
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure of olefin–imidacloprid and gas-phase fragmentation chemistry of its protonated form†
Roberto
Fusetto
,
Jonathan M.
White
,
Craig A.
Hutton
and
Richard A. J.
O'Hair
*
School of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, and ARC Centre of Excellence for Free Radical Chemistry and Biotechnology, University of Melbourne, Melbourne, Victoria 3010, Australia. E-mail: rohair@unimelb.edu.au
First published on 4th January 2016
Abstract
One of the major insect metabolites of the widely used neonicotinoid insecticide imidacloprid, 1 (1-[(6-chloro-3-pyridinyl)methyl]-N-nitro-1H-imidazol-2-amine), is the olefin 2. To better understand how the structure of olefin 2 relates to the gas-phase fragmentation of its protonated form, 2H+, X-ray crystallography, tandem mass spectrometry experiments and DFT calculations were carried out. Olefin 2 was found to be in a tautomeric form where the proton is on the N(1) position of the imidazole ring and forms a hydrogen bond to one of the oxygen atoms of the coplanar nitroamine group. Under conditions of low-energy collision-induced dissociation (CID) in a linear ion trap, 2H+, formed via electrospray ionization (ESI), fragments via a major loss of water, together with minor competing losses of HNO2 and 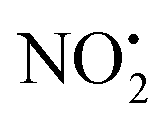 . This contrasts with 1H+, which mainly undergoes bond homolysis via
. This contrasts with 1H+, which mainly undergoes bond homolysis via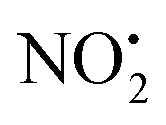 loss. Thus, installation of the double bond in 2 plays a key role in facilitating the loss of water. DFT calculations, carried out using the B3LYP/6-311G++(d,p) level of theory, revealed that loss of water was energetically more favourable compared to HNO2 and
loss. Thus, installation of the double bond in 2 plays a key role in facilitating the loss of water. DFT calculations, carried out using the B3LYP/6-311G++(d,p) level of theory, revealed that loss of water was energetically more favourable compared to HNO2 and 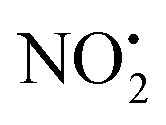 loss. Three multistep, energetically accessible mechanisms were identified for loss of water from 2H+, and these have the following barriers: (i) direct proton transfer from N(5) of the pyridine to O(1) on the NO2 group (119 kJ mol−1); (ii) rotation of the N(2)–N(4) bond (117 kJ mol−1); (iii) 1,3-intramolecular proton transfer between the two oxygen atoms of the NO2 group (145 kJ mol−1). Given that the lowest barrier for the losses of HNO2 and
loss. Three multistep, energetically accessible mechanisms were identified for loss of water from 2H+, and these have the following barriers: (i) direct proton transfer from N(5) of the pyridine to O(1) on the NO2 group (119 kJ mol−1); (ii) rotation of the N(2)–N(4) bond (117 kJ mol−1); (iii) 1,3-intramolecular proton transfer between the two oxygen atoms of the NO2 group (145 kJ mol−1). Given that the lowest barrier for the losses of HNO2 and 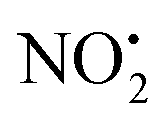 is 156 kJ mol−1, it is likely that all three water loss mechanisms occur concurrently.
is 156 kJ mol−1, it is likely that all three water loss mechanisms occur concurrently.
Introduction
The neonicotinoid insecticide imidacloprid, 1, is one of the most widely used insecticides worldwide.1 Its selective toxicity towards insects is a consequence of it binding more strongly to insect nicotinic acetylcholine receptors (nAChRs) than to their mammalian counterparts.2,3 Its use has generated considerable recent public attention4 due to a possible contribution to honey bee colony collapse disorder,5 ultimately leading to its ban in the EU.6 A range of studies has been carried out to better understand the biological consequences of exposure of insects to imidacloprid.7–9 For example, HPLC-MS based methods have been used to study the formation of its metabolites, including the olefin metabolite 2 and the hydroxyl metabolite 3 (Scheme 1) in vivo for: honeybees;10 termites;11 houseflies12 and the vinegar fly, Drosophila melanogaster.13 X-ray and molecular modelling docking studies, carried out on the binding of imidacloprid to nAChRs, have uncovered the molecular basis for the differences in binding sites in insect versus vertebrate nAChRs.14,15 Different docking studies on homology models for Apis mellifera16 and Lymnaea stagnalis acetylcholine binding protein (AChBP)17 and the in vivo electrophysiological studies on recombinant Drosophila SAD/chicken β2 hybrid nAChRs18 have highlighted the different binding properties and activity of these receptor subunits towards imidacloprid. In contrast, there has only been a single report on docking studies of 2 with different α/α and α/β subunits of nAChRs of Apis mellifera.16 An intriguing study on the neuroblocking potency of imidacloprid metabolites suggested that 2 and 3 (Scheme 1) are about ten times less potent than imidacloprid.19 The molecular basis for this effect is not understood, and while an X-ray structure for 3 has been reported,20 the X-ray structure of 2 has not been reported until now.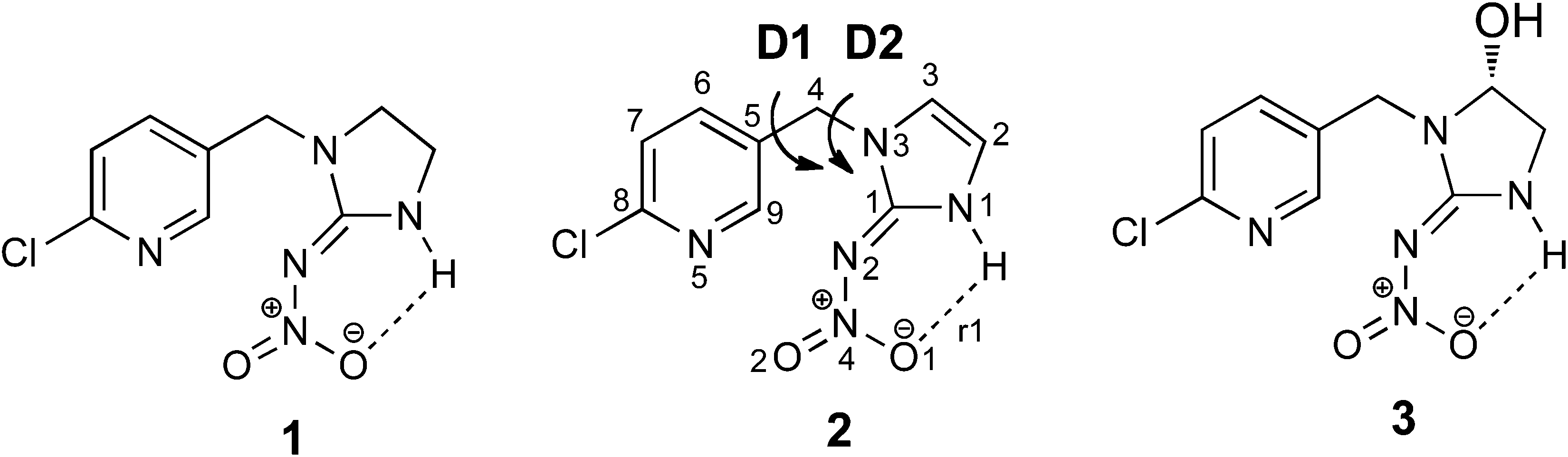 | ||
| Scheme 1 Imidacloprid, 1, and its metabolites: olefin–imidacloprid 2 and hydroxyl derivative 3. D1 and D2 represent the key bond rotations that describe conformations of 2. | ||
As part of a program aimed at examining the in vivo metabolism of 1 in Drosophila melanogaster,13 we required a sample of 2 to examine its potency in biological tests and as both an internal standard and an authentic standard for MS studies. The synthetic method of Novák et al.,21 was used to prepare olefin 2 to examine its structure via X-ray crystallography. Since tandem mass spectrometry (MS/MS) typically involves low energy collision-induced dissociation (CID) of protonated analytes and the full CID spectrum of 2H+ has not yet been reported, we studied the fragmentation mechanism of 2H+ in the gas phase and compared its gas-phase ion chemistry to the parent insecticide 1H+ discussed previously.22
Results
X-ray crystallography
Compound 2 was recrystallised from ether and suitable crystals for X-ray diffraction were collected. The X-ray structure of olefin–imidacloprid is reported in Fig. 1, while its key structural parameters are reported in Table S1.† The thermal ellipsoid plot for compound 2 highlights that the nitroamine group is coplanar with the imidazole ring.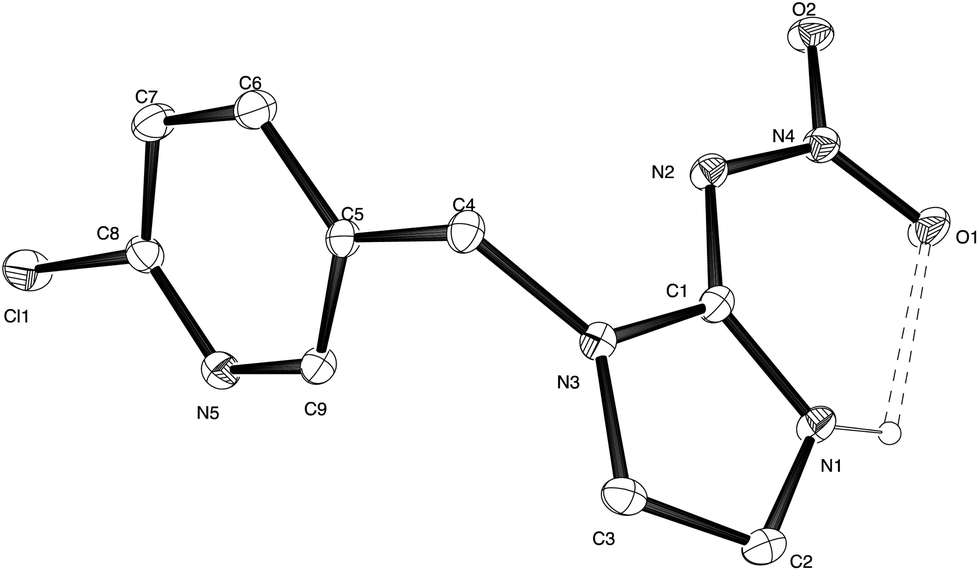 | ||
| Fig. 1 Thermal ellipsoid plot for X-ray structure of olefin–imidacloprid 2. Ellipsoids are at the 20% probability level. | ||
The formation of the N(1)–H⋯O(1) hydrogen bond in addition to extensive delocalization of electron density over the entire nitroamino imidazole moiety, constrains the nitroguanidinic moiety in this configuration. The N(1)–C(1)–N(2) bond angle is opened out to 133.7(3)° while the N(3)–C(1)–N(2) is closed to 119.1(2)° compared to the ca. 126° external bond angles expected for a substituted sp2 hybridized atom in this 5-membered ring. The crystal packing is characterised by a bifurcated intermolecular hydrogen bond between the imidazole N(1)–H donor and the nitro oxygen O(2) and the exocyclic guanidine nitrogen N(2) (ESI, Fig. S1†).
As shown in Scheme 2, the nitroguanidine group in compounds such as 2 can potentially adopt two tautomeric forms. A search of the Cambridge database23 for structures containing the nitroguanidine group located 151 structures of which 132 were represented by tautomer A. The remaining structures were either protonated salts or had the guanidine moiety embedded in a substituted 1,3,5-triazine derivative and were not representative of the fragment B.
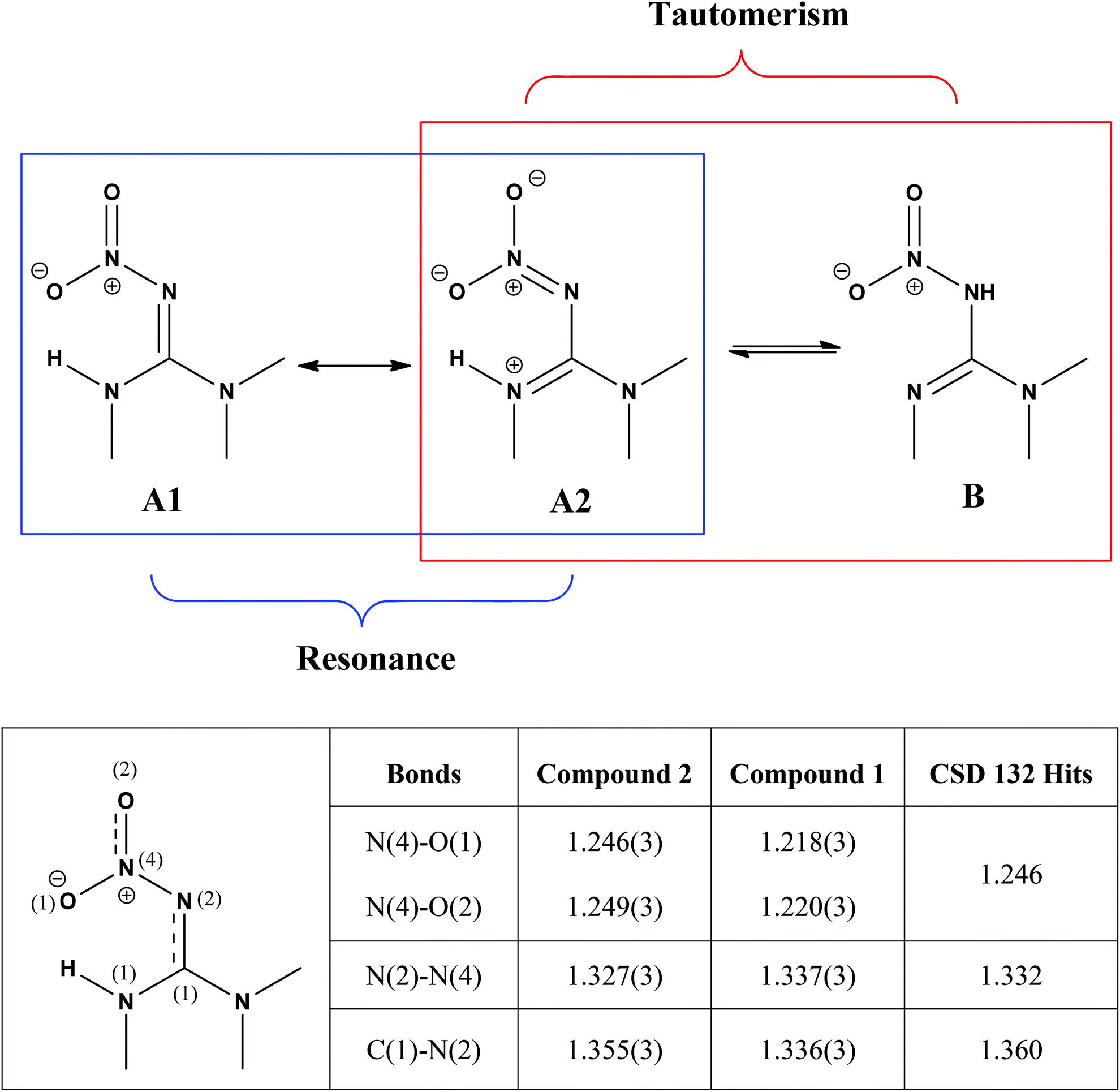 | ||
| Scheme 2 The two possible tautomeric forms of 2 and the resonance forms deriving from the tautomer A. A comparison among the selected structural parameters (values in Å) obtained from the Cambridge Structural Database using the search fragment A (table below) with olefin–imidacloprid (compound 2) and imidacloprid (compound 1)24 suggest that 2 is best represented by the resonance form A2, whereas resonance form A1 is more likely to represent 1. | ||
Tautomer A in turn has two main resonance forms (A1 and A2) generated by the delocalisation of the guanidinyl nitrogen lone pairs onto the oxygen atoms of the nitro group through the imine functional group (Scheme 2). The C–N and N–N bond distances provided information on which of these resonance forms predominates in a given structure. A scatter plot of N–N versus C–N bond distances from the 132 structures obtained from the Cambridge Structural Database revealed the presence of structures with differing contributions of resonance forms A1 and A2 (Fig. S2, ESI†). Selected structural parameters for olefin–imidacloprid, 2, and its saturated analogue imidacloprid, 1,24 were compared with the average distances obtained from the 132 structures collected (Scheme 2, table). The shorter N–N bond and the longer C–N bond of 2, suggested a significantly greater contribution to the A2 resonance form in compound 2 compared to that of 1.23
Gas-phase fragmentation of 2H+
There have been numerous LC-MS studies in which the concentration of 1 in complex biological matrices is determined via MS/MS experiments where selected ion fragments are monitored upon CID of 1H+ (the so called multiple reaction monitoring (MRM) technique).25–27 Fewer studies have used MRM to quantify the concentrations of imidacloprid metabolites. Two recent reports have used different fragment ions of 2H+ to quantify the concentrations of 2 in bee's wax28 and honeybee and honey samples.29 In the former study, the transitions m/z 254 → m/z 205 and m/z 254 → m/z 171 were used, while the later utilized m/z 254 → m/z 236 and m/z 254 → m/z 171. Given that the full CID spectrum of 2H+ does not appear to have been reported, we examined its fragmentation reactions under low energy CID conditions in ion trap and QTof mass spectrometers. Mass selection of 2H+ (m/z 254), formed in high abundance upon electrospray ionization of 2, followed by CID in the linear ion trap mass spectrometer, gave three main fragment ions (Fig. 2): m/z 236 corresponding to water loss (eqn (1)); m/z 208 corresponding to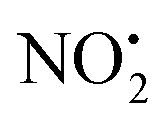 loss (eqn (2)) and m/z 207 corresponding to HNO2 loss (eqn (3)) (Fig. 2). These assignments were confirmed by High Resolution Mass Spectrometry (HRMS) on the fragment ions (ESI Table S2†). By varying the activation voltage, energy resolved CID experiments were carried out in a 3D ion trap to establish the relative onsets for these fragmentation channels. The onset for loss of water is less than that for
loss (eqn (2)) and m/z 207 corresponding to HNO2 loss (eqn (3)) (Fig. 2). These assignments were confirmed by High Resolution Mass Spectrometry (HRMS) on the fragment ions (ESI Table S2†). By varying the activation voltage, energy resolved CID experiments were carried out in a 3D ion trap to establish the relative onsets for these fragmentation channels. The onset for loss of water is less than that for 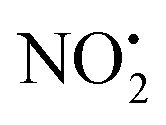 and HNO2 loss (Fig. 2B), suggesting it is energetically favoured. The loss of water must involve one of the oxygen atoms of the nitro group as well as both the ionizing proton and another proton derived from 2. In order to establish which protons are involved in this loss, H/D exchange was performed on 2 and the resultant solution was subjected to ESI/MS, giving rise to a mixture of [M + H]+, [M + D]+ and [M − H + 2D]+ of compound 2. The presence of 35Cl and 37Cl isotopes give rise to a cluster of ions from m/z 254 to m/z 258. CID of the selected [M − H + 2D]+ ion at m/z 256 gave rise to D2O loss (data not shown), highlighting that the N(1) proton of 2 is involved in water loss. Thus the likely structure of the ion arising from water loss is the hetero (aryl-)-N-nitroso-onium ion 4+ (Fig. 2D).30–32 The related loss of water from a nitro group that involves both the ionizing proton and a heterocyclic NH has been recently described for protonated 2-nitroimidazole.33 As the fragment ions at m/z 205 and m/z 171 have been used in MRM transition studies of 2H+,28,29 we used the multistage mass spectrometry capabilities of the linear ion trap mass spectrometer to isolate and study the fragmentation reactions of the product ion 4+ (m/z 236). The major fragments in the MS3 spectrum (ESI Fig. S3†) are ions at: m/z 206, which corresponds to NO˙ loss (eqn (4)); and m/z 205, which corresponds to HNO loss (eqn (5)). The low intensity fragment ion at m/z 171 likely arises from the loss of the chlorine atom from the m/z 206 product, a reaction that occurs when the ion at m/z 206 is mass selected and subjected to a further stage of CID in a MS4 experiment (data not shown). We also examined the CID spectra of 2H+ as a function of collision energy in a QTof mass spectrometer (ESI Fig. S4†), which confirmed that m/z 205 and m/z 171 can be formed under the multi collision conditions at higher CID energies.
and HNO2 loss (Fig. 2B), suggesting it is energetically favoured. The loss of water must involve one of the oxygen atoms of the nitro group as well as both the ionizing proton and another proton derived from 2. In order to establish which protons are involved in this loss, H/D exchange was performed on 2 and the resultant solution was subjected to ESI/MS, giving rise to a mixture of [M + H]+, [M + D]+ and [M − H + 2D]+ of compound 2. The presence of 35Cl and 37Cl isotopes give rise to a cluster of ions from m/z 254 to m/z 258. CID of the selected [M − H + 2D]+ ion at m/z 256 gave rise to D2O loss (data not shown), highlighting that the N(1) proton of 2 is involved in water loss. Thus the likely structure of the ion arising from water loss is the hetero (aryl-)-N-nitroso-onium ion 4+ (Fig. 2D).30–32 The related loss of water from a nitro group that involves both the ionizing proton and a heterocyclic NH has been recently described for protonated 2-nitroimidazole.33 As the fragment ions at m/z 205 and m/z 171 have been used in MRM transition studies of 2H+,28,29 we used the multistage mass spectrometry capabilities of the linear ion trap mass spectrometer to isolate and study the fragmentation reactions of the product ion 4+ (m/z 236). The major fragments in the MS3 spectrum (ESI Fig. S3†) are ions at: m/z 206, which corresponds to NO˙ loss (eqn (4)); and m/z 205, which corresponds to HNO loss (eqn (5)). The low intensity fragment ion at m/z 171 likely arises from the loss of the chlorine atom from the m/z 206 product, a reaction that occurs when the ion at m/z 206 is mass selected and subjected to a further stage of CID in a MS4 experiment (data not shown). We also examined the CID spectra of 2H+ as a function of collision energy in a QTof mass spectrometer (ESI Fig. S4†), which confirmed that m/z 205 and m/z 171 can be formed under the multi collision conditions at higher CID energies.
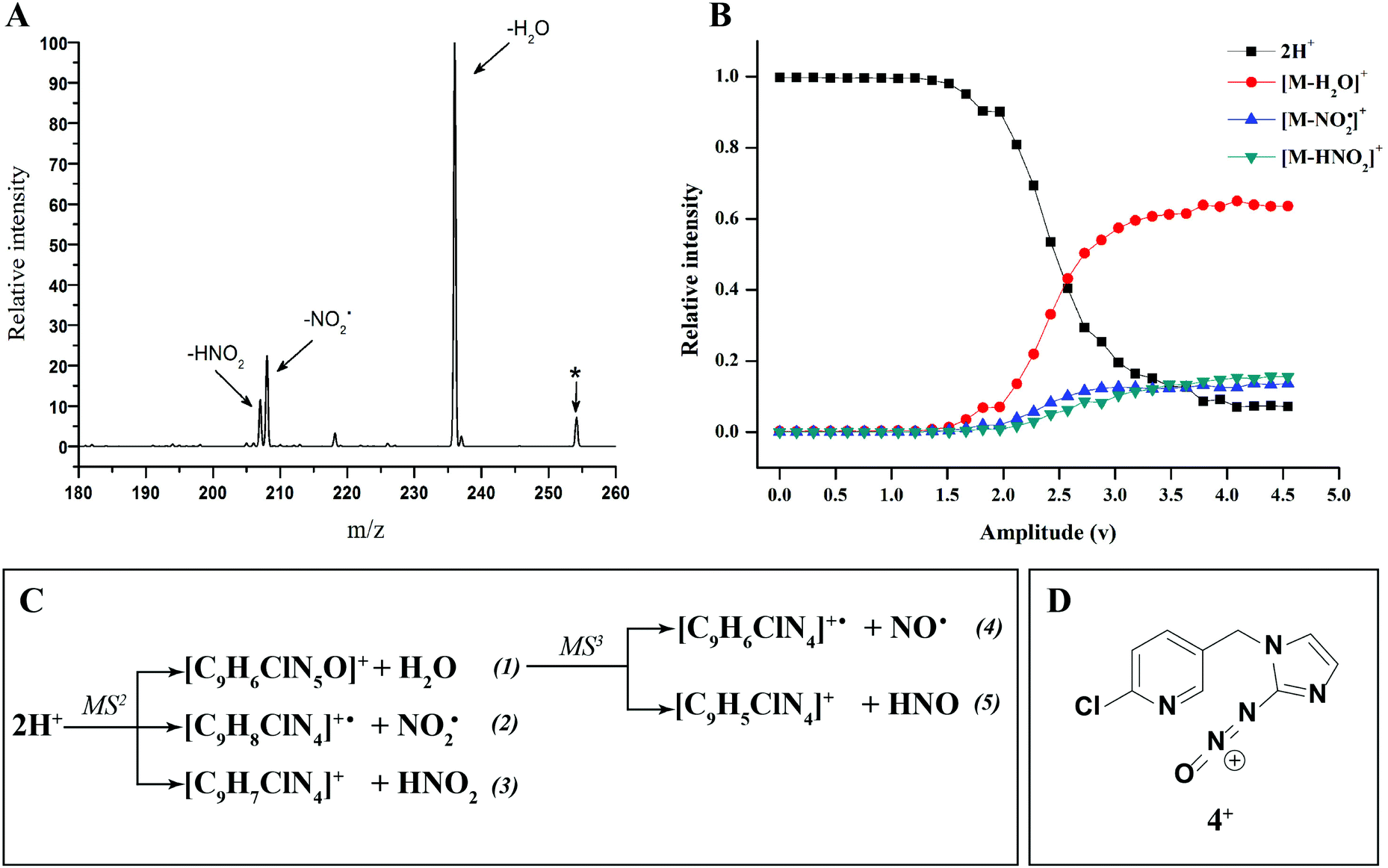 | ||
Fig. 2 (A) CID mass spectrum of 2H+ (m/z 254), obtained using the linear ion trap with normalized collision energy of 18% for 30 ms and a 1.0 m/z wide isolation window. The 35Cl isotope was mass selected and the precursor ion is indicated by an asterisk. (B) Energy Resolved CID diagram of 2H+. The relative intensity of produced water shows how this pathway is highly competitive against 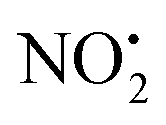 and HNO2 loss. (C) Equations reporting the MS2 and MS3 fragmentation products of compound 2H+. (D) The structure of the main product ion 4+. and HNO2 loss. (C) Equations reporting the MS2 and MS3 fragmentation products of compound 2H+. (D) The structure of the main product ion 4+. | ||
Density functional calculations on the fragmentation of 2H+
To better understand the role of the proton in the fragmentation reactions of 2H+, density functional theory calculations were carried out on a range of conformers for different structural isomers in which the proton is located at different basic sites of 2 and various potential mechanisms for the competing losses of H2O,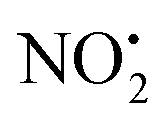 and HNO2 were investigated. The B3LYP/6-311G++(d,p) level of theory was chosen to allow direct comparison to the calculated fragmentation chemistry of 1H+.22
and HNO2 were investigated. The B3LYP/6-311G++(d,p) level of theory was chosen to allow direct comparison to the calculated fragmentation chemistry of 1H+.22
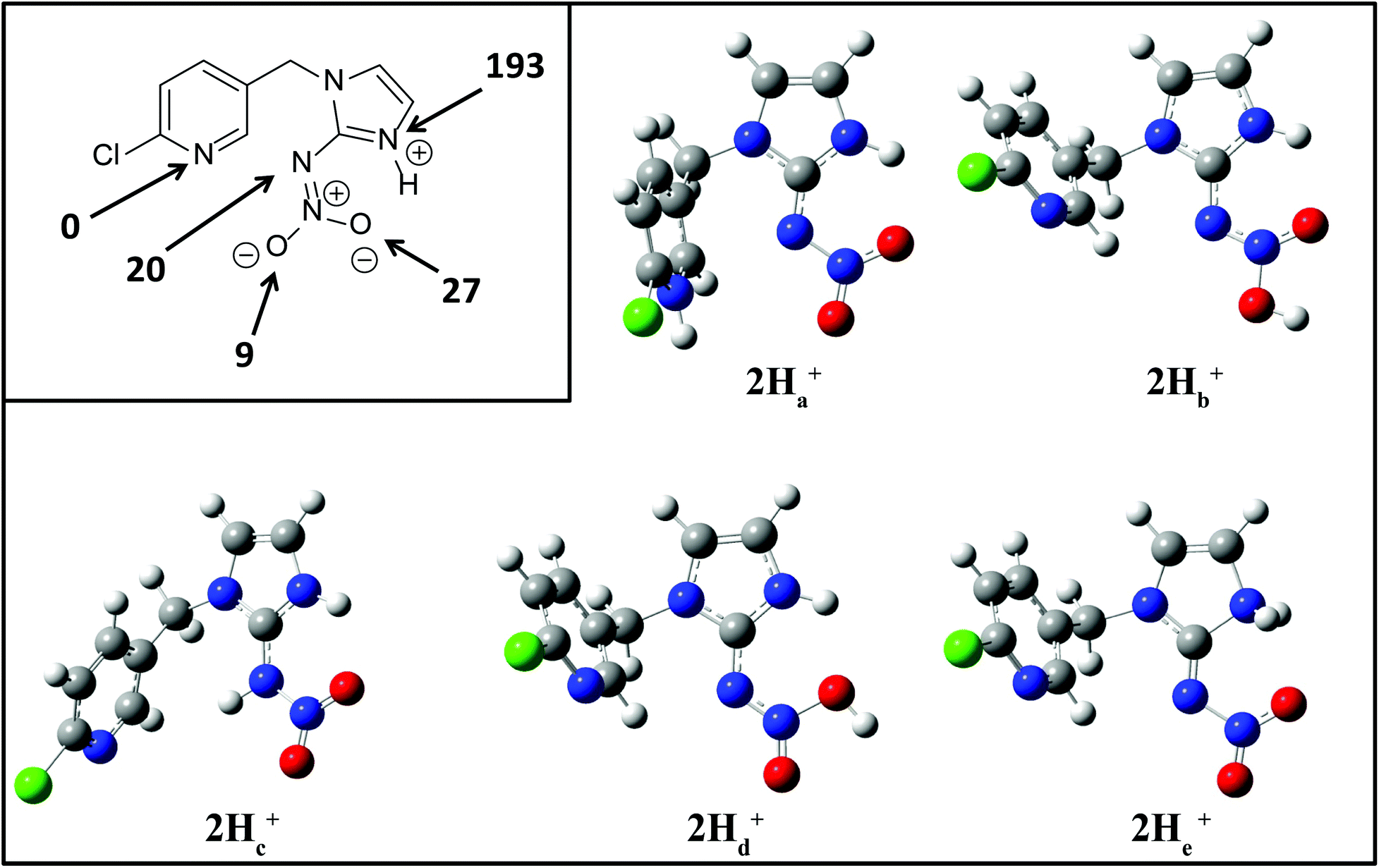 | ||
| Fig. 3 B3LYP/6-311G++(d,p) DFT calculations on the gas-phase protonation of 2. Inset shows relative local proton affinities in kJ mol−1. Optimized structures are for protonation at: N(5), O(2), N(2), O(1) and N(1) designated as 2Ha+, 2Hb+, 2Hc+, 2Hd+ and 2He+ respectively. | ||
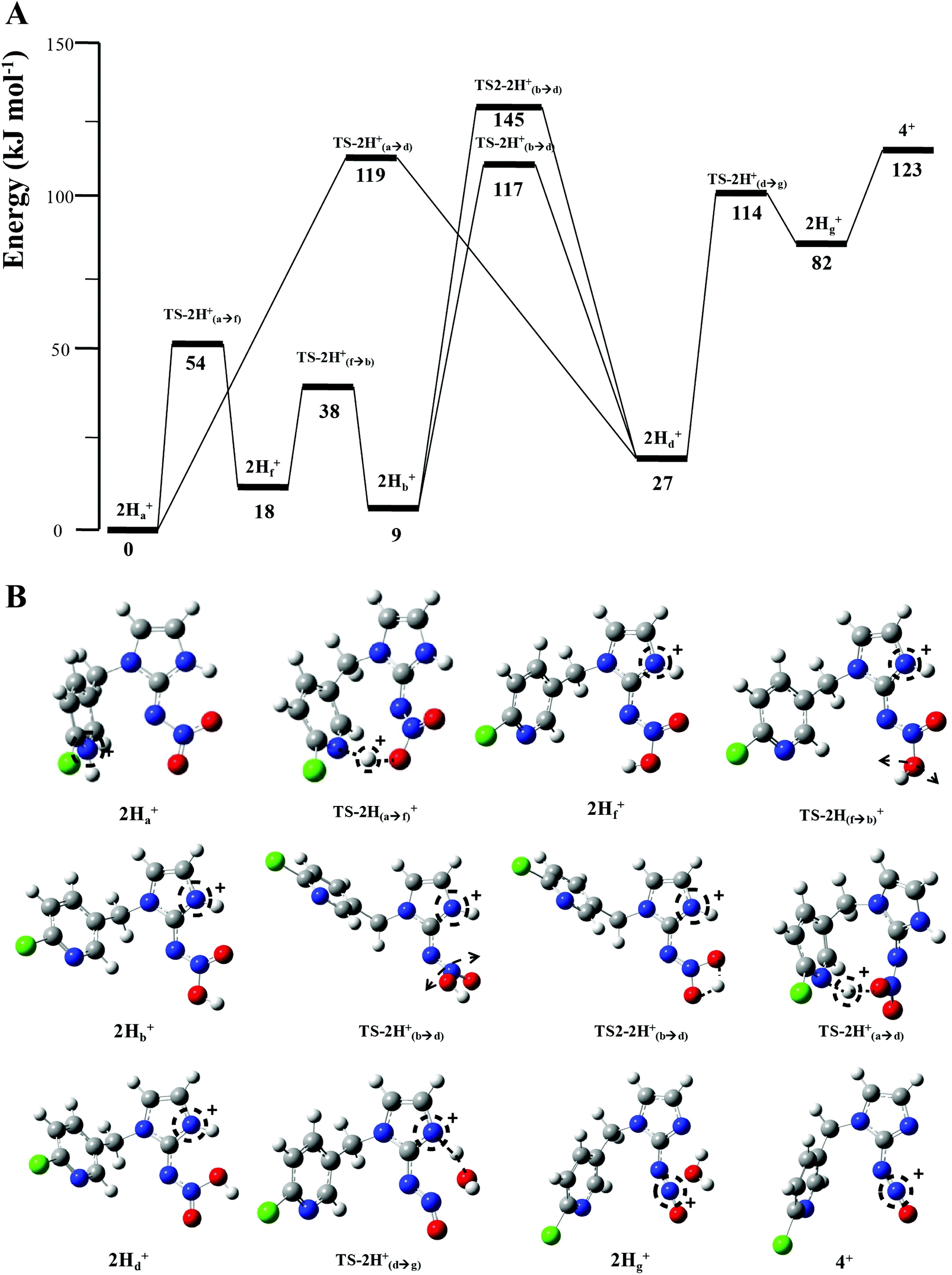 | ||
| Fig. 4 B3LYP/6-311++G(d,p) DFT calculated mechanisms for H2O loss from 2H+: (A) Potential energy diagram. (B) Structures of key species. | ||
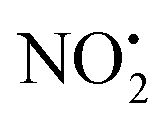 and HNO2.
The energy resolved CID reported in Fig. 2 and the DFT calculated potential energy diagram (Fig. 4) showed that the loss of water must involve lower energetic barriers compared to the loss of neutral HNO2 and the radical
and HNO2.
The energy resolved CID reported in Fig. 2 and the DFT calculated potential energy diagram (Fig. 4) showed that the loss of water must involve lower energetic barriers compared to the loss of neutral HNO2 and the radical 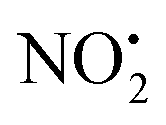 when fragmentation of 2Ha+ is induced in the gas-phase. Thus we used DFT calculations to examine the energetics associated with potential mechanisms for the losses of HNO2 and
when fragmentation of 2Ha+ is induced in the gas-phase. Thus we used DFT calculations to examine the energetics associated with potential mechanisms for the losses of HNO2 and 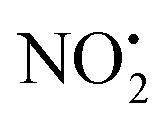 . The lowest energy mechanisms found are shown in Fig. 5 (all activation barriers and energies of intermediates are relative to 2Ha+ at 0 kJ mol−1). Two pathways are possible to reach the optimised state 2Hc+, the key intermediate in this mechanism. Pathway 1 involves the direct transfer of the proton from N(5) on the pyridine to N(2) on the nitroguanidine group through a single step transition state of 166 kJ mol−1 (TS-2H(a→c)+). Alternatively, in pathway 2, the proton is transferred to the oxygen O(2) as described in water loss mechanism (TS-2H(a→f)+, 54 kJ mol−1, Fig. 4) and is subsequently transferred to N(2) with an activation barrier of 156 kJ mol−1 (TS-2H(f→c)+). When 2Hc+ is formed (20 kJ mol−1),
. The lowest energy mechanisms found are shown in Fig. 5 (all activation barriers and energies of intermediates are relative to 2Ha+ at 0 kJ mol−1). Two pathways are possible to reach the optimised state 2Hc+, the key intermediate in this mechanism. Pathway 1 involves the direct transfer of the proton from N(5) on the pyridine to N(2) on the nitroguanidine group through a single step transition state of 166 kJ mol−1 (TS-2H(a→c)+). Alternatively, in pathway 2, the proton is transferred to the oxygen O(2) as described in water loss mechanism (TS-2H(a→f)+, 54 kJ mol−1, Fig. 4) and is subsequently transferred to N(2) with an activation barrier of 156 kJ mol−1 (TS-2H(f→c)+). When 2Hc+ is formed (20 kJ mol−1), 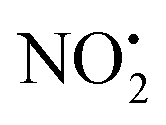 loss can occur through homolysis of the σ-bond between the two nitrogens, N(2)–N(4), with the formation of the final radical 2Hj+˙ (89 kJ mol−1). Alternatively, loss of neutral HNO2 can be achieved through a third process involving two-steps. The first step involves isomerization of 2Hc+ to 2Hh+. In principle this might involve two discreet transition states: (i) a 1,5-intramolecular proton transfer of the N(1) proton to the oxygen O(1); (ii) axial rotation on the bond N(4)–O(1). However, only a single transition state TS-2H(c→h)+ at 168 kJ mol−1 could be found, and IRC calculations connected this transition state to both 2Hc+ to 2Hh+. This either suggests a concerted process to isomerize 2Hc+ to 2Hh+, or that intramolecular proton transfer of the N(1) proton to the oxygen O(1) is barrierless. The second step involves heterolytic cleavage of the N(2)–N(4) bond in 2Hh+via transition state TS-2H(h→i)+ at 204 kJ mol−1 to generate the product ion 2Hi+, with an overall endothermicity of 112 kJ mol−1. The loss of neutral HNO2 and the radical
loss can occur through homolysis of the σ-bond between the two nitrogens, N(2)–N(4), with the formation of the final radical 2Hj+˙ (89 kJ mol−1). Alternatively, loss of neutral HNO2 can be achieved through a third process involving two-steps. The first step involves isomerization of 2Hc+ to 2Hh+. In principle this might involve two discreet transition states: (i) a 1,5-intramolecular proton transfer of the N(1) proton to the oxygen O(1); (ii) axial rotation on the bond N(4)–O(1). However, only a single transition state TS-2H(c→h)+ at 168 kJ mol−1 could be found, and IRC calculations connected this transition state to both 2Hc+ to 2Hh+. This either suggests a concerted process to isomerize 2Hc+ to 2Hh+, or that intramolecular proton transfer of the N(1) proton to the oxygen O(1) is barrierless. The second step involves heterolytic cleavage of the N(2)–N(4) bond in 2Hh+via transition state TS-2H(h→i)+ at 204 kJ mol−1 to generate the product ion 2Hi+, with an overall endothermicity of 112 kJ mol−1. The loss of neutral HNO2 and the radical 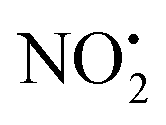 can occur from either 2Hf+ or 2Ha+, but formation of the final products 2Hl+ and 2Hk+˙ is very endothermic (250 and 200 kJ mol−1 respectively).
can occur from either 2Hf+ or 2Ha+, but formation of the final products 2Hl+ and 2Hk+˙ is very endothermic (250 and 200 kJ mol−1 respectively).
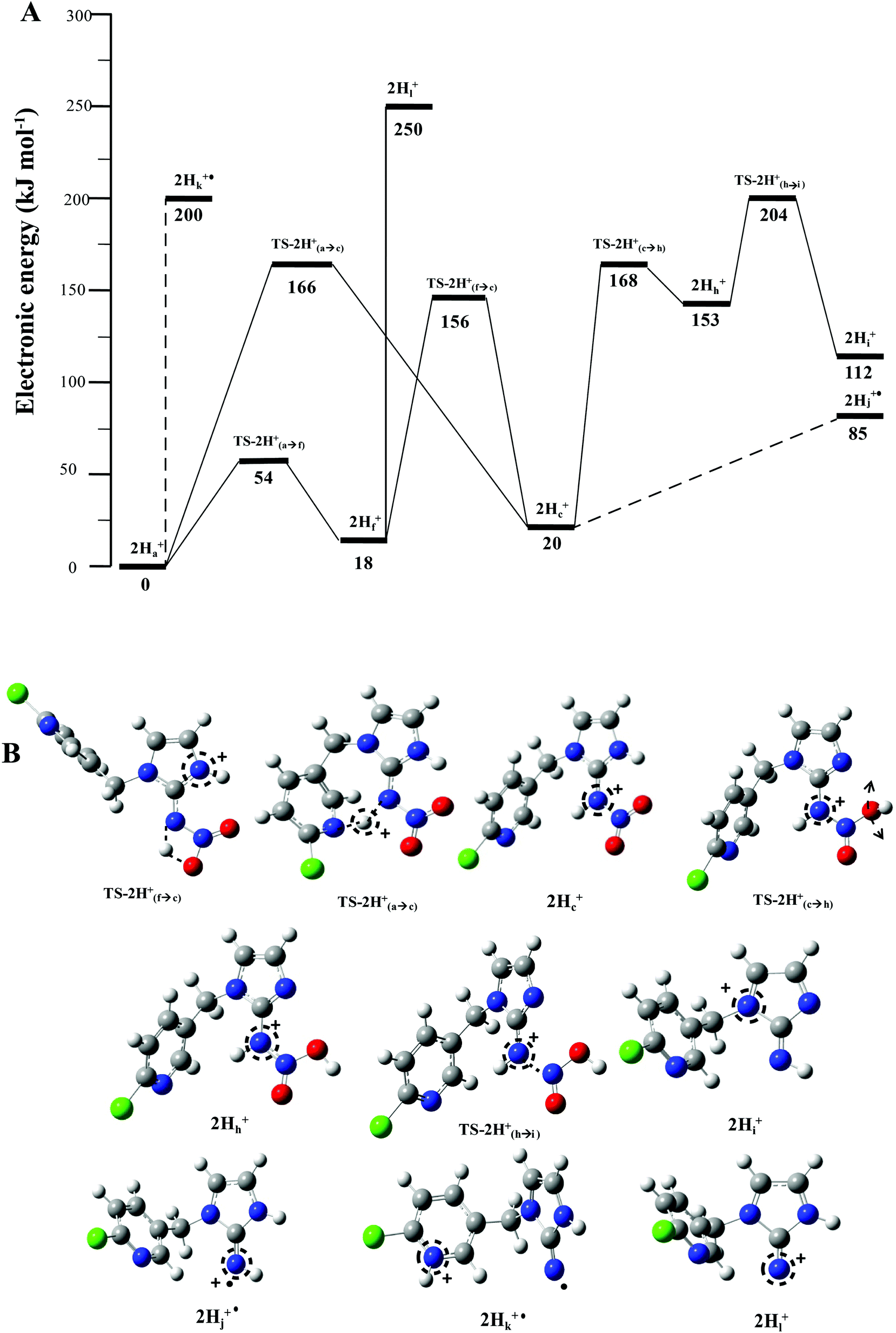 | ||
Fig. 5 B3LYP/6-311G++(d,p) DFT calculated mechanisms for competing 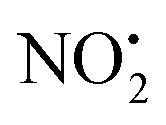 and HNO2 losses from 2H+: (A) potential energy diagram. (B) Structures of key species. Energetic levels connected by dash lines indicate bond homolysis reactions where it was not possible to optimize a transition state. and HNO2 losses from 2H+: (A) potential energy diagram. (B) Structures of key species. Energetic levels connected by dash lines indicate bond homolysis reactions where it was not possible to optimize a transition state. | ||
The alternative mechanism for HNO2 loss (Fig. S5†) follows the same intramolecular H-transfer from N(1) to O(1) described for the water loss mechanism given in Fig. S5,† but it involves a different transition state with an activation barrier of 253 kJ mol−1, and is thus is also unlikely to happen. Even if the final products 2Hi+ and 2Hj+˙ are located at lower energetic levels compared to 4+, the losses of neutral HNO2 and the radical 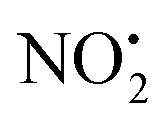 cannot readily occur under low energy CID conditions due to the higher barriers required.
cannot readily occur under low energy CID conditions due to the higher barriers required.
Discussion
How the increased contribution of resonance form A2 influences both the crystal structure of 2 relative to 1 as well as the gas phase chemistry of 2H+ compared to 1H+
| 1H+ | 2H+ | |
|---|---|---|
| Proton affinities | ||
| Relative PA for N(5) site | 0 | 0 |
| Relative PA for N(2) site | 24 | 20 |
| Relative PA for O(1) site | Not calculated | 27 |
| Relative PA for O(2) site | 32 | 9 |
| Relative PA for N(3) site | 109.8 | Not calculated |
| Relative PA for N(1) site | 122 | 193 |
ΔE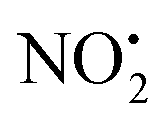 loss loss |
||
| Protonation site on N(5) | 182 | 200 |
| Protonation site on N(2) | 150 | 89 |
| Energy activation barriers | ||
| Proton transfer from N(5) to O(2) | 68 | 54 |
| Proton transfer from N(5) to N(2) | 175 | 166 |
| Proton transfer from O(2) to N(2) | 163 | 156 |
| Rotation N–N bond O(2)–H | Not calculated | 117 |
| Proton transfer from N(5) to O(1) | Not calculated | 119 |
| Proton transfer from O(2) to O(1) | Not calculated | 145 |
 (m/z 208) and HNO2 (m/z 207) being less important. Loss of water is unique to 2H+, with 1H+ fragmenting instead via losses of
(m/z 208) and HNO2 (m/z 207) being less important. Loss of water is unique to 2H+, with 1H+ fragmenting instead via losses of 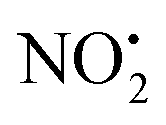 , N2O and NO˙.22 The fragmentation mechanism of nitroguanidine and its derivatives have been reported in the literature showing that these moieties lose predominantly
, N2O and NO˙.22 The fragmentation mechanism of nitroguanidine and its derivatives have been reported in the literature showing that these moieties lose predominantly 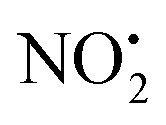 followed by H2O.36 A recent study on low energy CID of protonated 2-nitroimidazole showed that the major loss of water occurs: (i) from the position of the NO2 substituent on the imidazole; and (ii) via a series of 1,4-intramolecular proton transfer coupled by a 180° rotation of the NO2 functional group.33 The two hydrogen atoms involved in water loss from 2H+ are the ionizing proton located on N(5) and the N(1)–H, as confirmed by a H/D exchange reaction on compound 2. Thus, when the deuterated form [M − H + 2D]+ at m/z 256 was mass selected and fragmented with the same CID conditions applied for 2H+, the product ion at m/z 236 was regenerated demonstrating that [M − H + 2D]+ underwent loss of D2O in the gas-phase.
followed by H2O.36 A recent study on low energy CID of protonated 2-nitroimidazole showed that the major loss of water occurs: (i) from the position of the NO2 substituent on the imidazole; and (ii) via a series of 1,4-intramolecular proton transfer coupled by a 180° rotation of the NO2 functional group.33 The two hydrogen atoms involved in water loss from 2H+ are the ionizing proton located on N(5) and the N(1)–H, as confirmed by a H/D exchange reaction on compound 2. Thus, when the deuterated form [M − H + 2D]+ at m/z 256 was mass selected and fragmented with the same CID conditions applied for 2H+, the product ion at m/z 236 was regenerated demonstrating that [M − H + 2D]+ underwent loss of D2O in the gas-phase.
The other two possible competing mechanisms for loss of water involve alternative direct transfer of the ionizing proton from N(5) to O(2) via the transitions state TS-2H(a→d)+ with a barrier of 119 kJ mol−1 and the 1,3-intramolecular proton transfer via the TS2-2H(b→d)+ with a barrier of 145 kJ mol−1. Since all these three mechanisms possess barriers lower than those described for the competing losses of 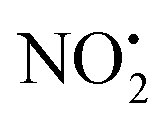 and HNO2 (Table 1), they are likely to operate simultaneously, which offers a reason why 4+ increases with the increase of the energy in the energy-resolved CID experiments (Fig. 2B), while the relative intensities of the product ions 2Hi+ and 2Hj+˙ are maintained.
and HNO2 (Table 1), they are likely to operate simultaneously, which offers a reason why 4+ increases with the increase of the energy in the energy-resolved CID experiments (Fig. 2B), while the relative intensities of the product ions 2Hi+ and 2Hj+˙ are maintained.
For 1H+, the losses of 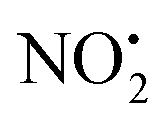 and HNO2 also only occur when the proton is transferred to the nitrogen N(2). The transfer of the proton from N(5) to N(2) (viaTS-2H(a→c)+ at 166 kJ mol−1) or from O(2) to N(2) (viaTS-2H(f→c)+ at 156 kJ mol−1) in 2H+ require significantly higher energies compared to all three mechanisms involved in water loss. Furthermore, the contributing resonance form A2 that characterizes compound 2 means that more energy is required to break the N(2)–N(4) bond to allow the final release of HNO2 (viaTS-2H(h→i)+ at 204 kJ mol−1).
and HNO2 also only occur when the proton is transferred to the nitrogen N(2). The transfer of the proton from N(5) to N(2) (viaTS-2H(a→c)+ at 166 kJ mol−1) or from O(2) to N(2) (viaTS-2H(f→c)+ at 156 kJ mol−1) in 2H+ require significantly higher energies compared to all three mechanisms involved in water loss. Furthermore, the contributing resonance form A2 that characterizes compound 2 means that more energy is required to break the N(2)–N(4) bond to allow the final release of HNO2 (viaTS-2H(h→i)+ at 204 kJ mol−1). 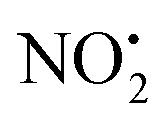 and HNO2 loss can occur directly from 2Ha+ and 2Hf+ but as for 1H+, the final products are energetically unfavourable. The CID spectrum of 1H+ also shows losses of N2O and NO˙ from the nitroguanidine group.
and HNO2 loss can occur directly from 2Ha+ and 2Hf+ but as for 1H+, the final products are energetically unfavourable. The CID spectrum of 1H+ also shows losses of N2O and NO˙ from the nitroguanidine group.
The challenge of assigning structures of small molecule natural products and metabolites from comparisons of CID spectra of related species
Since the late 1950s, mass spectrometry has played a key role in determining the structures of small organic molecules.37,38 It was rapidly recognized that the fragment ions formed under conditions of electron ionization (EI) provided a unique “structural fingerprint”, which led to the development of EI/MS libraries (databases)39,40 and the use of computer assisted structure elucidation, as pioneered by the late Carl Djerassi.41 The use of automated tools for the identification of small molecules from MS data is emerging as a key way of dealing with: (i) the vast amounts of data in metabolomics; and (ii) dereplication of “hits” in the field of natural products chemistry. Since spectral libraries only contain known reference compounds, they will always be incomplete. Thus, there is a push to seek alternative methods to replace searching in spectral libraries by searching molecular structure databases such as PubChem and KEGG (Kyoto Encyclopedia of Genes and Genomes) and using these to create in silico spectral libraries based on programs that use empirical “rules” to predict gas-phase fragmentation behavior.42 Often these programs predict fragmentation based on the similarities of substructures, but the differences in fragmentation behavior of 1H+ and 2H+ clearly highlight the challenges of such an empirical approach. Thus there is a continued need to adopt a “physical organic” approach to understand how structure dictates which fragmentation pathways are the lowest in energy.43 With ever increasing computer speeds, it may be possible to predict tandem mass spectra using computational chemistry approaches similar to those being developed by Grimme for EI mass spectra.44Experimental methods
Chemicals
All chemicals were used as purchased. Acetonitrile, ethyl acetate and hexane were obtained from Merck (Kenilworth, NJ, USA). Acetic acid (glacial) was obtained from BDH (Poole, Great Britain). Deuterated water (D2O), acetonitrile-d3 and acetic acid-d4 were purchased from Cambridge Isotopes Laboratories (Andover, MA, USA). 2,2-Diethoxyethanamine and 2-chloro-5-chloromethylpyridine were obtained from Sigma-Aldrich (Saint Louis, United States). S-Methyilisothiouronium sulphate salt was purchased from Alfa Aesar (Heysham, Great Britain). Analytic grade ether was purchased from Chem-supply (Port Adelaide, SA, Australia).Synthesis of 2
The synthesis of olefin–imidacloprid 2 (ESI, Scheme S1†) was reproduced using the same method described by Novák et al.14,21 Full details of the synthesis and the characterization of key intermediates are given in the ESI.†X-ray crystallography of 2
The crystal structure of the final product 2 was determined on an Oxford SuperNova CCD diffractometer using Cu-Kα radiation (graphite crystal monochromators λ = 1.54184 Å). The crystal was maintained at 130.0 K during data acquisition. The structure was solved by direct methods and difference Fourier Synthesis45 Thermal ellipsoid plots were generated using the program ORTEP-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 integrated within the WINGX suite of programs.47 Crystal data for 2 C9H8N5O2Cl, M 253.65, T = 130.0(2) K, λ = 1.5418 Å, monoclinic, space group P21/c, a = 4.7377(5), b = 21.5338(19), c = 10.5144(8)Å, β = 101.413(9)°, V = 1051.48(17) Å3, Z = 4, Dc = 1.602 Mg M−3μ(Cu-Kα) = 3.244 mm−1, F(000) = 520, crystal size 0.51 × 0.05 × 0.05 mm. θmax = 74.29°, 4222 reflections measured, 2081 independent reflections (Rint = 0.050) the final R = 0.0525 [I > 2σ(I), 1659 data] and wR(F2) = 0.1554 (all data) GOOF = 1.091.
46 integrated within the WINGX suite of programs.47 Crystal data for 2 C9H8N5O2Cl, M 253.65, T = 130.0(2) K, λ = 1.5418 Å, monoclinic, space group P21/c, a = 4.7377(5), b = 21.5338(19), c = 10.5144(8)Å, β = 101.413(9)°, V = 1051.48(17) Å3, Z = 4, Dc = 1.602 Mg M−3μ(Cu-Kα) = 3.244 mm−1, F(000) = 520, crystal size 0.51 × 0.05 × 0.05 mm. θmax = 74.29°, 4222 reflections measured, 2081 independent reflections (Rint = 0.050) the final R = 0.0525 [I > 2σ(I), 1659 data] and wR(F2) = 0.1554 (all data) GOOF = 1.091.
Mass spectrometry experiments
:30:0.1% acetonitrile:water:formic acid mixture was introduced into the electrospray source at a flow-rate of ∼3 μL min−1 using a syringe pump. Typical electrospray conditions were employed using a needle potential of ∼+4 kV and a heated capillary temperature of 300 °C. The 2H+ was mass selected with a window of 1 m/z and then subjected to CID using a corresponding normalized collision energy of 20% and an activation Q of 0.25 for a period of 30 ms. For high accuracy mass measurements, the fragment ions are formed and isolated in the linear ion trap, and transferred to the FT-ICR MS for measurement of the m/z values with a mass accuracy that is sub part-per-million, providing unambiguous assignment of the stoichiometry of the precursor and fragment ions.
:30:0.1% acetonitrile:water:formic acid mixture was introduced into the electrospray source at a flow-rate of ∼3 μL min−1via a syringe pump system. The conditions used to ionize the compound were set as follow: spray voltage +3.5–4.0 kV, capillary temperature 250 °C, capillary voltage 25 V, nitrogen sheath gas pressure 50 (arbitrary unites). A selection window of 1 m/z and an activation Q of 0.25 for a period of 30 ms were used. 2H+ was mass selected in the gas phase and the normalised collision energy (NCE) was increased stepwise by 1.0% starting from 0% (no product ion formed) to a maximum of 30% (the relative intensity of the precursor ion was less than 5%). The NCE was converted to an amplitude of the resonance excitation RF voltage (tick amp) using the follow equation: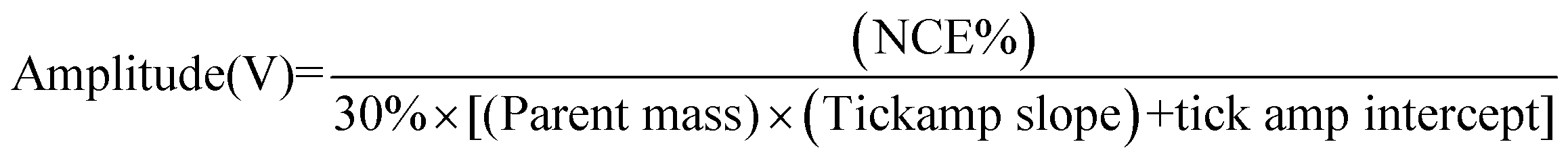
The tick amp slope and tick amp intercept are extracted from the normalised collision energy calibration file of the most recent calibration. The relative intensity of the product and precursor ions was plotted as a function of the increasing amplitude expressed in voltage.
:30:0.1% acetonitrile:water:formic acid mixture into a 100 μL HPLC injection loop using the Agilent 1100 autosampler. Sample was introduced in the mass spectrometer at the flow-rate of 0.3 mL min−1. Ionisation of 2H+ was obtained using a dual-nebulizer ESI source with the capillary voltage set at +3 kV, gas temperature (nitrogen) set at 300° C, dry gas at 12 L min−1 and nebulizer at 50 psig. The fragmentor and the skimmer were set at 120 and 65 V respectively to induce the ionisation of 2H+ avoiding ion source fragmentation. 2H+ was mass selected in the quadrupole using a selection window of 1.3 m/z to exclude the 37Cl-isotope pick at m/z 256. CID was performed applying increasing CID energy from 5 to 20% till 2H+ was less than 5% of its original intensity in the MS/MS spectrum.
:29:1 D2O:acetonitrile-d3:acetic acid-d4 mixture for 2 h at room temperature. The addition of acetic acid-d4 to the mixture helped the exchange of the N(1)–H with deuterium. Using the hybrid linear ion trap with the same settings described above, [M − H + 2D]+ was mass selected (m/z 256) and fragmented under identical CID conditions used for 2H+. Even though the H/D exchange was not complete in solution as evidenced by the presence of 2H+ in the mass spectrum, the HRMS information obtained from the FT-ICR helped to distinguish among the compounds derived from the fragmentation of [M − H + 2D]+ and the fragmentation of the 37Cl isotope of 2H+ (m/z 256.05648 and 256.04098 respectively) which cannot be distinguished and isolated in the ion trap.
DFT calculations
The coordinates from the crystal structure of 2 were used as a starting point for our computational analysis. We carried out molecular dynamic (MD) simulations using the Discovery Studio (DS) 2.5 program package and the method settings described in a previous work34 to provide stable structures for compound 2 in the gas-phase. At completed simulation, the 10 most stable structures were selected and further optimized in Gaussian 09 to refine the previous result and find the lowest energy structure. Rotations of 90° of the ‘nitroimine chain’ of the guanidine moiety were used to establish which tautomeric form was more stable and the role played by the hydrogen bond N(1)–H⋯O(1) in the stability of compound 2 in the gas-phase. A 360° scan on the dihedral angle D1 (Scheme 1) was carried out to confirm the position of the nitrogen N(5) on the pyridine in relation to the nitroguanidine moiety. The final optimised structure was used to determine the local PAs for the different heteroatom (N and O) and the barriers involved in the gas-phase fragmentation reactions of 2H+. DFT calculations were carried out using Gaussian 09 with the B3LYP level of theory and the 6-311G++(d,p) basis set to allow a direct comparison with compound 1H+.22 The vibrational frequencies of each DFT optimized structure were calculated to ensure they were either minima (all positive) or transition state structures (one negative frequency). Transition states were connected to reactants and products via intrinsic reaction coordinate (IRC) calculations. The Electronic energies reported are relative to the ground state structure of 2H+ (0 kJ mol−1). The transition state associated with bond homolysis (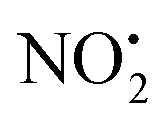 loss) was not calculated due to the challenges on moving from a singlet electronic ground state to doublet products. Thus dashed lines are used to connect 2H+ to the final energies of the radical products.
loss) was not calculated due to the challenges on moving from a singlet electronic ground state to doublet products. Thus dashed lines are used to connect 2H+ to the final energies of the radical products.
Conclusions
The structure/reactivity of olefin–imidacloprid 2 compared to the parent insecticide imidacloprid 1, was revealed using X-ray crystallography, tandem mass spectrometry and DFT calculations. A comparison of the X-ray structure of compounds 1 and 2 with 132 similar nitroguanidine containing compounds deposited in the Cambridge database, highlights that both compounds prefer tautomeric forms A. However, an analysis of the bond lengths at the nitroguanidine residue showed that the shorter N(3)–N(4) bond and the longer C(1)–N(2) bond of 2 best fit resonance form A2 while imidacloprid more closely resembles resonance form A1. These differences underpin the distinct gas-phase fragmentation reactions of 1H+ and 2H+. Thus the contribution of resonance form A2 makes (i) the oxygen atom O(2) more basic and (ii) the nitrogen N(1) more acidic. The increased basicity in O(2) is the reason why the 1,3-intramolecular proton transfer to N(2) or between the two oxygen atoms requires higher energies of 156 and 145 kJ mol−1 respectively compared to 117 kJ mol−1 required to rotate the NO2 group by 180°. With the oxygen positioned next to N(1)–H, a 1,5-intramolecular proton transfer can occur. As the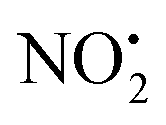 and HNO2 losses can only occur in 2H+ when the proton has been transferred on N(2), their relative abundance is minor (no more that 30% of total product ions). The use of multistage low-energy CID conditions established that: (i) water loss is the main fragmentation channel of 2H+; (ii) the product ions at m/z 205 and 171 used in MRM studies are likely to arise from sequential fragmentation reactions.
and HNO2 losses can only occur in 2H+ when the proton has been transferred on N(2), their relative abundance is minor (no more that 30% of total product ions). The use of multistage low-energy CID conditions established that: (i) water loss is the main fragmentation channel of 2H+; (ii) the product ions at m/z 205 and 171 used in MRM studies are likely to arise from sequential fragmentation reactions.
Acknowledgements
We thank the Australian Research Council for financial support via the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology (CE0561607). Roberto Fusetto thanks The University of Melbourne for a postgraduate scholarship and Michael Leeming and Athanasios Zavras for their support with the DFT calculations. We thank Dr W. Alex Donald and Huixin Wang from the University of New South Wales for their help with the molecular dynamic simulations. For generously providing computational resources high-performance computing facilities, we thank the Victorian Partnership for Advanced Computing and the School of Chemistry at the University of Melbourne.References
- P. Jeschke, R. Nauen, M. Schindler and A. Elbert, J. Agric. Food Chem., 2011, 59, 2897–2908 CrossRef CAS PubMed.
- M. Tomizawa and J. E. Casida, Acc. Chem. Res., 2009, 42, 260–269 CrossRef CAS PubMed.
- M. Tomizawa and J. E. Casida, J. Agric. Food Chem., 2011, 59, 2825–2828 CrossRef CAS PubMed.
- D. Carrington, Pesticides Linked to Honeybee Decline, in The Guardian, 2012 Search PubMed.
- P. R. Whitehorn, S. O'Connor, F. L. Wackers and D. Goulson, Science, 2012, 336, 351–352 CrossRef CAS PubMed.
- M. Gross, Curr. Biol., 2013, 23, R462–R464 CrossRef CAS PubMed.
- I. Yamamoto, Nicotinoid Insecticides and the Nicotinic Acetylcholine Receptor, Springer, Tokyo, 1999 Search PubMed.
- J. E. Casida, J. Agric. Food Chem., 2011, 59, 2923–2931 CrossRef CAS PubMed.
- M. A. Riaz, A. Chandor-Proust, C. Dauphin-Villemant, R. Poupardin, C. M. Jones, C. Strode, M. Régent-Kloeckner, J. P. David and S. Reynaud, Aquat. Toxicol., 2013, 126, 326–337 CrossRef CAS PubMed.
- S. Suchail, L. Debrauwer and L. P. Belzunces, Pest Manage. Sci., 2004, 60, 291–296 CrossRef CAS PubMed.
- M. Tomalski, W. Leimkuehler, C. Schal and E. L. Vargo, Ann. Entomol. Soc. Am., 2010, 103, 84–95 CrossRef CAS.
- H. Nishiwakl, K. Sato, Y. Nakagawa, M. Miyashlta and H. Miyagawa, J. Pesticide Sci., 2004, 29, 110–116 CrossRef.
- K. K. Hoi, P. J. Daborn, P. Battlay, C. Robin, P. Batterham, R. A. J. O'Hair and W. A. Donald, Anal. Chem., 2014, 86, 3525–3532 CrossRef CAS PubMed.
- T. T. Talley, M. Harel, R. E. Hibbs, Z. Radić, M. Tomizawa, J. E. Casida and P. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7606–7611 CrossRef CAS PubMed.
- S. L. Chao and J. E. Casida, Pestic. Biochem. Physiol., 1997, 58, 77–88 CrossRef CAS.
- A. Rocher and N. Marchand-Geneste, SAR QSAR Environ. Res., 2008, 19, 245–261 CrossRef CAS PubMed.
- M. Ihara, T. Okajima, A. Yamashita, T. Oda, K. Hirata, H. Nishiwaki, T. Morimoto, M. Akamatsu, Y. Ashikawa, S. Kuroda, R. Mega, S. Kuramitsu, D. B. Sattelle and K. Matsuda, Invertebr. Neurosci., 2008, 8, 71–81 CrossRef CAS PubMed.
- H. Nishiwaki, Y. Nakagawa, M. Kuwamura, K. Sato, M. Akamatsu, K. Matsuda, K. Komai and H. Miyagawa, Pest Manage. Sci., 2003, 59, 1023–1030 CrossRef CAS PubMed.
- S. Kagabu, C. Kato and K. Nishimura, J. Pesticide Sci., 2004, 29, 376–379 CrossRef CAS.
- Y. J. Dai, S. Yuan, F. Ge, T. Chen, S. C. Xu and J. P. Ni, Appl. Microbiol. Biotechnol., 2006, 71, 927–934 CrossRef CAS PubMed.
- L. Novák, G. Hornyánszky, I. Király, J. Rohály, P. Kolonits and C. Szántay, Heterocycles, 2001, 55, 45–58 CrossRef.
- W. A. Donald, M. G. Leeming and R. A. J. O'Hair, Int. J. Mass Spectrom., 2012, 316, 91–99 CrossRef.
- F. H. Allen, Acta Crystallogr., Sect B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- D. Chopra, T. P. Mohan, K. S. Rao and T. N. G. Row, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o2415–o2417 CAS.
- Y. Qin, P. Zhao, S. Fan, Y. Han, Y. Li, N. Zou, S. Song, Y. Zhang, F. Li, X. Li and C. Pan, J. Chromatogr., A, 2015, 1385, 1–11 CrossRef CAS PubMed.
- S. Fan, P. Zhao, C. Yu, C. Pan and X. Li, Food Addit. Contam., Part A Chemistry, Analysis, Control, Exposure and Risk Assessment, 2014, 31, 73–82 CrossRef CAS PubMed.
- S. H. Tseng, C. C. Liu, Y. J. Lin, H. C. Chen, S. C. Su, H. K. Chou, S. S. Chou and D. Y. C. Shih, J. Food Drug Anal., 2009, 17, 319–332 CAS.
- C. Jabot, M. Fieu, B. Giroud, A. Buleté, H. Casabianca and E. Vulliet, Int. J. Environ. Anal. Chem., 2015, 95, 240–257 CrossRef CAS.
- M. Gbylik-Sikorska, T. Sniegocki and A. Posyniak, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2015, 990, 132–140 CrossRef CAS PubMed.
- M. Eckert-Maksić, Z. Glasovac, H. Maskill and I. Zrinski, J. Phys. Org. Chem., 2003, 16, 491–497 CrossRef.
- L. G. Cabrini, M. Benassi, M. N. Eberlin, T. Okazaki and K. K. Laali, Eur. J. Org. Chem., 2007, 70–77 CrossRef CAS.
- A. Giordana, G. Ghigo, G. Tonachini, D. Ascenzi, P. Tosi and G. Guella, J. Chem. Phys., 2009, 131 Search PubMed.
- L. Feketeova, J. Postler, A. Zavras, P. Scheier, S. Denifl and R. A. J. O'Hair, Phys. Chem. Chem. Phys., 2015, 17, 12598–12607 RSC.
- H. Wang, B. Wang, Z. Wei, H. Zhang and X. Guo, J. Mass Spectrom., 2015, 50, 212–219 CrossRef CAS PubMed.
- R. A. J. O'Hair, P. S. Broughton, M. L. Styles, B. T. Frink and C. M. Hadad, J. Am. Soc. Mass Spectrom., 2000, 11, 687–696 CrossRef.
- V. Borrett, I. Dagley, M. Kony and T. Blumenthal, Eur. J. Mass Spectrom., 1995, 1, 59–64 CrossRef CAS.
- J. H. Beynon, Org. Mass Spectrom., 1968, 1, 739–740 CrossRef.
- K. Biemann, Mass spectrometry: organic chemical applications, McGraw-Hill, 1962 Search PubMed.
- Wiley Specialty Mass Spectral Libraries, http://www.sisweb.com/software/ms/wiley.htm.
- NIST 14 Mass Spectral Library, http://www.sisweb.com/software/ms/nist.htm.
- B. G. Buchanan, D. H. Smith, W. C. White, R. J. Gritter, E. A. Feigenbaum, J. Lederberg and C. Djerassi, J. Am. Chem. Soc., 1976, 98, 6168–6178 CrossRef CAS.
- F. Hufsky, K. Scheubert and S. Böcker, Nat. Prod. Rep., 2014, 31, 807–817 RSC.
- G. Bouchoux, J. Mass Spectrom., 2013, 48, 505–518 CrossRef CAS PubMed.
- S. Grimme, Angew. Chem., Int. Ed., 2013, 52, 6306–6312 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. Colorado and J. Brodbelt, J. Am. Soc. Mass Spectrom., 1996, 7, 1116–1125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis of olefin 2 and 1H-NMR and mass spectra information for each synthetic product, crystallographic analysis, HRMS measurements for key fragment ions, scatter plots for the two resonance forms, QTof CID mass spectra, alternative DFT calculated mechanisms for loss of water and HNO2. CCDC 1422950. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02371h |
| This journal is © The Royal Society of Chemistry 2016 |