
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent reactions of methyl substituted all-cis tetrafluorocyclohexane aldehydes†
Tetiana
Bykova
,
Nawaf
Al-Maharik
,
Alexandra M. Z.
Slawin
and
David
O'Hagan
*
School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: do1@st-andrews.ac.uk
First published on 1st December 2015
Abstract
This paper reports the preparation of methyl substituted all-cis tetrafluorocyclohexanes prepared from a Birch reduction of benzoic acid, worked up with a methyl iodide quench. The resultant methylcyclohexadiene carboxylic acid was reduced to the alcohol, protected as an ether and then a sequence of functional group manipulations carried out to introduce four fluorines. The cyclohexadienyl ring was then epoxidised and the C–O bonds sequentially converted through deoxyfluorination reactions to two sets of isomers of all-cis tetrafluorocyclohexane isomers. The blocking methyl group renders the ring safe to hydrogen fluoride elimination. Deprotection of the benzylic ether and then oxidation gave aldehydes which were then used in Ugi and Passerini multicomponent reactions, allowing this facially polarised cyclohexane to be incorporated into peptidic structural motifs.
1. Introduction
We have recently described the synthesis and properties of the all-cis 2,3,5,6-tetrafluorocyclohexane 1 ring system.1 The stereochemistry is such that in the chair conformation the four fluorine atoms are all on one face of the cyclohexane ring. Also two of the C–F bonds are 1,3-diaxial and align parallel to each other and this results in an orientated polarity, which is supplemented by the two equatorial C–F bonds which are also on the upper face of the cyclohexane ring (Fig. 1). The outcome is a large molecular dipole moment of 5.2 Dy for 1.1 The motif is polar hydrophobic and an interesting and unusual aspect of the cyclohexane ring system is that it has facial polarity, therefore it becomes attractive to access building blocks which might be used to introduce the motif into drug discovery and agrochemical research programmes. Derivatives of the ring system are relatively challenging to prepare, however recently we reported the preparation of phenyl derivative 2 and then access to a variety of aryl derivatives by standard electrophilic aromatic substitution reactions of 2.2–4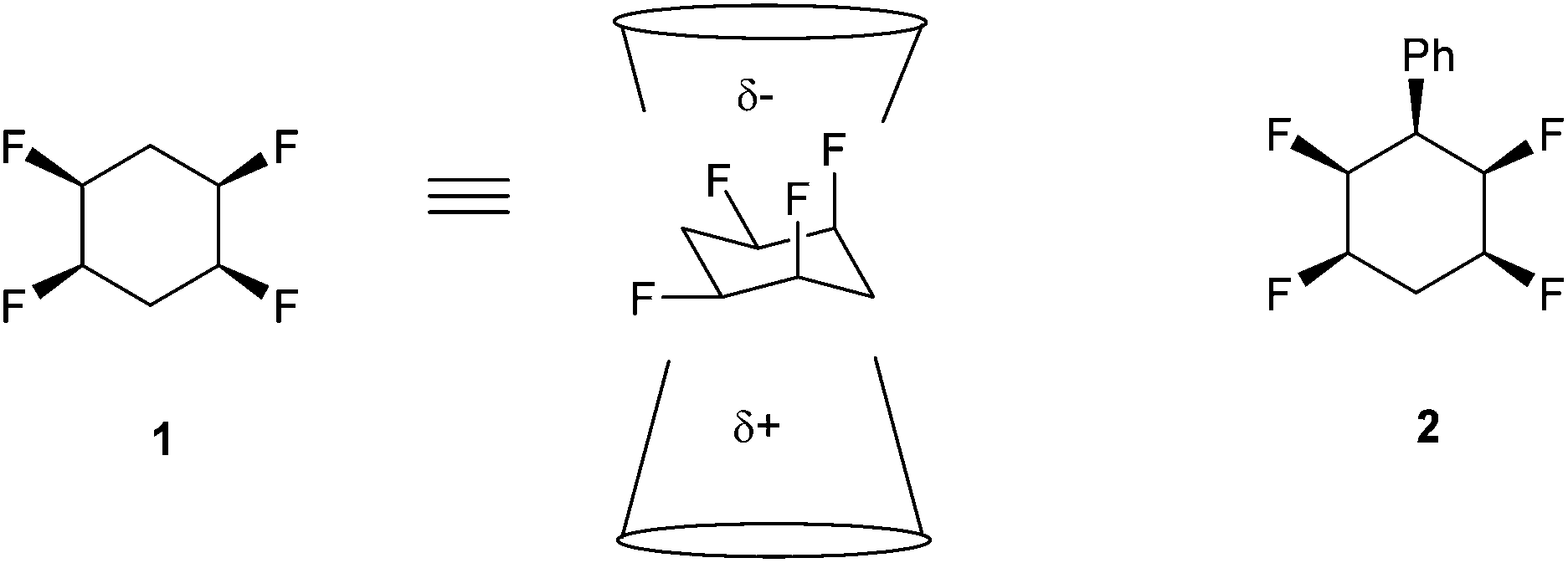 | ||
| Fig. 1 All-cis-tetrafluorocyclohexane 1 is facially polarised (μ = 5.2 D), with a more negative fluorine face and a more positive hydrogen face.1 | ||
In order to build further structural diversity around this motif, cyclohexane carboxaldehyde derivatives are reported, carrying a methyl group alpha to the carbonyl group. The methyl group is a design feature placed to protect against hydrogen fluoride elimination. The utility of the aldehydes in multicomponent reactions has allowed a library of peptidic analogues to be prepared.5,6
2. Results and discussion
At the outset a direct oxidation of phenyl derivative 2 was explored. The corresponding carboxylic acid 3 was the anticipated product, however it proved difficult to characterize in our hands as it is very vulnerable to hydrogen fluoride elimination to products related to 4, due to the relatively acidic hydrogen atom, alpha to the carboxylic acid derivative (Scheme 1).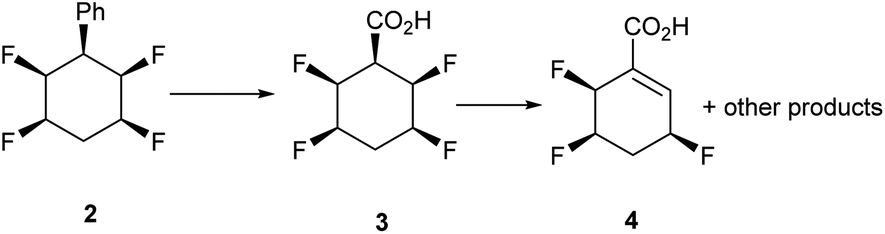 | ||
| Scheme 1 Carboxylic acid 3 is unstable to dehydrofluorination. | ||
In order to circumvent this problem it became an objective to replace the alpha hydrogen with a methyl group and generate a more stable motif. Following a literature procedure, the Birch reduction of benzoic acid 5 followed by in situ methylation afforded α-methylated carboxylic diene 6, which was then reduced with LiAlH4 to give alcohol 7 in good yield.7 Protection of the primary alcohol 7 with benzyl bromide furnished ether 8 (80% yield), which was then epoxidised using an excess of mCPBA.8,9 Three diastereoisomers of the diepoxide were generated, cis (9a and 9b) and trans (9c) in 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 ratio (Scheme 2).
:4:1 ratio (Scheme 2).
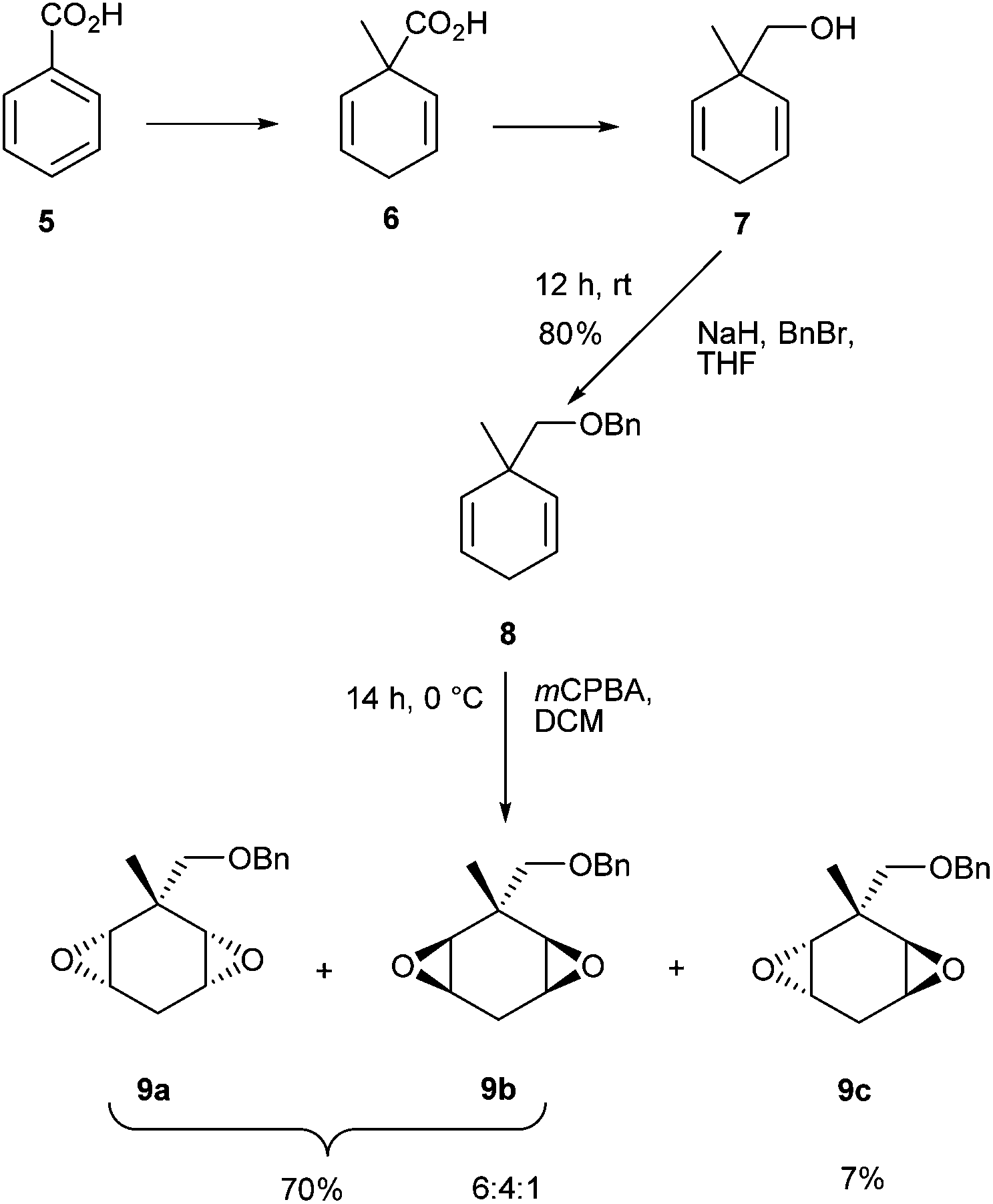 | ||
| Scheme 2 Synthesis of diepoxides 9a, b, c from benzoic acid 5. | ||
The trans-diepoxide stereoisomer 9c was separated (7%) from the inseparable cis-diepoxides 9a and 9b (70%). The configuration of the epoxides was determined by 1H NMR spectroscopy. The cis-diepoxides (9a and 9b) could be differentiated from the trans through the methylene hydrogen atoms H-4a and H-4b. For the trans-diastereoisomer, both hydrogens are in similar environments and appear as unresolved signals (δ 2.30 ppm), whereas for the cis-diepoxides (9a and 9b), these hydrogens are non-equivalent and resolve into a pair of multiplets (δ 2.77 ppm and δ 2.24 ppm).
Treatment of the diepoxide mixture 9a and 9b with Et3N·3HF at 140 °C, resulted in their full conversion to difluoro diols 10a and 10b as determined by NMR (Scheme 3). This mixture was then reacted with triflic anhydride which resulted in the diastereoisomeric triflates 11a and 11b. These products could be separated by chromatography and were recovered in yields of 30% (11a) and 28% (11b) respectively (Scheme 3).
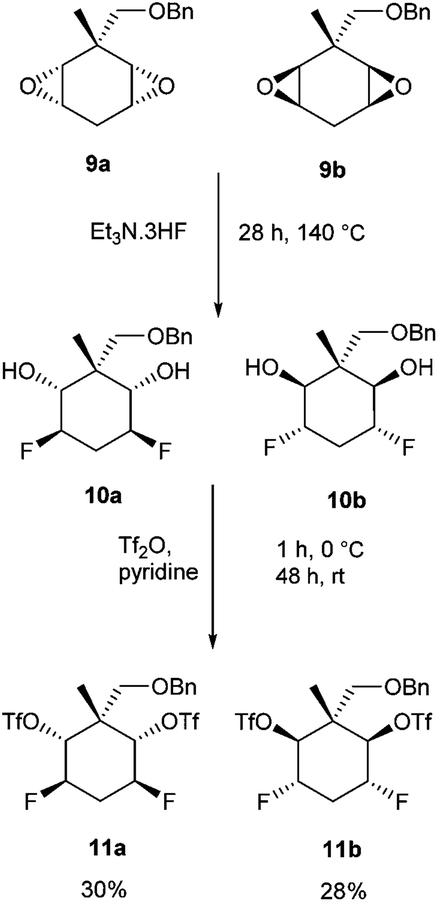 | ||
| Scheme 3 Ring opening fluorination of diepoxides 9a and 9b and then activation to ditriflates 11a and 11b. | ||
Diastereoisomer 11a was treated with Et3N·3HF at 110 °C for a prolonged period which generated tetrafluorocyclohexane 12a. O-Debenzylation (10% Pd/C/H2) generated alcohol 13a as illustrated in Scheme 4.10
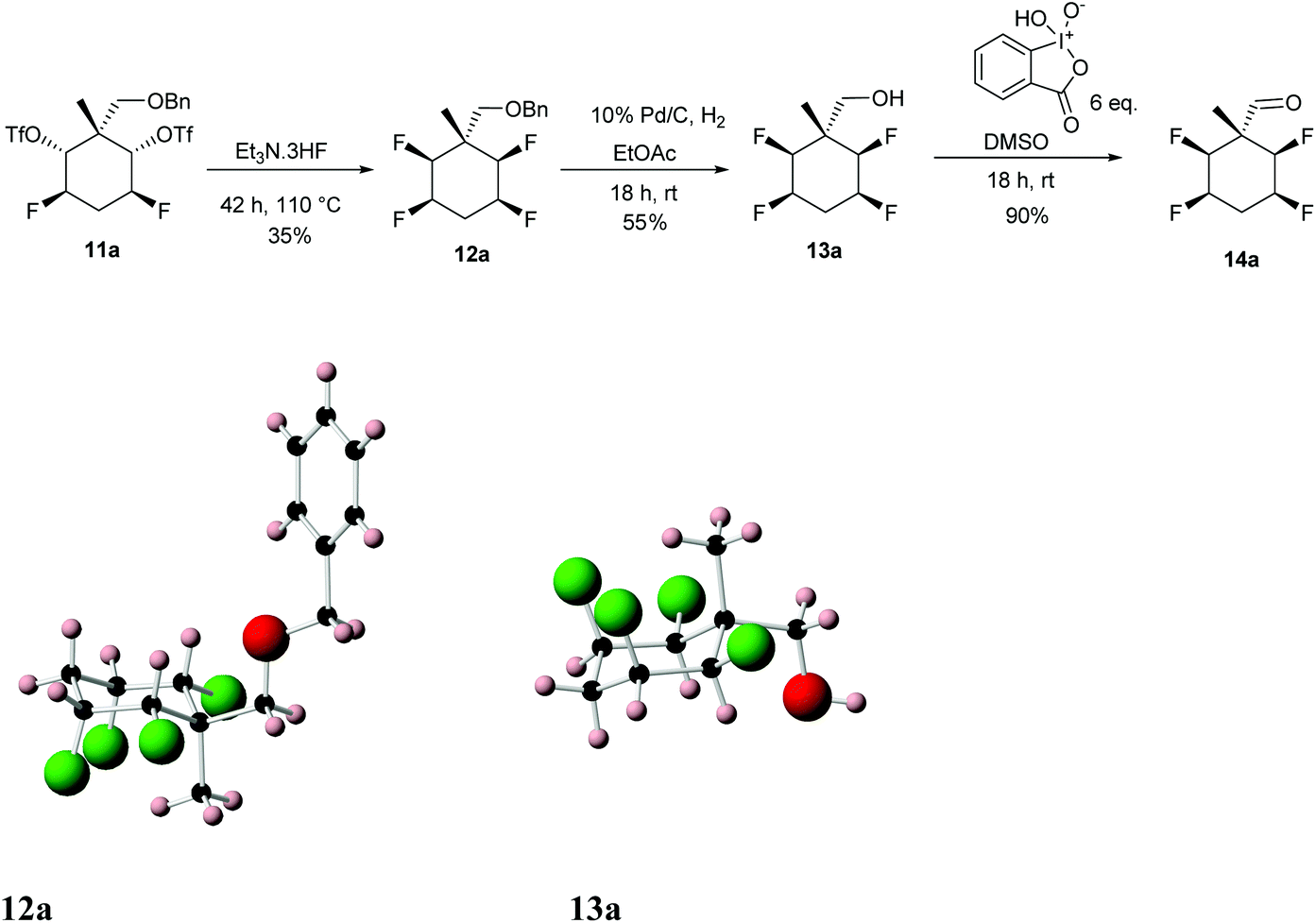 | ||
| Scheme 4 Preparation of aldehyde 14a from di-triflate 11a and showing the X-ray structures of 12a and 13a. | ||
The structures and stereochemistry of products 12a and 13a were confirmed by X-ray crystallography and are shown in Scheme 4. An analogous procedure was carried out on diastereoisomer 11b, to generate aldehyde diastereoisomer 14b. Again the X-ray structures of benzyl ether 12b and deprotected alcohol 13b were obtained to confirm stereochemistry and they are shown in Scheme 5. Finally oxidation of alcohols 13a and 13b using IBX in DMSO, gave aldehydes 14a (90%) and 14b (68%) respectively.11,12
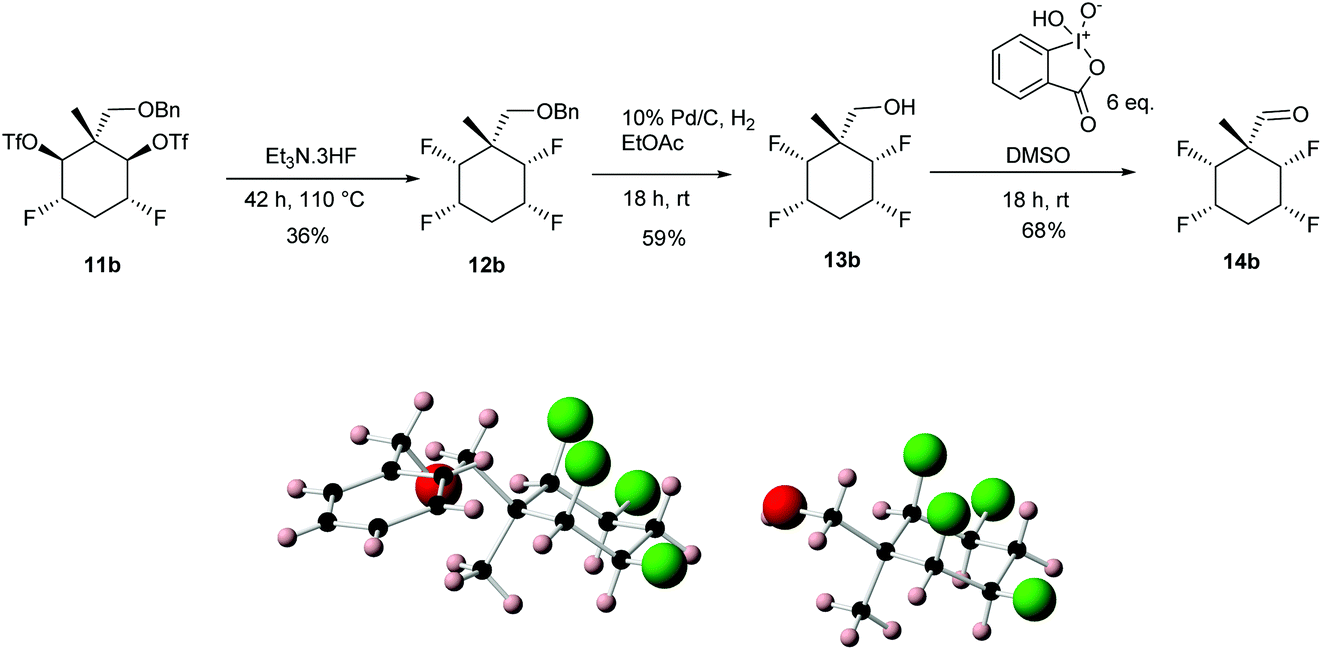 | ||
| Scheme 5 Preparation of aldehyde 14b from ditriflate 11b and showing the X-ray structures of 12b and 13b. | ||
The application of aldehydes 14a and 14b in Ugi four-component reactions was explored.5,6,13 In the first instance imines 15 were formed from the condensation of tetrafluoro aldehydes 14a/14b with a candidate amines A1/A2, and then individual isocyanides B and carboxylic acids C were added as illustrated in Scheme 6.
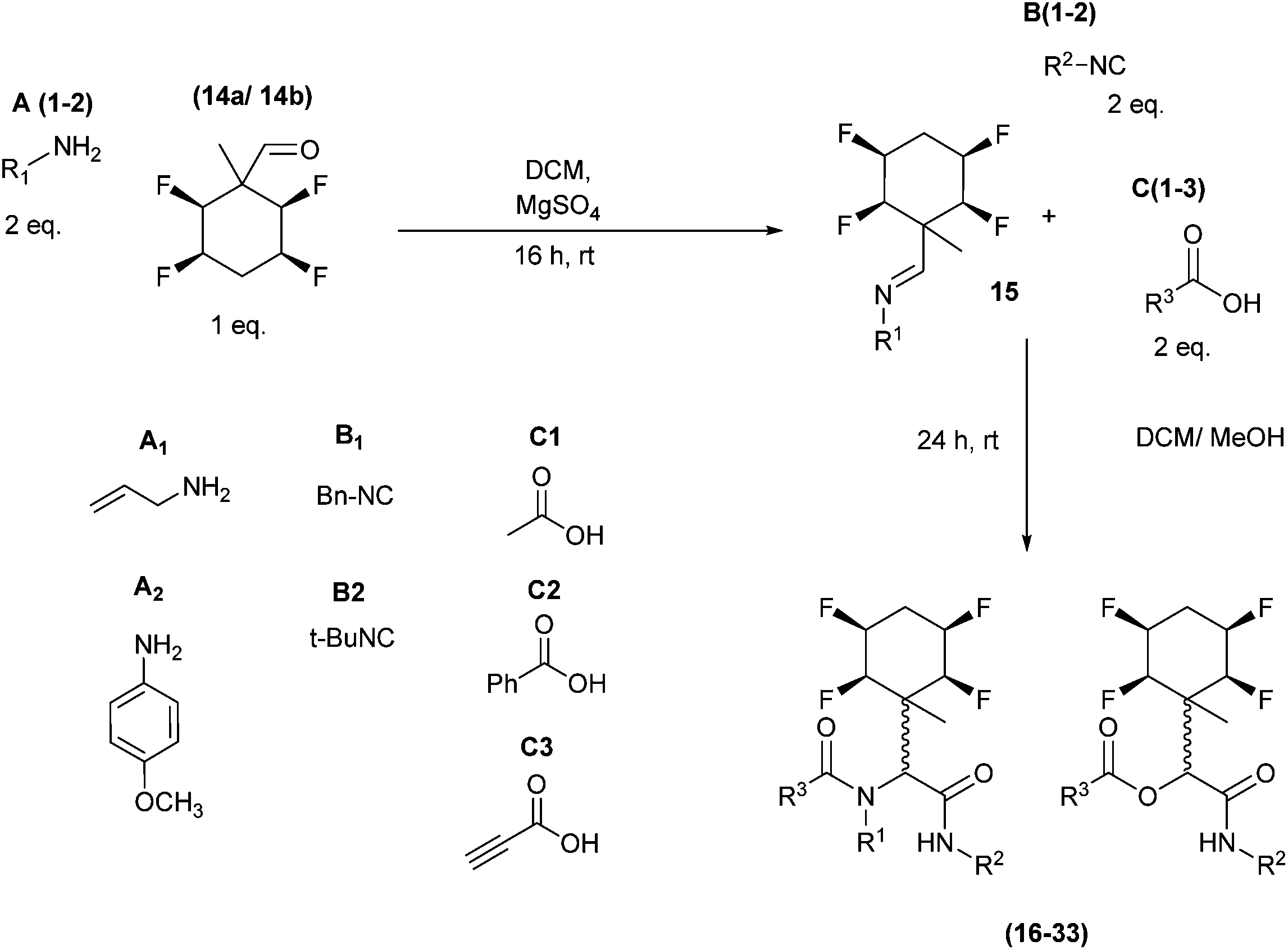 | ||
| Scheme 6 General Ugi procedure involving aldehydes 14a and 14b and a range of amines (A1–2), isocyanides (B1–2) and carboxylic acids (C1–3). | ||
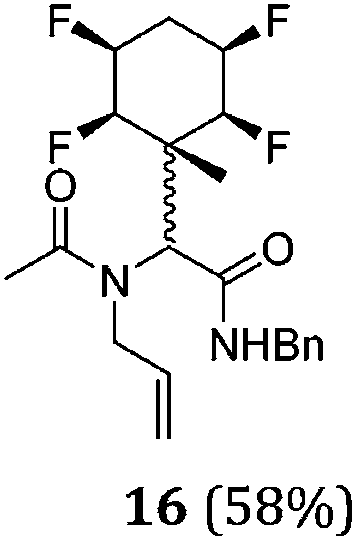
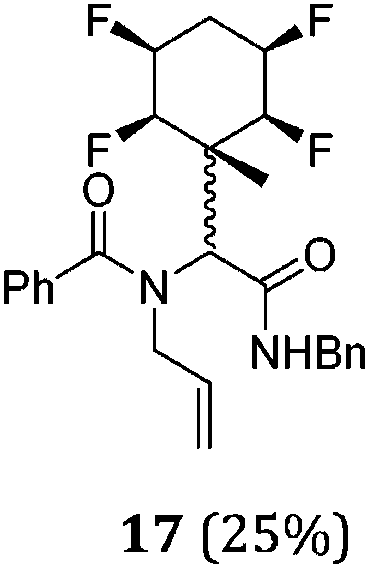
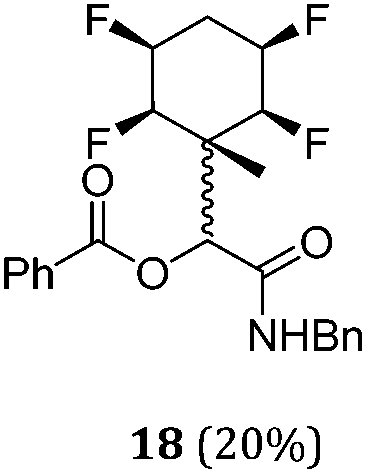
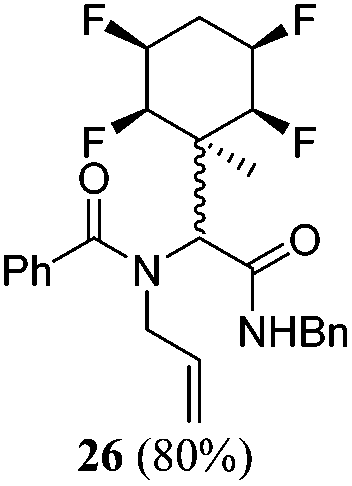
A series of reactions were conducted to generate a small chemical library of peptidomimetics 16–32 carrying the tetrafluorocyclohexyl ring motif. These compounds are all racemic as a new stereogenic centre is generated.
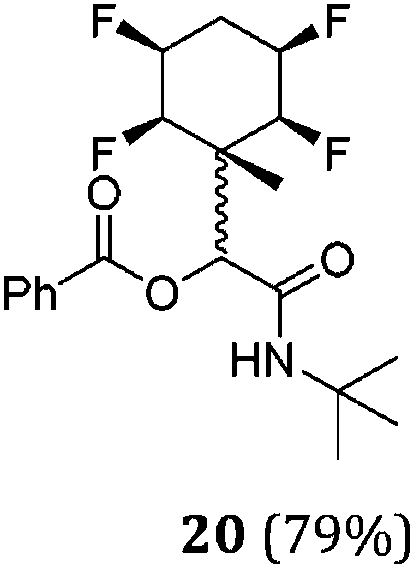
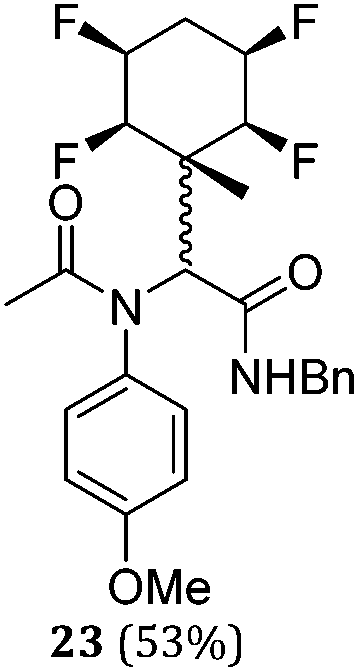
While some products were obtained by classic four component Ugi reactions, others derive from the Passerini reaction (Tables 1 and 2), through direct reaction of the free aldehyde 14 rather than the preformed imine 15, with the added isocyanides B and carboxylic acids C.13–17 In most cases the four component reactions were found to produce both, the α-aminoacyl amide Ugi derivatives and α-hydroxy carboxamide Passerini type products. The crystal structures of several Ugi products 16–17, 25–26, 32 and a Passerini α-acyloxy amide 20 are shown of Fig. 2. In each case compensating enantiomers of these racemic products are obvious in the unit cell of each crystal structure. The 19F{1H}-NMR spectra of these compounds merit some comment. For example the 19F{1H}-NMR spectra of aldehydes 4a and 4b have two sets of equivalent fluorines, which show clear AA′XX′ second order spectra, however for the multicomponent products a stereogenic centre is generated and the two sets of originally equivalent fluorines become diastereotopic and all four become chemically non-equivalent and they resolve to varying extents.
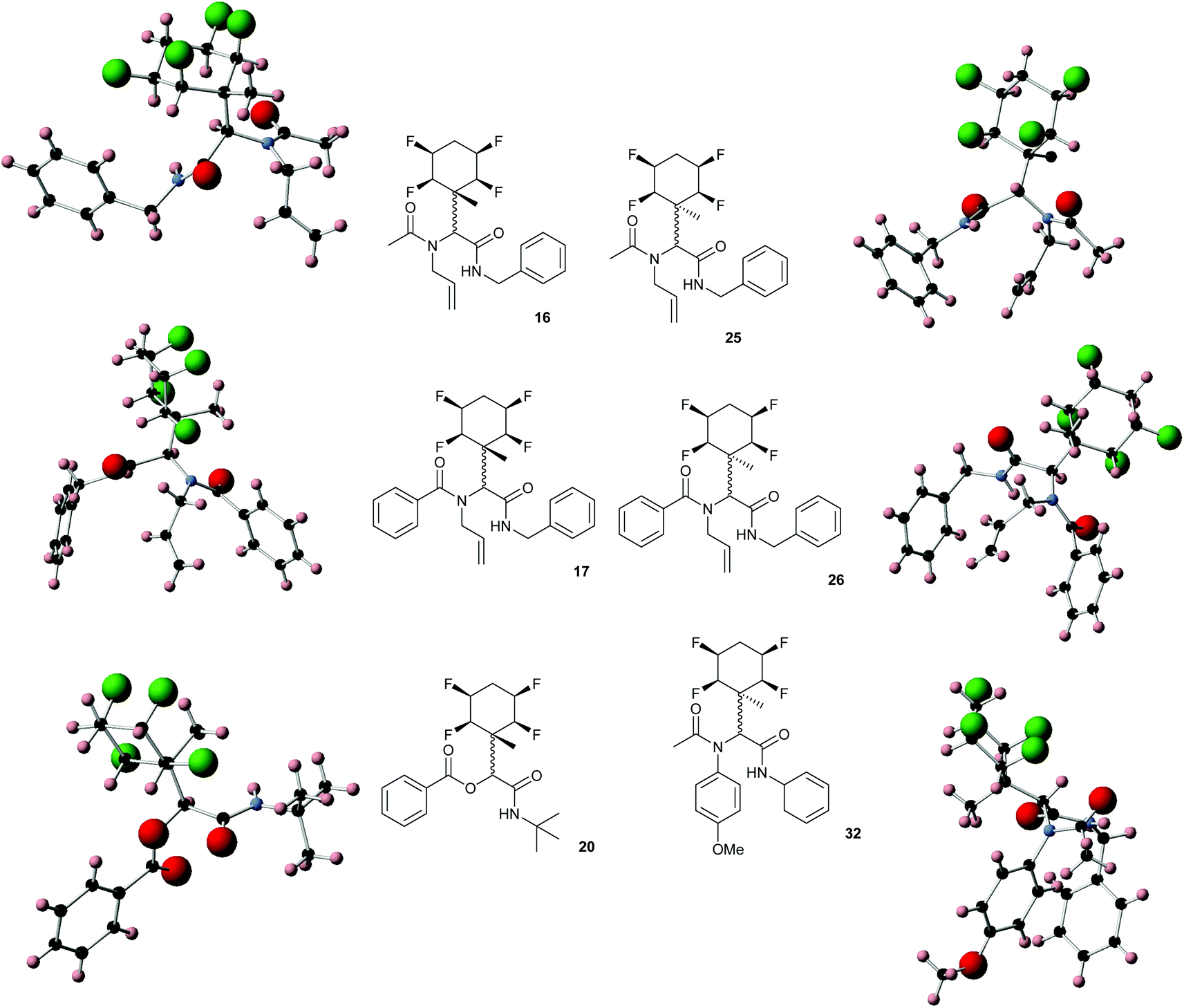 | ||
| Fig. 2 X-ray structures of a selected α-aminoacyl amides 16, 17, 25, 26, 32 which derive from reaction entries 1,2,8,9 and 14 in Table 1, and of the Passerini by-product 20 from reaction entry 3 of Table 1. | ||
| Entry | R1CNH2 | R2CN | R3COOH | Conditions (time, temp, solvent) | Ugi (yield) | Passerini (yield) | Ratio (1:2) |
|---|---|---|---|---|---|---|---|
| 1 | A1 | B1 | C1 | 40 h, 30 °C, DCM |
|
N/A | — |
| 2 | A1 | B1 | C2 | 40 h, 25 °C, DCM |
|
|
2:1 |
| 3 | A1 | B2 | C1 | 40 h, 25 °C, DCM | N/A |
|
— |
| 4 | A1 | B2 | C2 | 40 h, 25 °C, DCM | N/A |
|
— |
| 5 | A1 | B1 | C3 | 40 h, 25 °C, DCM |
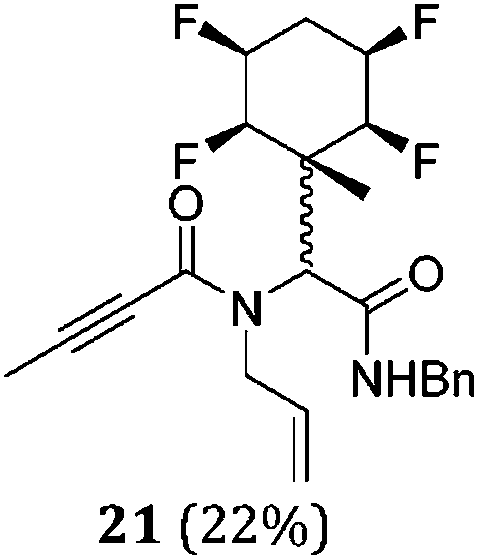
|
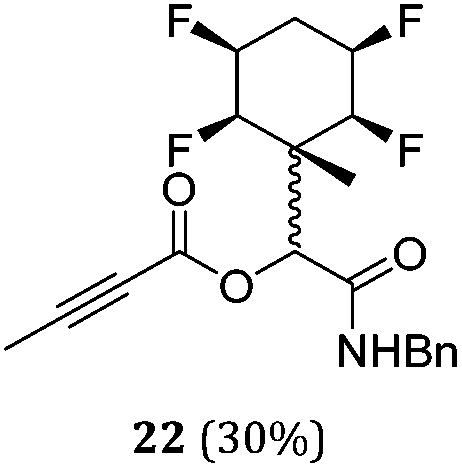
|
1:2.5 |
| 6 | A2 | B1 | C1 | 40 h, 25 °C, DCM |
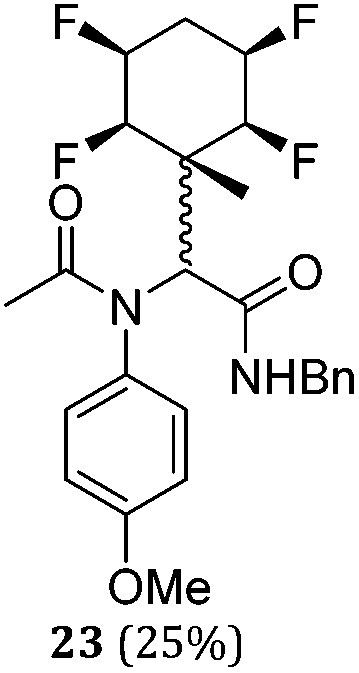
|
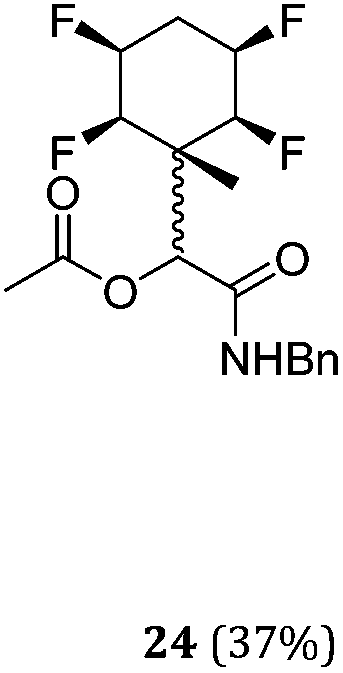
|
4:5 |
| 7 | A2 | B1 | C1 | 40 h, 25 °C, MeOH |
|
N/A |
| Entry | R1CNH2 | R2CN | R3COOH | Conditions (time, temp, solvent) | Product 1 (yield) | By-product (yield) | Ratio (1: 2) |
|---|---|---|---|---|---|---|---|
| 8 | A1 | B1 | C1 | 40 h, 30 °C, DCM |
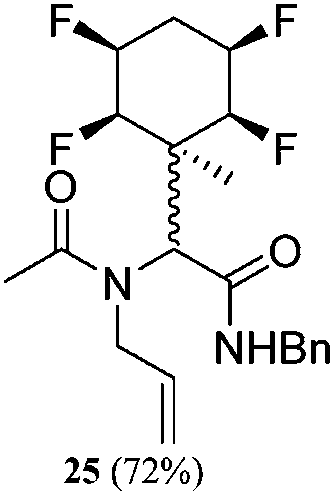
|
N/A | — |
| 9 | A1 | B1 | C2 | 40 h, 25 °C, DCM |
|
N/A | — |
| 10 | A1 | B2 | C1 | 40 h, 25 °C, DCM |
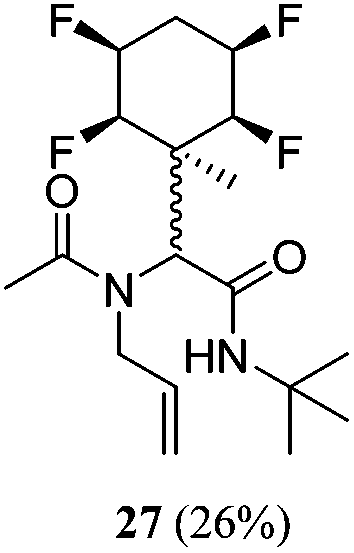
|
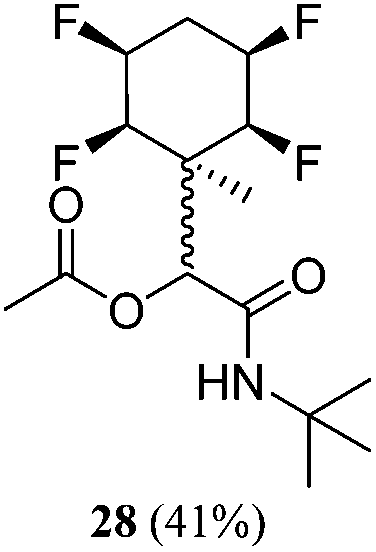
|
1:2 |
| 11 | A1 | B2 | C2 | 40 h, 25 °C, DCM |
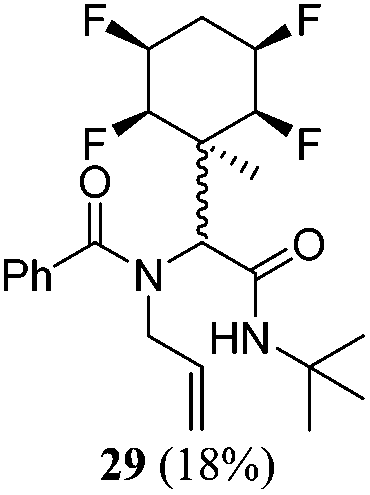
|
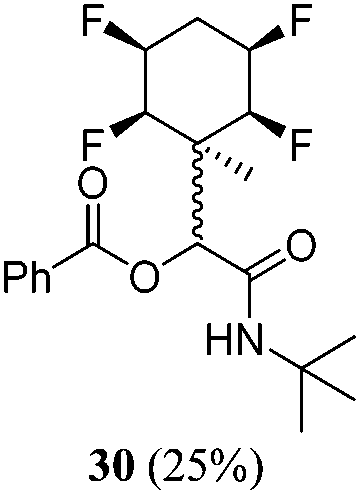
|
1:2 |
| 12 | A1 | B1 | C3 | 40 h, 25 °C, DCM |
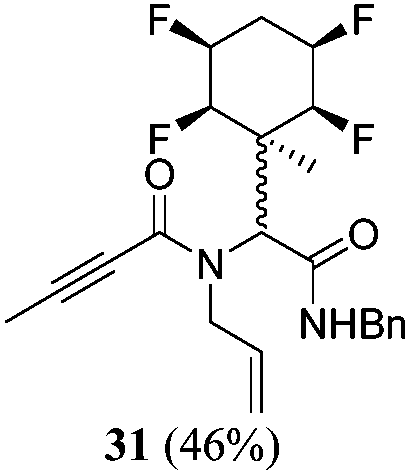
|
N/A | — |
| 13 | A2 | B1 | C1 | 40 h, 25 °C, DCM |
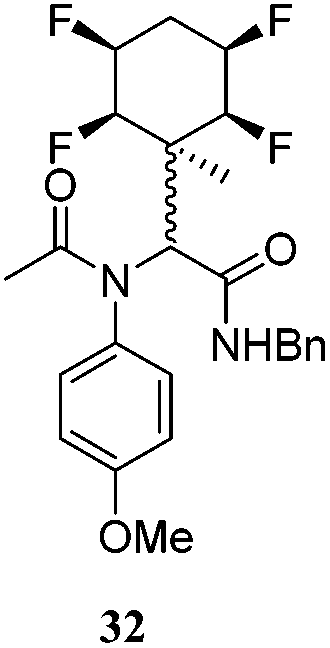
|
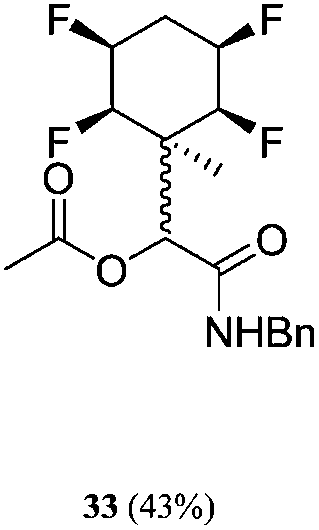
|
1:8 |
| 14 | A2 | B1 | C1 | 40 h, 25 °C, MeOH |
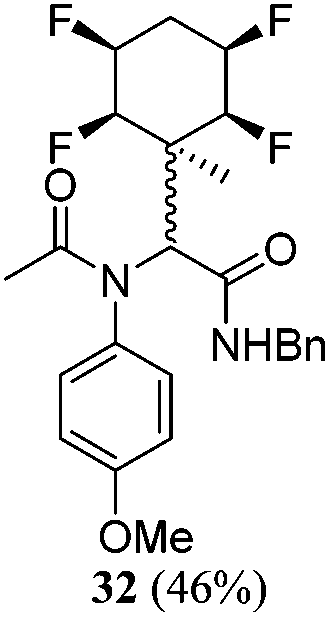
|
N/A | — |
3. Conclusion
In summary we report the synthesis of aldehyde diastereoisomers 14a and 14b, containing the all-syn 1,2,4,5-tetrafluorocyclohexane motif. These aldehydes are rendered stable to dehydrofluorination by having a blocking alpha methyl group. Aldehydes 14a and 14b were used as key components for Ugi four component reactions and in the event both α-aminoacyl amide and α-hydroxy carboxamide derivatives were generated. This approach allows the facially polarized all-cis tetrafluorocyclohexane motif to be introduced into peptidomimetic scaffolds for exploration in bioactivity screening programmes.Acknowledgements
We thank EPSRC for supporting this work and DO'H is grateful for a Royal Society Wolfson Research Merit Award. We also thank the EPSRC Mass Spectrometry Service at Swansea for analyses.References
- A. J. Durie, A. M. Z. Slawin, T. Lebl, P. Kirsch and D. O'Hagan, Chem. Commun., 2012, 48, 9643–9645, 10.1039/C2CC34679F.
- M. S. Ayoup, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Org. Biol. Chem., 2015, 13, 5621–5624, 10.1039/C5OB00650C.
- A. J. Durie, T. Fujiwara, R. Cormanich, M. Bühl, A. M. Z. Slawin and D. O'Hagan, Chem. – Eur. J., 2014, 20, 6259–6263, DOI:10.1002/chem.201400354.
- A. J. Durie, T. Fujiwara, N. Al-Maharik, A. M. Z. Slawin and D. O'Hagan, J. Org. Chem., 2014, 79, 8228–8233, DOI:10.1021/jo501432x.
- I. Ugi, A. Demharter, W. Hörl and T. Schmid, Tetrahedron, 1996, 52, 11657–11664, DOI:10.1016/0040-4020(96)00647-3.
- I. Ugi, B. Werner and A. Dömling, Molecules, 2003, 8, 53–66, DOI:10.3390/80100053.
- I. Usui, K. Nomura and B. Breit, Org. Lett., 2011, 13, 612–615, DOI:10.1021/ol1028546.
- N. N. Schwartz and J. H. Blumbergs, J. Org. Chem., 1964, 29, 1976–1979, DOI:10.1021/jo01030a078.
- H. Huang, C. G. Nelson and D. F. Taber, Tetrahedron Lett., 2010, 51, 3545–3546, DOI:10.1016/j.tetlet.2010.04.129.
- D. C. Johnson and T. S. Widlanski, Org. Lett., 2004, 6, 4643–4646, DOI:10.1021/ol048426w.
- M. Frigerio, M. Santagostino and S. Sputore, J. Org. Chem., 1999, 64, 4537–4538, DOI:10.1021/jo9824596.
- S. D. Meyer and S. L. Schreiber, J. Org. Chem., 1994, 59, 7549–7552, DOI:10.1021/jo00103a067.
- A. Dömling, Chem. Rev., 2006, 106, 17–89, DOI:10.1021/cr0505728.
- M. Passerini and L. Simone, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- M. Passerini and G. Ragni, Gazz. Chim. Ital., 1931, 61, 964–969 CAS.
- L. Banfi and R. Riva, Org. React., 2005, 65, 1–140, DOI:10.1002/0471264180.or065.01.
- L. Weber, K. Illgen and M. Almstetter, Synlett, 1999, 366–374, DOI:10.1055/s-1999-2612.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1436752–1436761. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02334c |
| This journal is © The Royal Society of Chemistry 2016 |