in FBDD including:
• Synthetic methodology that is not exemplified in the presence of heterocyclic aromatics and polar functional groups e.g. amines.
• Synthetic methodology that is not exemplified for water soluble compounds (we require low lipophilicity, Table 1).
• Requirement for multiple, synthetically accessible fragment growth positions (Fig. 3) and methodology that allows regio- and stereo-selective fragment growth in the presence of polar functionality.
Some of the chemistry methodology challenges for FBDD are similar to other drug discovery techniques including HTS, e.g. the desire to incorporate polar side chains and heteroatoms into lead molecules.17 Others, such as the small size and lipophilicity of fragments; the requirement for synthetic vectors in multiple directions; and the drive to use X-ray crystallography to design bespoke synthetic targets, are more evident in FBDD laboratories. We see a pressing need for even more synthetic organic chemists to engage with these challenges.
Experimental
General experimental
All reactions were carried out without exclusion of air or moisture unless otherwise stated. Commercial solvents and reagents were used without further purification. Reactions were monitored by HPLC-MS on an Agilent 1200 series instrument, coupled with an Agilent 1200 MWD SL UV detector and Agilent 6140 single quadrupole mass detector. Normal phase flash chromatography was performed using a Biotage Isolera and silica SNAP cartridges. Petrol refers to petroleum ether (b.p. 40–60 °C). Reverse phase chromatography was performed using a Biotage SP4 and Biotage C18 Ultra cartridges with an acetonitrile in water gradient, without use of modifiers. Preparative HPLC was performed on an Agilent 1260 instrument, using Waters prep columns (Atlantis prep T3 OBD 5 μm 100 × 19 mm, Sunfire prep C18 OBD 5 μm 100 × 19 mm or XBridge C18 5 μm OBD 100 × 19 mm). HRMS (ESI) spectra were recorded on an Agilent 6550 QTOF instrument.1H, 13C, 13C-Dept, COSY, HSQC and HMBC NMR spectra were recorded using Bruker 400 MHz or 500 MHz spectrometers (CDCl3 or d6-DMSO as solvent). Chemical shifts for 1H and 13C NMR spectra are reported as δ in units of parts per million (ppm) downfield from SiMe4 (δ 0.0) using the chemical shifts of the residual solvent peaks. Multiplicities are given as: s (singlet); brs (broad singlet); d (doublet); t (triplet); q (quartet); dd (doublets of doublet); td (triplet of doublet); m (multiplets) etc. Coupling constants are reported as J values in Hz. 13C chemical shifts were measured using direct 13C NMR or via 2D HSQC and HMBC spectra.
General procedure A. Step 1: to a suspension of the carboxylic acid (20 mmol, 1.0 eq.) in dry CH2Cl2 (20 mL) at room temperature under a nitrogen atmosphere was added a catalytic amount of dry DMF (4 drops). Oxalyl chloride (15 mL, 30 mmol, 1.5 eq., 2 M soln in DCM) was added dropwise over about 20 minutes with care as effervescence occurs. The reaction was allowed to stir at room temperature until completion which was judged to be when no further effervescence could be seen and in some cases, the solution became homogeneous. The solvent was then removed under reduced pressure to afford the corresponding crude acid chloride.
Step 2: hydroxylamine hydrochloride (1.39 g, 20 mmol, 1 eq.) was added to a biphasic mixture of K2CO3 (5.52 g, 20 mmol, 2.0 eq.) in a 2:1 mixture of EtOAc (40 mL) and H2O (20 mL). The resulting solution was cooled to 0 °C followed by dropwise addition of the crude acid chloride dissolved in a minimum amount of EtOAc. The flask containing the acid chloride was then rinsed with additional EtOAc. The reaction was allowed to warm to room temperature which in most cases resulted in formation of a thick white suspension of the hydroxamic acid. The reaction was stirred for 4 h to allow complete conversion of the acid chloride to hydroxamic acid then pivaloyl chloride (2.46 ml, 20 mmol) was added dropwise. The reaction can be monitored by TLC and HPLC and also the thick white precipitate dissolves as the reaction proceeds and the hydroxamic acid is consumed. Further pivaloyl chloride can be added if required. The phases were separated and the aqueous phase was extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and evaporated under reduced pressure. The crude products were purified by flash column chromatography using an appropriate solvent mixture of petrol/EtOAc as eluent.
Synthesis of 3,4 dihydroisoquinolones
:1 mixture of regioisomers (5a and 5b) that were inseparable by column chromatography. The mixture was a white solid, 93% combined yield (123 mg) from phenylformamido 2,2-dimethylpropanoate (4) (0.45 mmol, 100 mg, 1 equiv.). Partial separation of the regioisomers was possible by reverse phase chromatography, eluting 45–65% MeCN in water to give isomer 5a (18 mg) plus 5b (16 mg) in pure form with a mixed fraction (60 mg). δH (400 MHz, chloroform-d) 8.08 (1 H, dd, J 7.6, 1.5), 7.44 (1 H, td, J 7.6, 1.5), 7.39–7.30 (1 H, m), 7.22–7.16 (1 H, m), 6.25 (1 H, brs), 3.87–3.78 (1 H, m), 3.74 (1 H, dd, J 10.0, 1.0), 3.60 (1 H, dd, J 10.0, 8.4), 2.93–2.77 (2 H, m), 0.90 (9 H, s), 0.08 (3 H, s), 0.07 (3 H, s). δC (101 MHz, CDCl3) 165.74, 137.25, 132.26, 128.69, 128.14, 127.46, 127.08, 65.66, 52.68, 30.27, 25.83, 18.27, −5.45. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C16H25NO2Si 292.1728; found 292.1736. δ = 2.53 ppm.
:2 mixture of a:b as judged by 1H NMR. The mixture was a white solid in 52% combined yield (88 mg) from (4-bromothiophen-2-yl)formamido 2,2-dimethylpropanoate (18) (0.45 mmol, 137 mg, 1 equiv.). δH (400 MHz, DMSO-d6) 7.96 (1 H, s), 7.77 (1 H, d, J 2.9), 3.82–3.68 (1 H, m), 3.65–3.48 (2 H, m), 2.89 (1 H, dd, J 16.8, 6.6), 2.79 (1 H, dd, J 16.8, 5.5), 0.82 (9 H, s), 0.00 (6 H, s). δC (101 MHz, DMSO) −5.10, 17.78, 25.52, 26.15, 52.92, 64.84, 109.76, 128.97, 131.00, 141.64, 160.04. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C14H22BrNO2SSi 376.0397; found: 376.0391. δ = 1.60 ppm.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (Me)3 under chloroform resonances. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C13H17BrN2O3S 361.0216; found: 361.0214. δ = 0.55 ppm.
:4 mixture of a:b as judged by 1H NMR. The mixture was a white solid 36% yield (54 mg) from (2-chloro-1,3-thiazol-5-yl)formamido 2,2-dimethylpropanoate (25) (0.45 mmol, 118 mg, 1 equiv.). δH (400 MHz, DMSO-d6) 7.92 (1 H, d, J 2.8), 3.81–3.71 (1 H, m), 3.67–3.58 (2 H, m), 3.13 (1 H, dd, J 17.1, 7.1), 2.95 (1 H, dd, J 17.1, 5.4), 0.81 (9 H, s), 0.00 (6 H, s).
:2 mixture of a:b as judged by 1H NMR. The mixture was a white solid in 16% yield (22 mg) from (1,3-thiazol-5-yl)formamido 2,2-dimethylpropanoate (0.45 mmol, 102 mg, 1 equiv.). δH (400 MHz, DMSO-d6) 9.22 (1 H, s), 7.77 (1 H, d, J 2.7), 3.81–3.72 (1 H, m), 3.67–3.53 (2 H, m), 3.17 (1 H, dd, J 16.9, 6.8), 3.03 (1 H, dd, J 16.9, 5.8), 0.81 (9 H, s), −0.00 (6 H, s). δC (101 MHz, DMSO) 158.19, 157.76, 124.64, 51.95, 64.49, 26.57, 25.47, 17.78, −5.76. C
(Me)3 under chloroform resonances. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C13H17BrN2O3S 361.0216; found: 361.0214. δ = 0.55 ppm.
:4 mixture of a:b as judged by 1H NMR. The mixture was a white solid 36% yield (54 mg) from (2-chloro-1,3-thiazol-5-yl)formamido 2,2-dimethylpropanoate (25) (0.45 mmol, 118 mg, 1 equiv.). δH (400 MHz, DMSO-d6) 7.92 (1 H, d, J 2.8), 3.81–3.71 (1 H, m), 3.67–3.58 (2 H, m), 3.13 (1 H, dd, J 17.1, 7.1), 2.95 (1 H, dd, J 17.1, 5.4), 0.81 (9 H, s), 0.00 (6 H, s).
:2 mixture of a:b as judged by 1H NMR. The mixture was a white solid in 16% yield (22 mg) from (1,3-thiazol-5-yl)formamido 2,2-dimethylpropanoate (0.45 mmol, 102 mg, 1 equiv.). δH (400 MHz, DMSO-d6) 9.22 (1 H, s), 7.77 (1 H, d, J 2.7), 3.81–3.72 (1 H, m), 3.67–3.53 (2 H, m), 3.17 (1 H, dd, J 16.9, 6.8), 3.03 (1 H, dd, J 16.9, 5.8), 0.81 (9 H, s), −0.00 (6 H, s). δC (101 MHz, DMSO) 158.19, 157.76, 124.64, 51.95, 64.49, 26.57, 25.47, 17.78, −5.76. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O not determined. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C13H22N2O2SSi 299.1244; found 299.1247. δ = 1.02 ppm.
O not determined. HRMS (ESI-QTOF): m/z [M + H]+ Calcd for C13H22N2O2SSi 299.1244; found 299.1247. δ = 1.02 ppm.
Acknowledgements
The authors warmly thank our Astex colleagues, Stuart Whibley and Nicola Wilsher for providing the HRMS data.Notes and references
- (a) D. Erlanson, Introduction to Fragment-Based Drug Discovery, in Fragment-Based Drug Discovery and X-Ray Crystallography, ed. T. G. Davies and M. Hyvönen, Springer, Berlin Heidelberg, 2012, vol. 317, pp. 1–32 Search PubMed; (b) R. E. Hubbard and J. B. Murray, Chapter twenty - Experiences in Fragment-Based Lead Discovery, in Methods in Enzymology, ed. C. K. Lawrence, Academic Press, 2011, vol. 493, pp. 509–531 Search PubMed; (c) T. V. Magee, Progress in discovery of small-molecule modulators of protein–protein interactions via fragment screening, Bioorg. Med. Chem. Lett., 2015, 25(12), 2461–2468 CrossRef CAS PubMed; (d) C. W. Murray and D. C. Rees, The rise of fragment-based drug discovery, Nat. Chem., 2009, 1(3), 187–192 CrossRef CAS PubMed; (e) E. R. Zartler, Fragonomics: The -Omics with Real Impact, ACS Med. Chem. Lett., 2014, 5(9), 952–953 CrossRef CAS PubMed; (f) J. P. Burke, Z. Bian, S. Shaw, B. Zhao, C. M. Goodwin, J. Belmar, C. F. Browning, D. Vigil, A. Friberg, D. V. Camper, O. W. Rossanese, T. Lee, E. T. Olejniczak and S. W. Fesik, Discovery of Tricyclic Indoles That Potently Inhibit Mcl-1 Using Fragment-Based Methods and Structure-Based Design, J. Med. Chem., 2015, 58(9), 3794–3805 CrossRef CAS PubMed.
- M. Baker, Fragment-based lead discovery grows up, Nat. Rev. Drug Discovery, 2013, 12(1), 5–7 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Vemurafenib: the first drug approved for BRAF-mutant cancer, Nat. Rev. Drug Discovery, 2012, 11(11), 873–886 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Opportunity Knocks: Organic Chemistry for Fragment-Based Drug Discovery (FBDD), Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201506783 , published on line Nov 3, 2015.
- (a) M. Congreve, R. Carr, C. Murray and H. Jhoti, A ‘Rule of Three’ for fragment-based lead discovery?, Drug Discovery Today, 2003, 8(19), 876–877 CrossRef PubMed; (b) H. Jhoti, G. Williams, D. C. Rees and C. W. Murray, The ‘rule of three’ for fragment-based drug discovery: where are we now?, Nat. Rev. Drug Discovery, 2013, 12(8), 644–644 CrossRef CAS PubMed.
- (a) N. Baurin, F. Aboul-Ela, X. Barril, B. Davis, M. Drysdale, B. Dymock, H. Finch, C. Fromont, C. Richardson, H. Simmonite and R. E. Hubbard, Design and Characterization of Libraries of Molecular Fragments for Use in NMR Screening against Protein Targets, J. Chem. Inf. Comput. Sci., 2004, 44(6), 2157–2166 CrossRef CAS PubMed; (b) N. Blomberg, D. Cosgrove, P. Kenny and K. Kolmodin, Design of compound libraries for fragment screening, J. Comput. Aided Mol. Des., 2009, 23(8), 513–525 CrossRef CAS PubMed; (c) J. Fejzo, C. A. Lepre, J. W. Peng, G. W. Bemis, Ajay, M. A. Murcko and J. M. Moore, The SHAPES strategy: an NMR-based approach for lead generation in drug discovery, Chem. Biol., 1999, 6(10), 755–769 CrossRef CAS PubMed; (d) W. Lau, J. Withka, D. Hepworth, T. Magee, Y. Du, G. Bakken, M. Miller, Z. Hendsch, V. Thanabal, S. Kolodziej, L. Xing, Q. Hu, L. Narasimhan, R. Love, M. Charlton, S. Hughes, W. van Hoorn and J. Mills, Design of a multi-purpose fragment screening library using molecular complexity and orthogonal diversity metrics, J. Comput. Aided Mol. Des., 2011, 25(7), 621–636 CrossRef CAS PubMed; (e) A. Schuffenhauer, S. Ruedisser, A. Marzinzik, W. Jahnke, P. Selzer and E. Jacoby, Library Design for Fragment Based Screening, Curr. Top. Med. Chem., 2005, 5(8), 751–762 CrossRef CAS PubMed; (f) A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoelder, A. Jordan, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Fragment-based hit identification: thinking in 3D, Drug Discovery Today, 2013, 18(23–24), 1221–1227 CrossRef PubMed; (g) http://www.maggichurchouseevents.co.uk/bmcs/Downloads/Fragments%20-%20Mon%201115%20Brennan%20Paul.pdf .
- B. J. Davis and D. A. Erlanson, Learning from our mistakes: the ‘unknown knowns’ in fragment screening, Bioorg. Med. Chem. Lett., 2013, 23(10), 2844–2852 CrossRef CAS PubMed.
- (a) M. M. Hann, A. R. Leach and G. Harper, Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery, J. Chem. Inf. Comput. Sci., 2001, 41(3), 856–864 CrossRef CAS PubMed; (b) A. R. Leach and M. M. Hann, Molecular complexity and fragment-based drug discovery: ten years on, Curr. Opin. Chem. Biol., 2011, 15(4), 489–496 CrossRef CAS PubMed.
- N. Guimond, S. I. Gorelsky and K. Fagnou, Rhodium(III)-Catalyzed Heterocycle Synthesis Using an Internal Oxidant: Improved Reactivity and Mechanistic Studies, J. Am. Chem. Soc., 2011, 133(16), 6449–6457 CrossRef CAS PubMed.
- (a) J. R. Huckins, E. A. Bercot, O. R. Thiel, T.-L. Hwang and M. M. Bio, Rh(III)-Catalyzed C–H Activation and Double Directing Group Strategy for the Regioselective Synthesis of Naphthyridinones, J. Am. Chem. Soc., 2013, 135(39), 14492–14495 CrossRef CAS PubMed; (b) T. K. Hyster, D. M. Dalton and T. Rovis, Ligand design for Rh(III)-catalyzed C–H activation: an unsymmetrical cyclopentadienyl group enables a regioselective synthesis of dihydroisoquinolones, Chem. Sci., 2015, 6(1), 254–258 RSC; (c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, Rh(III)-Catalyzed Directed C–H Olefination Using an Oxidizing Directing Group: Mild, Efficient, and Versatile, J. Am. Chem. Soc., 2011, 133(8), 2350–2353 CrossRef CAS PubMed; (d) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C–H Activation, Science, 2012, 338(6106), 500–503 CrossRef CAS PubMed; (e) B. Ye and N. Cramer, Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C–H functionalization, Science, 2012, 338(6106), 504–506 CrossRef CAS PubMed.
- N. J. Webb, S. P. Marsden and S. A. Raw, Rhodium(III)-Catalyzed C–H Activation/Annulation with Vinyl Esters as an Acetylene Equivalent, Org. Lett., 2014, 16(18), 4718–4721 CrossRef CAS PubMed.
- B. Ye and N. Cramer, Asymmetric Synthesis of Isoindolones by Chiral Cyclopentadienyl-Rhodium(III)-Catalyzed C–H Functionalizations, Angew. Chem., Int. Ed., 2014, 53(30), 7896–7899 CrossRef CAS PubMed.
- D. Dou, P. Viwanathan, Y. Li, G. He, K. R. Alliston, G. H. Lushington, J. D. Brown-Clay, R. Padmanabhan and W. C. Groutas, Design, Synthesis, and In Vitro Evaluation of Potential West Nile Virus Protease Inhibitors Based on the 1-Oxo-1,2,3,4-tetrahydroisoquinoline and 1-Oxo-1,2-dihydroisoquinoline Scaffolds, J. Comb. Chem., 2010, 12(6), 836–843 CrossRef CAS PubMed.
- S. Wu, R. Zeng, C. Fu, Y. Yu, X. Zhang and S. Ma, Rhodium-catalyzed C–H functionalization-based approach to eight-membered lactams, Chem. Sci., 2015, 6(4), 2275–2285 RSC.
- N. Guimond, C. Gouliaras and K. Fagnou, Rhodium(III)-Catalyzed Isoquinolone Synthesis: The N–O Bond as a Handle for C–N Bond Formation and Catalyst Turnover, J. Am. Chem. Soc., 2010, 132(20), 6908–6909 CrossRef CAS PubMed.
- M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, Complementary Regioselectivity in Rh(III)-Catalyzed Insertions of Potassium Vinyltrifluoroborate via C–H Activation: Preparation and Use of 4-Trifluoroboratotetrahydroisoquinolones, Org. Lett., 2013, 15(7), 1528–1531 CrossRef CAS PubMed.
- A. Nadin, C. Hattotuwagama and I. Churcher, Lead-Oriented Synthesis: A New Opportunity for Synthetic Chemistry, Angew. Chem., Int. Ed., 2012, 51(5), 1114–1122 CrossRef CAS PubMed.
- Z. Zhang, Y. Yu and L. S. Liebeskind, N-Amidation by Copper-Mediated Cross-Coupling of Organostannanes or Boronic Acids with O-Acetyl Hydroxamic Acids, Org. Lett., 2008, 10(14), 3005–3008 CrossRef CAS PubMed.
- R. D. Farrant, J. C. Hollerton, S. M. Lynn, S. Provera, P. J. Sidebottom and R. J. Upton, NMR quantification using an artificial signal, Magn. Reson. Chem., 2010, 48(10), 753–762 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02461g |
| ‡ clogP is calculated from Daylight version 4.93 http://www.daylight.com/products/toolkit.html. Solubility measurements were carried out by 1H quantitative NMR at 500 MHz using the QUANTAS approach.19 Aliquots of fragments (100 mM in DMSO-d6) were diluted ×20 with buffer (40 mM phosphate, 100 mM TRIS, 15% D2O, pH = 7.1) to afford samples with nominal concentrations of 5 mM. A 2.5 mM sample of p-hydroxybenzoic acid, prepared by dilution of a 50 mM stock in DMSO-d6 ×20 with buffer was used as the qNMR standard. |
| This journal is © The Royal Society of Chemistry 2016 |