
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A practical method for the synthesis of peptoids containing both lysine-type and arginine-type monomers†
H. L.
Bolt
and
S. L.
Cobb
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: s.l.cobb@durham.ac.uk
First published on 8th December 2015
Abstract
Peptoids are a promising class of peptidomimetics that exhibit the key chemical and physical properties of peptides but without being hampered by susceptibility towards enzymatic degradation. Biologically active peptoids are often designed to be amphipathic in nature, consisting of hydrophobic monomers interspersed with either cationic lysine-type or arginine-type monomers. Access to amphipathic peptoids that contain both lysine-type and arginine-type monomers is highly desirable as it offers a route to further modulate the biological properties of this class of molecule. However, the lack of a suitable synthetic route to prepare mixed cationic peptoids has meant that their biological potential has remained almost largely unexplored. Herein, we present an efficient synthetic route that can be used to access novel cationic peptoids containing both lysine-type and arginine-type monomers within the same sequence.
Peptoids are peptidomimetic molecules that comprise of repeating poly-N-substituted glycine units.1 In terms of structure, peptoids differ from peptides in that their side-chain functionality is bonded to the nitrogen of the poly-amide backbone, rather than the α-carbon (Fig. 1a). This repeating N-alkyl amide backbone motif affords peptoids with an increased stability towards proteolytic degradation compared to analogous peptides.2 Peptoids can be easily accessed in a cost-effective and flexible manner via the efficient solid phase sub-monomer synthesis approach.1 Despite a lack of hydrogen-bond donor capacity in the peptoid backbone (i.e. no free NH groups) stable secondary structures such as peptoid helices can be designed and prepared.3–5 All of the aforementioned points have meant that peptoids are increasingly being investigated in the field of medicinal chemistry as a realistic option to replace peptides in the hunt for new therapeutics.6,7
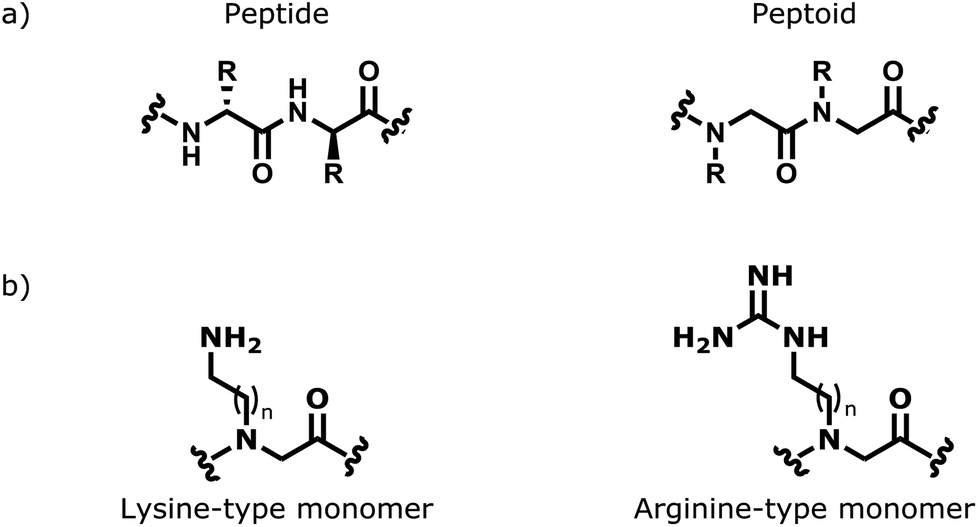 | ||
| Fig. 1 (a) A comparison of generic α-peptoid and peptide oligomers; (b) representation of the amino functionalised lysine-type monomers and guanido functionalised arginine-type monomers, where n = 1–6. | ||
Given the urgent need for the development of new antimicrobial treatments, both linear and cyclic peptoids are being increasingly investigated as anti-infective agents. They have been shown to have promising activity against a variety of microbial targets including both Gram-positive and Gram-negative bacteria,8a parasites,8b and fungi.8c In many cases, the anti-infective activity of peptoids has been found to be comparable to leading antimicrobial peptides (AMPs).7 Typically, antimicrobial peptoids have been designed to mimic simple, naturally occurring AMPs, and they contain amphipathic structures (i.e. mixture of hydrophobic and cationic residues). The presence of cationic side chains within the peptoid sequence helps to provide a degree of selectivity between zwitterionic mammalian cell membranes and the more negatively charged prokaryotic cell membranes. The cationic side chains in peptoids come from either the incorporation of lysine- or arginine-type monomers into the sequence (Fig. 1b). There are numerous examples within the literature of cationic anti-infective peptoids that contain either lysine-type or arginine-type monomers.8
Beyond anti-infective applications, amphipathic cationic peptoids have been studied in other areas of medicinal chemistry. For example, peptoids rich in arginine-type monomers have been shown to have applications as molecular transporters, cell-penetrating vehicles,9–13 heparin binding agents,14 and as analogues of Lung Surfactant Protein B.15
The differences between the inclusion of lysine-type and arginine-type side chains for cellular uptake has also been investigated by several groups and it was shown that guanidine containing peptoids can translocate into the cell more quickly than their amino analogues.13 Additionally, it has been reported that sequences including arginine-type monomers may have an increased biological activity, however this is typically accompanied by an increased toxicity towards mammalian cells.16 The aforementioned point highlights that it would be advantageous to study peptoids that contain mixtures of lysine- or arginine-type monomers in an effort to fine tune the balance between toxicity and activity. However, with the exception of one example,15 all of the peptoids investigated to date have sequences consisting exclusively of either the lysine-type (amino functionalised) or exclusively all arginine-type (guanido functionalised) monomers.
Polyarginine-type peptoids reported in the literature have generally been made using the method developed by Rothbard and co-workers.13 This approach uses pyrazole-1-carboxamide to transform lysine-type monomers into their arginine-type analogs (i.e. an amino to guanido transformation). This reaction can only be carried out after the peptoid has been cleaved from the resin and Boc protecting groups removed. Therefore, every lysine-type residue with the peptoid chain is transformed into an arginine-type residue and thus it is not possible to use this approach to synthesise sequences containing both amino and guanido functionalities.13
The Zuckermann group has previously described the synthesis of a PMC (2,2,5,7,8-pentamethylchroman-6-sulphonyl) protected guanidinopropyl amine monomer.17 Barron et al., attempted to utilise this type of PMC protected monomer in the on resin synthesis of a mixed guanido/amino linear peptoid.15 However, poor solubility (of the monomer) led to low coupling efficiency and the extended cleavage times necessary for the PMC group caused acid-induced degradation of the mixed peptoid (in particular, deterioration of the commonly used Nspe monomer). This prevented full isolation and purification of the required target mixed arginine/lysine-type peptoids.
Thus to the best of our knowledge an efficient generally applicable method for the synthesis of mixed arginine/lysine-type peptoids is yet to be reported (although peptoid–peptide hybrids have been made using the amino acids lysine and arginine).18,19 We were interested in combining arginine- and lysine-type monomer residues within the same peptoid so that we could begin to probe their biological activities, and, in particular the relationship between antimicrobial properties and cytotoxicity. Herein, we describe the development of an efficient synthetic approach that can be utilised to synthesise novel peptoids (linear and cyclic) containing both lysine-type and arginine-type monomers within the same sequence. Guanidinylation using pyrazole-1-carboxamide was optimised and orthogonal Dde protection developed to facilitate this reaction on resin. This approach benefits from being compatible with the sub-monomer solid-phase synthesis of peptoids, which is utilised by the majority of the scientific community. Both linear and cyclic peptoids have been synthesised to show the versatility and synthetic utility of the methodology developed.
Results and discussion
Currently, arginine-type monomers can be added to peptoid sequences using a post-synthetic modification of unprotected amino moieties. However, this transforms all amino functionalities, so mixed amino/guanido functionalised peptoids are not possible via this method. This procedure uses pyrazole-1-carboxamide, a convenient reagent for the guanidinylation of simple primary amines that leaves secondary amines unaffected, even when used in large excess.20In order to incorporate both amino and guanido functionalised side chains (lysine-type and arginine-type monomer residues respectively) within the same sequence, orthogonal protection is necessary. Commonly N-Boc protected amines are used in peptoid synthesis for the protection of amino groups, for example N-Boc 1,4-diaminobutane. For the other protecting group, Dde (N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl) was chosen.
Dde is commonly used to protect free primary amines in peptide synthesis and provides orthogonal protection to the acid cleavable N-Boc group.21,22 The Dde group is removed using 2% hydrazine in DMF which allows these residues to be selectively deprotected (leaving the Boc protection on the other lysine-type monomers intact). Following complete synthesis of the peptoid using the sub-monomer method, selective deprotection of the Dde amine on-resin enables the guanidinylation reaction at specific positions using pyrazole-1-carboxamide. Finally, cleavage from the resin under acidic conditions simultaneously deprotects the remaining Boc protected amino chains to yield a mixed peptoid, containing both lysine-type and arginine-type peptoid residues (see Fig. 2, steps (iv) to (vi)).
 | ||
| Fig. 2 The method used to synthesise mixed arginine/lysine peptoids, where x and y = 1–5; (i) standard displacement step in the sub-monomer method with diamine, 1.5 M in DMF, 60 minutes, RT; (ii) addition of Dde-OH, 10 eq. wrt resin in minimum volume DMF, 60 minutes at RT to protect free amine; (iii) further additions to extend the peptoid chain, using the normal sub-monomer procedure; (iv) deprotection of Dde using 2% hydrazine in DMF 4 × 3 min; (v) guanidinylation of free amine, on resin, using 6 eq. pyrazole-1-carboxamide per free amine and 6 eq. DIPEA per free amine in the minimum volume DMF, 60 minutes, RT; (vi) acidic cleavage from the resin and deprotection of Boc groups. | ||
A variety of mono-N-Boc protected amines are available to purchase from commercial sources, or can be made in high dilution syntheses using Boc anhydride and the required diamine.23 Initially, it was planned to synthesise a mono Dde-protected amine in a similar manner so it could be used directly in the sub-monomer procedure. However, in our hands, the major product of the reaction was a doubly protected amine under all reaction conditions investigated. Therefore, this procedure was deemed to be inefficient and an alternative route to mixed arginine/lysine peptoids was developed. Instead, the Dde protecting group was added to the free amine on resin using Dde-OH (2-acetyldimedone). A standard coupling was made during the displacement stage of the sub-monomer method with an unprotected diamine and one extra step added to protect the free amine using Dde-OH. This is shown in Fig. 2, steps (i) to (iii).
Adding the Dde group on resin has been used in peptide synthesis, but has never been used to make mixed arginine- and lysine-type peptoid residues as far as we are aware.21,22 Addition of Dde-OH (10 eq. wrt resin, in the minimum volume of DMF necessary to dissolve reagent fully) to the free amine on resin was complete after 60 minutes at room temperature. The sub-monomer procedure could then be resumed to lengthen the peptoid chain successfully. To illustrate all steps in this process, mass spectra from the syntheses have been shown in the ESI.†
The guanidinylation reaction with pyrazole-1-carboxamide as reported by Rothbard et al. must be carried out on fully deprotected peptoids in solution.13 Additionally, water was used as a solvent with sodium bicarbonate as a base. Our procedure uses reaction conditions (e.g. DMF with DIPEA) which are compatible with solid-phase synthesis. Carrying out the guanidinylation reaction on resin is preferable as the excess reagents used can simply be washed away at the end of the synthesis making the final purification easier. In addition, although not reported here, the methodology developed should be compatible with automated peptoid synthesisers as it adds only one step to the commonly used sub-monomer method and the Dde group is easily introduced under room temperature conditions.
To demonstrate the versatility of the methodology, a small library of linear peptoids containing both amino and guanido moieties within the same sequence were synthesised (see Table 1). Initially, ‘mixed’ lysine/arginine-type peptoids (Table 1, 1–4, 6) were prepared with monomers containing 4 carbons in the side chain (i.e. NLys and NhArg monomers derived from 1,4-diaminobutane). The Dde group was introduced at a variety of positions within the sequence. It was found that the Dde protection reaction on resin was efficient, it could be carried out near the resin linker at the C terminus and other Dde-protected residues in the same sequence were tolerated. The synthesis was also carried out using the shorter, 2 carbon amine monomer (Nae, Table 1, 5) to highlight that the methodology works with side chains of varying lengths (i.e. Nae and NnArg, derived from 1,2-diaminoethane).
| Peptoid | Sequence |
|---|---|
| 1 | (NLysNspeNspe)2(NhArgNspeNspe)2 |
| 2 | (NhArgNspeNspe)2(NLysNspeNspe)2 |
| 3 | (NLysNspeNspe)(NhArgNspeNspe)(NLysNspeNspe)2 |
| 4 | [(NhArgNspeNspe)(NLysNspeNspe)]2 |
| 5 | [(NnArgNspeNspe)(NLysNspeNspe)]2 |
| 6 | Cyclic (NLysNpheNhArgNpheNLysNphe) |
| 7 | (NhArgNspeNspe)4 |
| 8 | (NhArgNmfbNmfb)4 |
| 9 | [(NamyNspeNspe)(NhArgNspeNspe)]2 |
| 10 | (NnArgNspeNspe)4 |

|
|
Four all arginine-type peptoids were also synthesised on resin using the Dde protection strategy developed (Table 1, 7–10). Carrying out the guanidinylation reaction on resin significantly simplified the final purification of these peptoids. The preparation of 7–10 illustrated that the Dde methodology is compatible not just with Nphe and Nspe but also with other commonly used amine monomers (i.e. Nmfb and Namy).
A cyclic peptoid (6) with both lysine- and arginine-type residues was also synthesised via a Dde-protected linear precursor (Fig. 3). The linear peptoid bearing the Dde-group was successfully cleaved from the resin and cyclised in solution. Subsequent Dde-deprotection of the resultant cyclic peptoid allowed guanidinylation, prior to a final acidic deprotection of remaining Boc-protected amines. Despite the bulky nature of the Dde group, the cyclisation still occurred efficiently at room temperature.
 | ||
| Fig. 3 The synthesis of a cyclic peptoid (6) containing both arginine/lysine-type residues; (i) linear precursor made on 2-chlorotrityl chloride resin using sub-monomer method and cleaved under mildly acidic conditions (20% HFIP in DCM, 20 min); (ii) head-to-tail cyclisation of peptoid in solution (6 eq. PyBOP, 6 eq. DIPEA in DMF), 6 hours; (iii) deprotection of Dde (2% hydrazine in DMF, 4 × 3 min); (iv) guanidinylation in solution (6 eq. pyrazole carboxamide per free amine in the minimum volume of DMF, 6 eq. DIPEA, RT, 60 minutes); (v) deprotection of N-Boc groups (TFA, 60 minutes). | ||
Conclusions
We have developed a practical synthetic procedure that can be used to access linear and cyclic peptoids that contain both arginine- and lysine-type residues within the same sequence. This has enabled the preparation of peptoids (Table 1) that contain combinations of monomers not previously reported in the literature. The methodology developed utilises orthogonal N-Boc and N-Dde protection and pyrazole-1-carboxamide as a guanidinylation reagent. Significantly, this process is amenable for use with acid sensitive monomers (i.e. Nspe) and it is compatible with the commonly used sub-monomer method of peptoid synthesis. The synthesis of an extended library of mixed lysine/arginine type peptoids is now underway and the biological evaluation of these novel compounds will be reported in due course.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (HLB, Durham University, Doctoral Training Grant) for financial support.Notes and references
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS.
- C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 CrossRef CAS.
- H. M. Shin, C. M. Kang, M. H. Yoon and J. Seo, Chem. Commun., 2014, 50, 4465–4468 RSC.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg, C. K. Marlowe, D. C. Spellmeyer, R. Y. Tan, A. D. Frankel, D. V. Santi, F. E. Cohen and P. A. Bartlett, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS.
- J. A. Patch, K. Kirshenbaum, S. L. Seurynck, R. N. Zuckermann and A. E. Barron, The Many Roles of Peptoids in Drug Discovery, in Pseudopeptides in Drug Development, ed. P. E. Nielsen, Wiley-VCH, Germany, 2004, pp. 1–31 Search PubMed.
- (a) B. Mojsoska, R. N. Zuckermann and H. Jenssen, Antimicrob. Agents Chemother., 2015, 59, 4112–4120 CrossRef CAS PubMed; N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2794–2799 Search PubMed; J. A. Patch and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 12092–12093 Search PubMed; R. Kapoor, P. R. Eimerman, J. W. Hardy, J. D. Cirillo, C. H. Contag and A. E. Barron, Antimicrob. Agents Chemother., 2011, 55, 3058–3062 Search PubMed; R. Kapoor, M. W. Wadman, M. T. Dohm, A. M. Czyzewski, A. M. Spormann and A. E. Barron, Antimicrob. Agents Chemother., 2011, 55, 3054–3057 Search PubMed; M. L. Huang, M. A. Benson, S. B. Y. Shin, V. J. Torres and K. Kirshenbaum, Eur. J. Org. Chem., 2013, 3560–3566 Search PubMed; M. L. Huang, S. B. Y. Shin, M. A. Benson, V. J. Torres and K. Kirshenbaum, ChemMedChem, 2012, 7, 114–122 Search PubMed; (b) G. A. Eggimann, H. L. Bolt, P. W. Denny and S. L. Cobb, ChemMedChem, 2015, 10, 233–237 CrossRef CAS PubMed; (c) T. S. Ryge, N. Frimodt-Moller and P. R. Hansen, Chemotherapy, 2008, 54, 152–156 CrossRef PubMed.
- W. Huang, J. Seo, S. B. Willingham, A. M. Czyzewski, M. L. Gonzalgo, I. L. Weissman and A. E. Barron, PLoS One, 2014, 9, e90397 Search PubMed.
- C. Y. Huang, T. Uno, J. E. Murphy, S. Lee, J. D. Hamer, J. A. Escobedo, F. E. Cohen, R. Radhakrishnan, V. Dwarki and R. N. Zuckermann, Chem. Biol., 1998, 5, 345–354 CrossRef PubMed.
- W. Huang, J. Seo, J. S. Lin and A. E. Barron, Mol. BioSyst., 2012, 8, 2626–2628 RSC.
- D. K. Kölmel, D. Fürniss, S. Susanto, A. Lauer, C. Grabher, S. Bräse and U. Schepers, Pharmaceuticals, 2012, 5, 1265–1281 CrossRef PubMed.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef PubMed.
- B. K. Ford, M. Hamza and D. L. Rabenstein, Biochemistry, 2013, 52, 3773–3780 CrossRef CAS PubMed.
- S. L. Seurynck-Servoss, M. T. Dohm and A. E. Barron, Biochemistry, 2006, 45, 11809–11818 CrossRef CAS PubMed.
- B. Findlay, P. Szelemej, G. G. Zhanel and F. Schweizer, PLoS One, 2012, 7, e41141 Search PubMed; K. Andreev, C. Bianchi, J. S. Laursen, L. Citterio, L. Hein-Kristensen, L. Gram, I. Kuzmenko, C. A. Olsen and D. Gidalevitz, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2492–2502 CrossRef CAS PubMed.
- T. Uno, E. Beausoleil, R. A. Goldsmith, B. H. Levine and R. N. Zuckermann, Tetrahedron Lett., 1999, 40, 1475–147818 CrossRef CAS.
- M. Gobbo, M. Benincasa, G. Bertoloni, B. Biondi, R. Dosselli, E. Papini, E. Reddi, R. Rocchi, R. Tavano and R. Gennaro, J. Med. Chem., 2009, 52, 5197–5206 CrossRef CAS PubMed.
- C. A. Olsen, H. L. Ziegler, H. M. Nielsen, N. Frimodt-Moller, J. W. Jaroszewski and H. Franzyk, ChemBioChem, 2010, 11, 1356–1360 CrossRef CAS PubMed.
- M. S. Bernatowicz, Y. L. Wu and G. R. Matsueda, J. Org. Chem., 1992, 57, 2497–2502 CrossRef CAS.
- I. A. Nash, B. W. Bycroft and W. C. Chan, Tetrahedron Lett., 1996, 37, 2625–2628 CrossRef.
- B. Rohwedder, Y. Mutti, P. Dumy and M. Mutter, Tetrahedron Lett., 1998, 39, 1175–1178 CrossRef.
- D. Muller, I. Zeltser, G. Bitan and C. Gilon, J. Org. Chem., 1997, 62, 411–416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02279g |
| This journal is © The Royal Society of Chemistry 2016 |