
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tandem buildup of complexity of aromatic molecules through multiple successive electrophile generation in one pot, controlled by varying the reaction temperature†
Akinari
Sumita
,
Yuko
Otani
and
Tomohiko
Ohwada
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ohwada@mol.f.u-toko.ac.jp
First published on 24th December 2015
Abstract
While some sequential electrophilic aromatic substitution reactions, known as tandem/domino/cascade reactions, have been reported for the construction of aromatic single skeletons, one of the most interesting and challenging possibilities remains the one-pot build-up of a complex aromatic molecule from multiple starting components, i.e., ultimately multi-component electrophilic aromatic substitution reactions. In this work, we show how tuning of the leaving group ability of phenolate derivatives from carbamates and esters provides a way to successively generate multiple unmasked electrophiles in a controlled manner in one pot, simply by varying the temperature. Here, we demonstrate the autonomous formation of up to three bonds in one pot and formation of two bonds arising from a three-component electrophilic aromatic substitution reaction. This result provides a proof-of-concept of our strategy applicable for the self-directed construction of complex aromatic structures from multiple simple molecules, which can be a potential avenue to realize multi-component electrophilic aromatic substitution reactions.
Introduction
The assembly of multiple functionalized aromatic structures into a single molecule is of interest in drug design, as exemplified by the integration of multiple functionalized aromatic pharmacophores.1 For this purpose, the idea of sequentially connecting structurally simple aromatic compounds seems to be an attractive approach.2 Electrophilic aromatic substitution (SEAr) reactions can be used for individual functionalization of several different aromatic moieties in a single molecule (Chart 1). The conventional methodology would be convergent connection of several substituted aromatic compounds through electrophilic centers, as shown in Chart 1(a). However, this approach often requires workup/isolation processes to minimize side reactions or to remove excess reactants or by-products. For example, the reactivity of electrophiles is frequently insufficient to drive the reaction to completion, so an excess amount of aromatic compound(s) is often used to promote the intermolecular SEAr reaction (vide infra). On the other hand, one of the most interesting and challenging possibilities is one-pot build-up of a complex aromatic molecule from multiple starting components, i.e., ultimately multi-component electrophilic aromatic substitution reactions (Chart 1(b)). This strategy requires precise temporal control of bond formation via multiple sequential electrophilic aromatic substitution reactions in order to increase molecular complexity in a precisely defined manner. Several sequential electrophilic aromatic substitution reactions, known as tandem/domino/cascade reactions, have already been reported for the construction of aromatic single skeletons such as indoles,3 indenes,4 dihydroindenes,5 indanones,6 fluorenes,7 carbazoles,8 di- and triphenylmethane,9 naphthoquinones,10 and other skeletons.11 However, most of these reactions are limited to only two kinds of reactions, i.e., involving the formation of two bonds, often via intermolecular–intramolecular or intramolecular–intermolecular sequences (Chart 1(c)). The main reason for this is that most of the reactions require the use of an excess amount of aromatic compound(s) to effectively promote the intermolecular SEAr reaction, and thus the following SEAr reaction is inevitably restricted to a kinetically favorable intramolecular reaction (Chart 1(c-1)), or vice versa (Chart 1(c-2)). Low reactivity of electrophiles is also a reason why many electrophilic reactions are unsuitable for multi-component electrophilic aromatic substitution reactions: specifically, the second SEAr reaction can start before the first SEAr reaction is completed, and therefore the chemoselectivity is impaired. Thus, current methodology in this field is commonly restricted to the formation of two bonds, of which one is intramolecular. | ||
| Chart 1 (a) Conventional step-by-step build-up of aromatic molecules through electrophilic substitution. (b) Hypothetical multi-component aromatic electrophilic reactions in one pot. (c-1/c-2) Previous examples of two-step tandem reactions. | ||
One way to achieve multi-component electrophilic aromatic substitution reactions would be precise temporal control of the generation of highly reactive electrophiles, which can react rapidly with the target aromatic compound even when the target aromatic compound is present only in a stoichiometric amount.
An approach to the generation of such electrophiles has been to transform masked (deactivated or latent) electrophiles into active (unmasked) electrophiles. For example, ureas and carbamates can be regarded as masked isocyanate cations that can be activated via N–C and O–C bond cleavage.12 There have been a few reports on the application of masked isocyanate cations to SEAr reactions.12f–h These SEAr reactions of masked isocyanate cations have been used for intramolecular reactions, such as the construction of dihydroisoquinolones12g,h or isoindolin-1-one,12f,g but they are not suitable for intermolecular SEAr reactions due to the moderate yields, requirement of excess amounts of aromatic compounds, and the need for heating. These are obstacles for application in tandem/cascade/domino reactions. To our knowledge, there are few reports of sequential multiple unmasking reactions, mainly because the unmasking reaction rate is usually dependent on the substrate structure, not on the leaving group, resulting in severe limitations on suitable reactants.13a Thus, for the development of multi-component SEAr reactions, new chemical species of leaving groups are required, which can control the unmasking reaction rates.
In this paper, we report successive reaction-temperature-controlled generation of multiple unmasked electrophiles, particularly isocyanate cations and related cations, leading to autonomous sequential electrophilic aromatic substitution reactions. To achieve this, we focused on control of the bond strength of the leaving groups in carbamates as a strategy to obtain the differential leaving ability in order to control the generation of multiple electrophiles. We demonstrated autonomous formation of up to three bonds in one pot and the formation of two bonds arising from a three-component electrophilic aromatic substitution reaction. The present study provides a proof-of-concept of our strategy applicable for the self-directed construction of complex aromatic structures from multiple simple molecules, which can be a potential avenue to realize multi-component electrophilic aromatic substitution reactions, as illustrated in Chart 1(b).
Results and discussion
Tuning of leaving groups in aromatic amidation and acylation
We recently have shown that methyl salicylate is a good leaving group from carbamate (which is a stable functional group) under relatively strong acid-catalyzed conditions.13 The methyl ester group, located at the ortho position with respect to the phenolic oxygen atom, assists partial protonation of, or H-bonding to, the phenolic oxygen atom, which weakens the C–O (phenolic) bond, resulting in ready C–O bond cleavage to generate isocyanate cations at 20 °C (20 °C for 90 minutes: 1b, Fig. 1(a)).14 We have demonstrated previously that N-H and N-alkyl or N-aryl isocyanate cations show different reactivities: N-alkyl and N-aryl isocyanate cations are generated from the carbamates more slowly than N-H isocyanate cations.13a While multiple amidations can be achieved by using isocyanate cations with different reactivities, we found here that tuning of the leaving group ability to generate the isocyanate cations is a more effective strategy to achieve multiple amidations.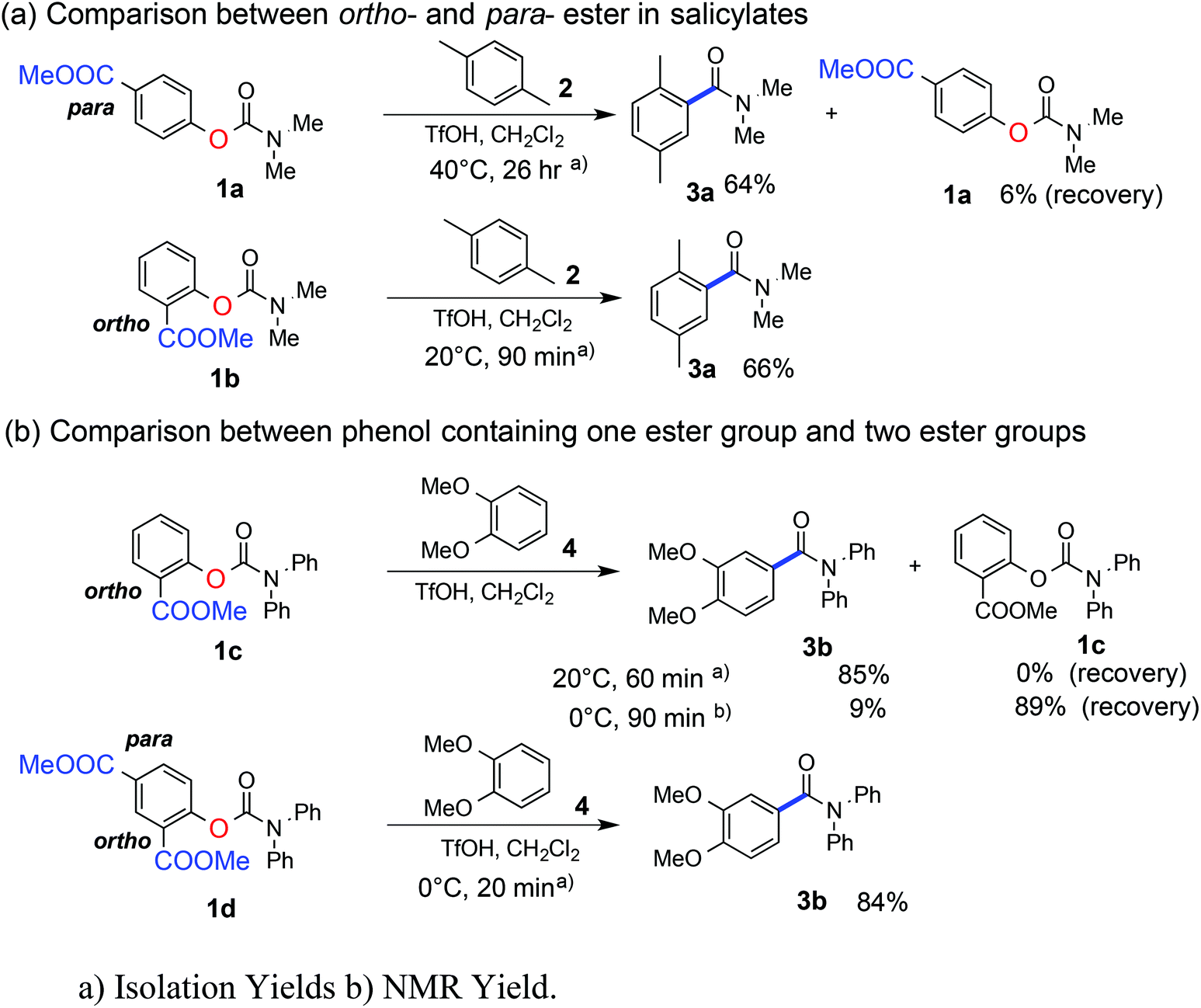 | ||
| Fig. 1 Control of amide formation based on different leaving abilities. | ||
Indeed, we found that when the ester group is substituted at the para-position with respect to the phenolic oxygen atom (1a, Fig. 1(a)), C–O (phenolic) bond cleavage occurs slowly even on heating (at 40 °C, 26 hours). Therefore, the change in the reaction temperature can be used to control the time of generation of even the same reactive electrophile, the isocyanate cation.
Because the substitution position of the ester groups dramatically changes the C–O bond cleavage reaction rates of carbamates bearing methyl salicylates, we expected that adding another ester group into the leaving group (methyl salicylate) would increase the unmasking reaction rate. It turned out that dimethyl 4-hydroxyisophthalate (1d), containing two ester groups at the ortho- and para-positions with respect to the phenolic oxygen atom, is a better leaving group than methyl salicylate (1c). In the case of 1d, the leaving group is cleaved to form isocyanate cations even at 0 °C, and the reaction is completed within 20 minutes (Fig. 1(b)). Thus, at 0 °C, the reaction of methyl salicylate (1c) can be differentiated from the reaction of diester 1d.
This concept is also applicable to differentiate between esters and carbamates, that is, between the generation of acyl cations and isocyanate cations. Because the C–O bond strength in esters 1e is weaker than that in carbamates 1c, the C–O bond in esters is cleaved faster than the C–O bond in carbamates: this enables temperature control of the generation of acyl cations (at 0 °C) and isocyanate cations (at 20 °C) (Fig. 2). Thus, we can control the speed of unmasking to generate different reactive electrophiles simply by changing the temperature.
 | ||
| Fig. 2 Comparison of amide formation rate and ketone formation rate. | ||
Activation of leaving groups through protonation and hydrogen bonding
We conducted kinetic studies to characterize the difference in the leaving ability between the monoester leaving group (see 1b) and diester leaving group (see 1f). Fig. 3 shows the dependence of the reaction profiles on the acidity of the medium. In the case of monoester leaving group (1b), as the acidity is increased, the reaction rate increases to a broad maximum, and then decreases in the strong acidity region. O,O-Diprotonation of salicyl carbamates (like TS-1b′, Fig. 3a) can be observed by proton NMR spectroscopy in a strong acid (−H0 = 14). Cleavage of the C–O bond of the diprotonated species will be difficult because of the generation of one neutral species (methyl salicylate) and one rather unstable dicationic species (O-protonated carbamate dication). Therefore, this means that the monocationic species (TS-1b, Fig. 3a) rather than the diprotonated species (TS-1b′, Fig. 3a) is involved in the bond dissociation transition state.13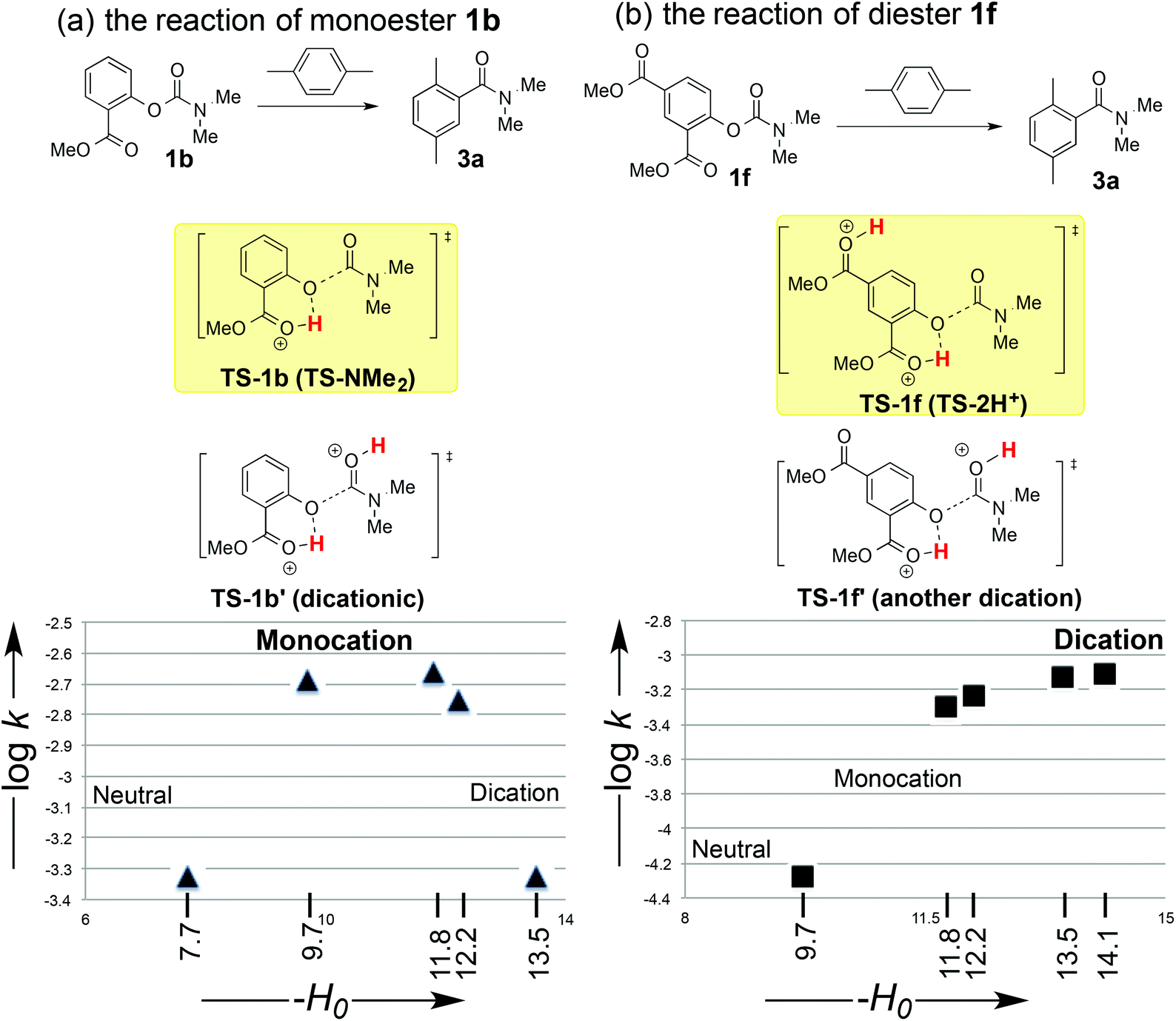 | ||
| Fig. 3 Acidity-dependence of the reaction profiles of different leaving groups (measured at 31 °C for (a), and at −6 °C for (b)). | ||
On the other hand, in the case of the diester leaving group (1f), as the acidity increased, the reaction rate increases monotonically to a broad maximum from a relatively weak acidic region (−H0 = 10) to a strong acidic region (−H0 = 14) (Fig. 3b). Because the ortho-monoester-substituted carbamate (1b) is diprotonated in the strong acidic region (−H0 = 14) (see TS-1b′, Fig. 3a), disubstituted carbamate (1f) also can be diprotonated in the strong acidic region (−H0 = 14). When the carbonyl oxygen atom of the carbamate functional group of diester-substituted carbamate (1f) is protonated (TS-1f′, Fig. 3), the C–O bond cleavage of this diprotonated species will be difficult because of the generation of one neutral species and one rather unstable gitonic (close) dicationic isocyanate cation species. Therefore, the C–O bond cleavage of the diprotonated species will take place through the alternative diprotonated state TS-1f rather than the dication state TS-1f′ (Fig. 3).
Computational support for leaving group activation
These kinetic results are supported by the results of calculations (Fig. 4). The calculated energy differences are shown in terms of ΔΔG at 25 °C (298 K), together with ΔΔH. | ||
| Fig. 4 Calculated energy diagram of C–O bond cleavage steps of 1f (CPCM-M06-2X/6-311++G(d,p)//CPCM-B3LYP/6-31+G(d)). | ||
In the continuum environment of TfOH, there are several possible stable conformers of diester-substituted carbamate 1f in protonated states, because the diester carbamate functionalities have several basic sites.13a,b The ester carbonyl oxygen atom(s), the most basic sites, is mono-protonated or are di-protonated, such cationic species, 1f-SM0-H+ and 1f-SM0-2H+, are the most stable (Fig. 4). On the other hand, a proton can switch the protonation site to the less basic phenolic oxygen atom, and such isomeric conformers, 1f-SM-H+ and 1f-SM-2H+, are destabilized as compared to the most stable conformers (1f-SM0-H+, 1f-SM0-2H+). Destabilization is similar in magnitude in both cases of the monocation and the dication, and the energy gaps are attainable probably because of the formation of intramolecular hydrogen bonding. Through the intervention of these equilibrating species, 1f-SM-H+ and 1f-SM-2H+, the transition states of C–O bond dissociation can be obtained (Fig. 4): the activation energy from the most stable conformer 1f-SM0-2H+ through the dicationic state (1f-TS-2H+) (ΔΔG298 K: 18.6 kcal mol−1; ΔΔH: 19.1 kcal mol−1) is lower than that from 1f-SM0-H+ through the monocationic state (1f-TS-H+) (ΔΔG298 K: 22.2 kcal mol−1; ΔΔH: 22.2 kcal mol−1). Other calculation levels (M06, M06-HF, B3LYP, B3PW91, MP2) also converged to similar results (ESI, Fig. SI1 and 2†). Thus, the observed acidity–rate relationship (Fig. 3) can be rationally explained. Moreover, the difference in energy between the dicationic pathway and monocationic pathway amounts to 3 kcal mol−1, which means that the reaction pathway of C–O bond dissociation through the dicationic state should be dominant.
The reaction rate of ester (1e) could not be measured due to the very rapid unmasking rate; however, the calculation results support a marked difference in reaction rates between the relevant carbamate and ester (Fig. 5). From the calculation result, the activation energy of C–O bond cleavage of the monoester-substituted carbamate 1b from the most stable conformer (SM0-1b) through the equilibrating minor monocationic species (SM-1b), ΔΔG298 K: 23.0 kcal mol−1; ΔΔH: 24.5 kcal mol−1, is higher than that of C–O bond cleavage of the ester from the more stable monocationic species (SM-1e), ΔΔG298 K: 13.1 kcal mol−1; ΔΔH: 13.7 kcal mol−1. This may be partially true because the carbamate is more stable than the ester due to Y-type conjugation,12h,15 and thus more energy is needed to break the stable C–O bond of the carbamate. In the case of ester, it is worth noting that when the intramolecular hydrogen bond is formed with the phenolic oxygen atom, the conformer SM-1e is more stable than the isomeric SM0-1e (Fig. 5). Therefore, an ester compound bearing the methyl salicylester group can easily cleave its O–C bond. These calculations are consistent with the experimental results: the reaction of ester (1e) is dramatically faster than that of monoester-substituted carbamate 1b, and 1e is distinctly faster than the diester-substituted carbamate 1f. Other calculation levels (M06, MP2) also support this conclusion (ESI, Fig. SI3 and 4†) and the observed relationship (Fig. 2).
 | ||
| Fig. 5 Calculated energy diagrams of C–O bond cleavage steps of 1b and 1e (CPCM-M06-2X/6-311++G(d,p)//CPCM-B3LYP/6-31+G(d)). | ||
Temperature control of multiple electrophile generation, enabling two-step buildup of complexity of aromatic molecules
Based on the above findings, we considered that temperature control for unmasking electrophiles could be applied to build-up aromatic molecular complexity in one-pot reactions (Fig. 6–8 and Tables 1 and 2). First, we explored the formation of two bonds. | ||
| Fig. 6 Formation of two amide bonds through temperature-controlled multiple electrophile generation: (a) overall reaction (b) individual steps. | ||
 | ||
| Fig. 7 Ketone and amide bond formations through temperature control of multiple electrophile generation: (a) overall reaction (b) individual steps. | ||
 | ||
| Fig. 8 Temperature control of multiple electrophile generation, enabling three-step buildup of complexity of aromatic molecules ((a) ketone–amide–amide; (b) amide–amide–amide). | ||
| Entry | Substrates | Reaction temperature 1 | Intermediate | Reaction temperature 2 and 3rd substrate | Final product |
|---|---|---|---|---|---|
| 1 |

|
0 °C |

|
20 °C |

|
| 2 |
|
0 °C |

|
20 °C |

|
| 3 |

|
0 °C |

|
20 °C |

|
| 4 |

|
0 °C |
|

|

|
| 5 |
|
0 °C |

|
|

|
| 6 |

|
0 °C |

|
20 °C |

|
| Entry | Substrates | Reaction temperature 1 | Intermediate | Reaction temperature 2 and 3rd substrate | Final product |
|---|---|---|---|---|---|
| 1 |

|
0 °C |

|
20 °C |

|
| 2 |
|
0 °C |

|
20 °C |

|
| 3 |

|
0 °C |

|
20 °C |

|
| 4 |

|
0 °C |

|
|

|
| 5 |

|
0 °C |
|

|
|
Double aromatic amidations
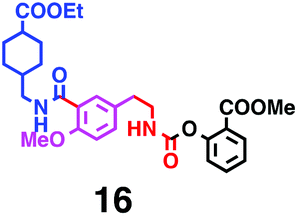
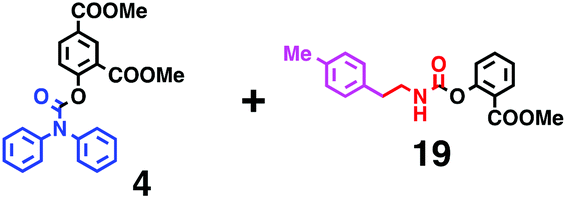
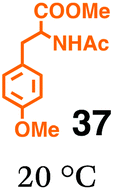
Fig. 6 shows a representative example of the build-up of two kinds of aromatic amide molecules. Mixing 4 and 5 in the presence of TfOH produced compound 7 in 86% yield (Fig. 6(a), in which the reaction temperature was changed from 0 °C to 20 °C). First, the corresponding isocyanate cation (4-cation) was generated from carbamate bearing ortho, para-disubstituted phenol at 0 °C, and then the electron-rich aromatic ring (magenta) of 5 reacted with the resulting isocyanate cation (blue) to form a C–C bond in the aromatic amide structure 6 (red bond) (Fig. 6(b)). Under cooling (0 °C), the carbamate containing para-monosubstituted phenol did not uncage to form the isocyanate cation (4-cation). When the reaction mixture was warmed to room temperature (20 °C), the second isocyanate cation (6-cation, red) was generated from the carbamate containing para-monosubstituted phenol (6), and the second aromatic compound (orange) reacted with the isocyanate cation (6-cation) to form a C–C bond in the aromatic amide structure, affording 7 in an intramolecular manner in this example (red bond) (Fig. 6(b)). The yield that is shown is over the two steps; thus, the average yield of each reaction is larger than 93%.The generality of this sequential dual amidation reaction was examined and the results are shown in Table 1, which supports the feasibility of multiple intermolecular or intramolecular amidation reactions. Because the reaction temperature (20 °C) is not so high, ethyl carbamate (8),12h the ester groups (14 and 16),16 and a methoxy group (15)17 can be tolerated; the former substituents can generate electrophile species, and the latter substituent can be demethylated. Examples of successive double intermolecular reactions were also obtained (entries 4 and 5, Table 1). After the first SEAr reaction was completed (the formation of 16 or 20), one aromatic molecule (17 or 21) was added and the reaction mixture was warmed to 20 °C to produce a double amidation product (18 and 22), respectively. Importantly, a three-component reaction to create two amide bonds can be realized (entry 6, Table 1): when TfOH was added into a mixture of three components (14, 23, 26) at 0 °C and then at 20 °C, the product 25 was obtained, though in moderate yield (overall yield for the two step reaction is 48%). One of the (equivalent) aromatic rings of the substrate 23 can react with the first electrophile (generated at 0 °C from 14), leading to the intermediate 24 smoothly, because we used stoichiometric amounts of the aromatic substrate (23) and the electrophile (14), to minimize side reactions. Then, the intermediate 24 reacted with the second electrophile, generated from 26 at 20 °C, to give compound 25. This result demonstrates that the strategy of tuning the leaving group ability of masked electrophiles, e.g., the phenolate derivatives, should be valid for real multi-component electrophilic aromatic substitution reactions. The average yield of each reaction ranged from 60% to 93% (Table 1).
Sequential aromatic acylation-amidation
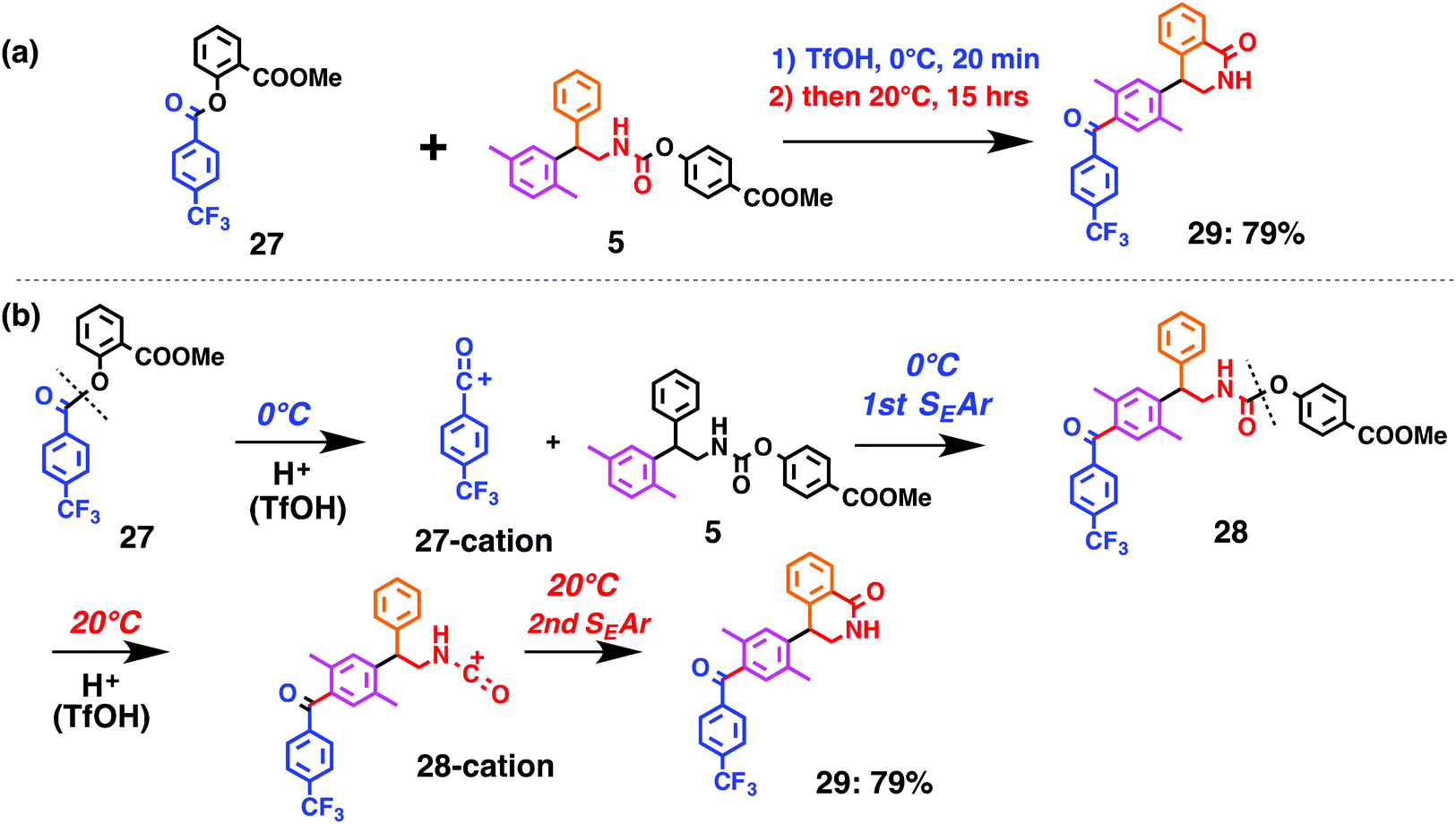
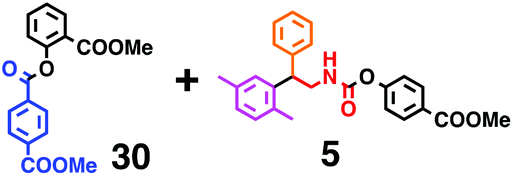
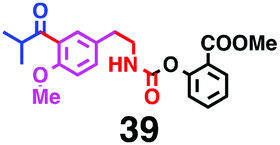
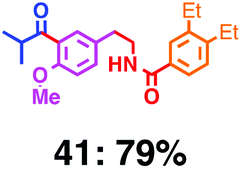
Fig. 7 shows a representative example of one-pot build-up of aromatic ketone and aromatic amide molecules. Mixing 27 and 5 in the presence of TfOH produced compound 29 in 79% yield, by the change in the reaction temperature from 0 °C to 20 °C (Fig. 7(a)). First, the corresponding acyl cation (27-cation) was generated from the carbamate bearing ortho-substituted phenol at 0 °C, and then the electron-rich aromatic ring (magenta) of 5 reacted with the resulting acyl cation (blue) to form a C–C bond in the aromatic ketone structure 28 (red bond; Fig. 7(b)). At 0 °C, the carbamate containing para-monosubstituted phenol did not decompose to isocyanate cation. However, on warming the reaction mixture to room temperature (20 °C), the second isocyanate cation (28-cation, red) was generated from the carbamate containing para-monosubstituted phenol (28), and the second aromatic compound (orange) reacted with the resulting isocyanate cation (28-cation) in an intramolecular manner to form an aromatic amide bond (red bond in product 29) (Fig. 7(b)). Again the yield shown is the two-step yield, and the average yield of each reaction was larger than 89%.The generality of this sequential acylation–amidation reaction was examined and the results are shown in Table 2, which supports the feasibility of multiple intermolecular or intramolecular acylation–amidation reactions. Substrates bearing a trifluoromethyl group (27)18 or an ester group (30)15 can produce electrophile species, but because of the low temperature (0 °C to 20 °C), acylation–amidation reactions are accomplished without interference from these functional groups. After the first SEAr reaction was completed (the formation of 36 or 39), the aromatic substrate (37 or 40) was added, and therefore three kinds of starting materials are combined sequentially into a single aromatic molecule (38 or 41) in one pot. The average yield of each reaction ranged from 77% to 89%.
Formation of three bonds in a one-pot reaction
Finally, we demonstrate the formation of three inter- and intramolecular bonds constituting ketone/amide functionalities in one pot (Fig. 8). The first electrophiles (blue) are generated from ester (27) containing ortho-mono-substituted phenol (methyl salicylate) (Fig. 8(a)) or carbamate (4) (Fig. 8(b)) containing ortho, para-disubstituted phenol at 0 °C, followed by generation of the second electrophiles (red) from carbamate (36 or 20) containing ortho-monosubstituted phenol at 20 °C for around 30 min. The third electrophiles (green) are generated very slowly (in around half a day) from carbamates (42 or 44) containing para-monosubstituted phenol at 20 °C. As described above, these combined reactions enable temporally controlled generation of highly reactive electrophiles (blue, red, and green), which react rapidly with one equivalent of the target aromatic compounds (pink, orange, and brown).The desired compounds were obtained in relatively good yields (43: 56%; 45: 53%) (these yields are three-step yields, so the average yield in reaction (a) is 82% and that in reaction (b) is 81%, respectively). Thus, we can control the unmasking reaction rates and the time of generation of highly reactive electrophiles.
Although the third electrophilic reaction was intramolecular and the third component (5) was added after the first SEAr reaction between the first and second components was completed, three components were combined into a single aromatic molecule in one pot with high regioselective formation of three bonds.
This reaction design is, therefore, a potential avenue to realize ultimate multi-component electrophilic aromatic substitution reactions (as shown in Chart 1(b)).
Conclusion
Tuning of the leaving group ability of phenolate derivatives from carbamates (1a–1d) and ester (1e) enables temporal control of the generation of multiple electrophiles (unmasking) simply by appropriate selection of the reaction temperature, so that the autonomous sequential electrophilic aromatic substitution reactions can proceed in one pot. This chemistry thus allows individual functionalization of several different aromatic moieties in one molecule by means of multiple electrophilic aromatic substitution reactions, affording complex aromatic assemblies in one pot. While the number of bonds that can be formed is unlimited in theory, in the present work, we demonstrated autonomous formation of up to three bonds in one pot, and we realized one example of the three-component reaction to make two amide bonds. In order to use this system for practical reactions applicable to the synthesis of libraries of compounds, it will be necessary to reduce the amount of acid and to design sophisticated leaving group systems. Nevertheless, in this work, we have demonstrated the conceptual validity of a one-pot build-up of a complex aromatic molecule from multiple starting components, ultimately leading to multi-component electrophilic aromatic substitution reactions (Chart 1(b)).Experimental procedure
General procedures
The melting points were determined with a Yanaco micro melting point apparatus without correction. 1H- (400 MHz) and 13C- (100 MHz) NMR spectra were recorded on a Bruker Avance 400. Chemical shifts were calibrated with tetramethylsilane as an internal standard or with the solvent peak, and are shown in ppm (δ) values, and coupling constants are shown in hertz (Hz). The following abbreviations are used: s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, dt = double triplet, dq = double quartet, h = hextet, m = multiplet, and brs = broad singlet. Electron spray ionization time-of-flight mass spectra (ESI-TOF MS) were recorded on a Bruker micrOTOF-05 to give high-resolution mass spectra (HRMS). All reagents were commercially available and used without further purification, unless otherwise noted. Flash column chromatography was carried out on silica gel (silica gel (40–63 μm)). The combustion analyses were carried out in the microanalytical laboratory of this department.Preparation of substrates
1H-NMR (CDCl3, 400 MHz) δ (ppm): 8.011 (2H, d, J = 8.4 Hz), 7.297 (2H, t, J = 7.6 Hz), 7.230–7.194 (3H, m), 7.135 (2H, td, J = 8.8, 2.0 Hz), 7.080–7.047 (2H, m), 6.990–6.971 (1H, m), 5.190–4.866 (1H, m), 4.419 (1H, t, J = 8.0 Hz), 3.878 (5H, m), 2.333 (3H, s), 2.229 (3H, s). 13C-NMR (CDCl3, 100 MHz) δ (ppm): 166.92, 155.23, 154.20, 141.77, 139.50, 136.17, 134.34, 131.55, 131.45, 129.23, 128.79, 128.08, 127.54, 127.31, 121.77, 52.60, 47.24, 46.00, 21.78, 19.80. HRMS (ESI-TOF, [M + Na]+): Calcd for C25H25NNaO4+: 426.1676. Found: 426.1652. Anal. Calcd for C25H25NO4 + 0.2H2O: C, 73.76; H, 6.29; N, 3.44. Found: C, 73.46; H, 6.02; N, 3.25.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 657.5 mg, 10.3 mmol) in CH2Cl2 (20 mL) and iPr2NEt (9.0 mL), 4-(trifluoromethyl)benzoyl chloride (1823.9 mg, 8.74 mmol) in CH2Cl2 (10 mL) was added at 0 °C. The whole mixture was stirred for 40 minutes at 0 °C. The reaction mixture was purified by column-chromatography on silica gel (eluent: EtOAc/n-hexane = 1/4) to afford methyl 2-((4-(trifluoromethyl)-benzoyl)oxy)benzoate (27) (1142.8 mg, 3.52 mmol, 40%) as a colorless solid. Mp. 68.7–69.2 °C (colorless plates, recrystallized from CH2Cl2/n-hexane). 1H-NMR (CDCl3, 400 MHz) δ (ppm): 8.339 (2H, d, J = 8.0 Hz), 8.091 (1H, dd, J = 7.8, 1.6 Hz), 7.788 (2H, d, J = 8.0 Hz), 7.627 (1H, td, J = 7.6, 2.0 Hz), 7.383 (1H, td, J = 7.8, 1.2 Hz), 7.252–7.230 (1H, m), 3.750 (3H, s). 13C-NMR (CDCl3, 100 MHz) δ (ppm): 165.33, 164.83, 151.14, 135.55 (q, J = 32 Hz), 134.60, 133.39, 132.60, 131.25, 126.98, 126.22 (q, J = 3 Hz), 124.39, 124.18 (q, J = 271 Hz), 123.73, 52.81. HRMS (ESI-TOF, [M + Na]+): Calcd for C16H11F3NaO4+: 347.0507. Found: 347.0496. Anal. Calcd for C16H11F3O4: C, 59.27; H, 3.42. Found: C, 59.08; H, 3.58.
657.5 mg, 10.3 mmol) in CH2Cl2 (20 mL) and iPr2NEt (9.0 mL), 4-(trifluoromethyl)benzoyl chloride (1823.9 mg, 8.74 mmol) in CH2Cl2 (10 mL) was added at 0 °C. The whole mixture was stirred for 40 minutes at 0 °C. The reaction mixture was purified by column-chromatography on silica gel (eluent: EtOAc/n-hexane = 1/4) to afford methyl 2-((4-(trifluoromethyl)-benzoyl)oxy)benzoate (27) (1142.8 mg, 3.52 mmol, 40%) as a colorless solid. Mp. 68.7–69.2 °C (colorless plates, recrystallized from CH2Cl2/n-hexane). 1H-NMR (CDCl3, 400 MHz) δ (ppm): 8.339 (2H, d, J = 8.0 Hz), 8.091 (1H, dd, J = 7.8, 1.6 Hz), 7.788 (2H, d, J = 8.0 Hz), 7.627 (1H, td, J = 7.6, 2.0 Hz), 7.383 (1H, td, J = 7.8, 1.2 Hz), 7.252–7.230 (1H, m), 3.750 (3H, s). 13C-NMR (CDCl3, 100 MHz) δ (ppm): 165.33, 164.83, 151.14, 135.55 (q, J = 32 Hz), 134.60, 133.39, 132.60, 131.25, 126.98, 126.22 (q, J = 3 Hz), 124.39, 124.18 (q, J = 271 Hz), 123.73, 52.81. HRMS (ESI-TOF, [M + Na]+): Calcd for C16H11F3NaO4+: 347.0507. Found: 347.0496. Anal. Calcd for C16H11F3O4: C, 59.27; H, 3.42. Found: C, 59.08; H, 3.58.
Mp. 61.2–61.5 °C (colorless cube, recrystallized from CH2Cl2/n-hexane). 1H-NMR (CDCl3, 400 MHz) δ (ppm) (presence of two amide conformers): 7.954 (1H, dd, J = 8.0, 1.2 Hz), 7.519 (1H, td, J = 7.8, 1.6 Hz), 7.268 (1H, t, J = 7.2 Hz), 7.192–7.128 (3H, m), 5.196 (0.93H, brs), 4.828 (0.14H, brs), 3.845 (3H, s), 3.792 (3H, s), 3.495 (2H, q, 6.8 Hz), 2.839 (3H, t, J = 7.2 Hz). 13C-NMR (CDCl3, 100 MHz) δ (ppm): 165.89, 158.94, 154.94, 151.21, 134.06, 132.01, 131.22, 130.35, 126.11, 124.67, 124.55, 114.70, 55.84, 52.64, 43.32, 35.68. HRMS (ESI-TOF, [M + Na]+): Calcd for C19H18NNaO5+: 352.1152. Found: 352.1165. Anal. Calcd for C19H18NO5: C, 65.64; H, 5.82; N, 4.25. Found: C, 65.32; H, 5.79; N, 4.11.
Multiple successive electrophile substitution reactions in one pot
Computational methods
We carried out computational studies by using the Gaussian 09 suites of programs.19 The geometries of the reactants (SM), transition states (TS), and products (PM) for dissociation steps were fully optimized using the CPCM (Complete Polarizable Continuum Model)-B3LYP/6-31+G(d) level.20 Harmonic vibrational frequency computations characterized the optimized structures. Intrinsic reaction coordinate (IRC) computations21 of the transition structures verified the reactants, intermediates, and products on the potential energy surface (PES). Bulk solvation effects (self-consistent reaction field, SCRF) were simulated by the CPCM method22 in trifluoromethanesulfonic acid as a solvent (eps = 77.4,23 rsolv = 2.5985274,23 density = 1.696,24 epsinf = 1.882384 (the value of acetic acid was employed)). Single point energies were calculated with CPCM-M06-2X/6-311++G(d,p) (and some other calculation levels) on the basis of the optimized structures.25 The zero-point vibrational energy corrections were done without scaling.Acknowledgements
This work was supported by the University of Tokyo. The computations were performed at the Research Center for Computational Science, Okazaki, Japan. We thank the computational facility for generous allotments of computer time.Notes and references
- P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- Y. Takayama, T. Yamada, S. Tatekabe and K. Nagasawa, Chem. Commun., 2013, 49, 6519 RSC.
- (a) H. Kheira, P. Li and J. Xu, J. Mol. Catal. A: Chem., 2014, 391, 168–174 CrossRef CAS; (b) X. Zhang, W. T. Teo and P. W. H. Chan, Org. Lett., 2009, 11, 4990–4993 CrossRef CAS PubMed; (c) M. Shi, L. Wu and J. Lu, Tetrahedron, 2008, 64, 3315–3321 CrossRef CAS; (d) J. A. Kozak, B. O. Patrick and G. R. Dake, J. Org. Chem., 2010, 75, 8585–8590 CrossRef CAS PubMed.
- (a) Y. Zhang, L. Chen and T. Lu, Adv. Synth. Catal., 2011, 353, 1055–1060 CrossRef CAS; (b) R. O. Iakovenko, A. N. Kazakova, V. M. Muzalevskiy, A. Y. Lvanov, I. A. Boyarskaya, A. Chicca, V. Petrucci, J. Gertsch, M. Krasavin, G. L. Starova, A. A. Zolotarev, M. S. Avdontceva, V. G. Nenajdenko and A. V. Vasilyev, Org. Biomol. Chem., 2015, 13, 8827–8842 RSC.
- (a) B. R. Park, S. H. Kim, Y. M. Kim and J. N. Kim, Tetrahedron Lett., 2011, 52, 1700–1704 CrossRef CAS; (b) B. V. Ramulu and G. Satyanarayana, RSC Adv., 2015, 5, 70972–70976 RSC; (c) R. Rendy, Y. Zhang, A. McElrea, A. Gomez and D. A. Klumpp, J. Org. Chem., 2004, 69, 2340–2347 CrossRef CAS PubMed.
- (a) F. Sun, M. Zeng, Q. Gu and S. You, Chem. – Eur. J., 2009, 15, 8709–8712 CrossRef CAS PubMed; (b) Q. Li, W. Xu, J. Hu, X. Chen, F. Zhang and H. Zheng, RSC Adv., 2014, 4, 27722–27725 RSC; (c) S. Wang, L. Han, M. Zeng, F. Sun, W. Zhang and S. You, Org. Biomol. Chem., 2012, 10, 3202–3209 RSC.
- (a) A. Suárez, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, Adv. Synth. Catal., 2014, 356, 374–382 CrossRef; (b) R. Sureshbabu, V. Saravanan, V. Dhayalan and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2011, 922–935 CrossRef CAS; (c) L. Bianchi, M. Maccagno, M. Pani, G. Petrillo, C. Scapolla and C. Tavani, Tetrahedron, 2015, 71, 7421–7435 CrossRef CAS; (d) A. Kulkarni, P. Quang and B. Török, Synthesis, 2009, 4010–4014 CAS.
- (a) J. Liu, T. He and L. Wang, Tetrahedron, 2011, 67, 3420–3426 CrossRef CAS; (b) M. Shiri, M. A. Zolfigol and R. Ayazi-Nasrabadi, Tetrahedron, 2010, 51, 264–268 CrossRef CAS; (c) J. Xu, J. Xia and Y. Lan, Synth. Commun., 2005, 35, 2347–2353 CrossRef CAS; (d) S. Saito, T. Ohwada and K. Shudo, J. Am. Chem. Soc., 1995, 117, 11081–11084 CrossRef CAS; (e) G. A. Olah, G. Rasul, C. York and G. K. S. Prakash, J. Am. Chem. Soc., 1995, 117, 1121–11214 CrossRef; (f) P. Thirupathi and S. S. Kim, J. Org. Chem., 2009, 74, 7755–7761 CrossRef CAS PubMed; (g) C. Huo, C. Sun, C. Wang, X. Jia and W. Chang, ACS Sustainable Chem. Eng., 2013, 1, 549–553 CrossRef CAS; (h) C. Huo, C. Wang, C. Sun, X. Jia, X. Wang, W. Chang and M. Wu, Adv. Synth. Catal., 2013, 355, 1911–1916 CrossRef CAS.
- (a) H. Ishida, H. Nukaya, K. Tsuji, H. Zenda and T. Kosuge, Chem. Pharm. Bull., 1992, 40, 308–313 CrossRef CAS; (b) H. Naeimi and S. S. Brojerdi, Polycyclic Aromat. Compd., 2014, 34, 504–517 CrossRef CAS; (c) A. P. Krapcho, Z. Getahun and K. J. Avery Jr., Synth. Commun., 1990, 24, 2139–2146 CrossRef; (d) G. A. Guerrero-Vásquez, C. K. Z. Andrade, J. M. G. Molinillo and F. A. Macías, Eur. J. Org. Chem., 2013, 6175–6180 CrossRef; (e) R. Huot and P. Brassard, Can. J. Chem., 1974, 52, 838–842 CrossRef CAS.
- (a) L. Bianchi, M. Maccagno, M. Pani, G. Petrillo, C. Scapolla and C. Tavani, Tetrahedron, 2015, 71, 7421–7435 CrossRef CAS; (b) G. Gangadhararao, A. Uruvakilli and K. C. K. Swamy, Org. Lett., 2014, 16, 6060–6063 CrossRef CAS PubMed; (c) L. Hao, Y. Pan, T. Wang, M. Lin, L. Chen and Z. Zhan, Adv. Synth. Catal., 2010, 352, 3215–3222 CrossRef CAS; (d) S. Sarkar, K. Bera and U. Jana, Tetrahedron Lett., 2014, 55, 6188–6192 CrossRef CAS; (e) X. Xie, X. Du, Y. Chen and Y. Liu, J. Org. Chem., 2011, 76, 9175–9181 CrossRef CAS PubMed; (f) A. C. Silvanus, S. J. Heffernan, D. J. Liptrot, G. Kociok-Köhn, B. I. Andrews and D. R. Carbery, Org. Lett., 2009, 11, 1175–1178 CrossRef CAS PubMed; (g) H. Li, R. Guillot and V. Gandon, J. Org. Chem., 2010, 75, 8435–8449 CrossRef CAS PubMed; (h) K. Fuchibe, H. Jyono, M. Fujiwara, T. Kudo, M. Yokota and J. Ichikawa, Chem. – Eur. J., 2011, 17, 12175–12185 CrossRef CAS PubMed.
- Up to the present, masked (blocked) electrophiles have mainly been limited to masked (blocked) isocyanates. For a review of masked (blocked) isocyanates, see: E. Delebecq, J. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed . Recent studies on application masked isocyanates to synthetic organic chemistry: (a) M. Hutchby, C. E. Houlden, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 8721–8724 CrossRef CAS PubMed; (b) C. Clavette, J. V. Rocan and A. M. Beauchemin, Angew. Chem., Int. Ed., 2013, 52, 12705–12708 CrossRef CAS PubMed; (c) H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 Search PubMed; (d) Y. Wei, J. Liu, S. Lin, H. Ding, F. Liang and B. Zhao, Org. Lett., 2010, 12, 4220–4223 CrossRef CAS PubMed; (e) C. Spyropoulos and C. G. Kokotos, J. Org. Chem., 2014, 79, 4477–4483 CrossRef CAS PubMed; (f) S. Adachi, M. Onozuka, Y. Yoshida, M. Ide, Y. Saikawa and M. Nakata, Org. Lett., 2014, 16, 358–361 CrossRef CAS PubMed; (g) E. K. Raja, S. O. N. Lill and D. A. Klumpp, Chem. Commun., 2012, 48, 8141–8143 RSC; (h) H. Kurouchi, K. Kawamoto, H. Sugimoto, S. Nakamura, Y. Otani and T. Ohwada, J. Org. Chem., 2012, 77, 9313–9328 CrossRef CAS PubMed.
- (a) A. Sumita, H. Kurouchi, Y. Otani and T. Ohwada, Chem. – Asian J., 2014, 9, 2995–3004 CrossRef CAS PubMed; (b) H. Kurouchi, A. Sumita, Y. Otani and T. Ohwada, Chem. – Eur. J., 2014, 20, 8682–8690 CrossRef CAS PubMed.
- In recent years, several reports have shown that isocyanates are useful chemical species to form aromatic amides. (a) G. Schäfer, C. Matthey and J. W. Bode, Angew. Chem., Int. Ed., 2012, 57, 9173–9175 CrossRef PubMed; (b) G. Schäfer and J. W. Bode, Org. Lett., 2014, 16, 1526–1529 CrossRef PubMed and ref. 12h.
- P. Gund, J. Chem. Educ., 1972, 49, 10–103 CrossRef.
- J. P. Hwang, G. K. S. Prakash and G. A. Olah, Tetrahedron, 2000, 56, 7199–7203 CrossRef CAS.
- M. Kozelj and A. Petric, Synlett, 2007, 1699–1702 CAS.
- A. Kethe, A. F. Tracy and D. A. Klumpp, Org. Biomol. Chem., 2011, 9, 4545 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. L. Lira, M. Zolotukhin, L. Fomina and S. Fomine, J. Phys. Chem. A, 2007, 111, 13606–13610 CrossRef CAS PubMed.
- R. Corkum and J. Milne, Can. J. Chem., 1978, 56, 1832–1835 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental sections, details of computation, and 1H and 13C NMR spectra. See DOI: 10.1039/c5ob02240a |
| This journal is © The Royal Society of Chemistry 2016 |