
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tetra-substituted 5-trifluoromethylpyrazoles via sequential halogenation/palladium-catalyzed C–C and C–N cross-coupling†
Carson
Wiethan
a,
Wilian C.
Rosa
a,
Helio G.
Bonacorso
*a and
Mark
Stradiotto
*b
aDepartamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria, Brazil. E-mail: helio.bonacorso@ufsm.br
bDepartment of Chemistry, Dalhousie University Halifax, NS, B3H 4R2 Canada. E-mail: mark.stradiotto@dal.ca
First published on 26th January 2016
Abstract
A mild and efficient protocol for the assembly of tetra-substituted 5-trifluoromethylpyrazoles is presented, involving halogenation at the 4-position of readily prepared tri-substituted 5-trifluoromethylpyrazoles to give 4-halo-1-phenyl-5-trifluoromethyl pyrazoles, and subsequent palladium-catalyzed Negishi or Buchwald–Hartwig cross-couplings to install carbon or nitrogen-based 4-substituents. Key to the success of these challenging cross-couplings is the use of XPhos and JosiPhos CyPF-tBu ligands, respectively.
Introduction
In recent years, N-aryl trifluoromethyl-substituted pyrazoles have emerged as important synthetic targets in both pharmaceutical and agricultural chemistry, owing to their unique biological properties.1The traditional condensation reaction between α,β-unsaturated carbonyl compounds provides an inexpensive and high-yielding route to tri-substituted trifluoromethylpyrazoles (Scheme 1A).2 Conversely, effective protocols for the assembly of related tetra-substituted structures are still lacking, mainly due to the difficulty in building the requisite synthons, and the low yields associated with such procedures (Scheme 1B and C).3 Furthermore, despite significant advances in palladium-catalyzed C–C and C–N cross-coupling methods, reactions involving five-membered heterocyclic electrophiles bearing more than one heteroatom have proven to be particularly challenging, with the majority of examples reported to date involving non-substituted pyrazoles (Scheme 1D), and in a few cases, involving 3-trifluoromethylpyrazoles.4 In this context, there exists a dearth of reports pertaining to the selective assembly of tetra-substituted 5-trifluoromethylpyrazoles. Given the potential utility of such compounds, in particular N-phenyl derivatives,1c,d we sought to develop protocols for the halogenation of the 3- substituted 1-phenyl-5-trifluoromethylpyrazoles,2 and subsequent elaboration into tetra-substituted heterocycles by use of palladium-catalyzed Negishi coupling or Buchwald–Hartwig amination (Scheme 1).
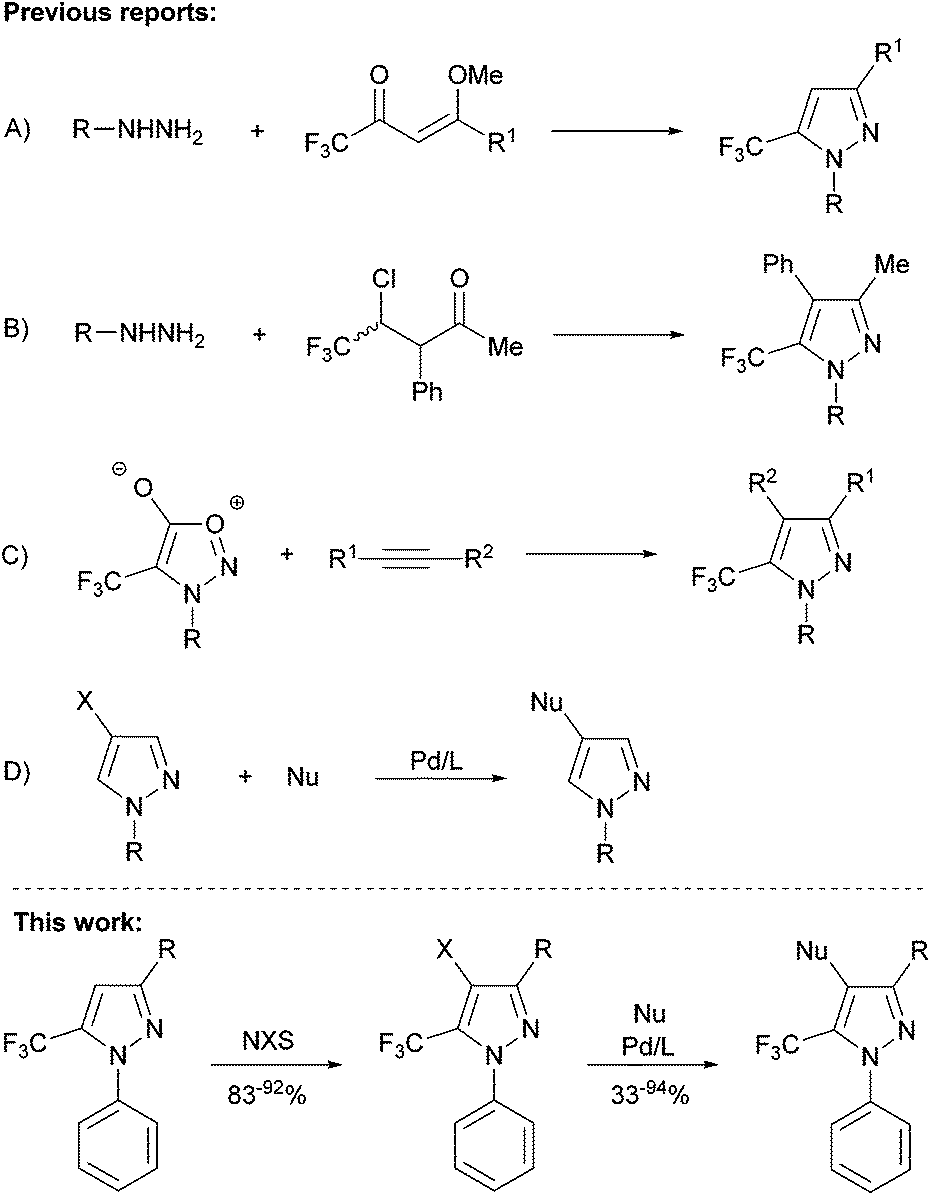 | ||
| Scheme 1 Synthesis of substituted trifluoromethylpyrazoles. | ||
We report herein on our successful development of such modular syntheses, in many cases leading to previously unreported tetra-substituted trifluoromethylpyrazoles, whereby the second step is enabled by the judicious selection of ancillary ligand for use in palladium catalysis.
Results and discussion
We commenced our investigations by targeting an expedient procedure for the halogenation of 3-substituted 1-phenyl-5-trifluoromethylpyrazoles at the 4-position (Table 1). Our initial efforts in this regard employing mild conditions reported by Zhao and Wang5 for the halogenation of non-CF3 containing pyrazoles were unsuccessful; this is likely due to the presence of the electron-withdrawing trifluoromethyl group at the adjacent position, which is known to decrease considerably the reactivity of the heterocycle. On the other hand, the use of more forcing conditions (i.e., high temperatures over long reaction times) resulted in the partial halogenation of the N-phenyl group. Given the similar polarity of the starting material and mono/poly-halogenated products, reaction conditions that afforded clean pyrazole monohalogenation selectivity were required. The 4-iodopyrazoles 2a,b were obtained in good yields by using an adapted protocol inspired by that described by Li and co-workers6 for the iodination of non-CF3 containing pyrazoles, employing NIS in TFA/AcOH medium (3 h, 80 °C), and using conventional rather than microwave heating. Whereas the same methodology when extended to NBS resulted in polyhalogenation, further optimization of the methodology described by Jeong and co-workers4e employing DMF as solvent furnished the 4-bromopyrazoles 2c,d in excellent yields after 2 h of stirring at 80 °C (Table 1).
a Reaction conditions: (i) iodinated pyrazoles (2a,b): 1a,b (5 mmol), NIS (6 mmol), TFA/AcOH (10 ml, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 80 °C, 3 h. (ii) Brominated pyrazoles (2c,d): 1a,b (5 mmol), NBS (12.5–15 mmol), DMF (10 ml), 80 °C, 2 h. :1), 80 °C, 3 h. (ii) Brominated pyrazoles (2c,d): 1a,b (5 mmol), NBS (12.5–15 mmol), DMF (10 ml), 80 °C, 2 h.
|
|---|
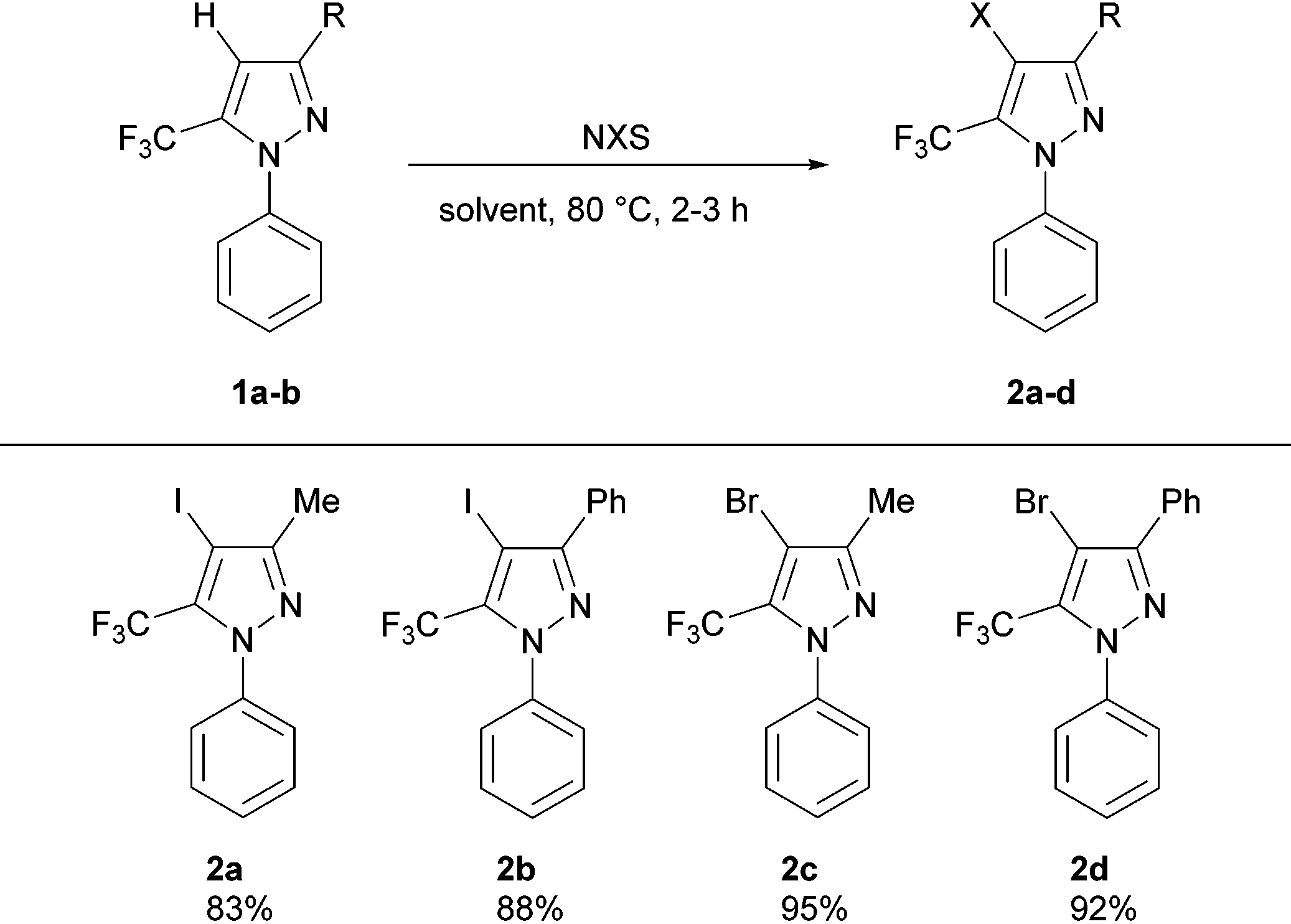
|
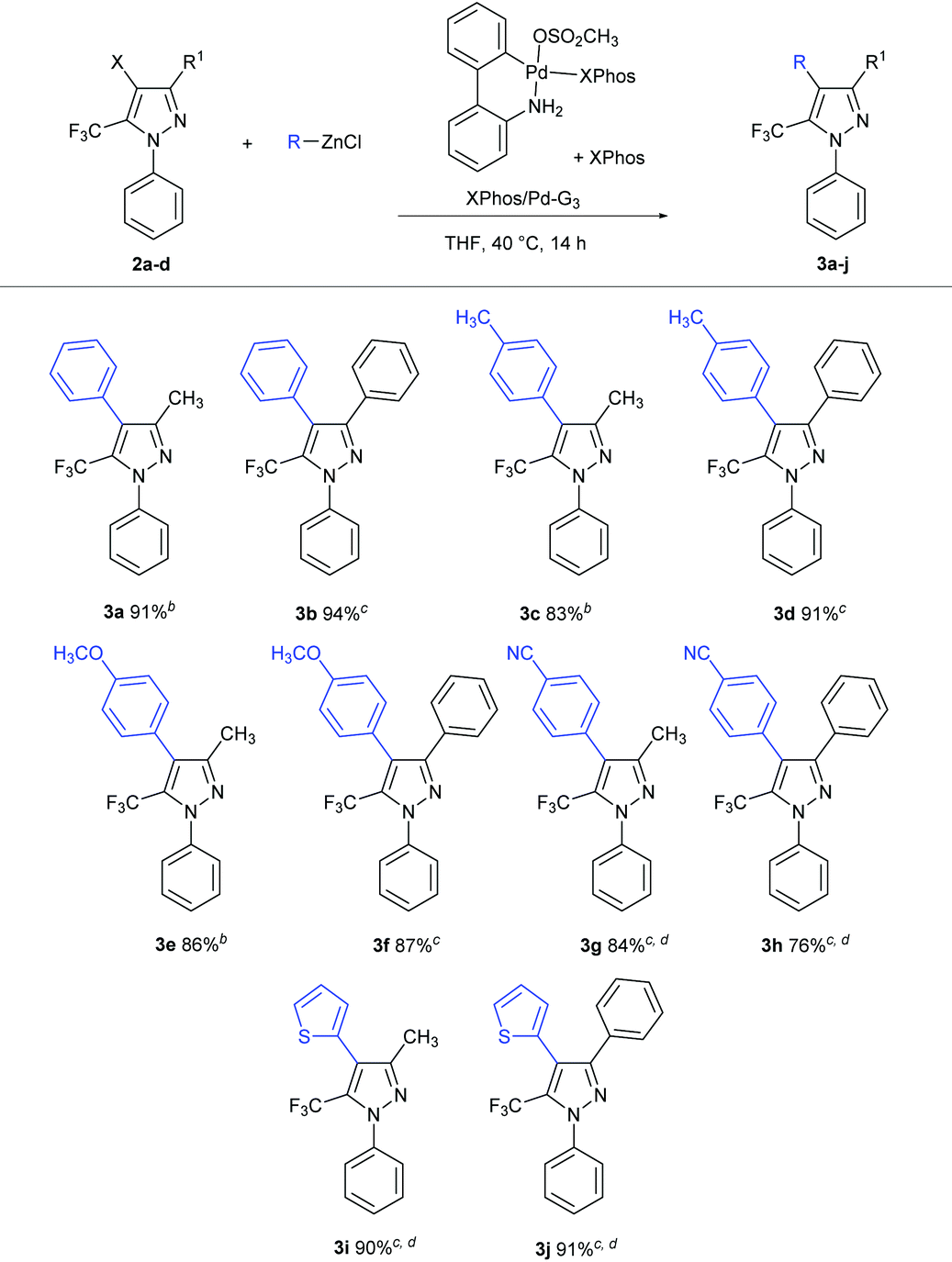
With the 4-halopyrazoles 2a–d in hand, we turned our attention to the application of relatively inexpensive and readily accessed organozinc reagents in Negishi cross-coupling chemistry as a means of (hetero)arylating the trifluoromethylpyrazole core. Initial screening involving the reaction between 2a and 3 equivalents of phenylzinc chloride (60 °C, 24 h) employing 10 mol% of Pd(PPh3)4 as catalyst provided a conversion rate of 45%. Further efforts to optimize the catalyst performance (i.e., longer reaction times, higher catalyst loadings) were ineffective, thereby underscoring the relative inefficiency of this simple catalyst for such challenging substrates.
In 2013 Buchwald and co-workers reported on Negishi cross-couplings involving various heterocycles, including a single example of a cross-coupling featuring a 5-chloro-3-trifluoromethylpyrazole, enabled by the use of biarylphosphine palladacycle pre-catalysts.7 Among the phosphines tested, XPhos exhibited optimal catalytic performance, providing excellent results under relatively low catalytic loadings (1 mol%). Encouraged by this report, we focused our attention on the use of the XPhos/Pd-G3 pre-catalyst; given the potential difficulties associated with purifying remaining pyrazole starting material from the products in such transformations, our main focus in optimizing was on achieving full conversion. While the original conditions described for such reactions with halopyrazoles (1.5 equiv. organozinc chloride, 1 mol% XPhos/Pd-G3 plus additional XPhos (1:1), room temperature, 12 h) were promising,7 we found that optimal yields for our substrates of interest (2a–d) were obtained by use of 1.8 equiv. organozinc and double the catalyst loading at 40 °C, over 14 h. Such conditions afforded excellent yields of the target C–C cross-coupling product when using electron-neutral or electron-rich arylzinc reagents (3a–f, 83–94%; Table 2), with little difference in reactivity observed between bromo and iodo pyrazoles. The use of 2.5 equiv. of the Negishi reagent derived from 4-bromobenzonitrile proved necessary to achieve the full conversion en route to products 3g,h. The stereoelectronic influence of the trifluoromethyl group was apparent in reactions involving heterocyclic Negishi reagents in which the heteroatom features non-shared electron pairs, as evidenced by the required use of 2.5 equiv. of the Negishi reagent derived from 2-chlorothiophene (3i,j). Notably, analogous reactions employing 2- or 3-pyridylzinc chlorides were unsatisfactory (<25% conversion on the basis of GC data).
We subsequently turned our attention to the use of Buchwald–Hartwig amination chemistry as a means of preparing nitrogen-functionalized trifluoromethylpyrazoles. The cross-coupling of the pyrazole 2c and n-octylamine was selected as a preliminary test reaction, in which a selection of ligands with established capability in amination chemistry were examined (Table 3). In contrast to the Negishi couplings discussed above, the application of XPhos (L1) proved ineffective under the Buchwald–Hartwig amination test conditions employed (entry 1). The use of BippyPhos (L2), a ligand that has been successfully employed in a range of challenging amination reactions,8 also did not provide any appreciable conversion (entry 2). We were surprised to find that tBu-BrettPhos (L3), which has been employed successfully in aminations involving pyrazole electrophiles,9 provided only modest conversion under the test conditions (entry 3). The ligand Mor-DalPhos (L4) has proven useful in a range of primary amine monoarylations,10 and displayed some competence in the test reaction; however, a high amount of dehalogenated product was also observed (entry 4). In contrast, we were pleased to observe that the use of the JosiPhos ligand variant L5 (CyPF-tBu)11 afforded high conversion to 4a, without significant accompanying hydrodehalogenation, and without the need for excess ligand (Pd: L5 = 1:1; entry 5).
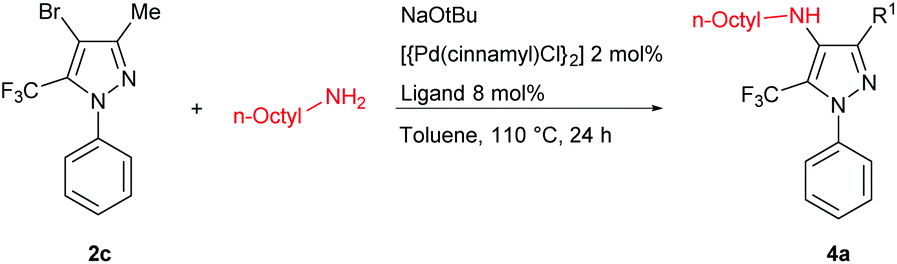
Having identified a suitable catalyst system for the amination of 2c, we then turned our attention to exploring the scope of reactivity (Table 4); for simplicity, we focused on the use of the bromides 2c,d and each proved to be a suitable substrate.
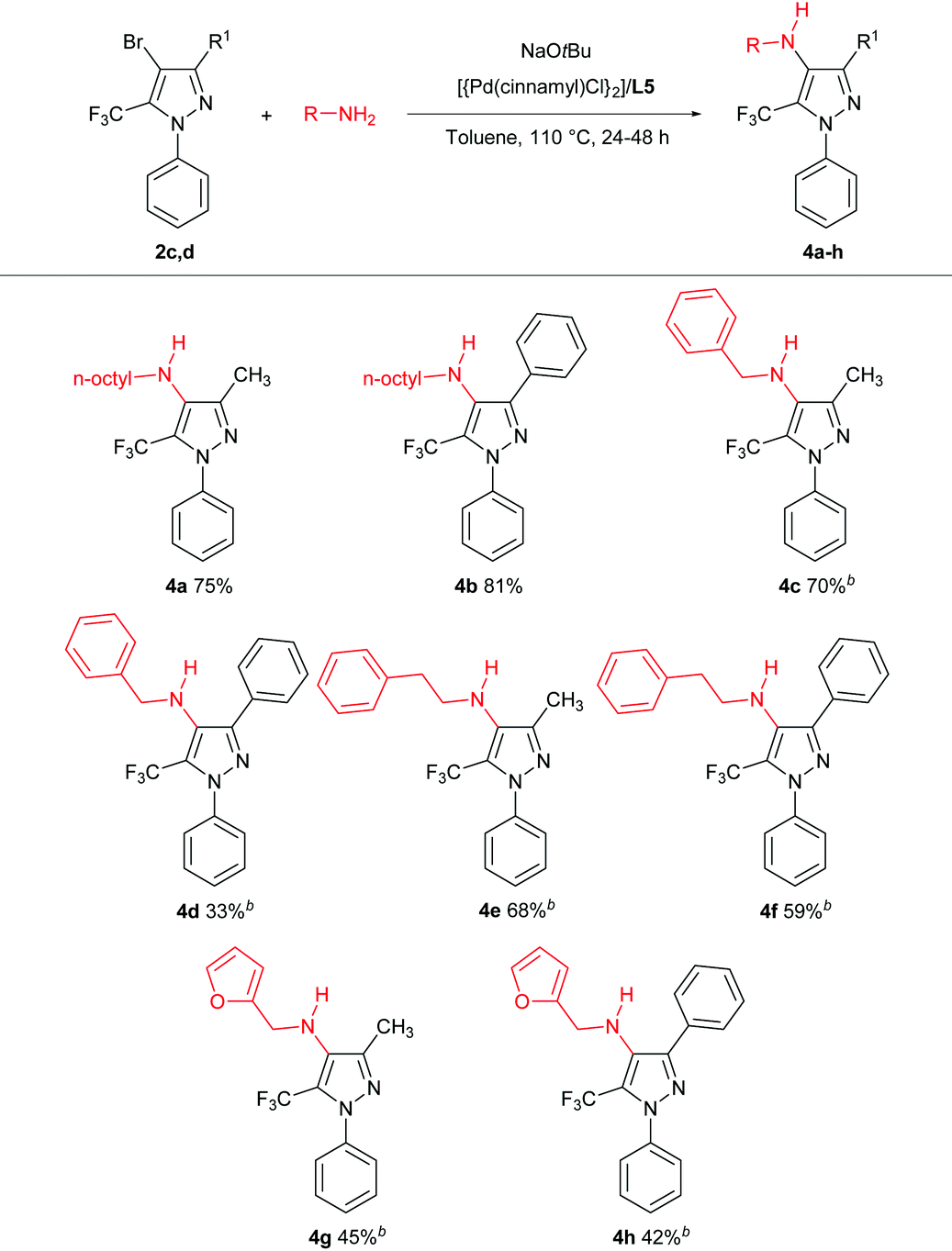
In keeping with the successful screening results using L5, the tetra-substituted trifluoromethylpyrazole 4a was obtained in 75% isolated yield and the analogous 3-phenyl derivative 4b was obtained in 81% isolated yield. Related cross-couplings using benzylamine required longer reaction times (48 h), and whereas the reaction involving 2c afforded 4c in 70% yield, the use of 2d proved more challenging, with the derived amination product 4d obtained in 33% isolated yield. Analogous transformations involving phenethylamine leading to 4e,f, and furfurylamine leading to 4g,h, also proved viable. Collectively, these observations highlight the feasibility and challenges associated with promoting such C–N cross-couplings within the highly substituted frameworks of 2c,d. Our efforts to effect the amination of such hindered trifluoromethylpyrazoles by use of (hetero)aryl amines were unsuccessful, as were transformations involving secondary amines, such as morpholine.
Conclusions
In summary, we have developed a synthetically useful methodology for the construction of tetra-substituted trifluoromethylpyrazoles involving halogenation of 3-substituted 1-phenyl-5-trifluoromethylpyrazoles, and subsequent palladium-catalyzed Negishi coupling or Buchwald–Hartwig amination employing (hetero)arylzinc chlorides and primary alkylamines, respectively. The presence of a trifluoromethyl group and the sterically encumbered nature of the requisite halopyrazole coupling partners make such transformations rather challenging; nonetheless, with the appropriate selection of ancillary co-ligand (XPhos and JosiPhos CyPF-tBu), such sought-after transformations were realized. Future work will involve applying such catalytic strategies toward the construction of alternative congested heterocyclic frameworks.Experimental section
General considerations
Unless otherwise indicated, all reagents and solvents used were obtained from commercial suppliers and were used without further purification. THF was purified by refluxing over Na/benzophenone followed distillation, all under nitrogen atmosphere. Toluene was deoxygenated by sparging with nitrogen followed by passage through an mBraun double column solvent purification system packed with alumina and copper-Q5 reactant and storage over activated 4 Å molecular sieves. All Buchwald–Hartwig cross-coupling reactions were setup inside a nitrogen-filled inert atmosphere glovebox and worked up in air using benchtop procedures. Column chromatography was performed on silica gel (230–400 mesh). All NMR spectra were acquired on a 400, 500 or 600 MHz spectrometer, employing tetramethylsilane or a residual non-deuterated solvent signal as an internal reference (CHCl3 = 1H 7.26 ppm, 13C 77.0 ppm). All results are reported as follows: chemical shift (δ) (multiplicity, coupling constant, integration). Splitting patterns are indicated as follows: s = singlet, d = doublet, t = triplet, q = quartet, qui = apparent quintet, m = multiplet, b = broad. All coupling constants (J) are reported in Hertz (Hz). GC-MS analyses were registered with a split-splitless injector, autosampler, capillary column (30 m, 0.32 mm internal diameter), using helium as the carrier gas. High resolution mass spectrometry spectrometric data (HRMS) were obtained using ion trap (ESI) instruments operating in positive ion mode.General procedure for the synthesis of 1-phenyl-5-(trifluoromethyl)-1H-pyrazoles (1a–b)
The pyrazoles 1a–b were prepared following the procedures described in the literature.12 A solution of phenyl hydrazine (5.5 mmol) in ethanol (20 mL) was added dropwise via pipette over the course of five minutes into a 100 mL round bottom flask containing a magnetic stir bar and a solution of the corresponding 1,1,1-trifluoro-4-methoxy-alken-2-one (5 mmol) in ethanol (50 mL) that was held at 0 °C under the influence of magnetic stirring. The system was subsequently left to warm to room temperature over 1 h. A reflux condenser was then affixed to the reaction flask, and the flask was submerged into an oil bath set to 78 °C with continued stirring for 24 h. The solvent was removed on the rotary evaporator under reduced pressure and the residual crude pyrazoles 1a–b were purified by use of column chromatography employing 5% ethyl acetate in hexanes solution as eluent.General procedure for the synthesis of 4-halo-1-phenyl-5-(trifluoromethyl)-1H-pyrazoles (2a–d)
General procedure for the synthesis of the organozinc reagents
Into a Schlenk-flask equipped with a septum and a magnetic stirrer bar were added magnesium turnings (3 equiv.) and LiCl (1.2 equiv.). The system was dried with a heat gun under vacuum for 10 minutes. Upon cooling to room temperature, the Schlenk-flask was backfilled with argon and charged with anhydrous THF (2 mL per mmol of organic halide to be added). The magnesium turnings were activated with iBu2AlH (0.2 mL 0.1 M in THF per 3 mmol of magnesium). After 5 minutes of stirring, the organic halide was added in one portion at 0 °C (25 °C for 4-bromobenzonitrile and 2-chlorothiophene). The mixture was stirred for 0.5 h (2 h for 4-bromobenzonitrile; 1 h for 2-chlorothiophene), and then was transferred via cannula to another Schlenk-flask (previously dried under vacuum and backfilled with argon) containing 1.1 equivalents of anhydrous ZnCl2 solution in THF, cooled in an ice bath. The mixture was stirred under this condition for 10 minutes prior to use.General procedure for the Negishi cross-coupling reactions with 4-halo-5-trifluoromethyl-1H-pyrazoles
An oven dried Schlenk-flask, equipped with a rubber septum and a stirrer bar, was charged with 4-halopyrazole (2a–d, 0.5 mmol), XPhos/Pd-G3 (2 mol%, 8.4 mg) and XPhos (2 mol%, 4.8 mg). The system was evacuated and backfilled with argon (operation performed three times). At room temperature THF (2 mL) and the freshly prepared organozinc reagent solution were transferred to the reaction flask via cannula. The reaction mixture was stirred magnetically at 40 °C for 14 h and then was quenched with 1 mL of aqueous saturated NH4Cl solution. The mixture was washed with ethyl acetate (3 × 15 ml), the organic layers were combined and dried over sodium sulfate and were filtered. The eluent was concentrated on the rotary evaporator under reduced pressure. The crude reaction products thus obtained were purified by use of column chromatography.General procedure for Buchwald–Hartwig cross-coupling reactions with 4-bromo-1-phenyl-5-(trifluoromethyl)-1H-pyrazoles
Inside a nitrogen-filled inert atmosphere glovebox, [Pd(cinnamyl)Cl]2 (2–3 mol%), JosiPhos CyPF-tBu (4–6 mol%), NaOtBu (96.1 mg, 1 mmol), 4-bromo pyrazole 2a–b (0.5 mmol), amine (1 mmol) and toluene (3 mL) were added to a screw capped vial containing a magnetic stir bar. The vial was sealed with a cap containing a PTFE septum, removed from the glovebox and placed in a temperature-controlled aluminum heating block set at 110 °C for the established time. The crude reaction mixture was filtered over a silica plug, followed by rinsing of the plug with 60 mL of CH2Cl2. The combined eluent was collected, concentrated on the rotary evaporator, and the crude product thus obtained was purified by use of column chromatography.13C{1H} (125 MHz, CDCl3, 25 °C) δ = 142.8, 139.9, 139.2, 132.0, 128.9, 128.6, 128.5, 127.9, 127.5, 125.5, 124.1, 120.9 (q, 1JC–F = 269, CF3), 120.9 (q, 2JC–F = 37), 53.6, 11.8. HRMS (ESI-TOF): C18H17F3N3 (M + H) requires 332.1375/Found: 332.1365.
Acknowledgements
We are appreciative of the generous financial support of this work from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Natural Sciences and Engineering Research Council of Canada (NSERC Canada).Notes and references
- (a) J. Elguero, P. Goya, N. Jagerovic and A. M. S. Silva, Targets Heterocycl. Syst., 2002, 6, 52 CAS; (b) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, John Wiley & Sons, New York, 2nd edn, 2004 Search PubMed; (c) R. S. Keri, K. Chand, T. Ramakrishnappa and B. M. Nagaraja, Arch. Pharm. Chem. Life Sci., 2015, 348, 1 CrossRef PubMed; (d) C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061 CrossRef CAS PubMed.
- (a) M. E. F. Braibante, G. Clar and M. A. P. Martins, J. Heterocycl. Chem., 1993, 30, 1159 CrossRef CAS; (b) L. P. Song, Q. L. Chu and S. Z. Zhu, J. Fluorine Chem., 2001, 107, 107 CrossRef CAS; (c) H. G. Bonacorso, M. S. Correa, L. M. F. Porte, E. P. Pittaluga, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2012, 53, 5488 CrossRef CAS.
- (a) M. Zouaoui and M. M. E. Gaïed, J. Chem. Res., 2003, 4, 242 CrossRef; (b) R. S. Foster, H. Jakobi and J. P. A. Harrity, Org. Lett., 2012, 14, 4858 CrossRef CAS PubMed.
- (a) M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 4710 CrossRef CAS PubMed; (b) M. Schnürch, R. Flasik, A. F. Khan, M. Spina, M. D. Mihovilovic and P. Stanetty, Eur. J. Org. Chem., 2006, 3283 CrossRef; (c) T. Zang, X. Gao and H. B. Wood, Tetrahedron Lett., 2011, 52, 3111 Search PubMed; (d) C. W. Cheung, D. S. Surry and S. L. Buchwald, Org. Lett., 2013, 15, 3734 CrossRef CAS PubMed; (e) S. L. Jeon, J. H. Choi, B. T. Kim and I. H. Jeong, J. Fluorine Chem., 2007, 128, 1191 CrossRef CAS; (f) K. M. Clapham, A. S. Batsanov, M. R. Bryce and B. Tarbit, Org. Biomol. Chem., 2009, 7, 2155 RSC.
- Z.-G. Zhao and Z.-X. Wang, Synth. Commun., 2007, 37, 137 CrossRef CAS.
- G. Li, R. Kakarla and S. W. Gerritz, Tetrahedron Lett., 2007, 48, 4595 CrossRef CAS.
- Y. Yang, N. J. Oldenhuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 615 CrossRef CAS PubMed.
- (a) G. J. Withbroe, R. A. Singer and J. E. Sieser, Org. Process Res. Dev., 2008, 12, 480 CrossRef CAS; (b) S. M. Crawford, C. B. Lavery and M. Stradiotto, Chem. – Eur. J., 2013, 19, 16760 CrossRef CAS PubMed.
- M. Su, N. Hoshiya and S. L. Buchwald, Org. Lett., 2014, 16, 832 CrossRef CAS PubMed.
- B. J. Tardiff, R. McDonald, M. J. Ferguson and M. Stradiotto, J. Org. Chem., 2012, 77, 1056 CrossRef CAS PubMed.
- Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586 CrossRef CAS PubMed.
- (a) M. Hojo, R. Masuda, Y. Kokuryo, H. Shioda and S. Matsuo, Chem. Lett., 1976, 5, 499 CrossRef; (b) F. Effenberger, R. Maier, K. H. Schonwalder and T. Ziegler, Chem. Ber., 1982, 115, 2766 CrossRef CAS; (c) F. Effenberger and K. H. Schonwalder, Chem. Ber., 1984, 117, 3270 CrossRef CAS; (d) A. Colla, G. Clar, M. A. P. Martins, S. Krimmer and P. Fisher, Synthesis, 1991, 483 CrossRef CAS; (e) A. F. C. Flores, G. M. Siqueira, A. R. Freitag, N. Zanatta and M. A. P. Martins, Quím. Nova, 1994, 17, 298 CAS; (f) Y. Kamitori, M. Hojo, R. Masuda, T. Fujitani and T. Kobuschi, Synthesis, 1986, 340 CrossRef CAS; (g) M. Hojo, R. Masuda and E. Okada, Synthesis, 1986, 1013 CrossRef CAS; (h) G. M. Siqueira, A. F. C. Flores, G. Clar, N. Zanatta and M. A. P. Martins, Quím. Nova, 1994, 17, 24 CAS; (i) H. G. Bonacorso, M. A. P. Martins, S. R. T. Bittencourt, R. V. Lourega, N. Zanatta and A. F. C. Flores, J. Fluorine Chem., 1999, 99, 177 CrossRef CAS.
- M. Rossato, C. Casanova, A. P. Lima, D. J. Emmerich, V. Oliveira, R. M. Dallago, C. P. Frizzo, D. N. Moreira, L. Buriol, S. Brondani, N. Zanatta, H. G. Bonacoro and M. A. P. Martins, ARKIVOC, 2014, ii, 224 Search PubMed.
- M. Schlosser, J. N. Volle, F. Leroux and K. Schenk, Eur. J. Org. Chem., 2002, 2913 CrossRef CAS.
- H. Alinezhad, M. Tajbakhsh and M. Zare, J. Mex. Chem. Soc., 2011, 55, 238 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra. See DOI: 10.1039/c5ob02390d |
| This journal is © The Royal Society of Chemistry 2016 |