
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future
Markus D.
Kärkäs
,
Bryan S.
Matsuura
,
Timothy M.
Monos
,
Gabriel
Magallanes
and
Corey R. J.
Stephenson
*
Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: crjsteph@umich.edu
First published on 14th December 2015
Abstract
The development of a sustainable, carbon-neutral biorefinery has emerged as a prominent scientific and engineering goal of the 21st century. As petroleum has become less accessible, biomass-based carbon sources have been investigated for utility in fuel production and commodity chemical manufacturing. One underutilized biomaterial is lignin; however, its highly crosslinked and randomly polymerized composition have rendered this biopolymer recalcitrant to existing chemical processing. More recently, insight into lignin's molecular structure has reinvigorated chemists to develop catalytic methods for lignin depolymerization. This review examines the development of transition-metal catalyzed reactions and the insights shared between the homogeneous and heterogeneous catalytic systems towards the ultimate goal of valorizing lignin to produce value-added products.
 Markus D. Kärkäs | Markus D. Kärkäs received his M.Sc. degree in Chemistry from Stockholm University in 2008, where he conducted undergraduate research in the laboratory of Prof. Åkermark. In the same year, he began his Ph.D. studies under the guidance of Professor Björn Åkermark and Professor Jan-Erling Bäckvall. In 2013, he obtained his Ph.D. degree after conducting research on the development of artificial water oxidation catalysts. He is currently working as a postdoctoral fellow at the University of Michigan with Professor Corey Stephenson where he is developing methods for valorization of lignin. |
 Bryan S. Matsuura | Bryan S. Matsuura received his B.A. from Boston University in 2009 and started his Ph.D. studies in the same year. In 2015 he obtained his Ph.D. at the University of Michigan working under the supervision of Professor Corey Stephenson where he studied the biomimetic synthesis of the resveratrol oligomers and the degradation of biomass derived polymers using visible-light photoredox catalysis. He is currently working as a postdoctoral fellow in the research group of Professor Dirk Trauner at the Ludwig-Maximilians-Universität München. |
 Timothy M. Monos | Timothy M. Monos received his B.Sc. in Chemistry from the University of Connecticut in 2013 and began his Ph.D. studies in the same year. He is currently a Ph.D. candidate at the University of Michigan under the direction of Professor Corey Stephenson, studying methodology development and applications of photoredox catalysis. |
 Gabriel Magallanes | Gabriel Magallanes received his B.Sc. in Chemistry from Purdue University in 2014. He started his Ph.D. studies at the University of Michigan in 2014 where he is currently working under the supervision of Professor Corey Stephenson. Gabriel is studying the degradation of biomass derived polymers using visible-light photoredox catalysis. |
 Corey R. J. Stephenson | Corey R. J. Stephenson earned his Ph.D. from the University of Pittsburgh under Professor Peter Wipf before pursuing his postdoctoral studies at ETH Zürich with Professor Erick M. Carreira. He began his independent career at Boston University in 2007 and moved to the University of Michigan in 2013. His research interests are broadly focused on biomass degradation, complex molecule synthesis and catalysis. |
1. Introduction
Petroleum is a critically important commodity resource that modern society is reliant on as a carbon feedstock for the production of fuel and fine chemicals. Aside from the environmental and political impact of its extraction and distribution, petroleum is a non-renewable resource with unmatched sustainable alternatives. Currently, biomass is the largest source of renewable energy, supplying 3% of the total consumption of energy in the United States, surpassing hydroelectric and wind power. The United States has the capacity to produce 190 million tons of biomass each year, with the potential to increase production to 1 billion tons by 2050.1,2 As the only available renewable carbon feedstock, utilization of biomass for this purpose is considered essential to reducing humanity's unsustainable consumption of non-renewable resources. Converting biomass into fuel or fine chemicals on a scale that would rival petroleum refining represents an enormous collection of challenges from a scientific, engineering, and economic standpoint that will take several years of research and development from all fronts. However, this is a chemistry problem at its core, and the development of novel chemical transformations for processing biomass is central to the realization of this ambitious goal.Although it is unlikely that biomass will replace the demand for petroleum outright, it possesses great potential as a replacement for crude oil as the carbon feedstock for the production of commodity chemicals, solvents, pharmaceuticals, and agrochemicals. Prior to 2006, biomass conversion largely escaped the attention of synthetic chemists, despite the great scientific and societal significance of this problem.3–6 Since then, biomass conversion-particularly lignin depolymerization has undergone rapid development from chemists and chemical engineers. The progress in lignin depolymerization chemistry has been reviewed by several groups, documenting the rapid advances in the liquefaction of lignocellulosic biomass,7,8 oxidative valorization,9,10 and hydrogenolysis.11 This review seeks to provide a comprehensive overview of current lignin depolymerization chemistry within the context of synthetic chemistry. The majority of the reactions described herein involve the cleavage of lignin model compounds using homogeneous catalysis, though stoichiometric reactions are discussed where applicable. For further reading we recommend the recently published review by Deuss and Barta.12
1.1 Structure and biosynthesis of lignin
Lignocellulosic biomass (dry plant matter) is the most abundant renewable carbon resource on earth. It is mainly composed of the carbohydrate polymers cellulose and hemicellulose and the aromatic polymer lignin. While technologies to process cellulose and hemicellulose are well developed, lignin processing constitutes a considerably more challenging task. Lignin is the second most abundant biopolymer on earth, comprising up to 20–35% of lignocellulosic biomass and greater than 40% of the energy content. Unlike polysaccharides or lipids, lignin is the only biopolymer possessing a high content of aromatic groups and is an ideal candidate as a renewable resource for aromatic commodity chemicals. Lignin's highly branched and irregular structure plays a vital role in providing the plant biomechanical support, aiding in pathogenic defense and water transport. Lignin is composed of three phenylpropanol alcohol precursors–coumaryl alcohol (1), coniferyl alcohol (2) and sinapyl alcohol (3) (Fig. 1)—collectively called monolignols. Although all monolignols are derived biosynthetically from the cinnamate pathway (Scheme 1), the monomer composition is species dependent.13–16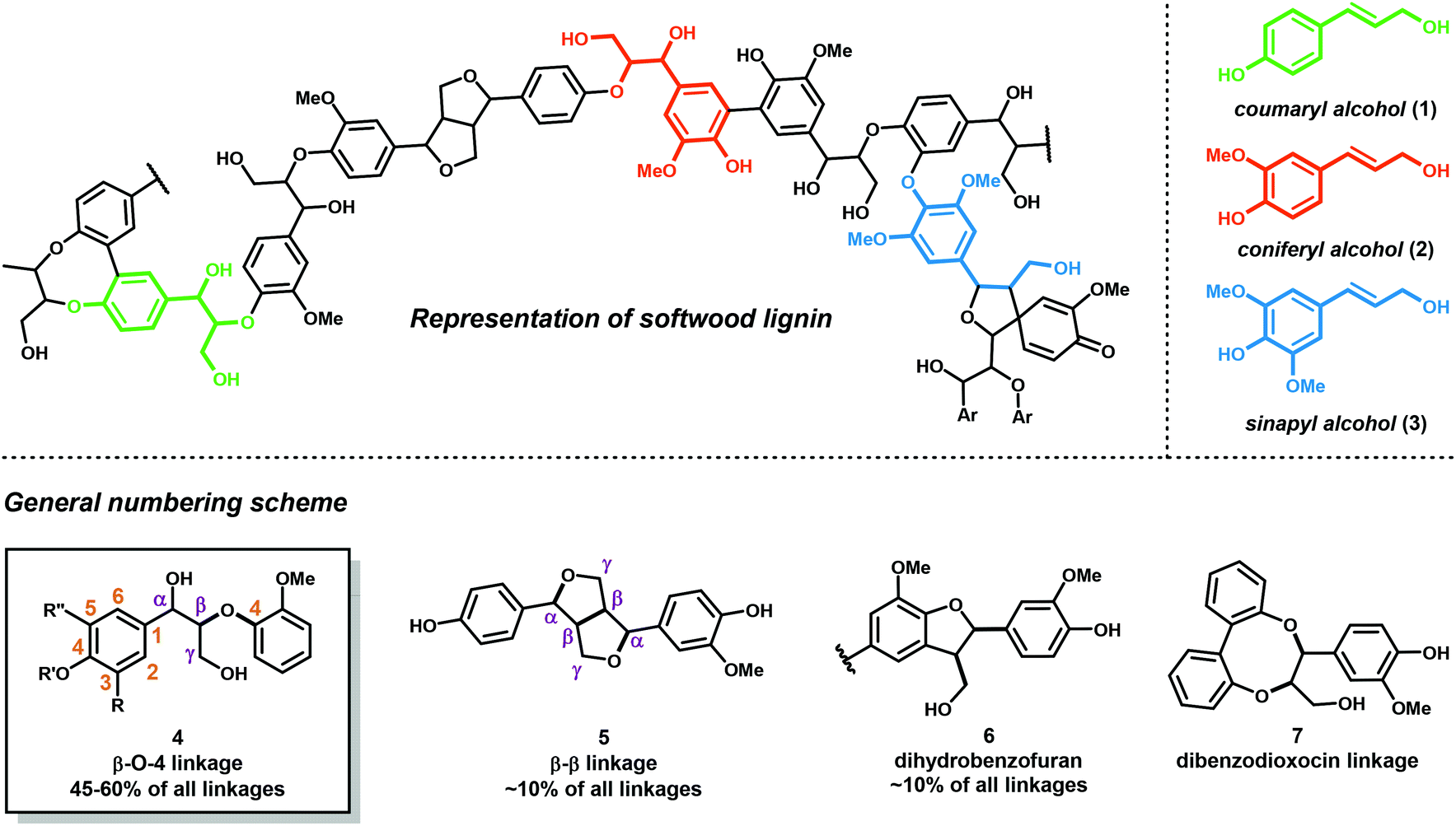 | ||
| Fig. 1 Structure of lignin and the main linkage motifs present in lignin. | ||
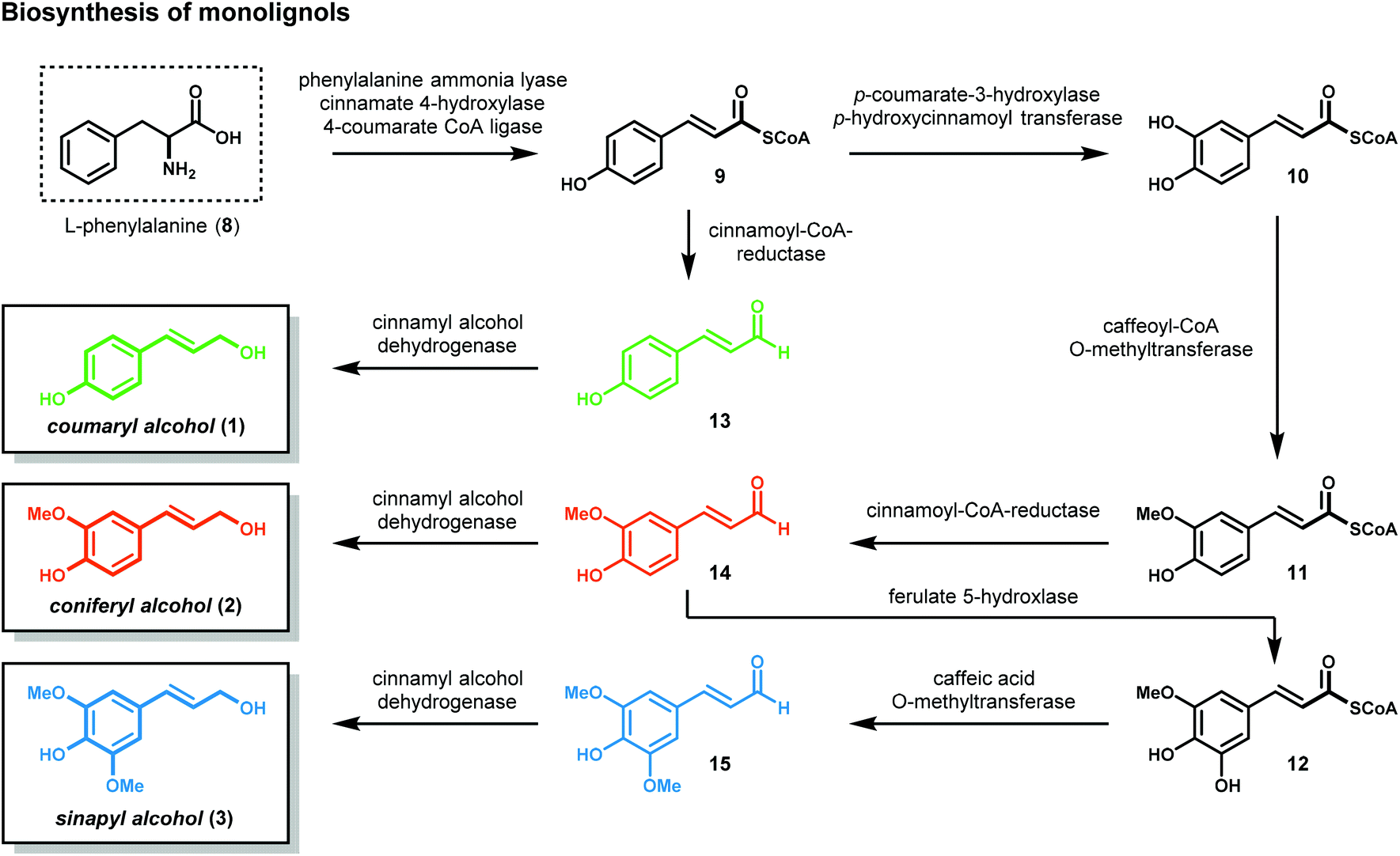 | ||
| Scheme 1 Biosynthesis of the monolignols present in lignin. | ||
After their biosynthesis, the monolignols are transported through the cell membrane to the cell wall, through an unknown mechanism, where co-polymerization occurs. It is generally accepted that polymerization is initiated by undirected oxidative radical polymerization of the cinnamyl alcohol precursors. For example, the oxidation of coumaryl alcohol (1) results in the formation of a highly stabilized phenoxyl radical (16) that is best described by the resonance hybrids A, B, and C (Fig. 2). Radical 16 can couple in a random, radical–radical combinatorial fashion, resulting in the formation of highly reactive quinone methide intermediates (17–19). Subsequent intermolecular nucleophilic substitution with water and other monolignols (causing cross-linking) or intramolecular cyclization reactions yields a structurally diverse array of linkage motifs (20–22) (Fig. 2).13,14 Consequently, the lignin polymer is random in both monomer sequence, linkage motif, and stereochemical configuration.17 Of the different linkages, the β-O-4 motif (see Fig. 1) is by far the most abundant, comprising 45–60% of all linkages found in lignin with the dihydrobenzofuran, β–β′ linkage, and the 5–5′ linkages present in significant but less abundant quantities.18,19
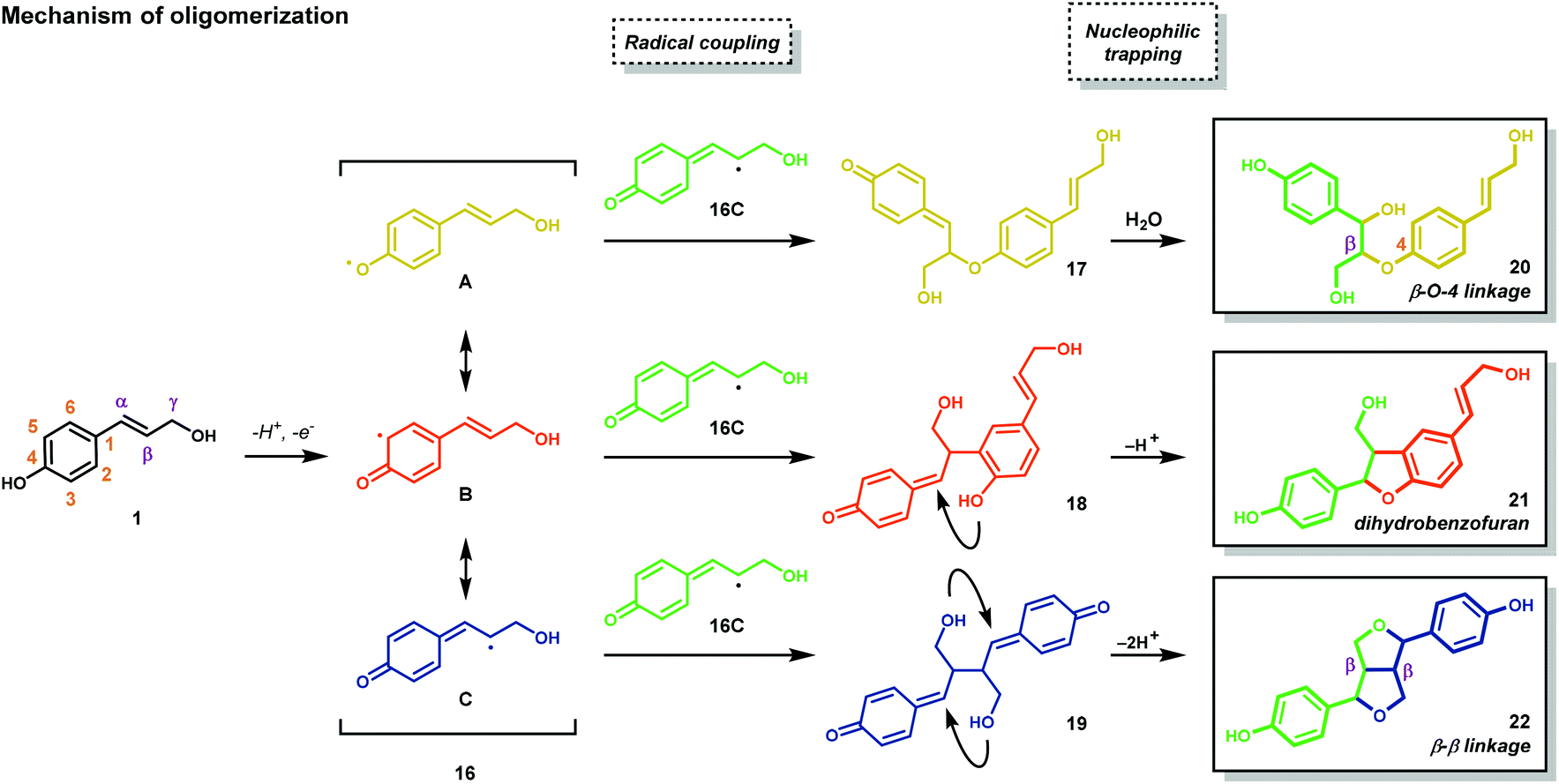 | ||
| Fig. 2 Mechanism of oligomerization. | ||
1.2 Valorization of lignin – a sustainable solution for producing value-added chemicals
In the hypothetical carbon-neutral biorefinery, biomass would be converted into fuel or fine chemicals over several steps in a process analogous to petroleum refining. The first, and perhaps the most critical step, is the mechanochemical separation of raw plant biomass, such as stover or bagasse, into its constituent parts in a process called ‘pulping’, which produces two significant fractions: a cellulosic fraction and lignin (Fig. 3). The former, which consists of cellulose and hemicellulose, comprises about 60–75% of biomass by weight. Current biomass processing research is primarily focused on processing cellulose as the main carbon source because it is amenable to both chemical and enzymatic processing techniques.20–26 Cellulose and oligosaccharides are typically broken down via acidic digestion and can be further processed by fermentation to yield important non-aromatic commodity chemicals, such as ethanol, n-butanol and hydroxy methylfurfural. These products can then be refined into saturated hydrocarbons, analogous to olefin–alkane distillates from petroleum refining.27,28 Although the technologies to convert alkanes and alkenes into aromatic compounds exist,29 these processes generally require high temperatures and are not energetically feasible to perform on a scale needed for commodity chemical synthesis. In contrast, the high aromatic content of lignin makes it an ideal feedstock for the renewable production of high value aromatics such as phenol, benzene, toluene and xylene (BTX), complementing the products obtained from cellulose or algal bio-oil processing. However, the highly complex and irregular structure of lignin has severely complicated the development of controlled methods for depolymerizing lignin for the production of fine chemicals. In the absence of any chemical processing technology, pyrolysis of pretreated lignin yields syngas (a mixture of CO and H2) in about 15–20% yield, plus an intractable char that is rich in polyaromatic hydrocarbons.30 This process, which is inefficient and highly energy intensive could be used to generate synthetic alkanes through the Fischer–Tropsch process,31 but destroys the aromatic backbone of lignin. The development of more suitable lignin conversion chemistry that preserves the aromatic structure is considered an essential objective in establishing the sustainable, carbon-neutral bio-refinery of the future (Fig. 3).32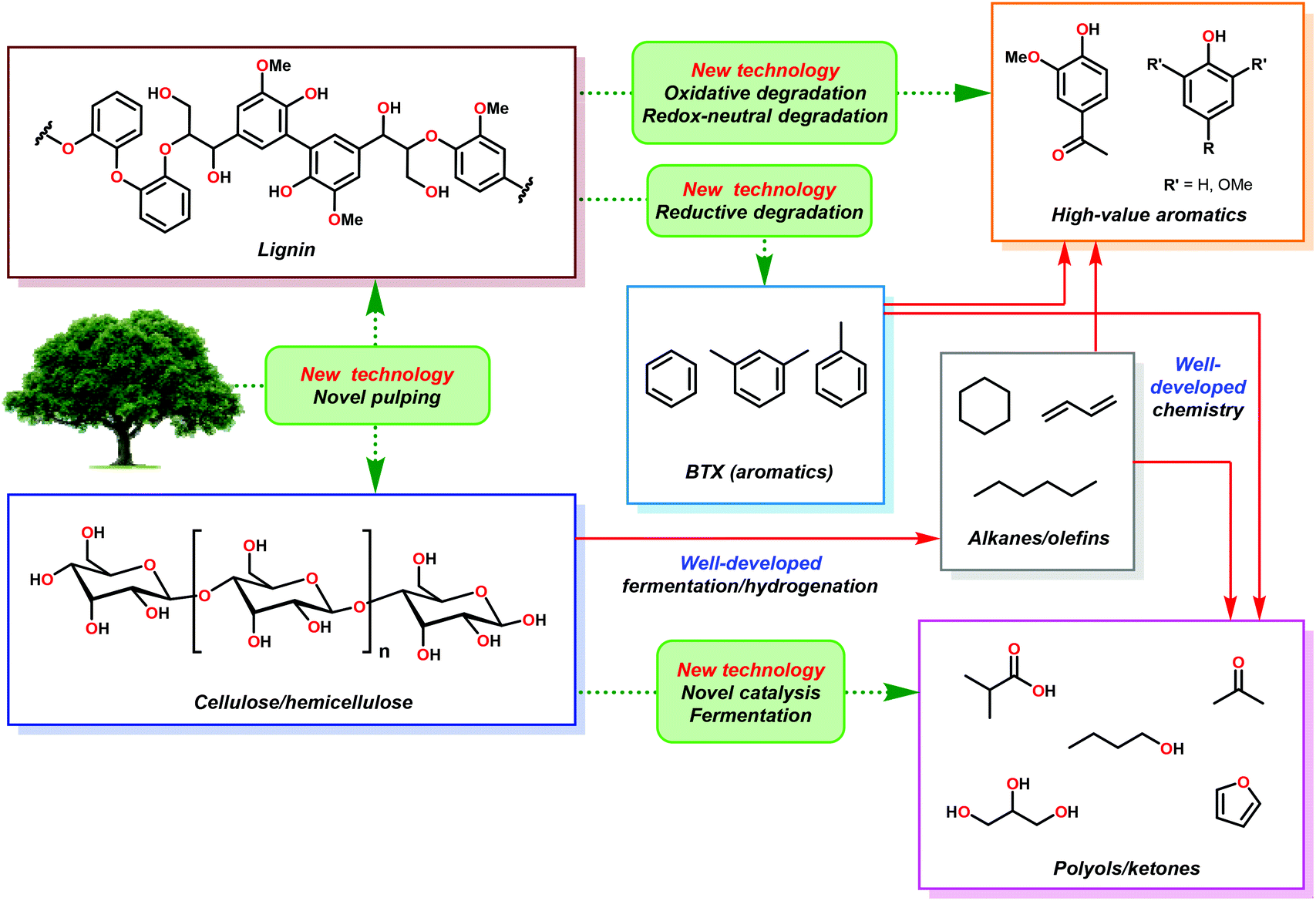 | ||
| Fig. 3 The hypothetical biorefinery and its fundamental challenges. | ||
1.3 Preprocessing and isolation of lignin
Lignin is separated from wood during the wood pulping by three industrial processes: the Kraft process, sulfite process, and the organosolv processes (Fig. 4).18,33,34 In the Kraft35 and sulfite processes,36,37 wood chips, corn stover, or plant bagasse are digested under high heat in either basic or acidic conditions to delignify cellulose and solubilize the lignin byproduct in water. Under these harsh conditions, lignin undergoes structural rearrangement, displacement of aryl ethers and benzylic ethers with sulfides and sulfites, while simultaneously promoting other C–O and C–C bond cleavage reactions. The resulting Kraft or sulfite lignin is generally a highly intractable, water-soluble dark brown powder of unknown structure. In addition, these processes incorporate up to 4 wt% of sulfur into the lignin backbone. This adds additional difficulties in designing post-processing reactions since sulfur is a well-known poison for transition metal catalysts. These pulping processes, which were designed solely to delignify cellulose, render lignin unsuitable for further processing. Nearly 40 million tons of lignin are produced from these pulping processes, whereby 95% of it is burned as a low value fuel. In select cases, people have designed lignin-based newspaper, composite boards and plastic bags, whereby lignin is a component of a polymeric mixture, giving the biosourced consumables.38 Attempts to valorize this waste stream led to the development of a short-lived chemical process for the synthesis of vanillin from the oxidation of Kraft lignin in 7–10% yield, which was eventually abandoned due to high cost and low efficiency.9,39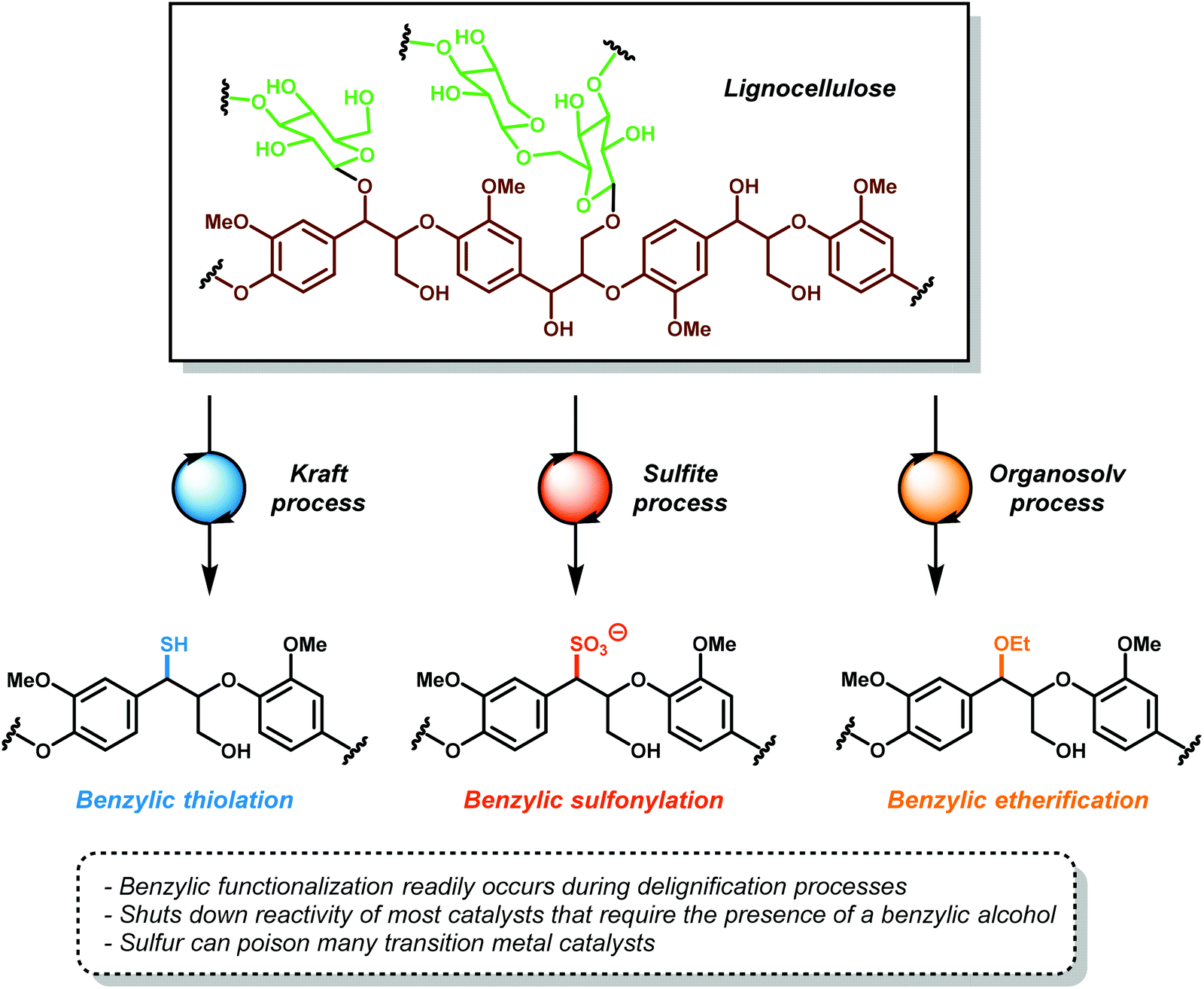 | ||
| Fig. 4 The lignin pulping processes. | ||
The organosolv process is a milder delignification process that occurs through the organic extraction of lignin with organic solvents, such as ethanol, dioxane, or acetone.40,41 Like the Kraft and sulfite processes, organosolv pulping significantly alters the native structure of lignin. All three processes modify the labile β-O-4 linkages, rendering the polymer even more intractable for further degradation. These types of processed lignin are unsuitable for studying the depolymerization of the β-O-4 linkage motif, which is estimated to compose 45–60% of the linkages in native lignin.18 Utilization of these lignins as a carbon feedstock is hampered by a poor understanding of their structure, which is not only difficult to depolymerize but also likely to produce a large number of inseparable compounds instead of a discrete, easily purified set of products. More advanced pulping technologies such as ball milling42 or the recently developed γ-valerolactone liquefaction43 are milder and preserve the overall lignin structure and produce product streams that are more amenable to postprocessing. The development of more advanced preprocessing techniques is an essential challenge to address for the development of a sustainable biorefinery. However, it is unlikely that these techniques will gain any widespread adoption in industry until it is economically viable to do so.
1.4 Strategies towards catalytic depolymerization of lignin
From a topical standpoint, the depolymerization of lignin appears to be a collection of engineering challenges rather than chemical, especially considering the scale by which biomass processing must occur. However, this perception could not be further from the truth—fundamental aspects surrounding lignin's structure, chemoselectivity and general reactivity are poorly understood. The principle challenge of lignin depolymerization arises from its complex chemical structure. This highly branched polymer is not only composed of three different monomers, but the linkage motifs between each monomer are structurally different and non-uniform in sequence. The ideal depolymerization process should thus be able to selectively degrade lignin into discrete, high-value small molecule products. In order to address these issues, a depolymerization catalyst not only has to be able to tolerate the functional groups found in lignin, but also react selectively and resist catalyst deactivation. It is highly probable that the depolymerization of lignin will need to occur over a series of steps to adequately engage the several linkage motifs found in the polymer backbone. The depolymerization strategies that have been exploited thus far can be categorized into three groups: oxidative, reductive, and redox-neutral catalysis and will be further discussed throughout this review.2. Reductive methods for C–O bond reduction in lignin model systems and lignin
The history of reductive lignin depolymerization began in the late 1930s when scientists experimented with hydrogenating the insoluble biomass isolated from tree sawdust. The central question in these studies was two-fold: how much hydrogen gas could lignin absorb, and what were the products of the hydrogenation? Analysis of the reaction products by distillation and elemental analysis revealed mixtures of alcohols resulting from the hydrogenation and hydrogenolysis of phenol and alkyl phenol derivatives. These early studies greatly contributed towards the understanding of the lignin monomeric constituents, and demonstrated the central chemical challenge for lignin valorization: selective hydrogenolysis over hydrogenation. Formally, hydrogenolysis converts lignin to valuable oxygenated aromatics such as polymethoxylated phenols, benzyl alcohols and phenylpropanols. Alternatively, hydrogenation yields methoxylated carbohydrate derivatives. In most methods, hydrogenation is a result of over-reduction and renders cyclohexyl ether products, which are more easily produced from cellulosic biomass. This section, which covers the development of reductive depolymerization methods, is focused on reports that demonstrated hydrogenolysis selectivity as well as processes that yielded valuable lignin reduction products.2.1 Early lignin precedent and structure elucidation
Selective hydrogenolysis over hydrogenation was identified by Sauer and Adkins in 1937.44 At that time, it was known that hydrogenation of alkenes occurred in the presence of Ni, Pd or Pt catalysts with atmospheric pressures of hydrogen gas. Yet, the corresponding ester reduction over platinum group metals required higher reaction temperatures (>200 °C) and hydrogen pressures (>100 atm).45 This distinct difference in reactivity between alkenes and esters motivated their study of selective hydrogenolysis of unsaturated esters using bimetallic oxide catalysts. Based on reports by Kraft, metallic oxides effectively reduced esters to the corresponding alcohols.46 Sauer and Adkins then applied this to the reduction of unsaturated esters of butyl oleate and butyl erucate. By screening various bimetallic catalysts, they identified two heterogeneous catalysts—zinc–chromium oxide (ZnCrO) and copper–chromium oxide (CuCrO)—that were capable of selectively reducing unsaturated esters to unsaturated alcohols in good yields (Fig. 5). While selective hydrogenolysis of unsaturated esters to unsaturated alcohols was achieved, Sauer and Adkins noted that this only occurred at a minimum of 300 °C with a 50 mol% metal–oxide loading.44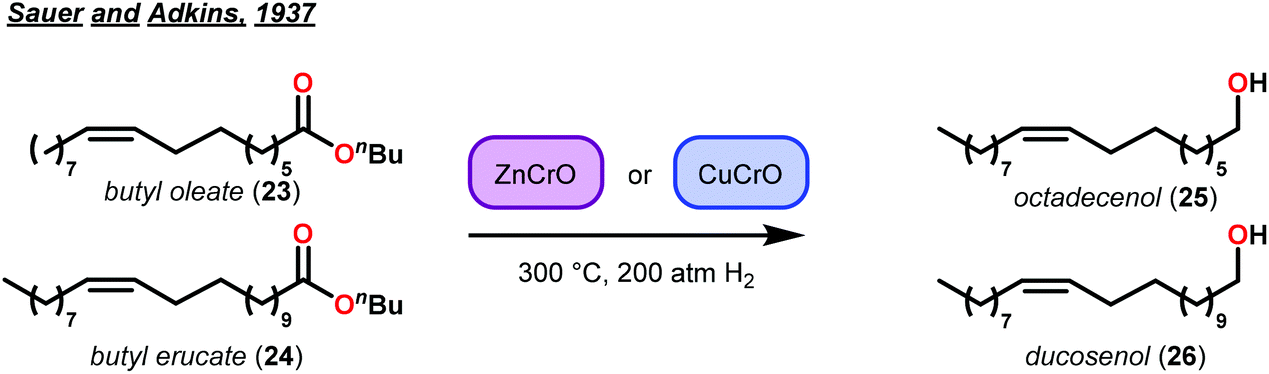 | ||
| Fig. 5 Sauer and Adkins’ selective ester hydrogenolysis in the presence of an alkene. | ||
The importance of Sauer and Adkins’ findings were realized a year later by Harris, D'Ianni and Adkins when they used a copper–chromium oxide catalyst (Cu–Cr–O) for the reduction of 80 g of hardwood lignin.47 While selectivity for hydrogenolysis was not observed, the reduction of lignin led to the isolation of a few key structure-elucidating products. From the 80 g lignin sample, methanol (22 g), 4-propylcyclohexan-1-ol, (9 g, 27), 4-(3-hydroxypropyl)cyclohexan-1-ol (20 g, 28) and 4-propylcyclohexan-1,2-diol (3 g, 29) were isolated. This report represented the highest recoveries of lignin depolymerization products, at that time and provided support to the hypotheses that lignin was composed of phenyl propanoid units of either C8 or C9 length (Fig. 6).
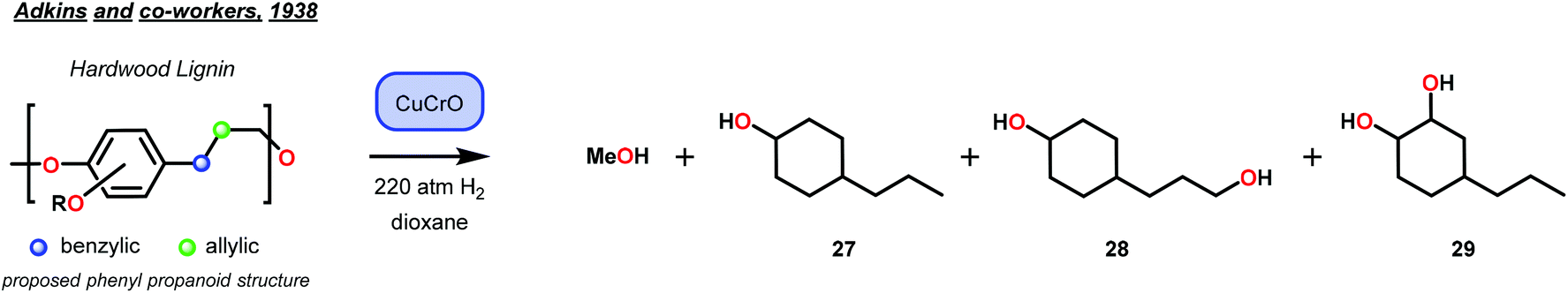 | ||
| Fig. 6 Isolation of lignin products by Adkins and co-workers, supporting the presence of the phenyl propanoid structure in lignin. | ||
About ten years later, Hibbert and co-workers confirmed the phenyl propanoid monomer structure by isolating 4-(3-hydroxypropyl)-2,6-dimethoxyphenol (30), 4-(propyl)-2,6-dimethoxyphenol (31) and 4-(3-hydroxypropyl)-2-methoxyphenol (32) in a combined yield of 3% from lignin biomass.48 Hibbert and co-workers applied a reduction procedure using RANEY®-Ni (previously developed by Adkins49), whereby maple woodmeal was reacted at 165–170 °C for 4 hours.48 The products were subsequently isolated from the unwanted cellulose and hemicellulose biomass by filtration and precipitation, respectively. Furthermore, Hibbert and co-workers reduced the generated products (30–32) with Adkins’ copper–chromium catalyst to yield the same diol products (27–29) that Adkins observed. These studies by Hibbert demonstrated the selective hydrogenolysis of unprocessed lignin, albeit in low yield (Fig. 7).48
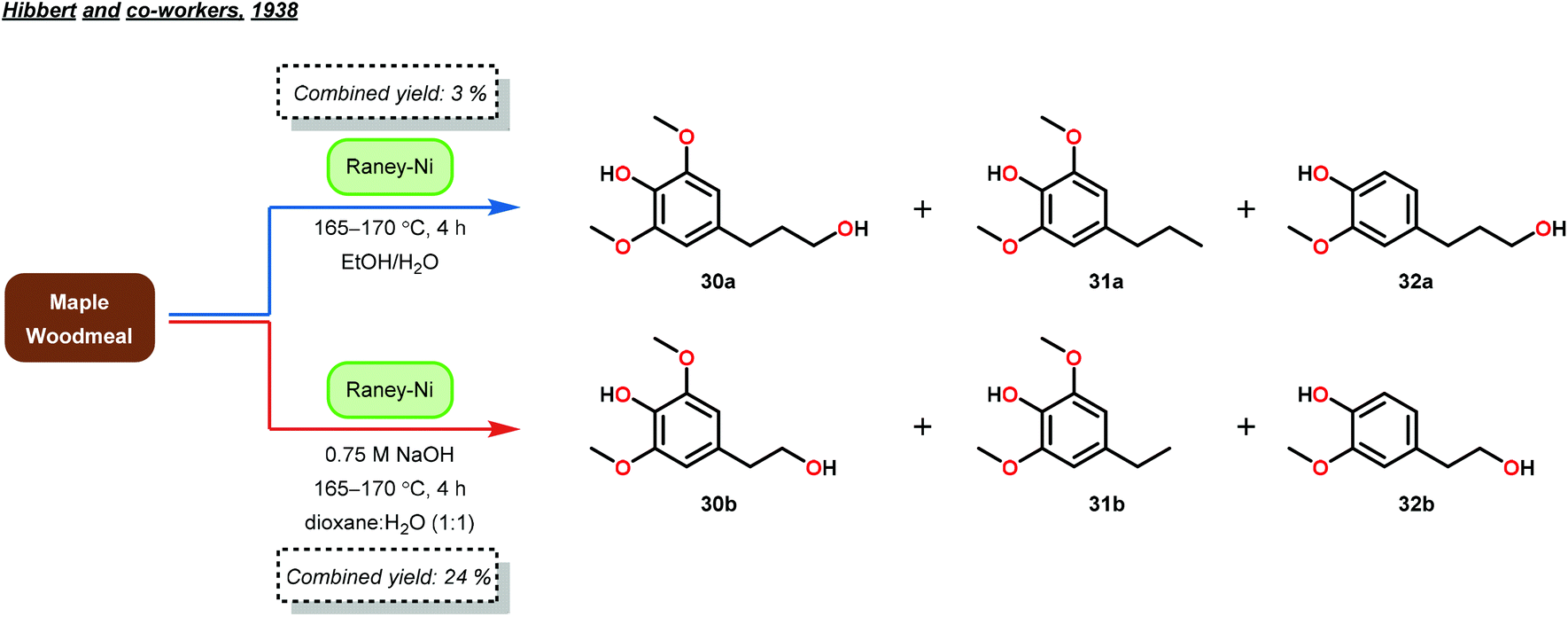 | ||
| Fig. 7 Reduction of lignin and initial isolation of phenyl propanoids by Hibbert and co-workers. | ||
While the isolation of the actual guaiacyl (32) and sinapyl products (30) was a major achievement for elucidating the structure of lignin, the isolated yields for this reaction were low (3%). Hibbert further optimized his reduction conditions by studying the reaction pH. Hibbert found that a mixture of ethanol, water and lignin naturally exhibited a slightly acidic pH of 5.5. Highly acidic conditions proved to be detrimental to the reduction of lignin as acid promoted cross-linking and Ni-catalyst poisoning occurred, while basic conditions promoted Ni reduction and prevented polymerization. Interestingly, these alkaline conditions yielded phenylethanoid monomers in a slightly higher yield of ∼24%, suggesting the existence of a β-O-4 lignin linkage and a selectivity basis for hydrogenolysis. Variations in the lignin source (hardwood vs. softwood) also afforded different ratios of guaicyl to syringyl monomers.50
It is clear from the seminal work of Adkins and Hibbert and their predecessors, that the structure of lignin is highly complex, with source dependent ratios of monomers. Since then, the advancement of nuclear magnetic spectroscopy (NMR) experimentation, including quantitative 13C NMR, 1H NMR, and two-dimensional (2D) NMR, have contributed towards a general understanding of the molecular composition of lignin.51,52 Ultimately, the body of work describing the chemical structure of lignin has attracted chemists to apply tangentially developed synthetic methodologies to the depolymerization of lignin.
2.2 Hydrodeoxygenation using homogeneous Ni catalysis
Recently, the use of homogeneous transition metal catalysts has been employed for the selective hydrogenolysis of model lignin substrates. The development of this chemistry arose from a fundamental understanding of the mechanism of traditional transition metal-catalyzed cross-coupling chemistry53–59 (Fig. 8). In this sense, arene C–O bonds are activated by a Pd or Ni catalyst for replacement by a transmetallating hydride. While the ultimate goal of lignin depolymerization has yet to be realized due to catalyst inefficiencies and the complexity of lignin, a variety of metal complexes and hydride sources have been demonstrated for the reductive cleavage of C–O bond frameworks in lignin models.60–62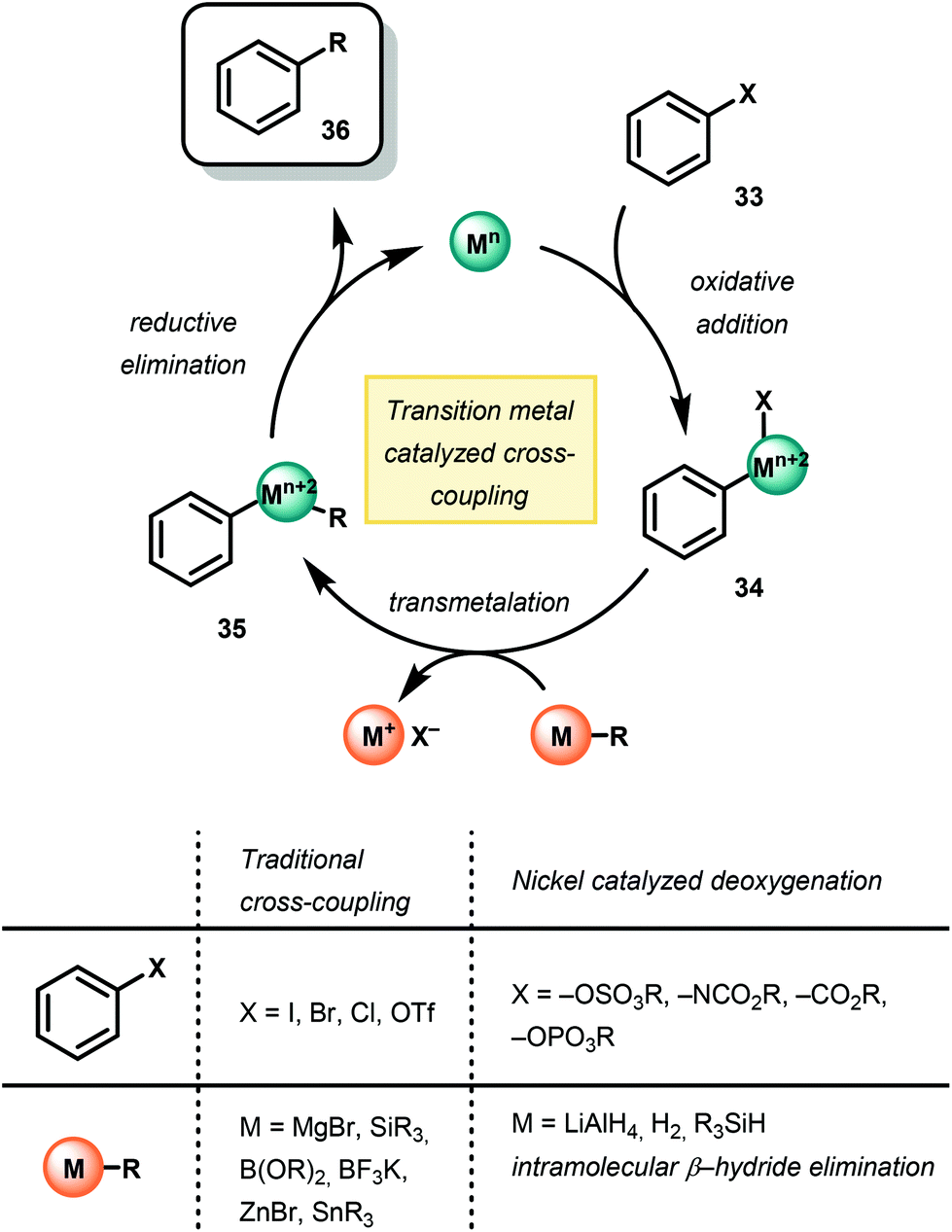 | ||
| Fig. 8 Comparison of traditional cross-coupling methodology and Ni-catalyzed arene deoxygenation. | ||
Observation of C–O bond oxidative insertion using Ni was first reported by Wenkert in 1979.63 Using Ni(PCy3)2Cl2 as a precatalyst, Wenkert and co-workers demonstrated Ni-catalyzed Kumada cross-coupling using phenylmagnesium bromide and a variety of methoxy napthelene and anisole derivatives, demonstrating that methoxy arenes are competent substrates for oxidative additions to Ni. Comparing the reactivity of the methoxy naphthalene to anisole, Wenkert noted a significantly faster reduction of methoxy napthelene substrates opposed to anisole derivatives. This result suggested an initial π-coordination of the NiII complex to the substrate prior to C–O insertion, in which the polycyclic aromatic structure of the methoxynaphthalenes, compared to the methoxybenzenes, was more amenable for reactivity. Overall, the success of this reaction was an early example of a selective Ni-catalyzed Caryl–O bond insertion over the Calkyl–O insertion and affect overall deoxygenation.
Inspired by Wenker's report, Dankwardt and co-workers investigated a greater Kumada cross-coupling scope using a more electron-rich Ni catalyst.64 Using a Ni(PCy3)2Cl2 precatalyst, Dankwardt and co-workers successfully demonstrated the Ni-catalyzed cross-coupling on a large variety of methoxyarenes with phenyl magnesium bromide. This Ni–phosphine precatalyst proved more efficient and versatile than Wenkert's catalyst (Ni(PPh3)2Cl2), tolerating a number of nucleophilic functionalities, including hydroxy, phenol, amine, enamine and N-heterocyclic moieties. More relevant to lignin valorization, Dankwardt and co-workers focused on dimethoxy-benzenes and naphthalanes to explore the scope of arene substitution tolerance (Fig. 9). Regardless of the functional group tolerance observed, the poor yields of the Kumada-coupling products with dimethoxynaphthalene and dimethoxybenzene suggested poor compatibility with guaiacyl and syringyl-type substrates.
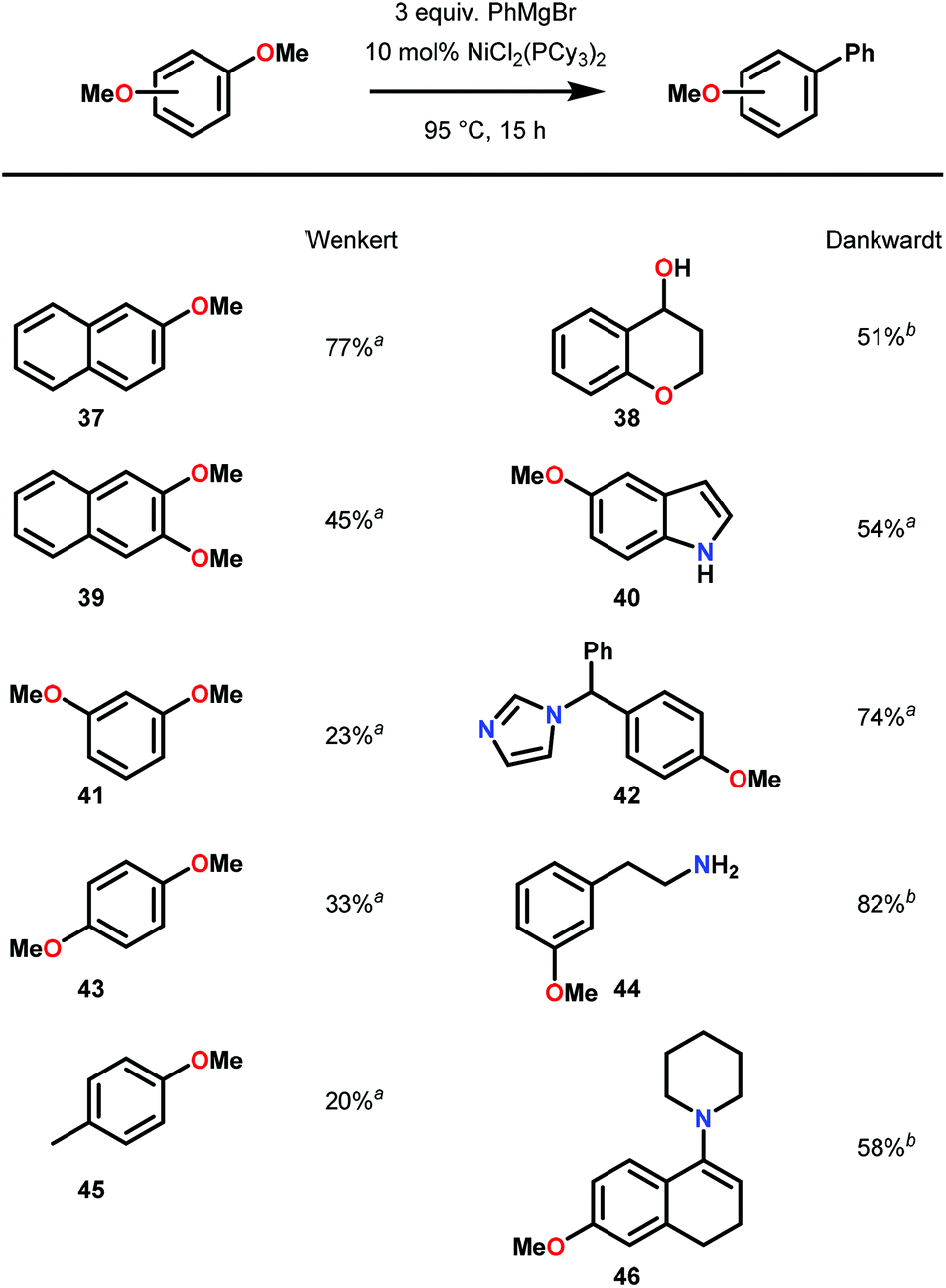 | ||
| Fig. 9 Ni-catalyzed Kumada cross-coupling of methoxy arenes. a Yields of isolated products. b Conversions were determined by GC using tridecane as internal standard. | ||
Since Wenkert's intial report, Ni-mediated C–O bond activation strategies have been reported for a large variety of activated oxyarene bonds including sulfonates, sulfates, mesylates, vinyl esters, carbamates and carbonates for C–C bond synthesis.65–67 In search of a general hydrogenolysis methodology of oxyarenes, Martin and co-workers screened a number of hydride reagents including silanes, borohydride salts, alkyl zinc and alkyl aluminum reductants. Optimally, they found the combination of Ni(COD)2 with 1,1,3,3-tetramethyldisiloxane (TMSDO) was an efficient method for reductive demethoxylation of arenes (Fig. 10).68 Importantly, methoxy arenes were reduced in highest yield in comparison to other oxygen functionalities, such as OEt, OAc, OPiv, OTs and OMs.
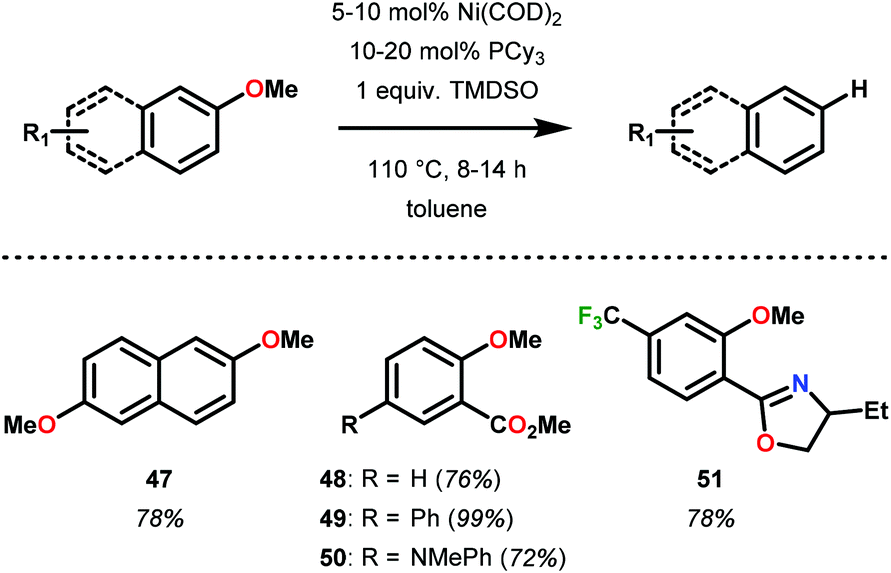 | ||
| Fig. 10 Ni-catalyzed C–O bond reduction of aryl ethers. TMSDO = 1,1,3,3-tetramethyldisiloxane. | ||
Soon after Martin's report, Hartwig and Sergeev reported the Caryl–O bond cleavage of a variety of alkyl aryl ether substrates (Fig. 11). Hartwig and Sergeev accomplished these challenging reductions via a Ni–NHC (N-heterocyclic carbene, SIPr) complex that is uniquely resistant towards irreversible reductive elimination to a heterogeneous solid, a common issue in homogeneous cross-coupling chemistry. Impressively, the Ni-catalyst formed in situ was compatible with a number of hydride sources (including DIBAL-H, LiAl(OiBu)3H, Et3SiH), however, the system garnered the interest of the catalysis community by engaging H2 as the terminal reductant. In practice, Hartwig demonstrated a general substrate tolerance of Ar–OAr (52), Ar–OMe (53) and ArCH2–OMe (54), showcasing the ability of Ni to reduce both Csp2–O and Csp3–O bonds. Notably, this catalyst selectively reduced aryl–aryl ethers in the presence of aryl–alkyl ethers, showing promise for a selective 4-O-5 depolymerization of lignin and lignin model systems (Fig. 11).69 Despite the high selectivity and mild reaction conditions in comparison to classic lignin hydrogenolysis reactions, this system employs high loadings of potassium tert-butoxide as an additive and an expensive NHC ligand (∼$66/g Sigma Aldrich70).
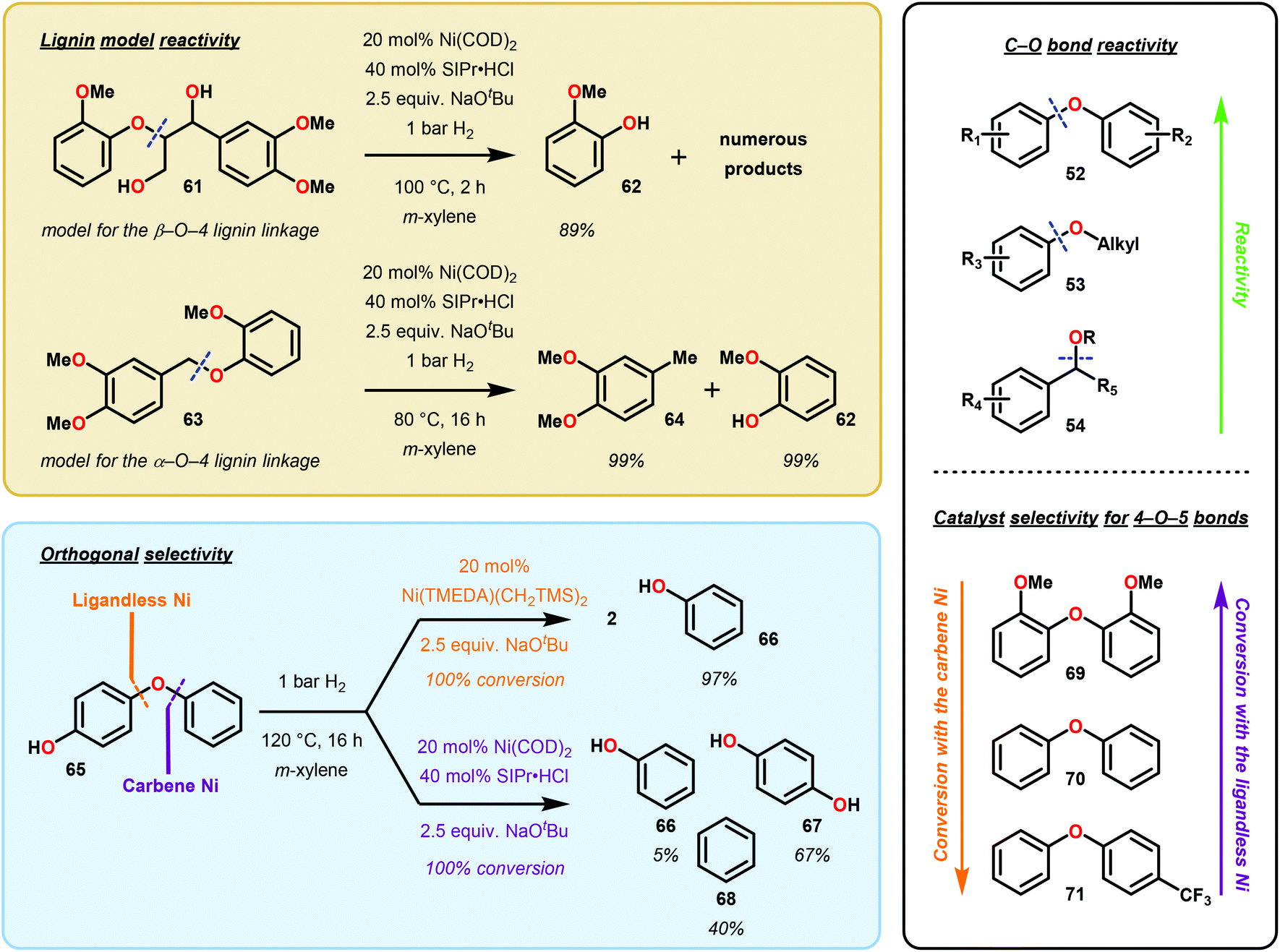 | ||
| Fig. 11 Ni-catalyzed hydrogenolysis of aryl ethers. | ||
About two years later, Hartwig and co-workers refined their Ni catalyzed deoxygenation by excluding the carbene ligand (SIPr) after discovering heterogeneous conditions capable of similar C–O reductions.71 This second generation system solely relied upon NaOtBu and 1 atm of H2 to achieve the wanted C–O hydrogenolysis. Further pre-catalyst screening uncovered that the use of Ni(CH2TMS)2(TMEDA) (in a ten times lower catalyst loadings than the first generation conditions), could achieve similar C–O bond reduction. Lastly, the first and second generation conditions exhibited orthogonal efficiency in aryl ether reductions of electron-rich and electron-deficient arenes.
Overall, the impressive reactivity of Martin's and Hartwig's Ni-based systems represents orthogonal reactivity amongst reductive methodologies. Martin's approach exemplifies a homogeneous cross-coupling method in which hydride transmetalates and reductively eliminates to yield a deoxygenated arene. The reactivity observed under Hartwig and co-workers’ conditions is intriguing and indicates that slight changes in reaction conditions can significantly affect the selectivity. While Hartwig's first generation “Ni carbene” approach mechanistically mimics that of Martin's, the second generation “ligandless Ni” system is heterogeneous and likely mechanistically unrelated, allowing milder reaction temperatures and H2 pressures.
In a subsequent mechanistic study,72 Agapie and co-workers studied the hydrogenolysis of aryl ethers by Ni. By using a specially designed phosphine-methoxyarene ligand, the authors directly observed the Ni0 complex 73 as a dark red solid where the Ni center interacts with two carbon centers of the central arene. Additionally, after C–O bond insertion, the NiII complex (74) readily underwent β-hydride elimination to afford a NiII aryl hydride species (75) and the corresponding carbonyl compound. Subsequent reductive elimination and decarbonylation afforded an isolable NiII complex (76). While the stoichiometric studies provided isolable intermediates, Agapie and co-workers further studied the β-hydride elimination in a Ni catalytic reaction. Applying Hartwig's deoxygenation conditions to trideuteromethoxy naphthalene (78) and 2-((hexyl-1,1-d2)oxy)naphthalene (79), 90% deuterium incorporation from deoxygenation was observed. These studies suggested that β-hydride elimination is a competent pathway and explained why other methods are capable of using mild atmospheres of H2. Furthermore, the obtained results also illustrated that the selectivity for Caryl–O insertion over the weaker Calkyl–O insertion was due to π-coordination of the arene to the Ni center, which promoted insertion of Ni into the Caryl–O bond (Scheme 2).
 | ||
| Scheme 2 Ni-mediated hydrogenolysis of Caryl–O bonds. | ||
Prompted by Agapie's work, Martin and co-workers investigated the catalytic mechanism of Ni catalyzed arene deoxygenation, under the pretence that the system did not operate via a β-hydride elimination mechanism.73 Martin found that Agapie's C–O inserted complexes, such as complex 74, were not reactive towards exogenous silane sources for deoxygenation and were not indicative of an intermolecular reaction between hydride reductants and Ni inserted species. Yet, empirical evidence for a Ni0/NiII cross-coupling mechanism for Martin's first generation Ni(COD)2 arene deoxygenation using TMSDO was not proven. Lastly, the following experimental observations were gleaned from their initial study:68 (1) Ni(COD)2 was the only Ni precatalyst capable of deoxygenating methoxy arenes, (2) control experiments performed in the absence of silane indicated that silane was necessary for the reduction to occur, (3) napthalene substrates consistently afforded higher yields compared to their anisole derivatives, and (4) PCy3 was the critical ligand.
The mechanistic experiments performed by Martin and co-workers suggest that the formation of a NiI-SiR3 species is necessary for arene deoxygenation (Fig. 12). Firstly, they found that preparation of a napthyl-Ni–O species was not possible by simple oxidative insertion with the Ni(COD)2 precatalyst (Fig. 12A). Alternative synthesis by means of oxidative insertion into 2-napthylchloride, followed by sodium methoxide transmetalation was indeed feasible (Fig. 12A). However, this complex decomposed at room temperature to yield naphthalene (from H/arene reductive elimination) and the Ni(PCy3)2(CO) complex. In order to successfully synthesize a napthyl-Ni–O complex, [Ni(PCy3)2]2N2 was needed for direct C–O oxidative insertion, to give naphthalene in 54% yield. Lastly, they found that silane reduction outcompeted intramolecular β-hydride elimination, even when using the activated precatalyst [Ni(PCy3)2]2N2. The experiments are highly suggestive that the β-hydride elimination pathway is merely a minimally contributing pathway.73
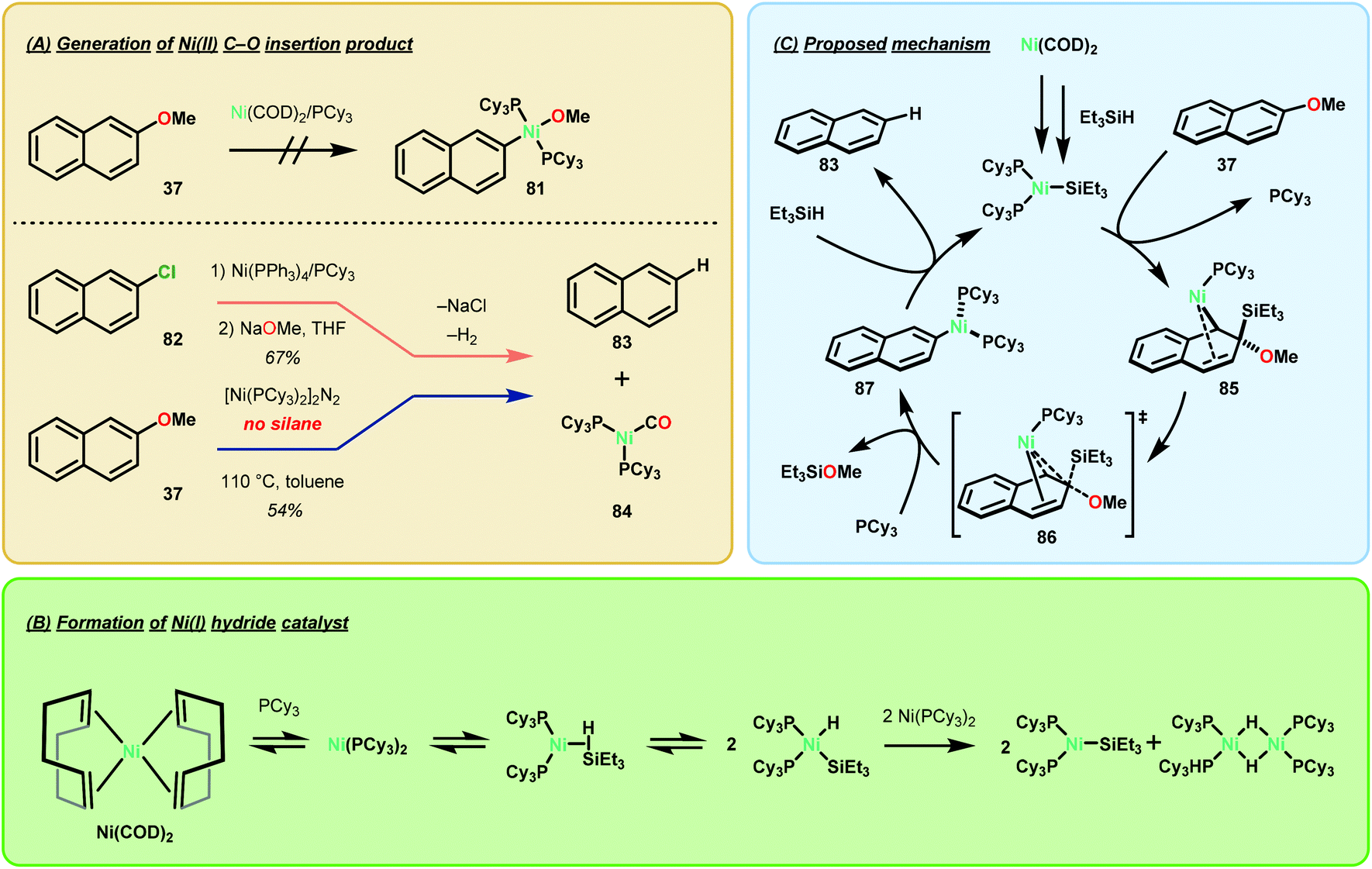 | ||
| Fig. 12 Mechanistic considerations of Ni-catalyzed reductive C–O bond cleavage. | ||
The authors subsequently determined the kinetic dependence of each of the reaction components and found that all three components—substrate, catalyst, and the silane—exhibited a first-order dependence. Analysis of silane consumption by GC indicated an induction period which was further exasperated by a kinetic isotope effect when Et3SiD was used. Rapid consumption of the silane was observed by 1H NMR in the presence of a stoichiometric amount of Ni, even in the absence of naphthalene production. Support for a NiIH–Si insertion product was given by the observation of a 1H NMR signal at −15.8 ppm, indicative of a Ni–H intermediate, and a characteristic NiI EPR spectrum. These results together with computational modeling suggested the formation of a catalytically active NiI species as shown in Fig. 12C.73 Overall, this study concluded that the mechanism in which Ni deoxygenates arenes is greatly dependent upon the exogenous reductant employed. While intramolecular β-hydride elimination from the napthyl-Ni–OCH3 intermediate is precedented,74 it is not the dominant process when stoichiometric amounts of hydride (R3SiH, LiAlH4) or H2 are used.
2.3 Heterogeneous Ni-catalyzed deoxygenation
A major limitation of homogeneous Ni catalysis is the incompatibility of most Ni precursors with aqueous solvents. To address this concern, Lercher and co-workers developed a heterogeneous Ni–SiO2 catalyst capable of hydrogenolysing α-O-4 and β-O-4 linkages in lignin model systems. For the synthesis of the catalyst, Ni was deposited on silica using urea as the hydrolysis reagent, where the particle size of Ni was controlled by the calcination temperature (400–800 °C). Catalyst activity, as measured by turnover frequency (TOF), peaked at 26 h−1 when the particle size was 5.9 nm. Using the heterogeneous catalyst and 6 bar of H2, the reduction of simple lignin model ethers was demonstrated (Fig. 13), with moderate selectivity for hydrogenolysis in the α-O-4 and β-O-4 model systems versus over reduction to cyclohexanols. For the hydrogenolysis, a kinetic distinction between the α-O-4, 4-O-5 and β-O-4 systems was observed, consistent with an increasing order of activation energy (Ea).75 The development of a heterogeneous catalyst that utilizes relatively low hydrogenation pressures of H2 (6 bar) in aqueous media was a substantial step towards the reductive valorization of lignin. Although the Ni/SiO2 was shown to reduce simple lignin model systems, more complex systems containing free alcohol moieties, which are known to deactivate transition metal catalysts, were not evaluated.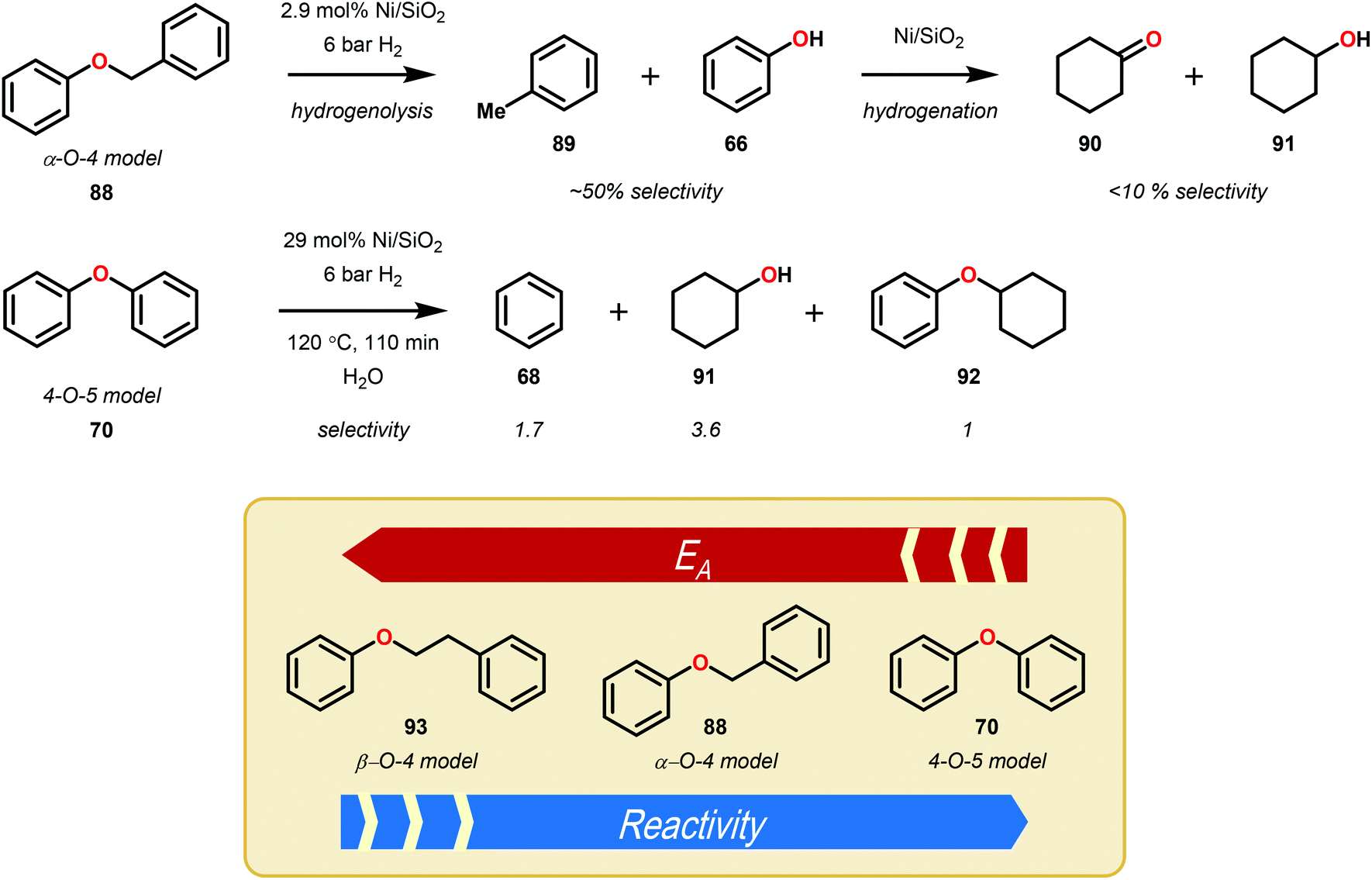 | ||
| Fig. 13 Hydrogenolysis of simple lignin model systems utilizing a heterogeneous Ni/SiO2 catalyst. | ||
A variety of heterogeneous catalysts have been prepared and shown to reduce lignin and associated models. Design aspects of these catalysts include: immobilization on a solid phase (typically carbon or silica), doping with another metal, or synthesized as the metal or bimetallic oxide.76 In certain cases, these catalysts were demonstrated to perform selective hydrogenolysis over hydrogenation.77–80 The results from these studies highlight the fact that the isolated lignin monomer units greatly vary and a controlled depolymerization has been difficult to accomplish. Despite this fact, bimetallic catalysts and heterogeneous catalysts are the subject of intense research for reductive methods towards the valorization of lignin. The key effects exhibited by bimetallic catalysts in the reduction of lignin feedstocks have been summarized by Dumesic and co-workers.81
2.4 Pd and Ni mediated hydrogen transfer methods for reduction of C–O bonds
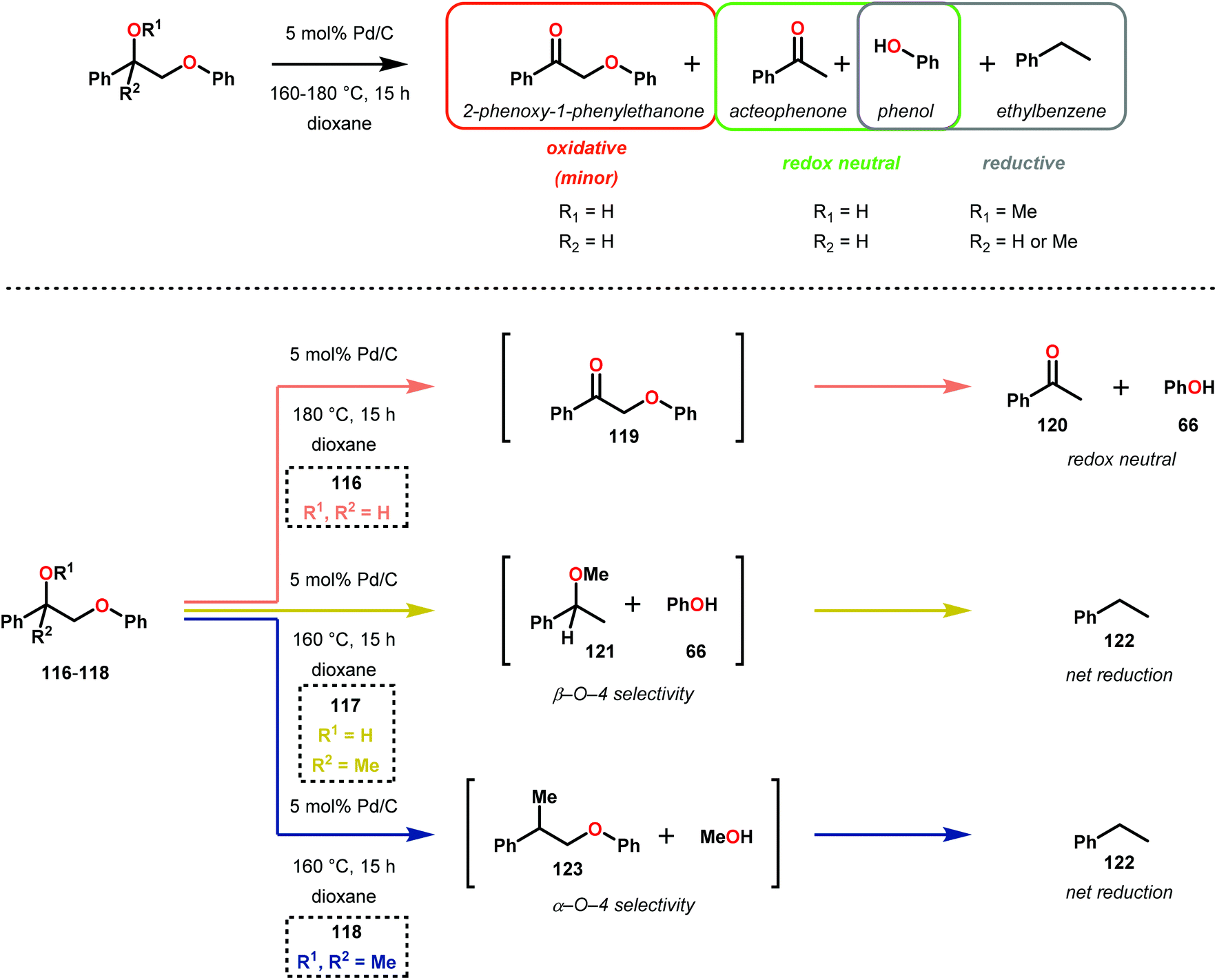
In this section, details on the work of Rinaldi,82 Samec83–85 and Rauchfuss86 are described as it highlights the C–O bond reduction through a formal redox-neutral hydrogen shuttle from an alcohol, ether or formic acid. Transfer hydrogenation chemistry that utilizes lignin as the hydrogen source is detailed in the redox-neutral section (section 4). Both exogenous and endogenous transfer hydrogenation methods have been directly studied for depolymerizing native lignin feedstocks and exemplify valorization through the isolation of monomeric or dimeric units, suggesting that these methods are the closest to industrialization.Recent development in catalytic lignin reduction through hydrogen transfer reduction was achieved by Rinaldi and co-workers, who showed the hydrogenation and hydrogenolysis of a number of sinapyl and guaiacyl models using RANEY®-Ni and 2-propanol at moderate temperatures (120 °C). While reduction between 80–150 °C using 2-propanol as the reductant is a notable achievement, each reaction produces an array of partially reduced products (Fig. 14). The selectivity is biased towards hydrogenation over hydrogenolysis; however, hydrogenolysis occurs in both the absence and presence of alkene and arene functionalities. The authors found that the selectivity of arene hydrogenation could be diverted when using bulky tert-butyl substituted substrates, albeit these are not recognized functionalities in the monomer constituents of native lignin. More importantly, the RANEY®-Ni catalyzed reduction on a mixture of benzaldehyde, benzofuran, diethyl maleate and guaiacyl acetone was evaluated. Transfer hydrogenation transformed the starting mixture to 2,3-dihydrobenzofuran, diethyl succinate, 4-(2-hydroxypropyl)-2-methoxyphenol and cyclohexane-1,2-diol, and showcased the reductive capability of hydrogen transfer in the presence of a mixture of oxygenated functionalities. Unfortunately, during their investigation they recognized that the presence of primary alcohol moieties inhibited the transfer hydrogenation when using 2-propanol as the hydrogen source. Although primary alcohols were not destructive for the catalyst system, they were shown to inhibit catalyst turnover. Overall, reaction efficiency was measured by starting material consumption, thus proving transfer hydrogenation with alcohols as a possible biomass conversion process for further applications.82
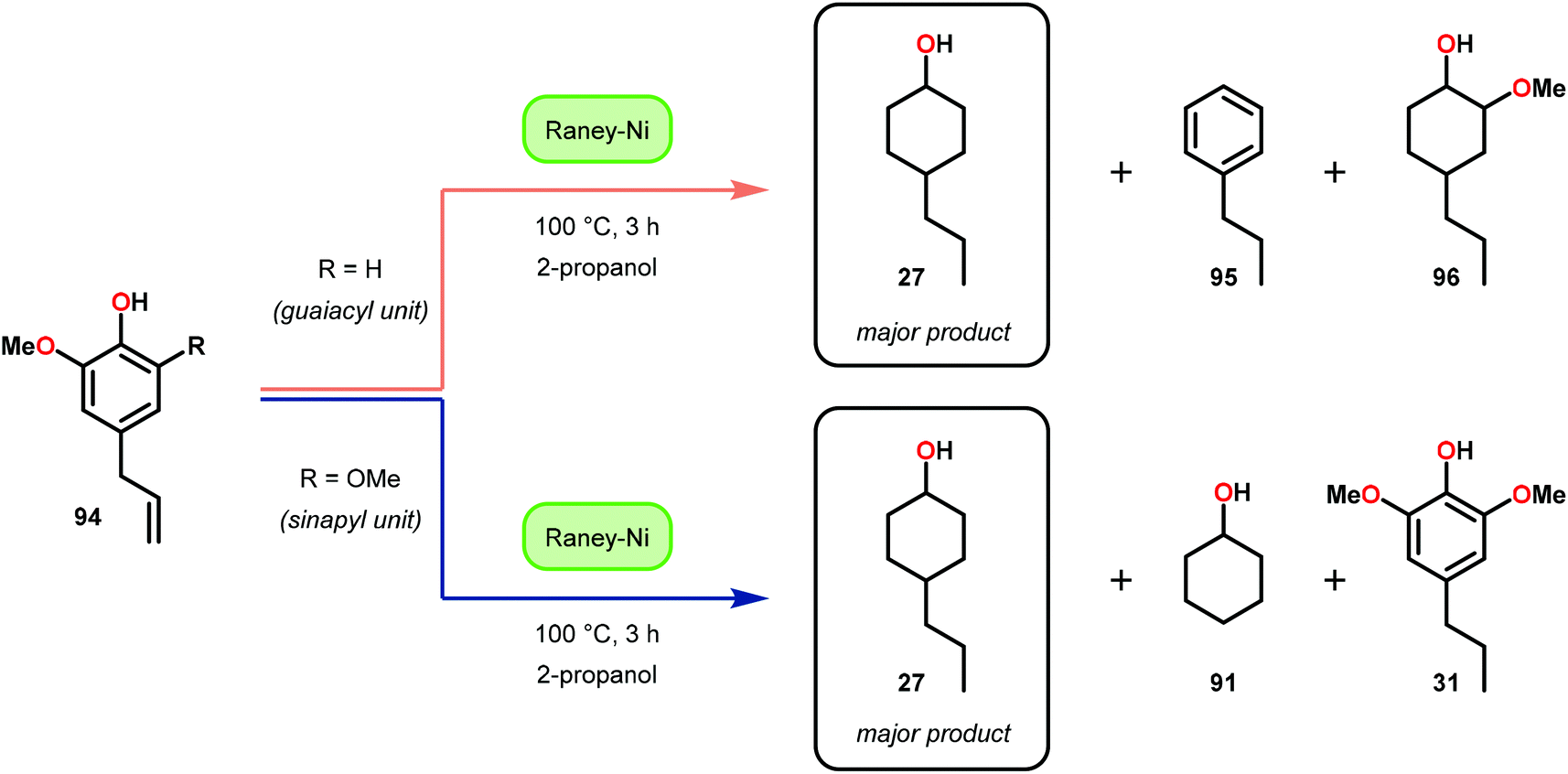 | ||
| Fig. 14 RANEY®-Ni catalyzed transfer hydrogenation of lignin model substrates. | ||
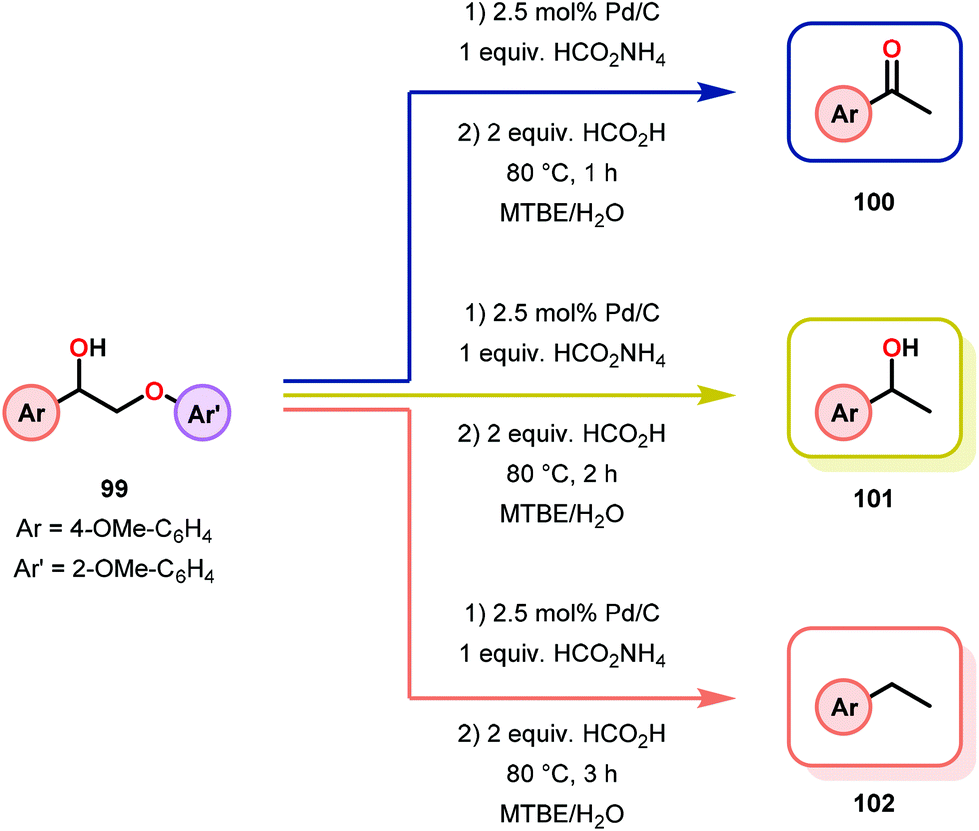
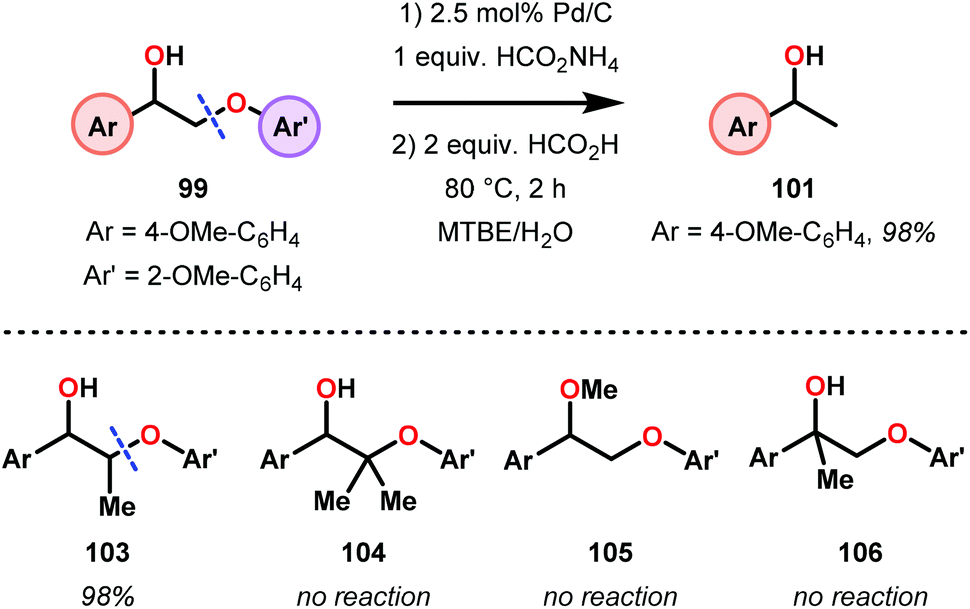
Samec and co-workers advanced reductive hydrogen transfer catalysis by demonstrating the reduction of β-O-4 lignin model systems using Pd/C and transfer hydrogenation. These conditions were developed by screening a variety of transition metals and hydrogen donors. A comparison of Pd/C, Rh/C, Ir/C, Re/C, Ni/C, revealed that Pd was unique in reducing β-O-4 model compounds to the corresponding acetophenone and phenol. Addition of a stoichiometric equivalent of amine (ammonia, ethyl amine, allylamine), directed the selectivity of this reaction towards β-O-4 cleavage and not the disproportionation reaction (to yield 100 and 102). Lastly, formic acid was found to efficiently facilitate reduction as compared to H2 or 2-propanol. Using these conditions, the authors demonstrated the reduction of a variety of β-O-4 model systems (97) to afford the cleaved products (98) in excellent yields (Scheme 3). The extent of the reduction was controlled by time and stoichiometric addition of formic acid (Scheme 4). The authors demonstrated that the β-O-4 cleavage was specific to benzylic alcohols containing at least one β-hydrogen (Fig. 15). These observations, in combination with an observed kinetic isotope effect of the benzylic C–H bond, suggested that the operating mechanism involves dehydrogenation and deprotonation to afford a Pd enolate species as the operative mode of transition metal activation (cf. Scheme 45). Further reduction by formic acid subsequently yields the acetophenone and phenol β-O-4 cleavage products. Finally, applying the reduction system on a β-O-4 model polymer resulted in almost quantitative formation of the acetophenone (109) or ethylbenzene product (110) depending on the reaction conditions (Scheme 5). Encouraged by these results, the authors applied the catalytic system on organosolv lignin from Pinus sylvestris. GPC analysis showed a moderate change towards lower molecular weight fragments.84
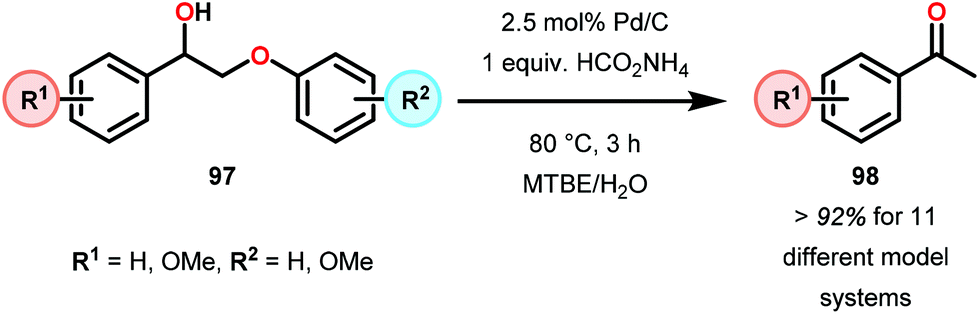 | ||
| Scheme 3 Heterogeneous Pd-catalyzed cleavage of β-O-4 model systems. | ||
 | ||
| Scheme 4 Pd-catalyzed selective transformation of lignin model systems. | ||
 | ||
| Fig. 15 Reactivity pattern for heterogeneous Pd-catalyzed cleavage of lignin model systems. | ||
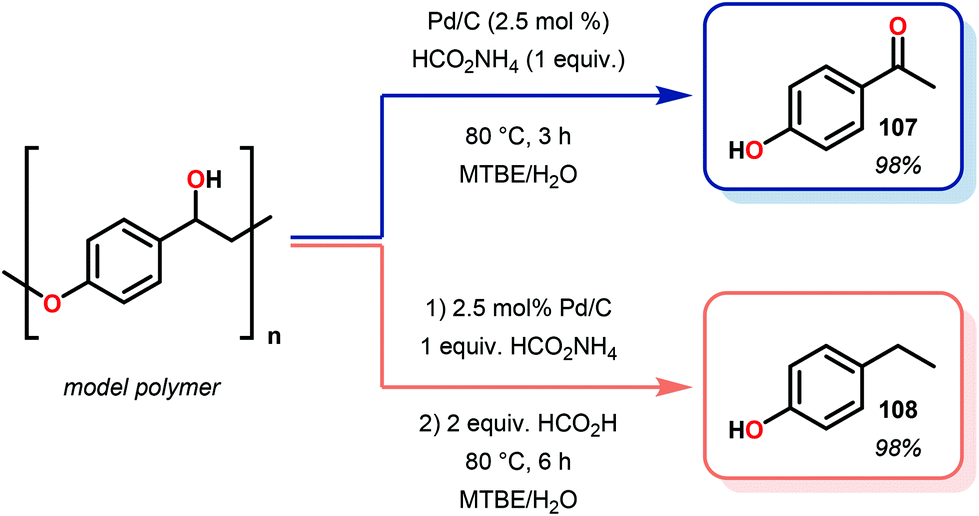 | ||
| Scheme 5 Depolymerization of lignin model polymer by Pd/C. | ||
Soon after this publication, Samec and co-workers reported on a key insight regarding traditional organosolv processing. Realizing that formic acid is generated in the organosolv lignin pulping process,87 the authors developed a tandem organosolv-hydrogen transfer protocol that depolymerized lignin. Specifically, pine sawdust was processed using ethanosolv conditions with Pd/C (195 °C, 1 h, EtOH/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). From the resulting biomass mixture, cellulose was filtered off, hemicellulose is removed by precipitation and a clear lignin bio-oil was obtained. In a comparison between an organosolv process with and without Pd reduction, it was found that the reduced feedstock had an average molecular weight half of the non-reduced stock, as well as 58% monomeric composition of the reduced material. 2D NMR spectroscopy additionally revealed that the targeted β-O-4 bonds were reduced. Interestingly, the major monomeric component from the pine feedstock was 3-methoxy-4-hydroxyphenyl-2-butene (109), isolated in 23% yield. This was proposed to be produced by sequential dehydration–hydrogenation, which ultimately produced arylbutenes 109 (Fig. 16).85 This study of lignin degradation is unique since the tandem organosolv transfer hydrogenolysis (TOTH) process reduced actual lignin to an isolable monomer and that the presence of an alkene functionality in this monomer indicates that the TOTH process is highly selective for C–O bond reduction over C
:1). From the resulting biomass mixture, cellulose was filtered off, hemicellulose is removed by precipitation and a clear lignin bio-oil was obtained. In a comparison between an organosolv process with and without Pd reduction, it was found that the reduced feedstock had an average molecular weight half of the non-reduced stock, as well as 58% monomeric composition of the reduced material. 2D NMR spectroscopy additionally revealed that the targeted β-O-4 bonds were reduced. Interestingly, the major monomeric component from the pine feedstock was 3-methoxy-4-hydroxyphenyl-2-butene (109), isolated in 23% yield. This was proposed to be produced by sequential dehydration–hydrogenation, which ultimately produced arylbutenes 109 (Fig. 16).85 This study of lignin degradation is unique since the tandem organosolv transfer hydrogenolysis (TOTH) process reduced actual lignin to an isolable monomer and that the presence of an alkene functionality in this monomer indicates that the TOTH process is highly selective for C–O bond reduction over C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond reduction.
C bond reduction.
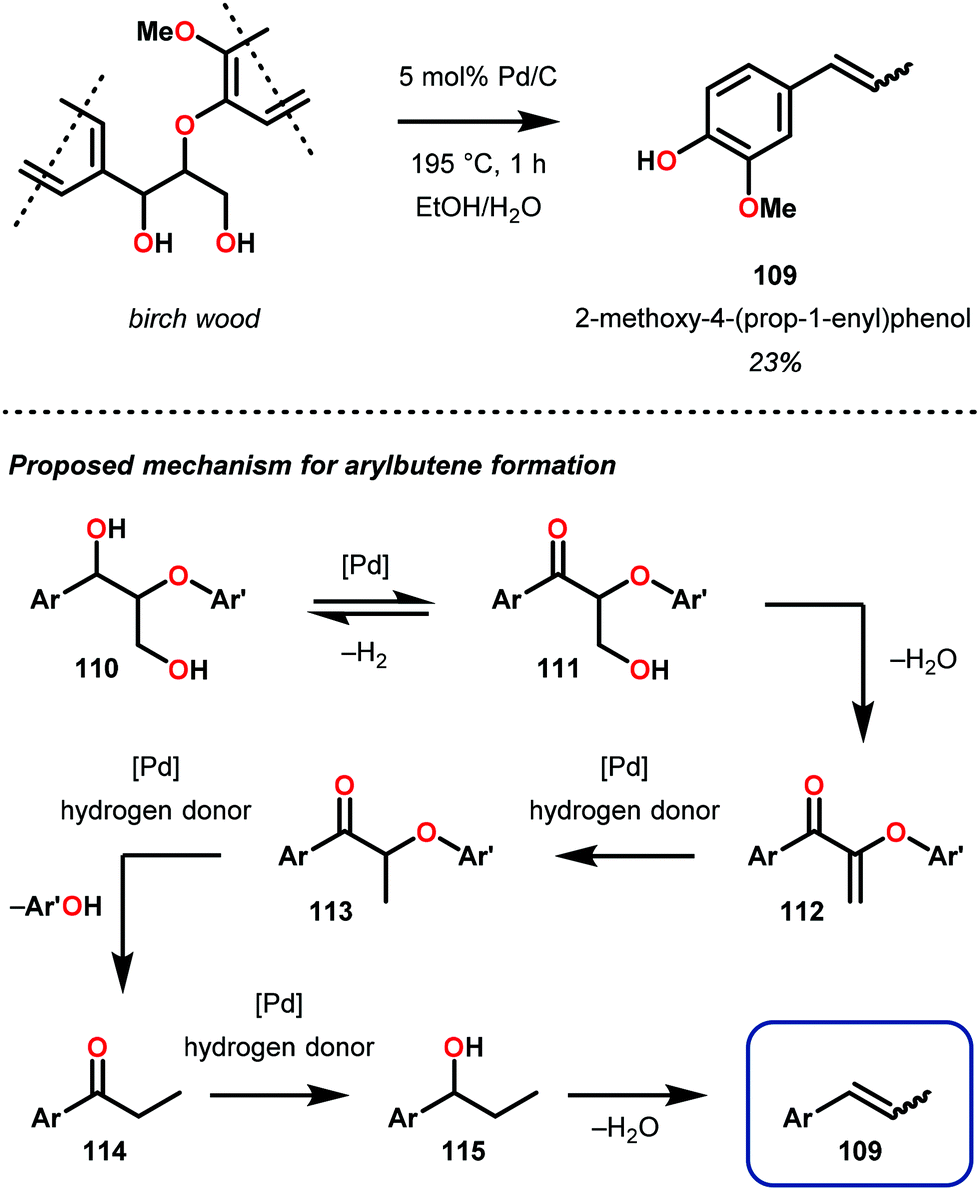 | ||
| Fig. 16 (Top) Pd/C catalyzed tandem organosolv transfer hydrogenolysis (TOTH) of birch wood and (bottom) proposed mechanism of aryl propene formation. | ||
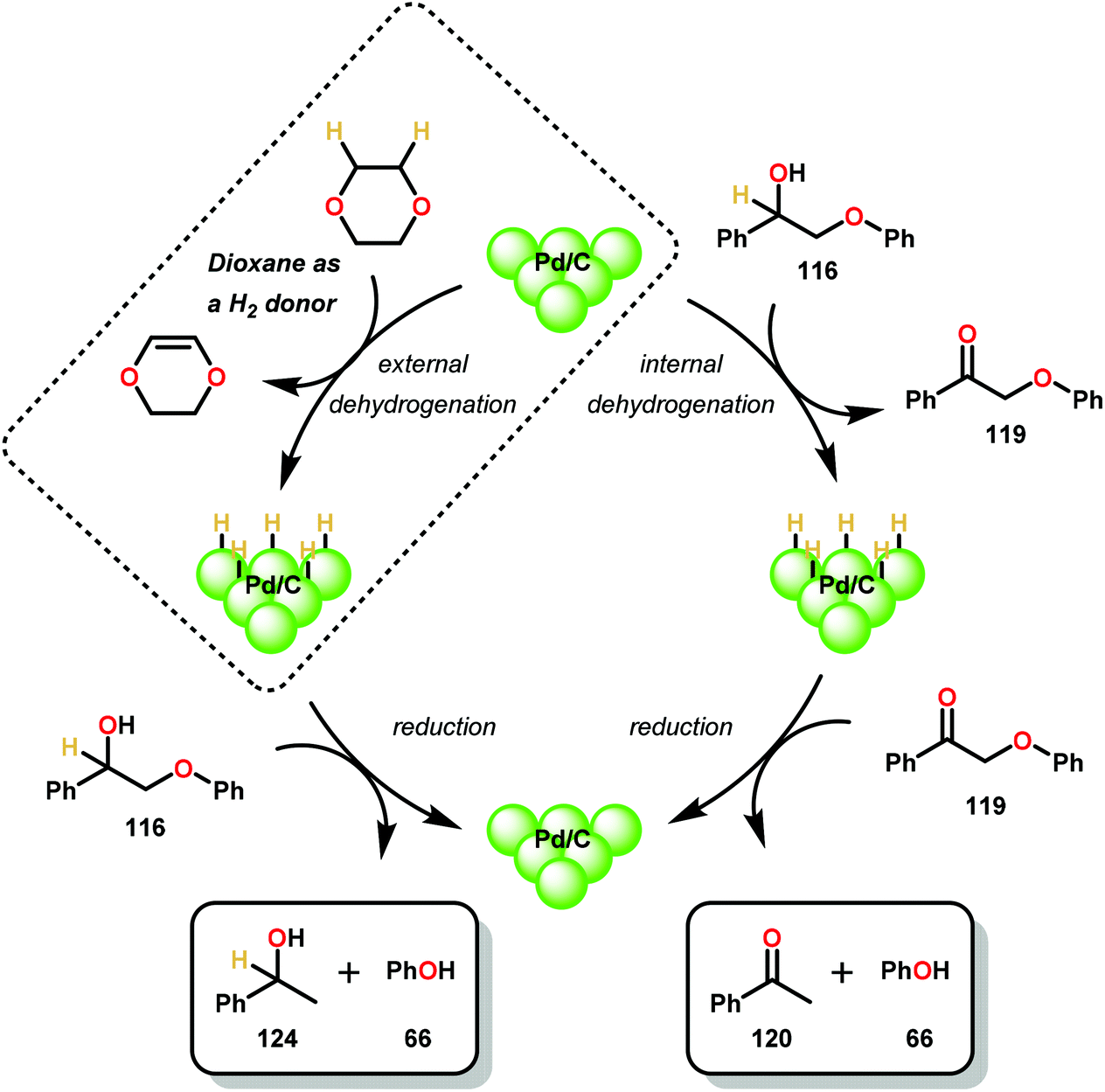
Similar approaches towards TOTH have been investigated by Rauchfuss and co-workers.86 Using Pd/C reduction, Rauchfuss and co-workers demonstrated the ability of ethereal solvents to donate hydrogen analagous to Rinaldi and co-workers.82 Yet, Rauchfuss found contradictory mechanistic results to Samec. For lignin motifs containing a primary benzylic alcohol unit, β-O-4 reduction was proposed to occur in one of two ways (Fig. 17). Firstly, the redox-neutral pathway of dehydrogenation and reduction could potentially occur, furnishing acetophenone (120) and phenol (31). Additionally, the same pathway could be reversed, whereby direct β-O-4 cleavage could occur by a Pd–H species (to produce phenol 31), followed by oxidation of 1-phenylethanol (124) to yield acetophenone (120). Unique to this investigation was the observance of the reduction of benzylic ethers. Conversion of these motifs proceeded in a net reductive fashion because the benzylic ether cannot initially be oxidized to the ketone. In this sense, Rauchfuss and co-workers observed selective β-O-4 reduction over α-O-4 reduction in the case of benzylic ethers containing a benzylic hydrogen, and selective α-O-4 reduction merely when the benzylic ether substrate contained an additional alkyl group at the benzylic position. Regardless of this, the lignin model systems were fully reduced to the corresponding phenol and alkyl arene components in these reactions.86 By showing that the reduction proceeded without the need of external hydrogen donors, the non-innocent role of dioxane in the reduction of lignin model systems containing both benzylic alcohol and benzylic ether motifs were demonstrated (Scheme 6).
 | ||
| Fig. 17 Pd/C catalyzed C–O bond cleavage using dioxane as hydrogen source. | ||
 | ||
| Scheme 6 Plausible mechanisms for Pd/C catalyzed C–O bond cleavage. | ||
2.5 Other metal-mediated methods for conversion of lignin related systems
While Ni and Pd have mainly been investigated with great promise towards conversion of lignin related systems through a combination of C–O bond activation or direct hydrogenolysis, other homogeneous metal catalysts have been probed for aryl ether C–O bond reduction. One such metal is Ti, which has been demonstrated to stoichiometrically cleave β-O-4 bonds through an enolate arlyoxide complex (127) of a model lignin substrate. Smith and co-workers designed this approach, targeting Ti for its oxophilicity and realizing that the vicinal oxygen functionalities of lignin could ligate and promote a Ti-mediated method. Using a TiCp2(BTSMA) complex (125; Cp = cyclopentadienyl; BTSMA = bis(trimethylsilyl)acetylene), Smith and co-workers observed the α-aryloxy ketone 126 to ligate and fragment at the β-O-4 bond. The identity of this TiCp2(enolate)(aryloxy) complex (127) was confirmed by a separate de novo synthesis and structural data was affirmed through X-ray crystal structure determination. Despite these results, the oxophilicity of TiIV proved too great to allow for catalytic turnover to be realized. Strong acids such as triflic acid were tested to promote ligand dissociation and generation of a TiCp2(OTf)2 complex, yet the aryloxy ligand was ultimately unwilling to undergo ligand exchange (Scheme 7).88 Although this method did not afford a catalytically viable procedure, further development in this area could address the issue of aryloxy disassociation and yield a TiIV catalyst capable of fragmenting lignin related compounds in a catalytic fashion. This scenario could perhaps be realized by identifying an ancillary ligand framework that can address the reactivity challenges identified in this study.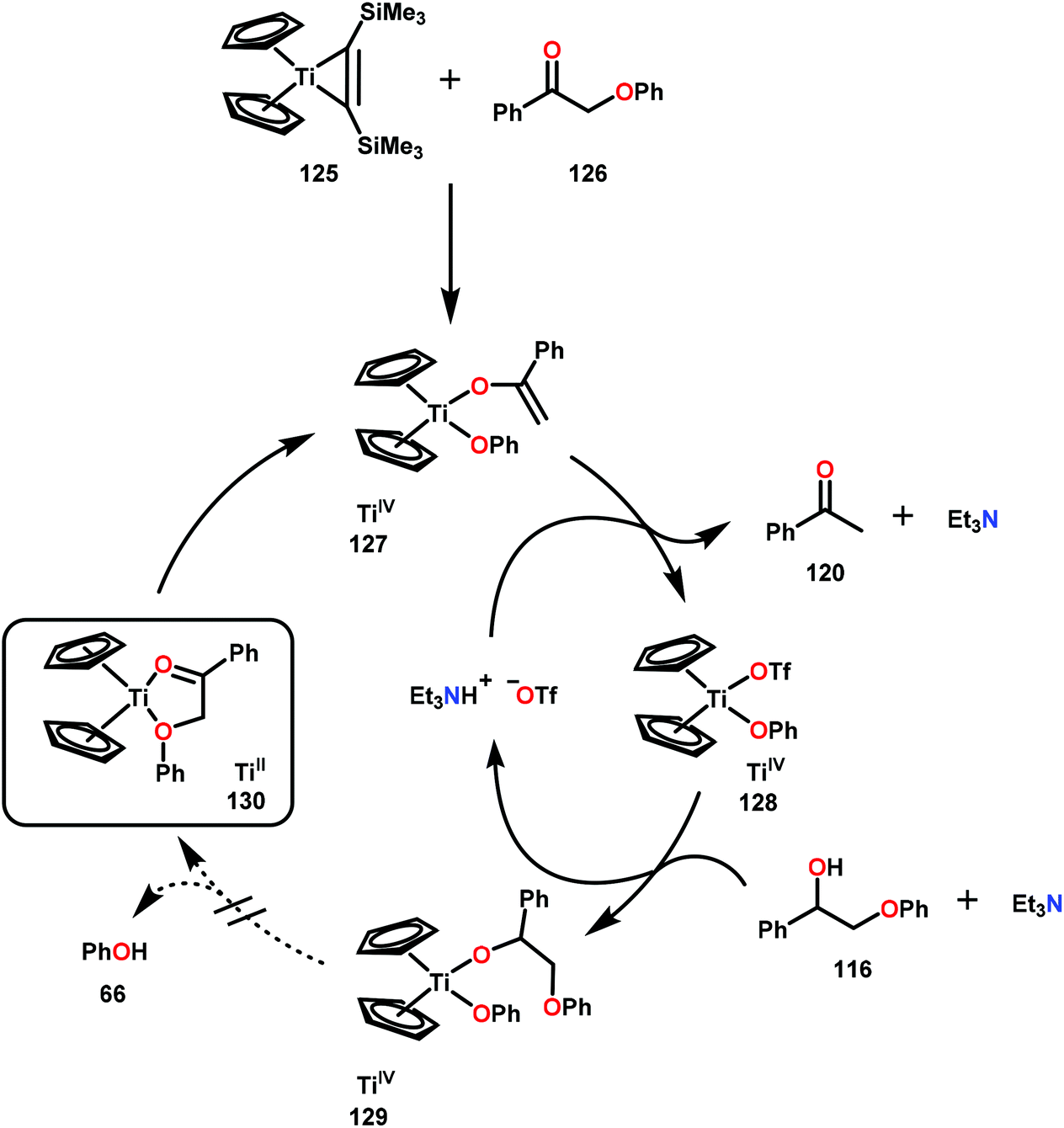 | ||
| Scheme 7 Titanium catalyzed C–O bond cleavage. | ||
Another abundant transition metal shown to reduce lignin model systems is Fe. Yao and co-workers recently demonstrated the reduction of diaryl ethers using Fe(acac)3 (acac = acetylacetonate) in combination with either LiAlH4 or H2 as a terminal reductant. Ligand selection proved crucial in the identification of an effective catalyst for this transformation. Yet, the active catalyst was found to be heterogeneous, as filtration of the reaction mixture ceased reaction progress.89 Additional support for a heterogeneous operating catalytic entity was determined from a mercury test,90 which poisoned the catalyst. Regardless, a combination of 20 mol% of Fe(acac)3 with 1 atm of H2 under basic conditions was sufficient to cleave the diaryl ether bond.89
The catalytic methods summarized thus far are all dependent upon inner-sphere coordination and transformation. A uniquely, outer sphere catalyst was developed by Nozaki and co-workers to avoid potentially insurmountable ligation issues encountered in inner-sphere catalysis. In doing so, Nozaki and co-workers identified a dihydridoiridium complex (131) capable of catalyzing arene deoxygenation, proceeding through an outer sphere mechanism. Utilizing the hydroxycyclopentadienyl dihydridoiridium complex 131, the authors observed this catalyst to facilitate the reduction of phenols and hydroxy napthalenes at high temperatures using low H2 pressures via metal–ligand cooperative hydrogen transfer (Fig. 18). Excellent yields were obtained for the more reactive naphthenol substrates, such as substrate 136, and the “conventional” reduced reactivity was observed with the phenol derivatives 132–135. This catalyst was proposed to operate through an intricate hydrogen-bonding network to effectively activate the aryl C–O bond for hydride reduction to yield benzene and water (Scheme 8).91 The relatively mild reaction conditions associated with this protocol together with a cooperative metal–ligand catalytic cycle can be of importance for future conversion of lignin to value-added products.
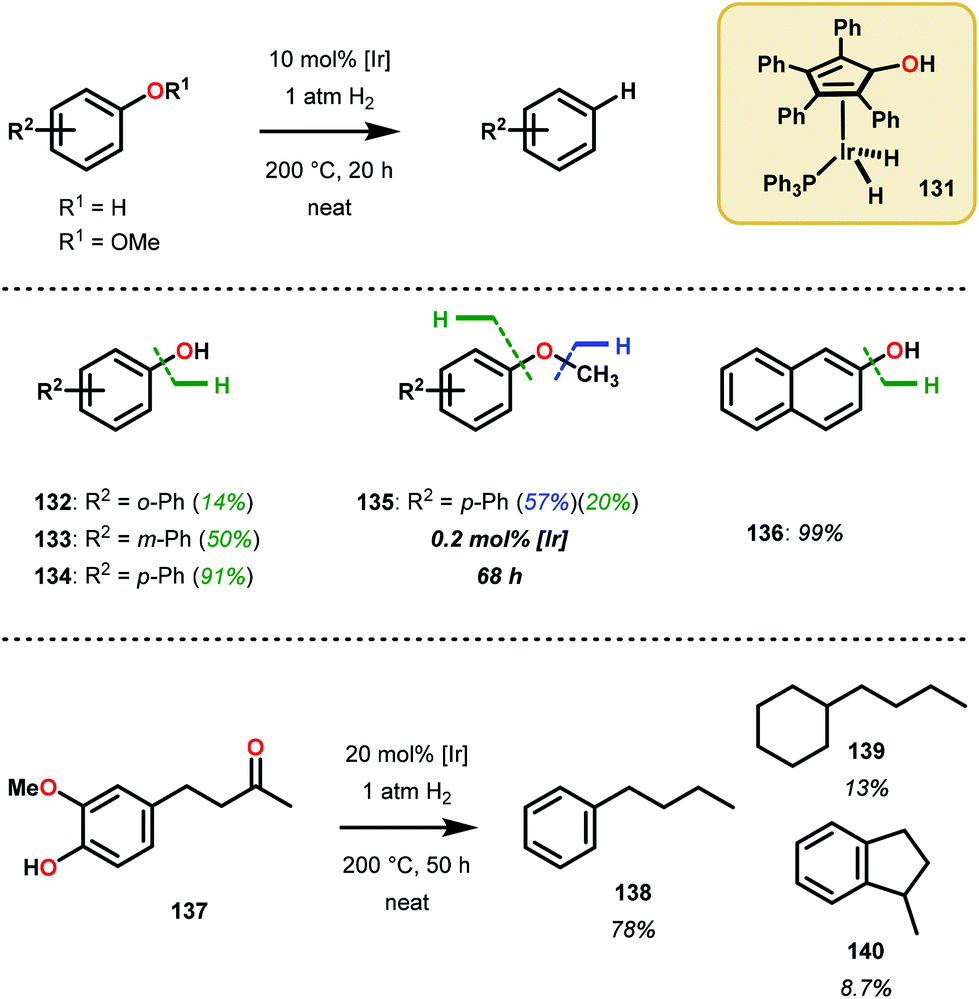 | ||
| Fig. 18 Ir-catalyzed hydrogenolysis of arenols and aryl methyl ethers. | ||
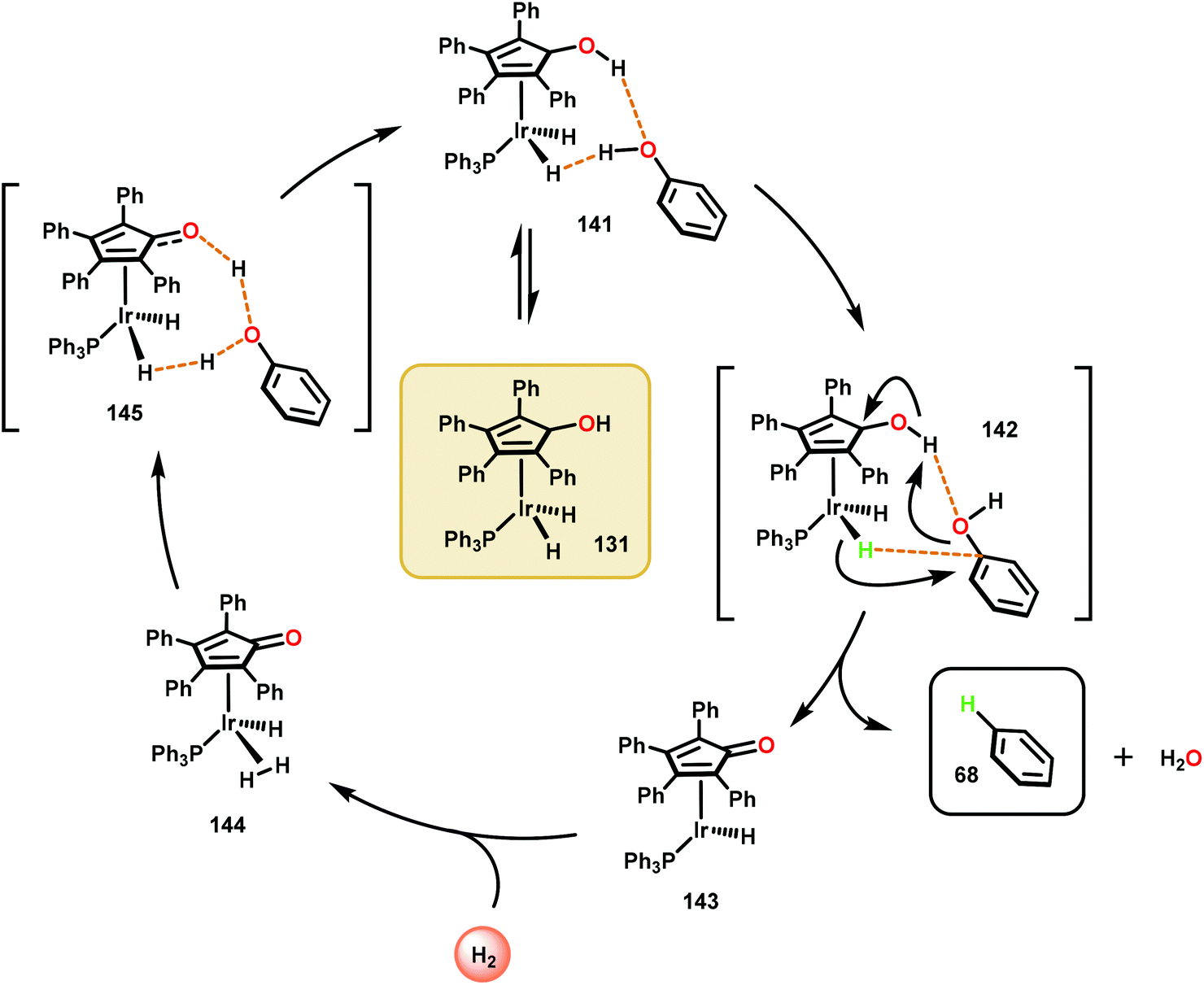 | ||
| Scheme 8 Proposed mechanism for the metal–ligand cooperative hydrogenolysis of phenol. | ||
2.6 Reductive cleavage of lignin model systems through photoredox catalysis
Recently, the emergence of photosensitive metal catalysts, and organic dyes, for organic redox transformations has attracted significant attention. One of the fascinating properties of photoredox catalysis (PRC) is its ability to engage in both oxidation and reduction chemistry from a visible light promoted photoexcited state (PC*, Fig. 19).92–97 Visible light-mediated PRC presents a number of advantages that makes it an attractive method of catalysis for lignin depolymerization: (1) PRC is operable at room temperature, compatible with aqueous–organic solvent mixtures, and insensitive to oxygen and water in select reactions, (2) PRC utilizes organic substrates for electron transfer, thus negating the need for stoichiometric transition metal additives traditionally used to reduce C–O bonds, and (3) PRC reacts via outer-sphere electron transfer; thereby avoiding detrimental ligation issues observed in other catalytic systems (see section 4). In this regard, PRC possesses the unique capacity to approach all three lignin depolymerization strategies (oxidative, reductive and redox-neutral) using a unified reaction design logic.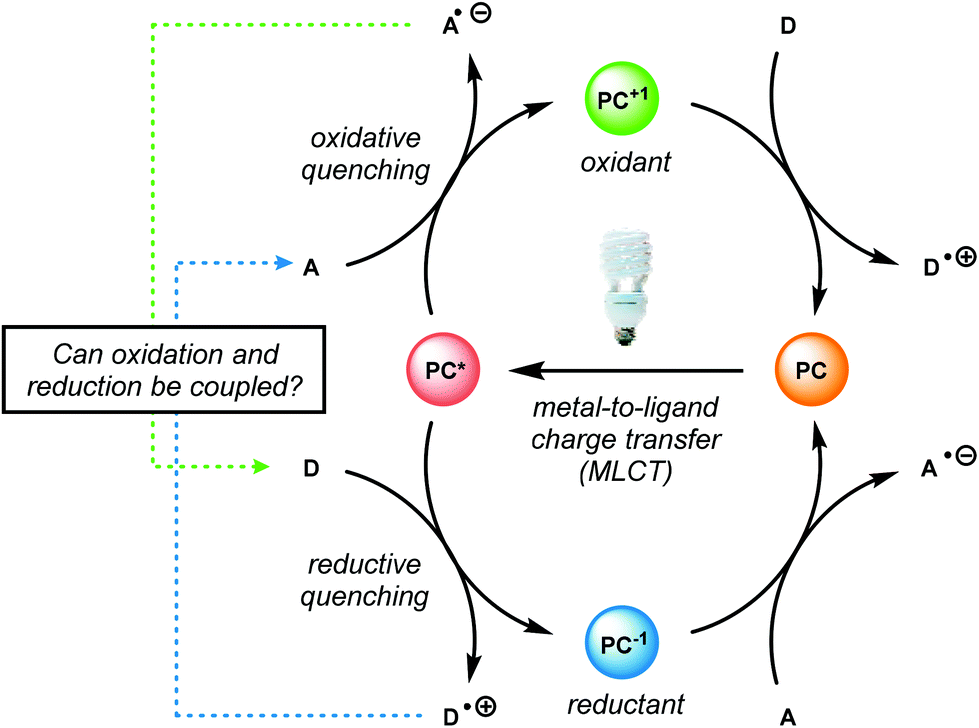 | ||
| Fig. 19 Oxidative and reductive quenching cycle of photocatalysts (PCs). A = acceptor. D = donor. | ||
While the polyether structure of native lignin does not present reactivity opportunity with PRC, Stephenson and co-workers drew inspiration from the works of Hasegawa98 and Ollivier99 to develop a β-O-4 reduction. In these reports, simple phenyl keto-epoxides were reduced to afford β-hydroxy ketones. In order to employ this reactivity on model lignin substrates, Stephenson and co-workers identified a selective benzylic oxidation using 4-(acetylamino)-2,2,6,6-tetramethyl-1-oxo-piperidinium tetrafluoroborate (Bobbitt's Salt,100 [4-NHAc–TEMPO]BF4) to obtain the necessary phenyl ketone motif.101 Interestingly, the benzylic oxidation concomitantly formed a reducible PRC functionality (phenyl ketone), decreasing the β-O-4 bond strength by ∼10 kcal mol−1 (see Fig. 35).102 While this approach was not a one-pot redox neutral sequence, batch oxidation using Bobbitt's salt was shown to oxidize the lignin model systems in an efficient and operationally simple manner. Additionally, Bobbitt's salt proved ideal, as it is recyclable by stoichiometric oxidation with store bought bleach.100
Subsequently, the β-O-4 reduction was realized with catalytic [Ir(ppy)2(dtbbpy)]PF6 (146), three equivalents of N,N-diisopropylethylamine (DIPEA) and formic acid. Under these conditions a variety of model lignin substrates were observed to fragment (Fig. 20). This substrate scope supported the mechanistic hypothesis of fragmentation to give a carbon radical (157) and an oxyanion (158), as the substrate reactivity correlated with the pKa values of the leaving group. Additionally, this method was selective for phenyl ketones and aldehydes, as the corresponding aliphatic ketones and esters could not be reduced under the employed reaction conditions (see Fig. 20). Interestingly, the commercially available benzyloxyacetaldehyde (152) underwent efficient reductive cleavage, yielding benzyl alcohol in 83% isolated yield. Not only does this demonstrate that aldehydes were sufficient electrophiles for this reaction but also that these reaction conditions are amenable to the reduction of stronger C–O bonds of unactivated ethers. This result suggests that this chemistry has potential application in the degradation of other biomass derived polymers such as polysaccharides. Substrates 147, 148, and 149 are model compounds for coumaryl, coniferyl, and sinapyl β-O-4 linkage motifs commonly found in lignin. These substrates were found to effectively undergo reductive fragmentation yielding the β-hydroxyl ketones in 85%, 85%, and 90% yield, respectively. These generated hydroxyketones were stable under the reaction conditions and did not undergo significant side reactions such as retro-aldol, pinacol coupling or oxidation.101 They are also unique fragmentation products which had only previously been observed in minute quantities.
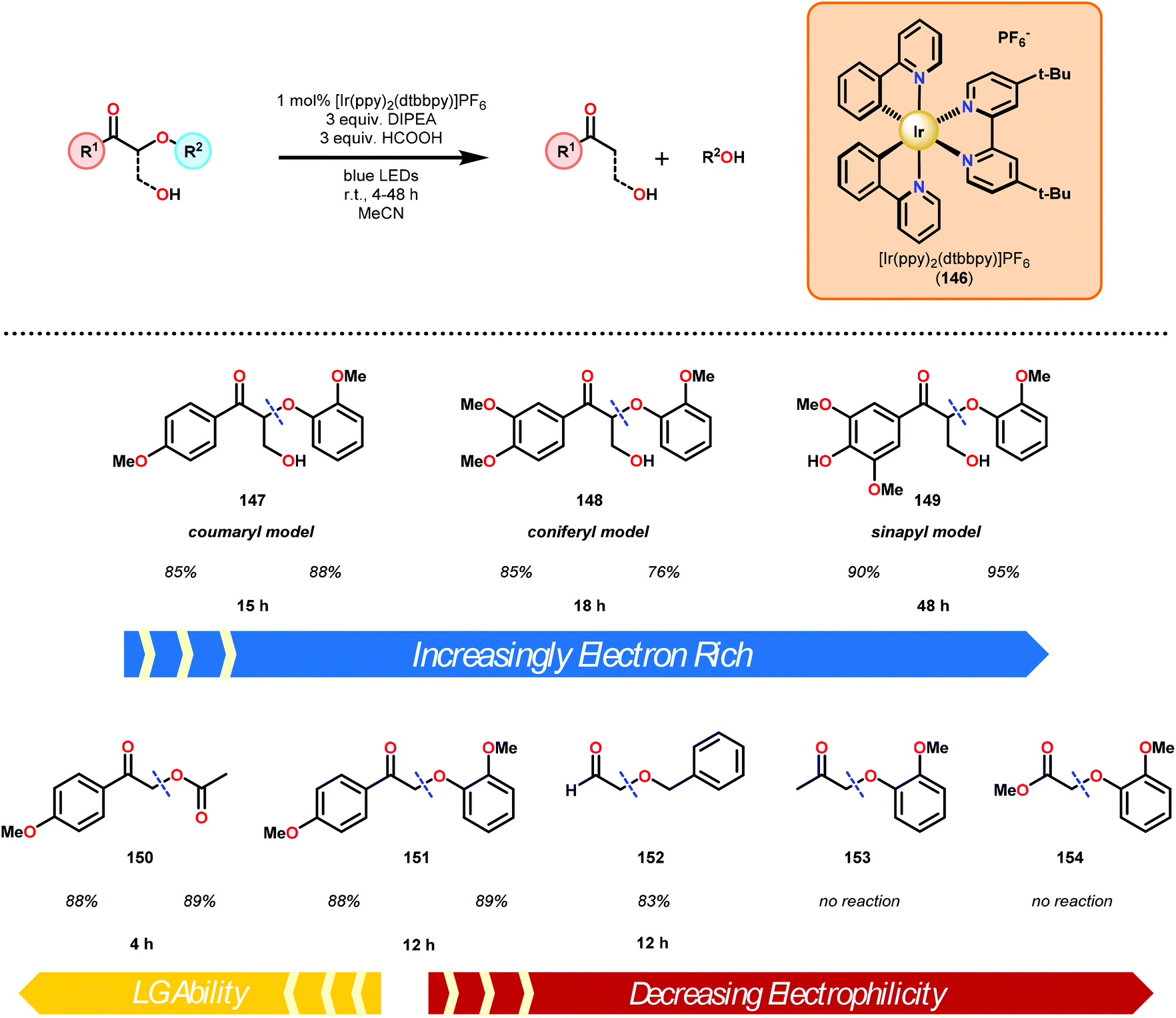 | ||
| Fig. 20 Visible-light-mediated C–O bond cleavage of oxidized lignin model systems. | ||
Consistent with the corresponding electron transfer reduction literature,103–109 Stephenson and co-workers proposed a reaction mechanism in which the reaction is initiated by visible-light excitation of the IrIII photocatalyst (146) to generate an excited state, IrIII*. From this excited state, the photocatalyst is reductively quenched by the single-electron oxidation of DIPEA. The generated IrII species is now an excellent reductant and reduces the oxidized lignin model substrates. Electron transfer most likely proceeds through the carbonyl functionality, generating an anionic ketyl radical (156) which can intramolecularly fragment to afford the corresponding α-keto radical (157) and phenoxide anion (158). Subsequent hydrogen atom transfer (HAT) to the α-keto radical generates the desired ketone (159) and completes the catalytic cycle (Scheme 9).
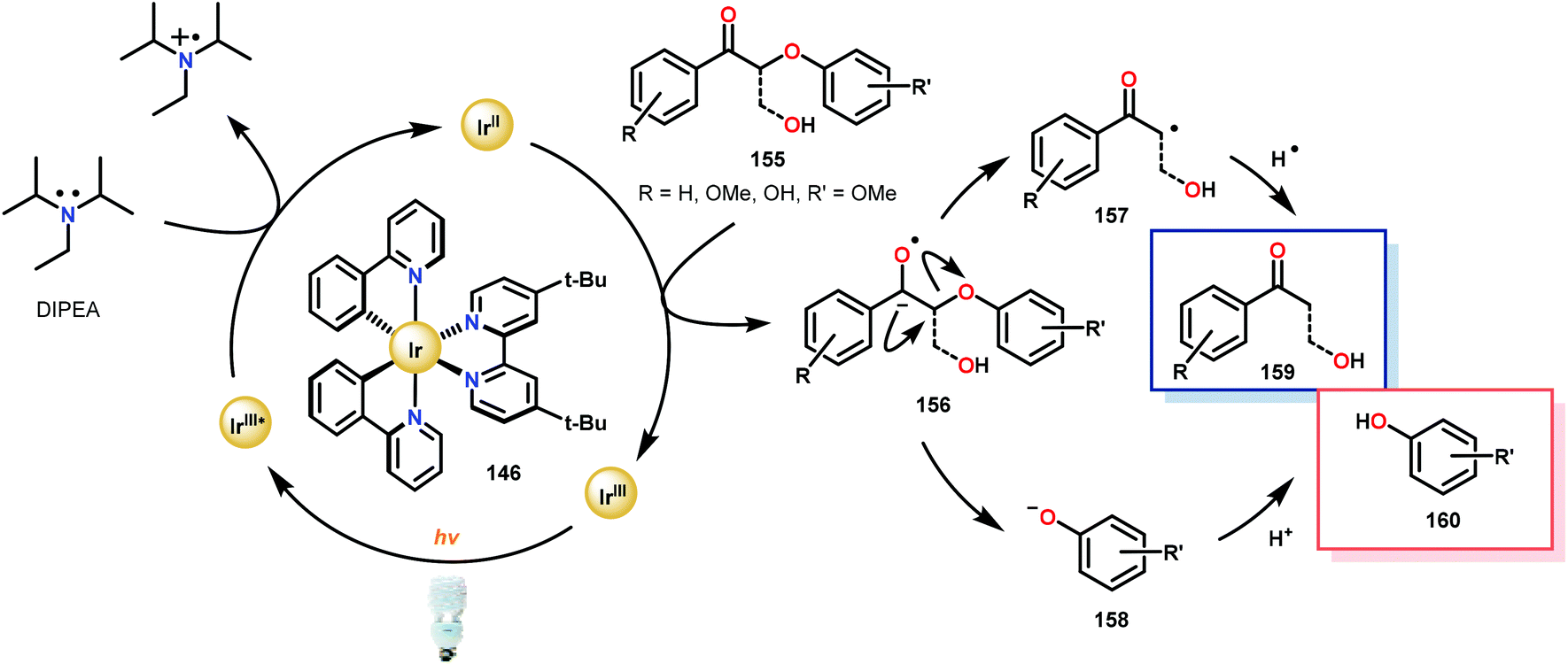 | ||
| Scheme 9 Proposed mechanism for the photocatalytic reductive cleavage of oxidized lignin model systems. | ||
With the success of the two-step batch-to-batch process, Stephenson and co-workers were interested in exploring the reaction within a continuous flow reactor. Indeed a simple batch-to-flow protocol where the oxidation occurred in batch and the photochemical reduction occurred within a continuous flow reactor effectively caused the β-O-4 cleavage to occur.101 Previous reports by the groups of Stephenson and Seeberger had demonstrated that the use of a flow reactor can greatly accelerate photocatalyzed reactions due to an efficient irradiation of the reaction solution.110,111 Thus, performing the reaction in the flow reactor greatly accelerated the reaction rate when compared to the analogous batch-to-batch reaction.101 Collectively, this batch-to-flow processing of lignin model substrates demonstrated the potential for future photoredox technologies for biomass conversion applications.
3. Oxidative conversion of lignin
From the previous chapter it is obvious that several methods have been investigated for the reductive transformation of lignin and lignin model systems. Another approach that has been widely explored is the oxidative conversion. In nature, there are several species of white-rot fungi that are capable of degrading lignin through the action of peroxide dependent ligninase or laccase.112,113 These enzymes work through the single-electron oxidation of the aromatic groups found in lignin, generating arene radical cations, such as radical 162 (Scheme 10) upon oxidation of 161. This reactive intermediate can either undergo α–β cleavage, causing a break in the polymeric backbone yielding the corresponding aldehyde 166 and radical 167. Alternatively, deprotonation of Hα from intermediate 162 generates ketyl radical 163, which can undergo β-O-4 fragmentation to yield β-hydroxyketone 164 or undergo direct oxidation to the benzylic ketone 165. Under these oxidative conditions, the cleaved products readily undergo further oxidation chemistry yielding carboxylic acid (169) and quinones (170). The relatively stable radical cations formed during arene oxidation by enzymes can also undergo reversible intramolecular or intermolecular electron transfer. This charge transfer phenomena allows the ligninase to initiate degradation at multiple sites within the polymer matrix despite having low surface contact with the substrate. The overall delignification process is relatively slow, taking many months to fully degrade higher molecular weight lignin, since the depolymerization process occurs on the heterogeneous surface of the wood itself. Despite the low contact surface area the fungi have to degrade lignin, especially in the early stages of wood decay, the depolymerization of lignin occurs efficiently.114–116 | ||
| Scheme 10 Depolymerization mechanism of lignin by delignifying enzymes. Nu = nucleophile. | ||
3.1 Biomimetically inspired catalysts for oxidative fragmentation of lignin model systems
Early reports on the oxidative degradation of lignin model systems centered on the use of biomimetic catalysts, such as metal porphyrins.117 Analogous to the hemo-protein ligninase, isolated from the white-rot fungus, which are enzymes that catalyze the oxygenative C–C bond cleavage of lignin model systems,112,113 Shimada and co-workers reported on the first example of an unprecedented C–C bond cleavage reaction of a lignin model compound.118 It was found that employing tetraphenylporphyrinatoiron(III) chloride (177) as a biomimetic catalyst, 1,2-bis(4-ethoxy-3-methoxyphenyl) propane-1,3-diol (171) was oxidatively cleaved to produce a variety of products where 4-ethoxy-3-methoxybenzaldehyde (172) was found to be the major product (Scheme 11).118 Although this study showed that it was possible to mimic the function of the lignin degrading enzymes, the oxidation of model system 171 was not selective and resulted in an array of products.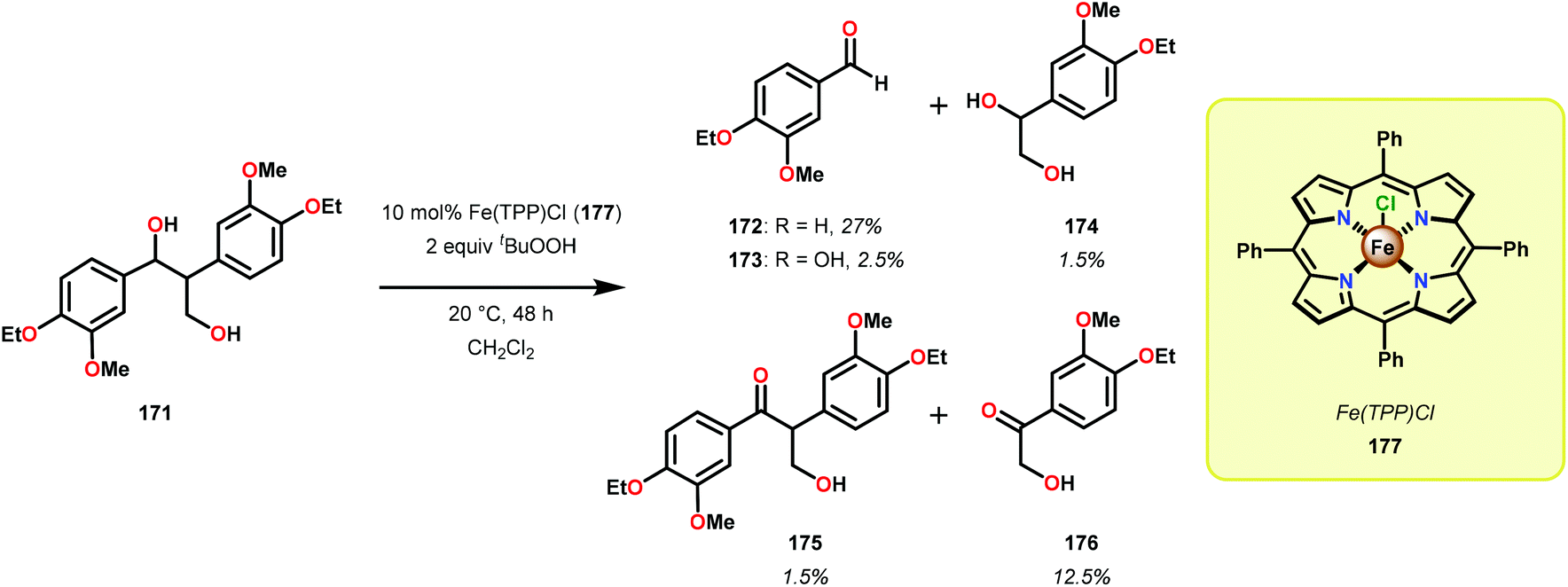 | ||
| Scheme 11 Oxidation of lignin model system 171 by Fe(TPP)Cl (177). | ||
Further studies by Shimada, Higuchi and co-workers explored the use of hemin, a Fe–porphyrin complex, to study the Cα–Cβ bond cleavage of model compound 178.119 It could be established that hemin (181) efficiently degraded model system 178 at ambient temperature in the presence of 5 equivalents tBuOOH to afford aldehyde 179 as the major product, along with the hydroxylated product 180 (Scheme 12). Diol 180 was also shown to undergo Cα–Cβ bond cleavage to yield aldehyde 179.
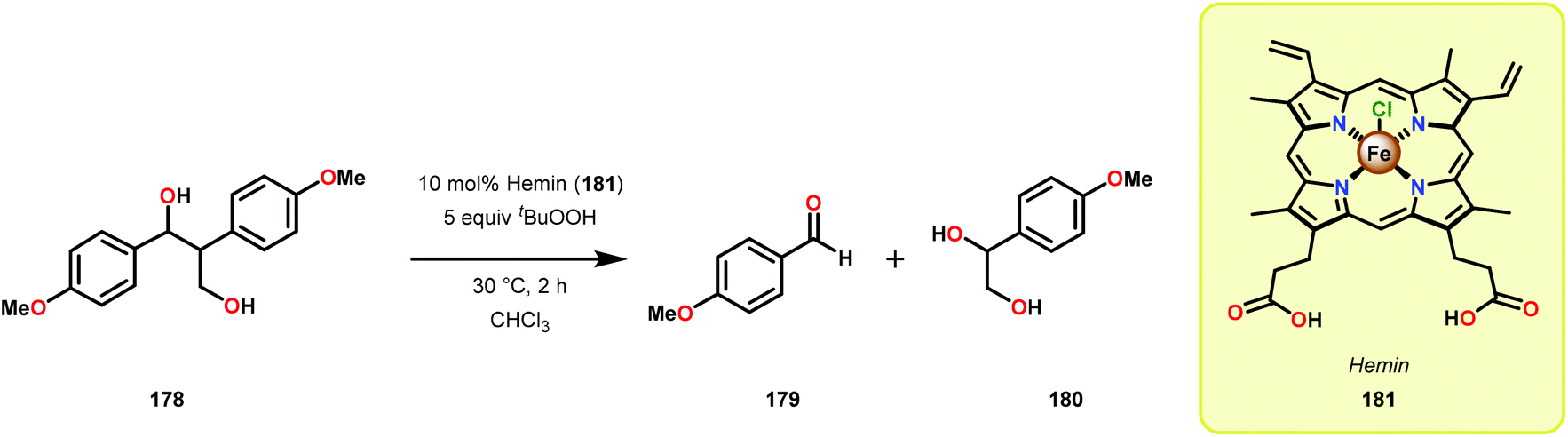 | ||
| Scheme 12 Oxidative Cα–Cβ bond cleavage of lignin model system 178 by hemin 181. | ||
A one-electron transfer mechanism was invoked in these Cα–Cβ bond cleavage reactions where the catalytically active intermediate was proposed to be an FeV-oxo species. It was proposed that a one-electron abstraction by FeV-oxo from the substrate generates a radical cation, which fragments in a manner analogous to 162 (cf. Scheme 10).119,120 The proposed mechanism is in good accordance with earlier C–C bond cleavages observed for electrochemical oxidations121 and ligninase.122 A related report by Crawford and co-workers made use of a variety of FeIII porphyrins to study the delignification of wood pulps. It could be demonstrated that the treated wood chips were extensively delignified and examination of the treated chips by transmission electron microscopy revealed morphological alterations similar to that of white rot fungi.123
In a related study, Meunier and Labat studied the conversion of 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol (61) for both homogeneous and resin-immobilized Fe and Mn derivatives of sulfonated tetraphenylporhyrins (TPPS) (182–186, Fig. 21). It could be established that the homogeneous catalysts displayed higher catalytic activity compared to the corresponding immobilized catalysts. Here the influence of pH was also investigated and the authors found that the Fe catalysts were more efficient at pH < 3 while an opposite effect was observed for the Mn(TPPS) catalyst (183), which showed an optimum between pH 4.5 and 6.0. For the Fe(TPPS) catalysts, it was reasoned that the acidic medium favored the cleavage of the inactive μ-oxo Fe porphyrin dimers, which are known to be generated during the oxidations, and represent a deactivation pathway for these Fe complexes.124 Crestini and co-workers,125 and Dolphin and co-workers126–128 have also used cationic and anionic water soluble metal porphyrin complexes (Fig. 21) for the degradation of lignin model systems. Additional investigations on immobilized metal porphyrins as lignin-peroxidase biomimetic catalysts include the studies by Sanjust129,130 and Crestini.131,132
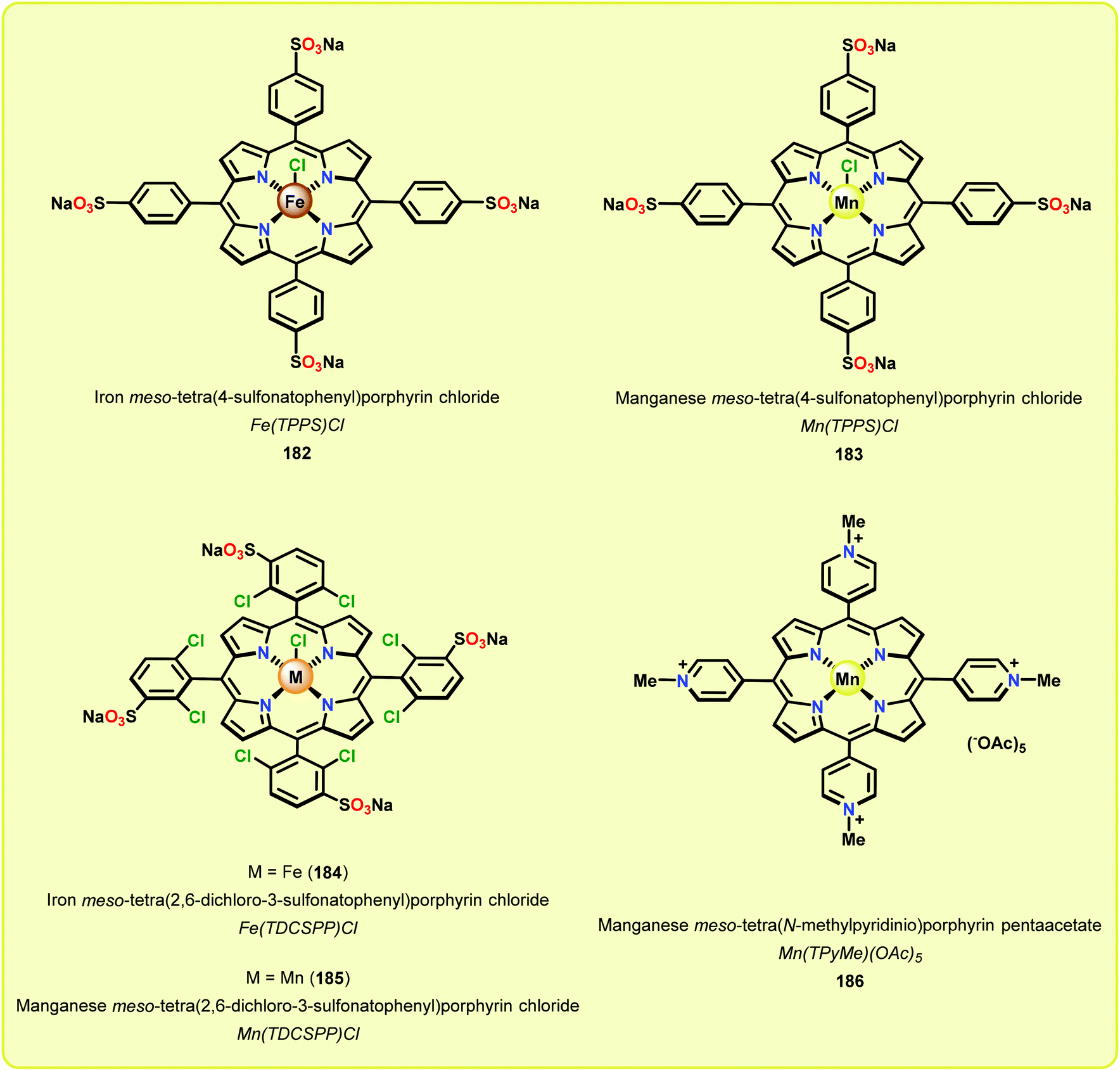 | ||
| Fig. 21 Water soluble porphyrin complexes 182–186 employed for degradation of lignin model systems. | ||
Although the early examples on the use of metal porphyrins resulted in unselective degradation of lignin model systems and rather low yields of the desired products, a recent study by Ying and co-workers highlighted that further progress in this area is achievable. Ying and co-workers employed metallo-deuteroporphyrins (Fig. 22) as biomimetic catalysts for the oxidative depolymerization of lignin model systems. Although the corresponding Fe- and Mn-based deuteroporphyrin complexes were not efficient in catalyzing the oxidation of the lignin model system, the Co-deuteroporphyrins were shown to efficiently convert the lignin model system in the presence of Oxone® (2 KHSO5·KHSO4·K2SO4).133
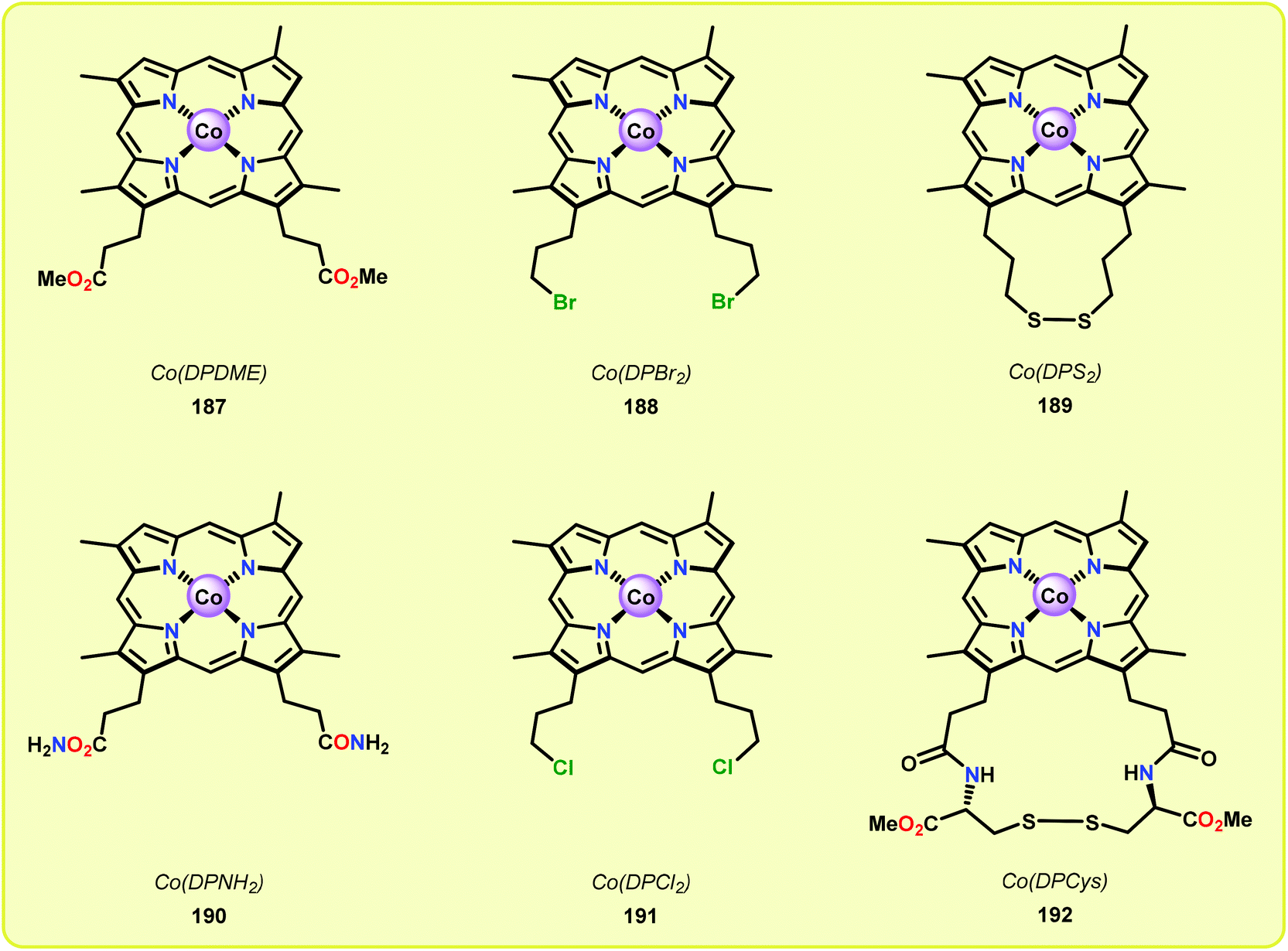 | ||
| Fig. 22 Structure of Co deuteroporphyrins 187–192. | ||
When using the Co(DPCys) catalyst 192 on phenolic lignin model substrate 161, full conversion was observed within 15 min at room temperature. This reaction yielded the three compounds 193, 194 and 62, where product 193 was obtained in 71% yield (see Scheme 13). In the absence of the Co-catalyst, 91% of the starting material was recovered, highlighting the need for the catalyst. The proposed mechanism for the formation of products 194 and 62 is depicted in Scheme 14. These products were proposed to form through the activated CoIV-oxo species, which initially abstracts the benzylic hydrogen atom of substrate 161, resulting in a CoIII-bound ketyl radical species (195). Elimination of a phenoxyl radical (197) generates CoIII-hydroxo intermediate 196, which is subsequently dehydrated to liberate product 194 and a CoIII-hydroxo species. This CoIII-hydroxo is concomitantly oxidized by the phenoxyl radical (197), which regenerates the high-valent CoIV-oxo catalyst and closes the catalytic cycle.133
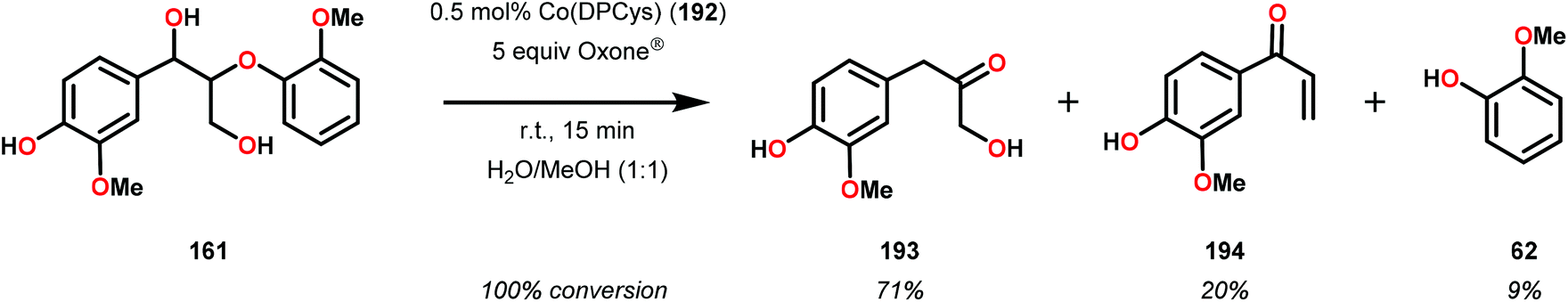 | ||
| Scheme 13 Oxidative degradation of lignin model system 161 by the Co deuteroporphyrin catalyst Co(DPCys) (192). | ||
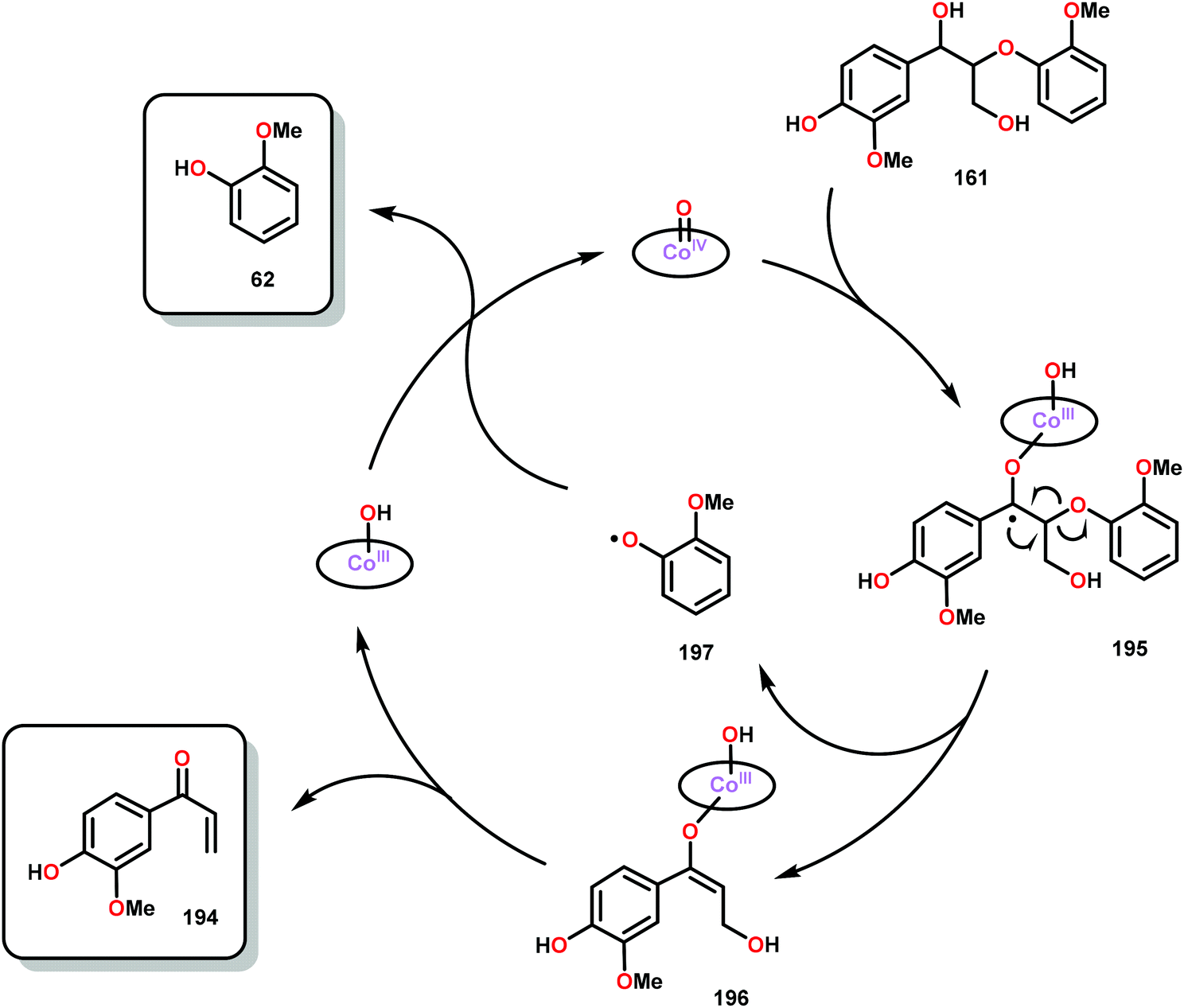 | ||
| Scheme 14 Proposed mechanism for the Co deuteroporphyrin catalyzed formation of products 194 and 62 from substrate 161. | ||
Attempts of employing the Co(DPCys) catalyst 192 on enzymolysis lignin (i.e. lignin obtained after enzymatic hydrolysis) revealed that almost no conversion occurred at room temperature. The temperature had to be increased to 120 °C to significantly improve the conversion and under optimized conditions, 31.2 wt% of discrete monomeric aromatic products could be obtained. The authors also investigated if the catalytic procedure could be applied on other types of lignin. When applying the catalytic protocol on organosolv lignins, such as ethanosolv lignin from bagasse, dioxasolv lignin from bagasse and ethanosolv lignin derived from pine sawdust, it was found that the source of the lignin had a dramatic effect on the conversion and the distribution of products. Although the solvent employed in the organosolv derived lignin from bagasse did not significantly alter the outcome of the Co-catalyzed degradation, the use of grass biomass lignin and ethanosolv lignin derived from pine only resulted in low yield of monomeric aromatics.133 These findings strongly imply that the source of the lignin has a crucial impact on the studied catalytic system.
It should also be mentioned that water soluble metallophthalocyanines (Fig. 23) have also been evaluated as lignin peroxidase models for the oxidative conversion of simple lignin model systems.134,135 However, these complexes were in general found to suffer from oxidative degradation and have therefore not received any widespread attention.
 | ||
| Fig. 23 Water soluble metallophthalocyanines (198–202) employed for studying oxidative conversion of lignin model systems. | ||
3.2 Cobalt-Schiff base complexes for catalytic oxidative degradation of lignin model systems
Analogous to metalloporphyrins and metallophthalocyanines, Co-Schiff base complexes are well-known to activate O2136,137 and have been used to catalyze a plethora of transformations.138 Co-Schiff base complex (203) and related complexes reversibly react with O2 to give an equilibrium mixture of the Co-superoxo species (204) and the dimeric peroxo species (205) (Fig. 24), where the equilibria of these species is dependent on the nature of the Schiff-base complex and the specific reaction conditions.139 The efficiency of activating O2 by Co-Schiff base complexes may be strongly dependent on the coordination environment around the Co center, where the four-coordinated Co–salen complex 208 (Fig. 25) binds O2 poorly at room temperature, thus resulting in a low concentration of the Co-superoxo species 204. On the other hand, the presence of a donor ligand in the axial position stabilizes the Co–O2 bond, making the five-coordinated Co-Schiff complexes, such as 206 and 207 (Fig. 25), more efficient in binding O2.140
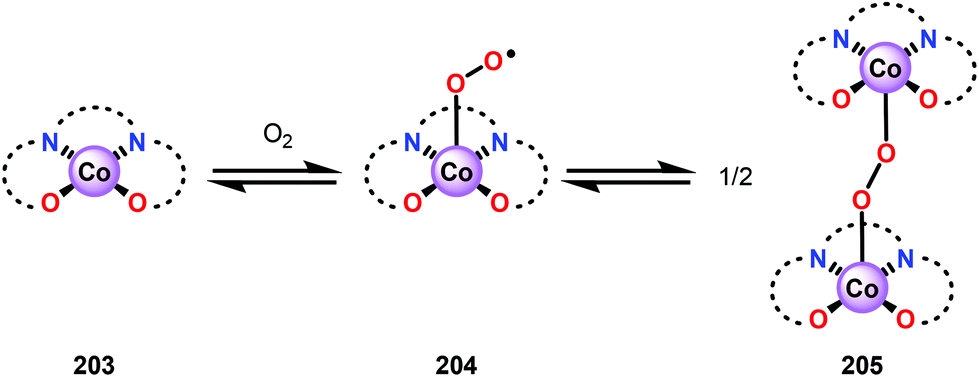 | ||
| Fig. 24 Activation of O2 by Co-Schiff base complexes. | ||
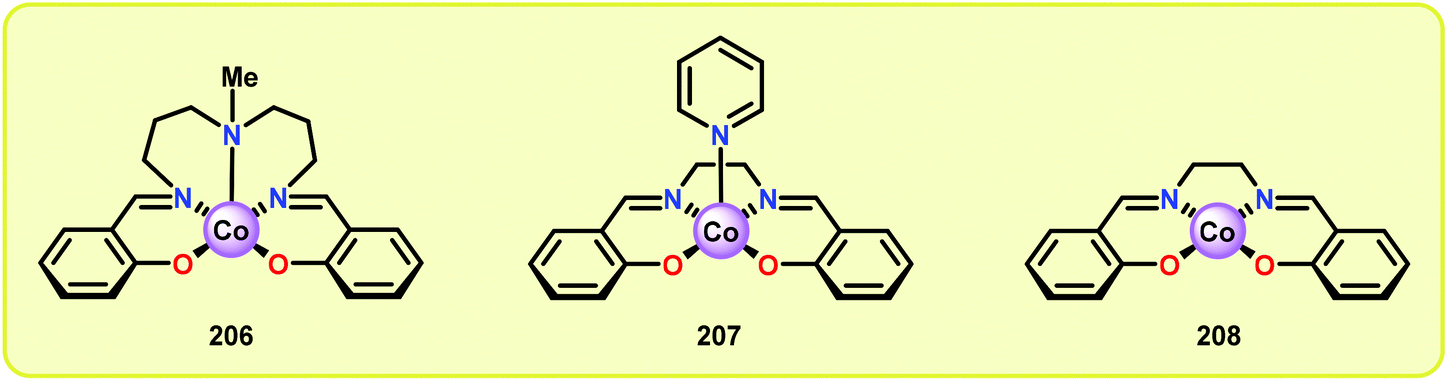 | ||
| Fig. 25 Co-Schiff base complexes used for oxidative degradation of lignin model systems. | ||
Early support suggested that Co-Schiff base complexes could be used for the oxidative conversion of lignin-like model systems.141 Bozell and co-workers found that treatment of syringyl alcohol (209) with 10 mol% of the five-coordinate Co-Schiff base complexes 206 and 207 (Fig. 25), in the presence of O2, afforded 2,6-dimethoxybenzoquinone (210) in 71% and 86% yield, respectively (Scheme 15). Competitive deactivation of the Co catalyst was found to occur with substrates of lower reactivity. It was also established that guaiacyl-type model substrates only afforded <20% of the methoxybenzoquinone product, probably because of the reduced ability of these model substrates to generate the essential intermediary phenoxyl radical.142 Related studies have also been conducted by Canevali,143 Sippola144 and Repo145–147 where Co(salen)-type catalysts such as complex 208 was employed as the oxygen-activating catalyst. Heterogeneous systems where Co–salen was immobilized on SBA-15 have also been employed for the oxidative conversion of lignin model systems.148,149
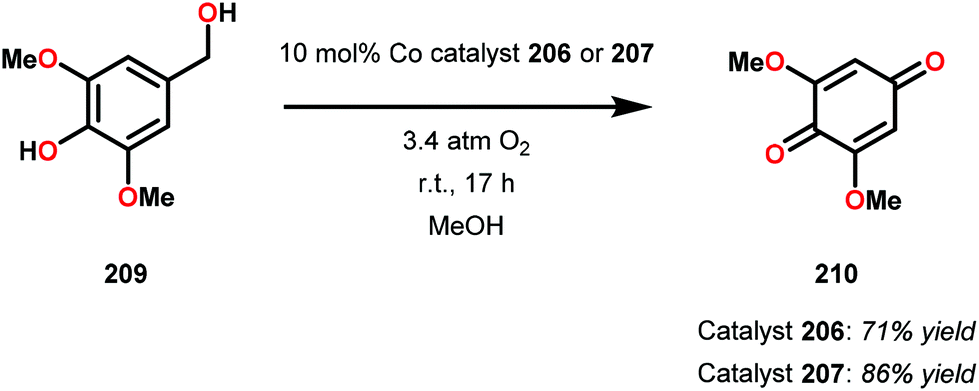 | ||
| Scheme 15 Aerobic oxidation of syringyl alcohol 209 to 2,6-dimethoxybenzoquinone (210) catalyzed by Co-Schiff base complexes 206 and 207. | ||
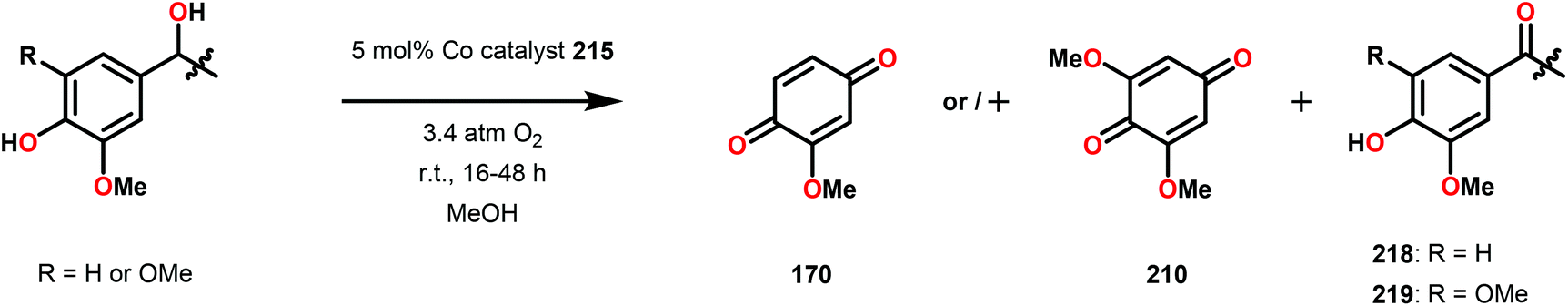
Bozell and co-workers subsequently discovered that the stoichiometric addition of a sterically encumbered aliphatic nitrogen base could yield the methoxybenzoquinone product in 51% yield.150 The authors therefore designed a new family of unsymmetrical Co-Schiff base catalysts that housed a sterically hindered base within the ligand scaffolds (Fig. 26).151,152 Several of the unsymmetrical catalysts were capable of oxidizing syringyl alcohol 209 to afford 2,6-dimethoxybenzoquinone 210 in good yield. It was established that the unsymmetrical Co-Schiff base complexes did not require the addition of an external axial ligand. Of the synthesized catalysts, N-benzyl catalyst 215 displayed the highest activity, giving 2,6-dimethoxybenzoquinone 210 in 74% yield after only 1 h. After identifying catalyst 215 as the optimal catalyst, several lignin model systems containing a phenolic OH were evaluated under the optimal conditions (Table 1). In general, high yields of the corresponding benzoquinone products were obtained through conversion of the studied lignin models.151
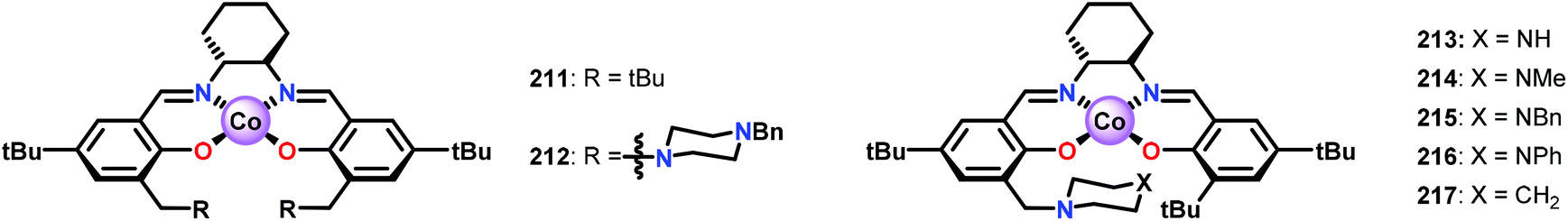 | ||
| Fig. 26 Symmetrical (211 and 212) and unsymmetrical (213–217) Co-Schiff base complexes developed for studying the oxidative conversion of lignin models. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (h) | Yielda (%) | ||
| 170 | 210 | 218/219 | |||
| a Isolated yields. b 10 mol% of Co catalyst used. c Yields relative to 2 equivalents of product formed. d 14% 2,5-dimethoxybenzoquinone was isolated. e 30% aldehydes was observed. | |||||
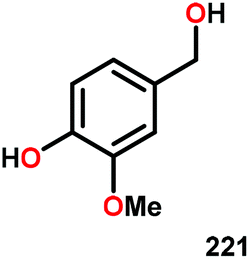
| 1 |
|
16 | — | 72 | 11 |
| 2 |
|
16 | 83 | — | — |
| 3b |

|
24 | 51 | — | 22 |
| 4c |
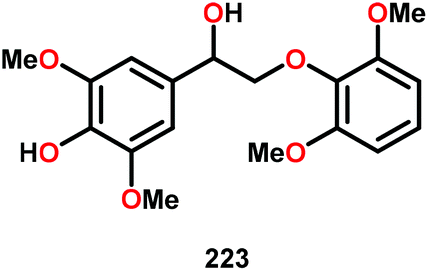
|
16 | — | 81 | Traces |
| 5 |
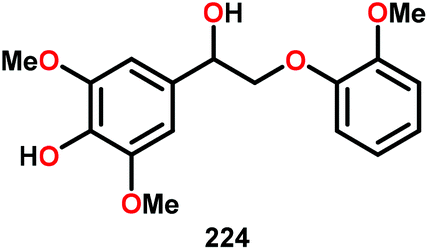
|
16 | 17 | 86 | Traces |
| 6b |
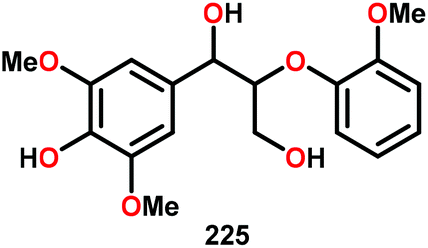
|
48 | 21 | 64 | Traces |
| 7b |
|
48 | 0d | 10 | 0e |
The Co-based system was subsequently applied on organosolv lignin, derived from tulip poplar. Treating this lignin with N-benzyl catalyst 215 and O2 for 72 h at room temperature produced 2,6-dimethoxybenzoquinone 210 as well as benzaldehydes.151 However, the absolute yields of monomeric aromatic species from this reaction were rather low, merely 3.5%, highlighting the difficulties when attempting to implement the developed catalytic systems on native lignin.
3.3 Vanadium-catalyzed aerobic oxidation of lignin model systems
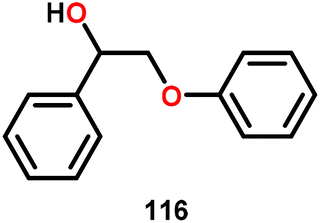
Vanadium complexes have proven to be useful catalysts in various oxidative transformations. Oxidative cleavage of C–C bonds in 1,2-diols has been investigated with various transition metals including V,153 Ru,154,155 and Mn.156,157 However, less is known about the oxidative C–C bond cleavage of 1,2-hydroxyethers. A variety of V complexes are able to affect oxidative transformations, and their reactivity can be greatly modified depending on the substrate and ligand or if reacted under photoirradiation, aerobic or anaerobic conditions.158–160Inspired by the reactivity associated with V complexes, Baker and co-workers investigated the aerobic oxidation of four different 1,2-hydroxyether model systems (116 and 227–229, Fig. 27) using a base metal V catalyst, VV(pda)(O)(OiPr) (230; H2pda = pyridine-2,6-dicarboxylic acid). These substrates allowed the authors to elucidate the effects and reactivity of the β-O-4 and β-1 lignin motifs, and the effects of the substituted backbones.161
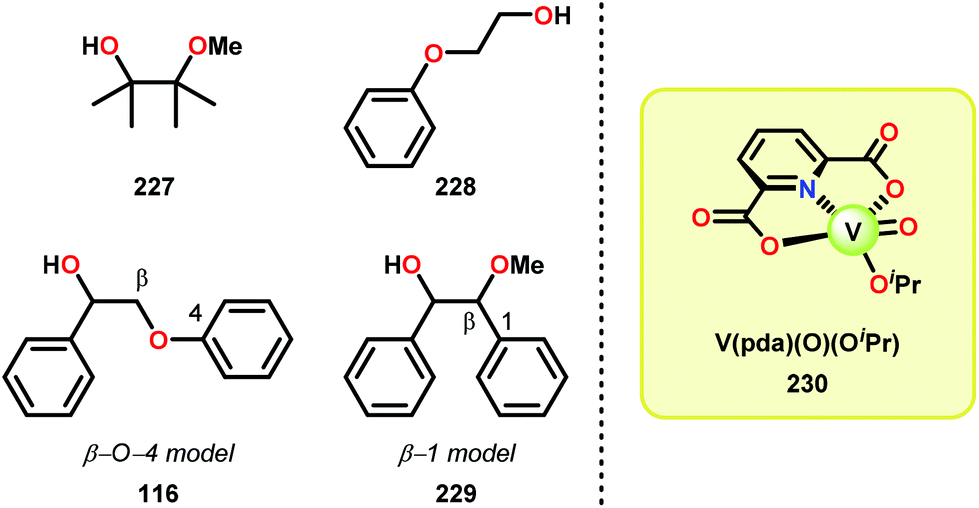 | ||
| Fig. 27 (Left) Model systems 116 and 227–229 and (right) V complex 230. | ||
Initial studies consisted of using stoichiometric amounts of V complex 230. When complex 230 was reacted with the different model systems, new VV complexes were isolated in good yields in which the alcohol–ether ligand was bound in a chelating fashion.161 Heating the pinacol monomethyl ether complex 231 in pyridine-d5 solution (6 h at 100 °C, or 3 weeks at 25 °C) the previously reported VIV complex (232) was generated,162 alongside acetone (233), 2-methoxypropene (234), and pinacol monomethyl ether (227) (Scheme 16).161
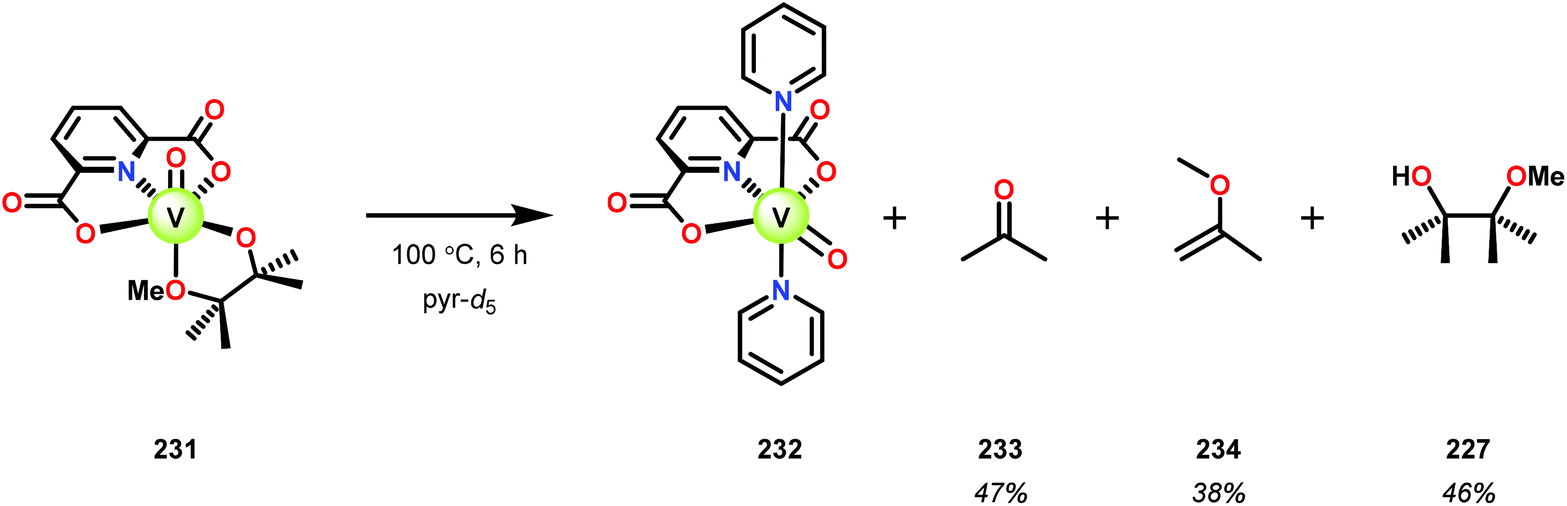 | ||
| Scheme 16 Stoichiometric C–C bond cleavage by V complex 231. | ||
When substrates with secondary C–H bonds were subjected to these conditions, C–H bond cleavage was observed when using stoichiometric amounts of the V complex. Heating V complex 235 in pyridine-d5, afforded aldehyde 236 and the original lignin model (228) (Scheme 17a). Analogous to complex 235, complexes 237 and 238 also underwent C–H bond cleavage; however, for these complexes the cleavage occurred at room temperature (Scheme 17b and c).161
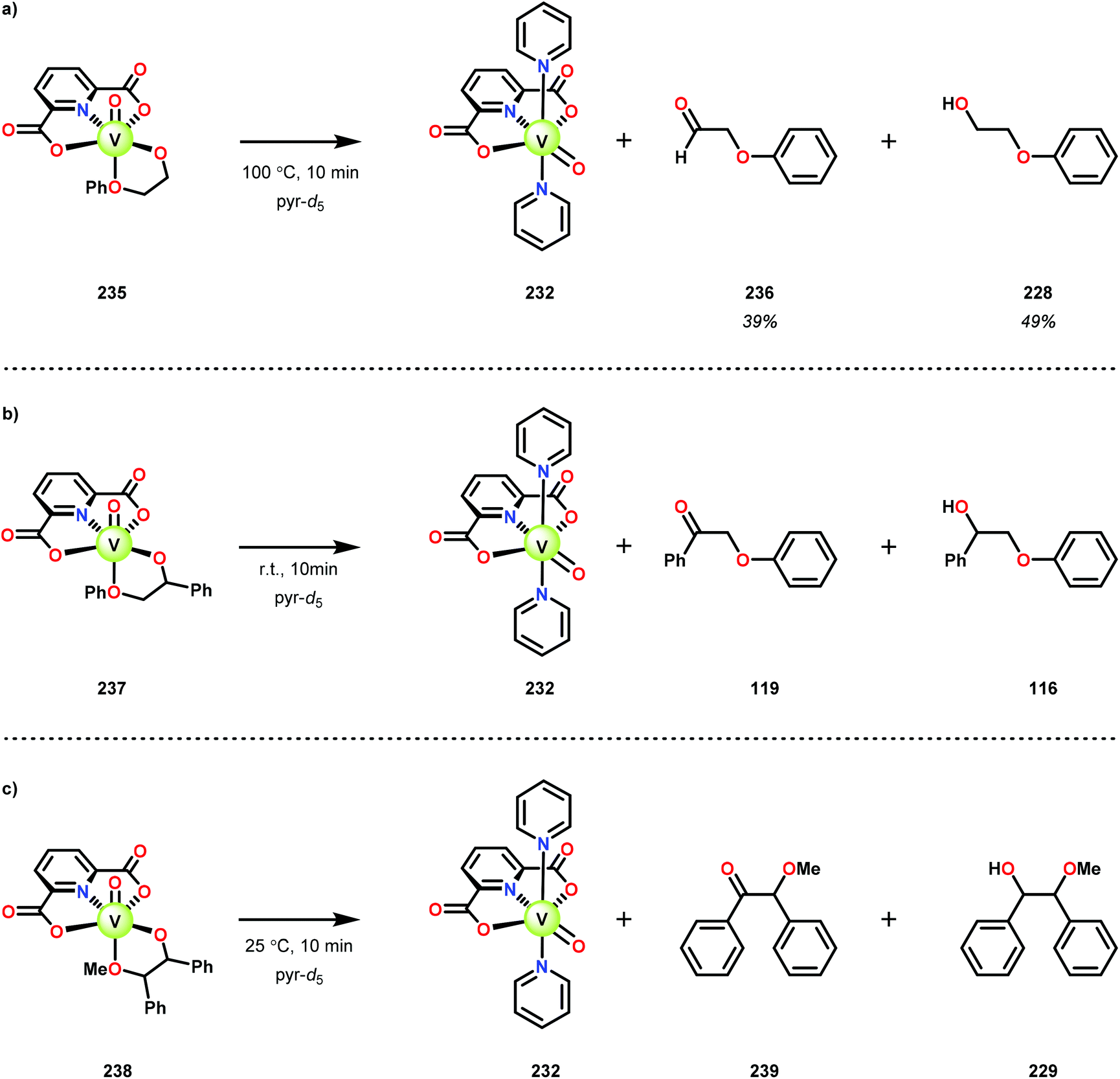 | ||
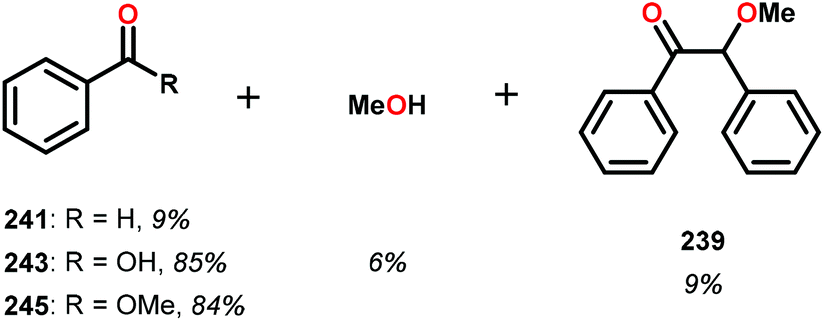
| Scheme 17 Alcohol oxidation by V complexes (a) 235, (b) 237 and (c) 239. | ||
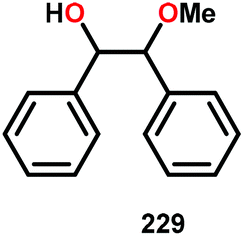
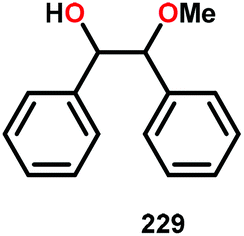
In the absence of pyridine, V complex 238 exhibited different reactivity. Not only did the complex mediate C–H bond cleavage, it also carried out C–C bond cleavage (Scheme 18). The products observed from heating complex 238 in DMSO included a VIV complex (240), benzoin methyl ether (239) together with methanol, benzaldehyde (241) and 1,2-diphenyl-2-methoxyethanol (229).161
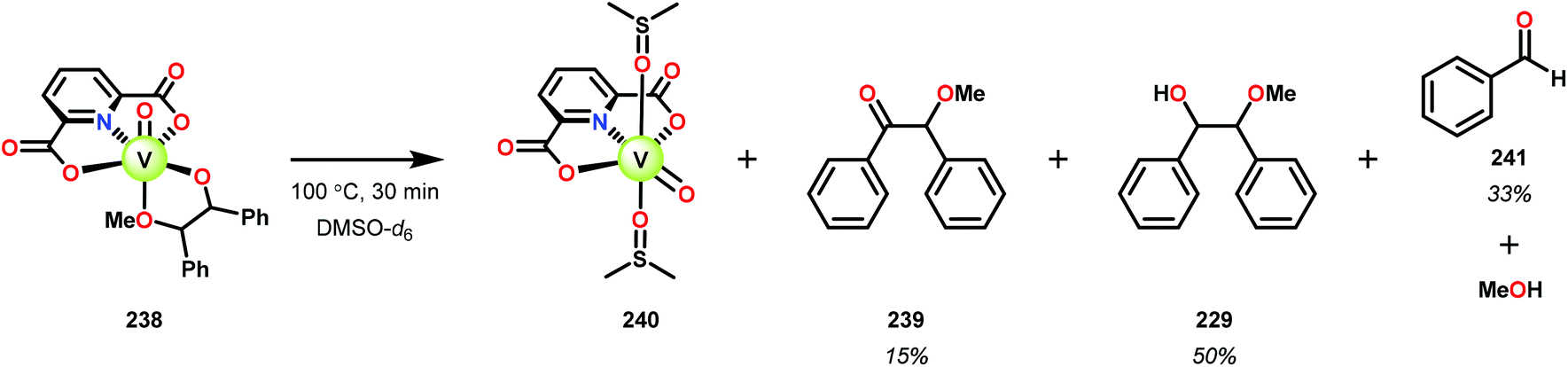 | ||
| Scheme 18 Heating of V complex 238 in DMSO-d6. | ||
The mechanism of the C–C bond cleavage is not fully understood, but the authors proposed that V complex 238 reacts to produce an aldehyde and a methoxybenzyl radical. The methoxybenzyl radical can subsequently react with water and a second equivalent of complex 238 to generate methanol, a second equivalent of benzaldehyde (241), with the release of 1,2-diphenyl-2-methoxyethanol (229). Attempts were made to evaluate the proposed mechanism by reacting V complex 238 in the presence of a radical trap, BHT (2,6-di-tert-butyl-4-methylphenol), and triphenyl phosphine. However, this reaction was inconclusive and the products could not be characterized. When adding 9,10-dihydroanthracene to the reaction mixture only trace amounts of anthracene (<1%) was detected, with the reaction course being unaffected. With a basic understanding of the reactivity of the V-based catalysts, attempts were made to run the reactions with catalytic amounts of VV(pda)(O)(OiPr) (230). These results are summarized in Table 2.161
| Entry | Substrate | Conversion (%) | Products (%) |
|---|---|---|---|
| a Reactions were carried out at 100 °C under an atmosphere of air in d6-DMSO if not otherwise stated. b 10 mol% V(pda)(O)(OiPr) (230), 7 days. c 5 mol% V(pda)(O)(OiPr) (230), 20 h. d 10 mol% V(pda)(O)(OiPr) (230), 6 days. Reaction was carried out in pyridine-d5. | |||
| 1b |
|
20% |
|
| 2b |
|
95% |
|
| 3c |
|
94% |

|
| 4d |
|
99% |
|
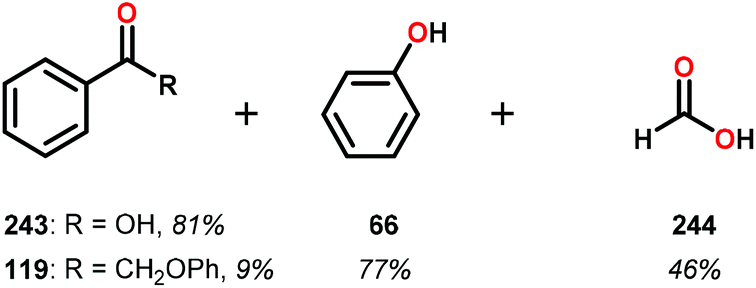
Consistent with the results from the stoichiometric oxidations, the substrates with substituted phenyl groups on the carbon backbone facilitated oxidation more efficiently. However, substrate 229 showed different product distributions when changing the solvent from DMSO to pyridine (cf. Table 2, entries 3 and 4). The generated benzoic acid (243) and methyl benzoate (245) are most likely oxidation products derived from the intermediate benzoin methyl ether (239).161
Control experiments were run under anaerobic conditions with DMSO to investigate the possible role of DMSO as an oxidant. A mixture of VIV(pda)(O)(pyr)2 and 1,2-diphenyl-2-methoxyethanol (229) in d6-DMSO after 1 week at 100 °C showed no turnover, suggesting that DMSO does not act as an oxidant in the catalytic cycle. Performing the reactions with VIV(pda)(O)(DMSO)2 (240) and 2-phenoxyethanol (228) or 1-phenyl-2-phenoxyethanol (116) as substrates also displayed no conversion after heating at 100 °C for 1 week in DMSO-d6. The selectivity of the stoichiometric reactions compared to the catalytic reactions with the VIV complexes is not well understood and is still the subject of ongoing experimental and computational investigations. The stoichiometric reactions afforded a mixture of C–H and C–C bond cleavage products while the catalytic reactions afforded only products derived from C–C bond fragmentation.161 Collectively, this method provides a proof of principle for the oxidation and cleavage of lignin models. The homogeneous nature of the catalyst provides opportunities for exploring new ligand designs in order to optimize the activity and selectivity for the oxidative disassembly of lignin model systems and native lignin.
Inspired by previous work on using 8-quinolinate ligands for the oxidation of benzylic, allylic, and propargylic alcohols in air,163,164 Hanson and co-workers explored the electron-donating nature of the ligand scaffolds on the catalytic oxidative cleavage of lignin models. They envisioned that a tridentate scaffold with phenolate donors could provide a more electron-rich environment and an additional open coordination site to facilitate the reaction of VIII or VIV with O2. A bis(phenolate)-pyridine ligand (246; H2BPP = 2,6-(HOC6H2-2,4-tBu2)2NC5H4), and a bis-(phenolate)amine ligand (247; H2BPA = N,N-bis(2-hydroxy-4,5-dimethylbenzyl)propylamine) (Fig. 28A) were identified as promising ligands and the corresponding V complexes (248 and 249) are depicted in Fig. 28B.165
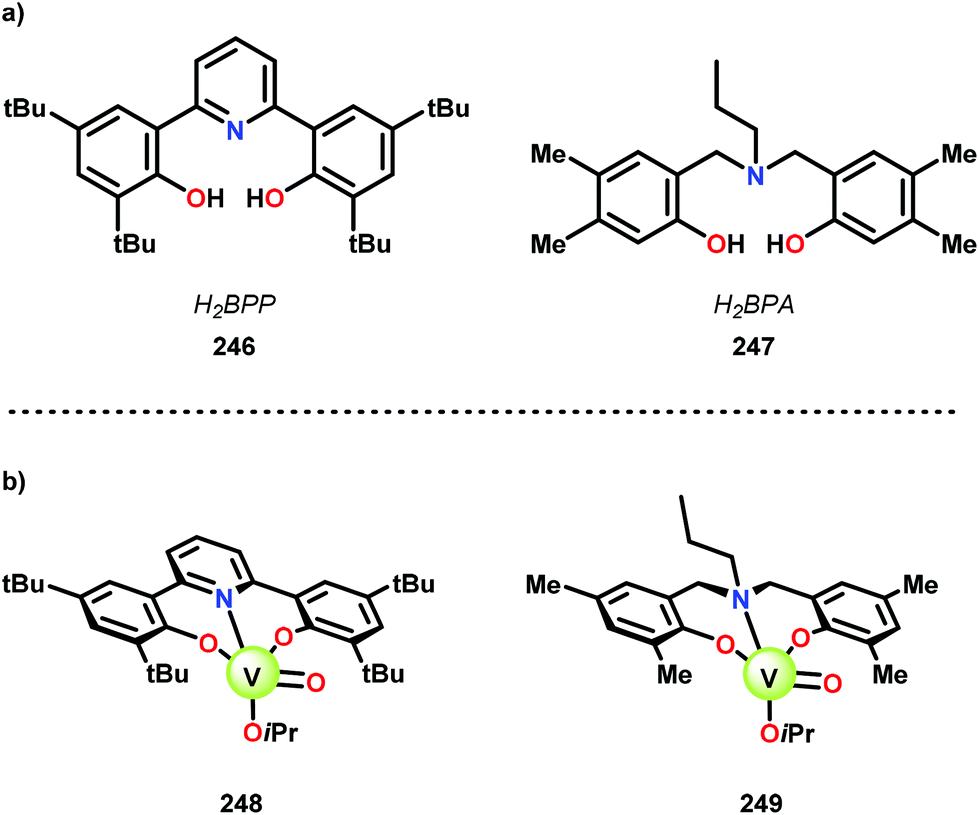 | ||
| Fig. 28 Structure of (a) ligands H2BPP (246) and H2BPA (247), and (b) the corresponding V complexes (248 and 249). | ||
In initial experiments, complexes 248 and 249 were tested stoichiometrically with pinacol to probe the reactivity of the complexes. These stoichiometric reactions were shown to afford the reduced VIV species in 95% and 97% yield from complexes 248 and 249, respectively. Acetone was also established as a product in these reactions. The VIV complexes were therefore isolated and exposed to air in CD2Cl2 to produce the corresponding VV complexes, an essential element that is required for the catalytic oxidative cleavage of C–C and C–O bonds. These initial reactions were optimized for the catalytic aerobic oxidation of 4-methoxybenzyl alcohol. Reacting V complex 248 (2 mol%) with 4-methoxybenzyl alcohol at 60 °C under air in 1,2-dichloroethane did not result in significant oxidation. Increasing the temperature to 80 °C under air, with Et3N (10 mol%) as an additive, in toluene afforded 4-methoxybenzaldehyde in 99% yield after 65 h. Complex 249 was also able to oxidize 4-methoxybenzyl alcohol but afforded lower yields (80%) of the oxidized product. After determining the optimized catalytic conditions, the oxidation of lignin model compounds was investigated using complexes 248 and 249. When non-phenolic lignin model 250 was subjected to V catalyst 248 (10 mol%) in toluene at 100 °C for 48 h, 84% conversion occurred with the formation of various oxidized products (Scheme 19).165
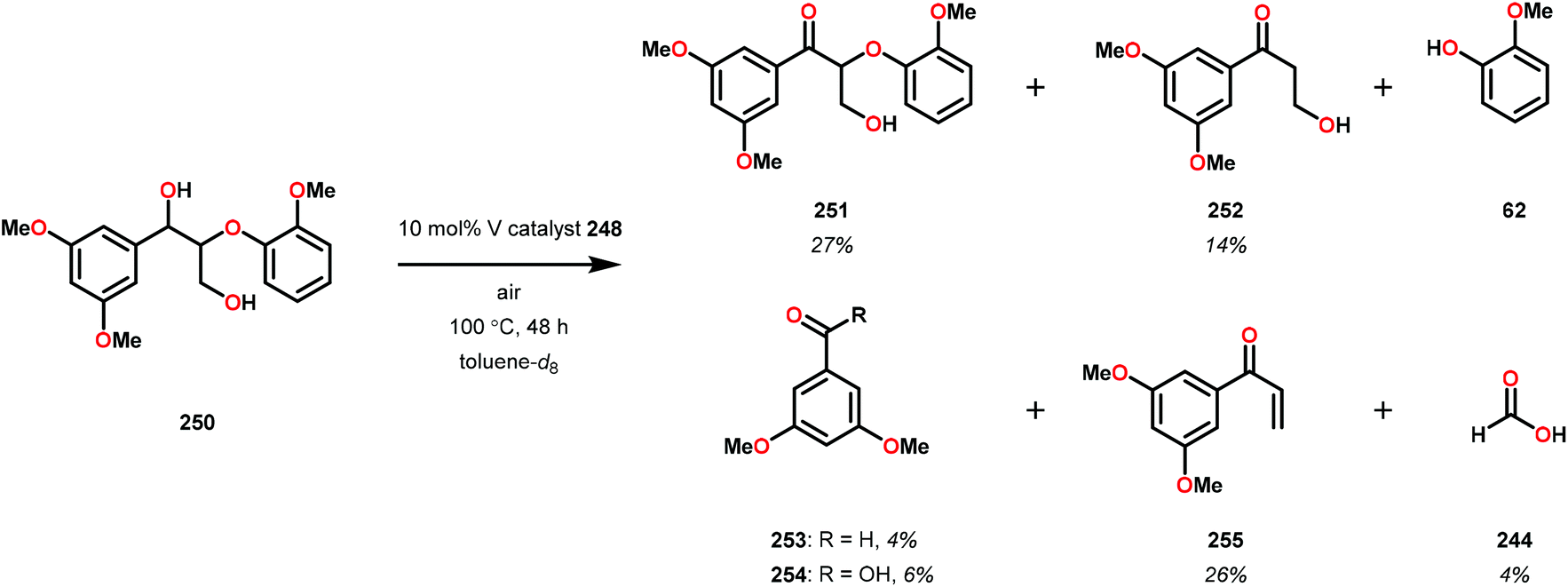 | ||
| Scheme 19 Catalytic aerobic oxidation of lignin model system 250 with V complex 248. | ||
The product mixture obtained was similar to that obtained from the previously reported V catalyst 230, consisting mostly of C–O and C–H bond cleavage products.165 It should be noted that these results contrast the results observed by Son and Toste using V catalyst 256 (Fig. 29) in which high C–O bond cleavage selectivity was observed.166 These results also contrast the C–H bond cleavage selectivity for the previously reported 8-quinolinate catalyst 257 (Fig. 29).167 In Son and Toste's work, it was proposed that the selectivity for C–O vs. C–H bond cleavage depends on the bite angle of the ligand framework.166 However, the preference for C–O vs. C–H cleavage is still not fully understood and the two cleavage pathways most likely go through different mechanisms as noted by the different selectivities for the different V catalysts.167
 | ||
| Fig. 29 (a) Previously reported V catalyst 256 that mediates C–O bond cleavage of non-phenolic lignin models and (b) the previously reported 8-quinolinate-based V catalyst 257 (VV(HQ)2(O)(OiPr)) that promotes C–H bond cleavage products of non-phenolic lignin model systems. | ||
V complex 248 was also used catalytically in the oxidation of substrate 250 at 100 °C for 48 h but afforded low conversion. Decomposition of the catalyst was suggested by the appearance of NMR signals corresponding to 4,5-dimethylsalicylaldehyde. The presence of 4,5-dimethylsalicylaldehyde could also be confirmed by GC-MS analysis, showing a peak with an expected m/z = 150. Phenolic lignin model systems have proven to be more difficult substrates to perform chemoselective reactions on, meriting separate investigation of reactivity with aerobic oxidations. Attempts to oxidize various phenolic lignin model systems with V complex 248 afforded low conversions (∼20%) and yielded mostly the oxidized ketone product with only trace amounts of cleavage products (Scheme 20). These results were surprising since complex 248 afforded C–O bond cleavage products of the non-phenolic lignin model but did not efficiently mediate C–O bond cleavage when employing the phenolic model system.165
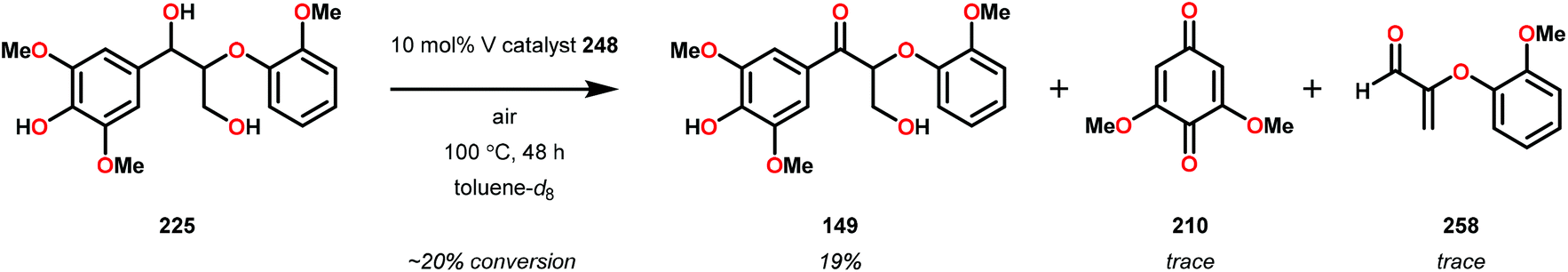 | ||
| Scheme 20 Aerobic oxidation of phenolic lignin model 225 catalyzed by V complex 248. | ||
These results are also perplexing considering that the 8-quinolinate V catalyst 257 was previously reported to afford the alkyl–phenyl bond cleavage products 210 and 258 as major products together with ketone 149 when reacted with the phenolic substrate 225.167 Realizing how different ligands give rise to different selectivities highlights the complexity involved in devising a reasonable mechanism for these V-catalyzed oxidations.
Having made these observations, Hanson and co-workers subsequently went on to explore the reactivity of V catalysts 256 and 257 with diastereomerically pure erythro and threo phenolic and non-phenolic β-1 lignin models 259 and 263. Aerobic oxidation of threo lignin model system 259 (259T) afforded the various oxidation products shown in Table 3, entry 1. Additional screening of the V-catalyzed cleavage of the erythro isomer (259E) with complex 257 was also carried out, as summarized in Table 3.168
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Conversion (%) | Yieldb (%) | |||||
| 260 | 261 | 253 | 179 | 254 | 262 | ||||
| a Reaction conditions: V complex 257 (10 mol%), substrate, air, 100 °C, 48 h. b Yield determined by 1H NMR using internal standard. c Reaction run in the presence of Et3N (15 mol%). | |||||||||
| 1 | 259T | Pyridine-d5 | 100 | 34 | 57 | 3 | 3 | 3 | 4 |
| 2 | 259T | DMSO-d6 | 91 | 66 | 6 | 3 | 3 | 4 | 11 |
| 3c | 259E | Toluene | 100 | 72 | <1 | 7 | 2 | <1 | <1 |
The phenolic substrate 263T was also reacted but afforded an incomplete mass balance (see Scheme 21).168 Mass balance inconsistencies have been previously reported on the oxidation of lignin models and are most likely attributed to polymerization or other reactions involving radical intermediates.169,170 To elucidate possibilities for loss of mass, aerobic oxidation of 2,6-dimethoxyphenol was carried out with the VV(HQ)2(O)(OiPr) catalyst (257) in the presence of Et3N (10 mol%). After 48 h of heating at 80 °C, 98% of the 2,6-dimethoxyphenol had reacted to produce a dark brown solid. An aliquot was removed and analyzed by NMR to show no soluble organics in the sample, consistent with polymerization or other reactions to afford recalcitrant byproducts.168
 | ||
| Scheme 21 Aerobic oxidation of the threo isomer of phenolic lignin model system 263 by V complex 257. | ||
It is apparent that this catalyst system mediates the benzylic oxidation preferentially over other oxidized products. However, the phenolic containing substrates afford slightly different product distributions and selectivities.168 This is consistent with the oxidative experiments of the phenolic and non-phenolic β-O-4 lignin models using the VV(HQ)2(O)(OiPr) catalyst (257).167
Based on the aforementioned results, it is evident that tuning the selectivity and reactivity with V catalysts is possible, although the outcome is rather difficult to predict. There are no established mechanisms for these oxidations but the selectivity and reactivity are greatly affected by the choice of ligand. The homogeneous nature of the various V catalysts has made them an attractive target for studying the oxidative degradation of lignin model systems. However, the evaluated V catalysts afford mainly oxidized ketone products in contrast to Cu and TEMPO-mediated oxidations that form mostly C–C bond cleavage products, implying that different mechanisms are operating (see section 3.4).
3.4 TEMPO-based catalytic system for oxidation and degradation of lignin models
N-Oxyl radicals have provided great utility for the oxidation of numerous functional groups. Commonly used oxidants are TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxyl) and ABNO (ABNO = 9-azabicyclo[3.3.1]nonane N-oxyl) derived reagents. N-Oxyl based oxidants are widely applicable in the field of organic oxidations and have proven to be excellent choices for the selective oxidation of primary alcohols, secondary alcohols, primary amines and aldehydes.171–176As previously stated, the primary pathway for degradation of lignin in nature is believed to occur via oxidative single-electron transfer (see Scheme 10).114,115 Inspired by nature, several research groups have turned their attention to developing methods for the oxidative degradation of lignin models using TEMPO-mediated oxidation protocols. Coupling such protocols with aerobic procedures is desirable since O2 is a bountiful, inexpensive and an environmentally benign terminal oxidant. Hanson and co-workers therefore compared the aerobic oxidation of lignin models using homogeneous Cu- and V-based catalysts. The Cu catalysts used in this study consisted of a Cu/TEMPO system,177 originally developed by Sheldon and co-workers, which was shown to mediate the aerobic oxidation of primary alcohols at room temperature.178,179
Hanson and co-workers began their investigations with a simple β-1 containing lignin model (229) where the catalytic system consisted of O2, catalytic amounts of CuCl (20 mol%) and catalytic amounts of TEMPO (30 mol%) in pyridine at 100 °C for 48 h. In the Cu/TEMPO systems, Cu is believed to activate O2, and Cu and TEMPO achieves the oxidation of the alcohol. This system was shown to afford benzaldehyde (241) and methyl benzoate (245) in good yields (Scheme 22).177
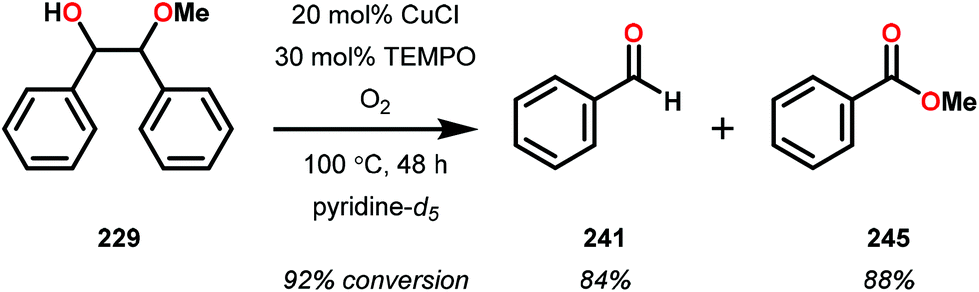 | ||
| Scheme 22 Aerobic oxidation of 1,2-diphenyl-2-methoxyethanol (229). | ||
The authors found that the active Cu species was not stable under the reaction conditions, which prompted for portion-wise additions of CuCl and TEMPO to achieve 92% conversion after 48 h.177 In contrast to the CuCl/TEMPO system, the V-catalyzed aerobic oxidation afforded benzoin methyl ether (239), which could subsequently undergo C–C bond cleavage and further oxidation to benzoic acid (243) and methyl benzoate (245) (Table 2, entry 4). With the V system, benzaldehyde (241) (9%), methanol (6%), benzil (<5%), and benzoin (<5%) could also be observed as minor products.161 To investigate the intermediacy of benzoin methyl ether (239) with the CuCl/TEMPO system, benzoin methyl ether (239) was subjected to the catalytic CuCl/TEMPO conditions for 18 h. The reaction afforded methyl benzoate (245) (85%), benzoic acid (243) (76%), and benzaldehyde (241) (5%) (Scheme 23), implying that ketone 239 is indeed an intermediate.177
 | ||
| Scheme 23 CuCl/TEMPO catalyzed oxidation of benzoin methyl ether (239). | ||
With the success of oxidizing 229, the authors subsequently applied the Cu/TEMPO system on more complex lignin model systems. Considering the instability and limited lifetime of the CuCl/TEMPO system, the authors decided to use stoichiometric amounts of the CuCl/TEMPO system on lignin model system 250 (Scheme 24).177
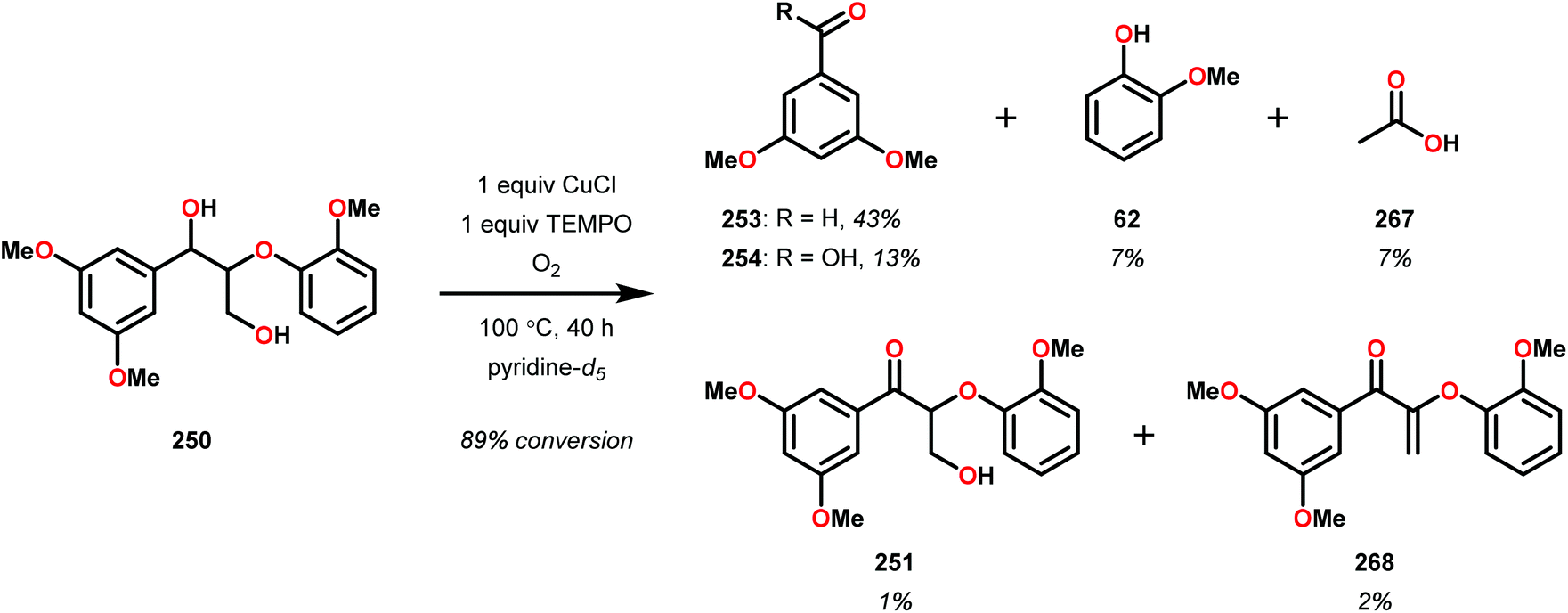 | ||
| Scheme 24 Stoichiometric oxidation of lignin model system 250 with CuCl/TEMPO. | ||
The stoichiometric reaction afforded a mixture of products including C–C bond cleavage products (253 and 254), the oxidized ketone (251), the dehydrated ketone (268), and C–O bond cleavage products (62 and 267). The authors also observed the formation of condensation products by NMR. This was later confirmed using LC-MS analysis after observing higher molecular weight compounds in the reaction mixture. In order to determine whether ketone 251 was an intermediate, substrate 250 was treated with 10 mol% CuCl/TEMPO at 100 °C under an O2 atmosphere for 18 h. This resulted in 48% conversion along with the dehydrated ketone 268 (18%) and other unidentified products; however, the aldehyde products could not be detected. When subjecting model system 229 to ceric ammonium nitrate, a strong single-electron oxidant, a similar product distribution was observed as when compound 229 was subjected to the CuCl/TEMPO system, implying a single-electron mechanism followed by direct C–C bond cleavage.177
Subsequently, Hanson and co-workers proceeded to further investigate a Cu(OTf)/2,6-lutidine/TEMPO catalyst system for the degradation of β-1 lignin models. Non-phenolic lignin models were initially investigated with the Cu(OTf)/2,6-lutidine/TEMPO system. Substrate 259T, a non-phenolic β-1 lignin model, was heated at 100 °C in the presence of 10 mol% Cu(OTf)/TEMPO, O2 and 2,6-lutidine (10 equiv.) for 48 h to afford 3,5-dimethoxybenzaldehyde (253) (81%) and 4-methoxybenzaldehyde (179) (69%) as major products (Scheme 25, top).168 These conditions afforded C–C bond cleavage products, similar to the previous CuCl/TEMPO system.177 This mode of reactivity resembles the observed reaction pattern from Stahl and co-workers in which a Cu(OTf)(MeCN)4/bpy/TEMPO/N-methylimidazole system afforded veratryl aldehyde (269) as the major product, resulting from initial oxidation of the primary alcohol followed by a retro-aldol reaction to cleave the Cα–Cβ bond (Scheme 25, bottom).180
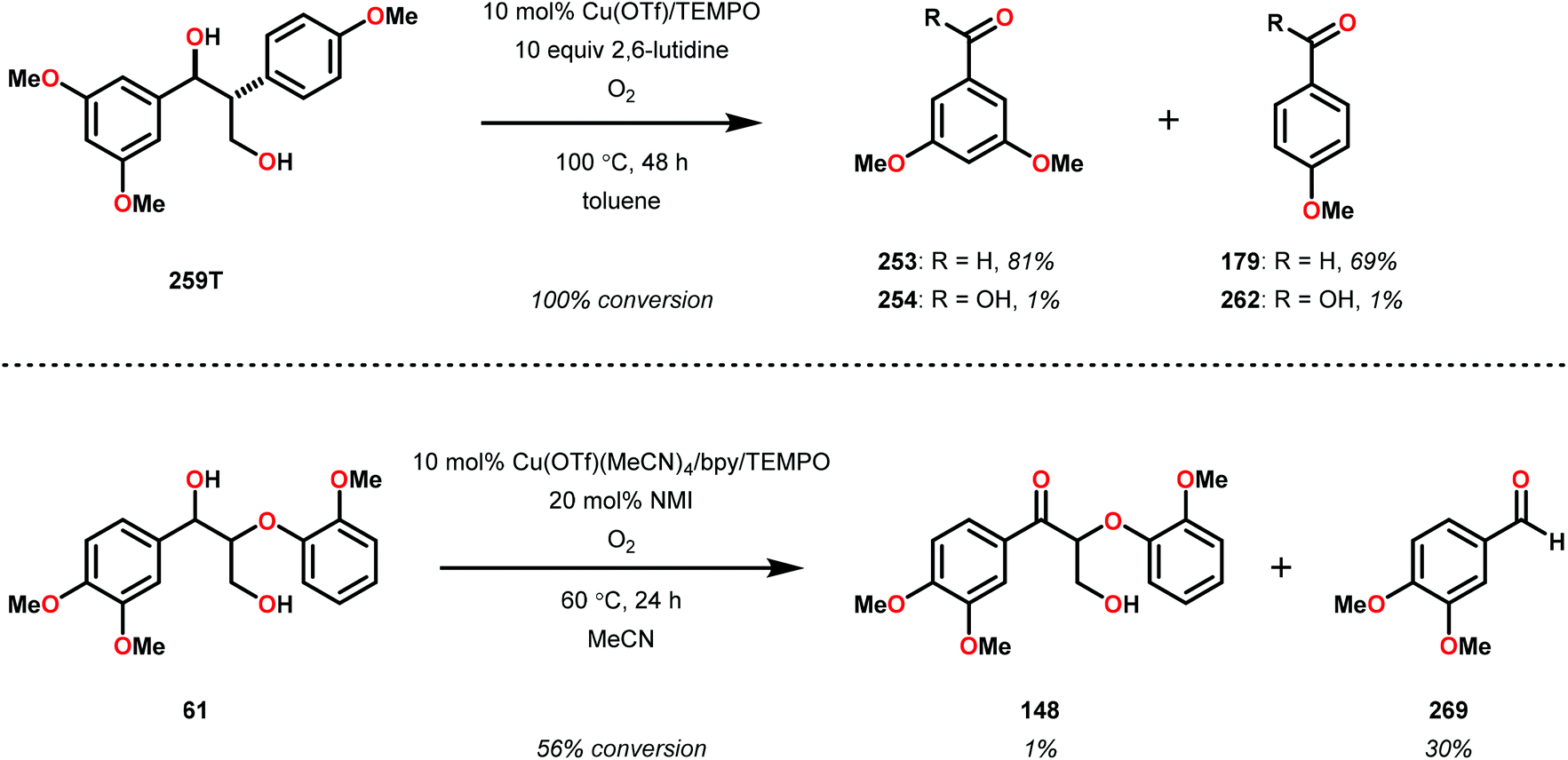 | ||
| Scheme 25 (Top) Oxidation of the non-phenolic lignin model 259T with the Cu(OTf)/2,6-lutidine/TEMPO system. (Bottom) Oxidation of non-phenolic lignin model 61 with the Cu(OTf)(MeCN)4/bpy/TEMPO/NMI system. | ||
Following the Cu(OTf)/TEMPO results on the non-phenolic lignin model (259T), Hanson and co-workers proceeded to apply these conditions on a phenolic lignin model system (263T), still containing the β-1 connectivity. To investigate the selectivity of the Cu system, a catalytic reaction using 10 mol% of Cu(OTf)/TEMPO and a stoichiometric reaction was carried out on the phenolic lignin model (Scheme 26).168 These results highlight that the aerobic oxidation of the phenolic β-1 lignin models afford mostly products derived from cleavage of the Caryl–Cα bond, whereas oxidation of non-phenolic β-1 lignin models afford Cα–Cβ bond cleavage products. This change in selectivity additionally emphasizes the challenges in catalyst development for the degradation of lignin models.
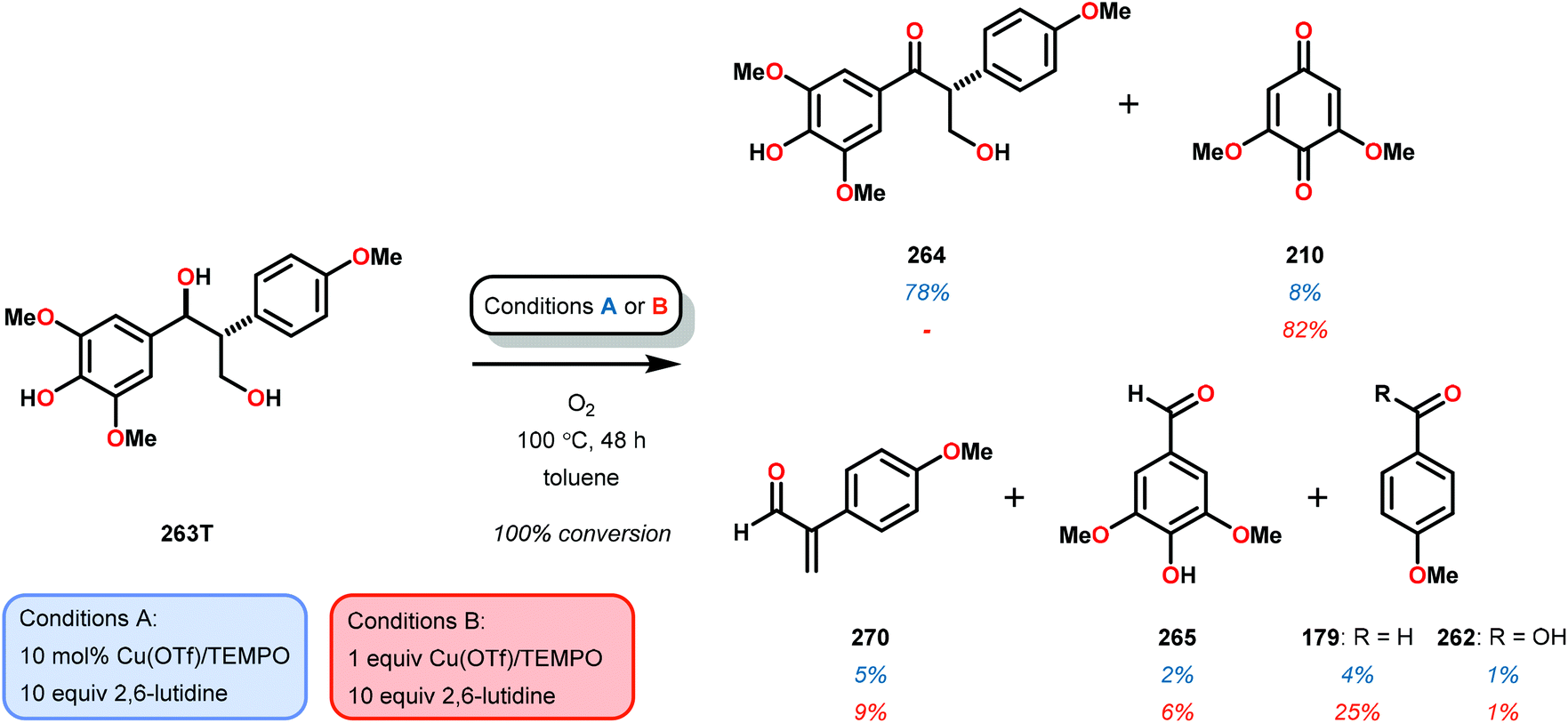 | ||
| Scheme 26 Catalytic and stoichiometric oxidation of lignin model system 263T by the Cu(OTf)/TEMPO system. | ||
Baker and co-workers continued working with the Cu/TEMPO system and applied it to β-O-4 lignin model systems. The authors first focused on optimizing the catalyst system with non-phenolic substrate 250 in order to maximize the yield of the aldehyde product. Optimization began using stoichiometric amounts of Cu and TEMPO where a variety of N-donor ligands, Cu catalysts, time, and solvents were screened. It was noted that applying bulkier N-donor ligands generally increased the selectivity and the yield of the aldehyde product (253). Using pyridine and 2,6-lutidine as solvents showed moderate yields but were not as efficient as when using toluene with 2,6-lutidine as an additive (10 equivalents). The authors finally settled on a Cu(OTf)/TEMPO system, 2,6-lutidine (10 equivalents) in toluene. Heating this reaction mixture at 100 °C gave 99% conversion and 62% yield of aldehyde 253 (Scheme 27).181
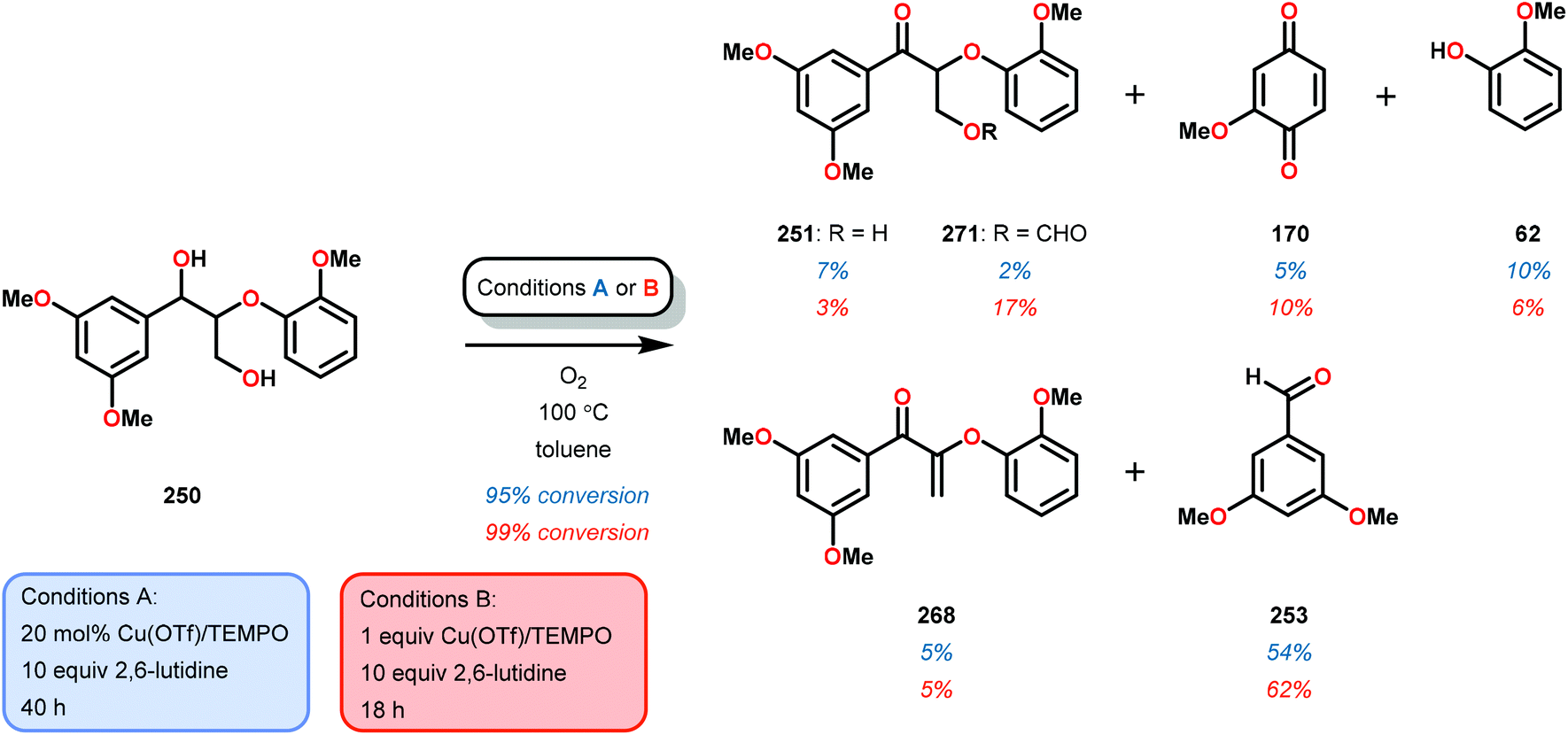 | ||
| Scheme 27 Stoichiometric and catalytic oxidation of non-phenolic β-O-4 lignin model 250 by the Cu(OTf)/TEMPO/2,6-lutidine system. | ||
After determining suitable conditions for the stoichiometric oxidation of non-phenolic lignin model system 250, the authors attempted to devise a catalytic procedure. A plethora of oxidation products were generated, with 3,5-dimethoxybenzaldehyde (253) being the major product (Scheme 27). Gratifyingly, improved selectivity was observed when applying the catalytic conditions to the phenolic β-O-4 lignin model 225 (Scheme 28). However, the reason for this change in selectivity when changing from non-phenolic to phenolic models is not fully understood.181
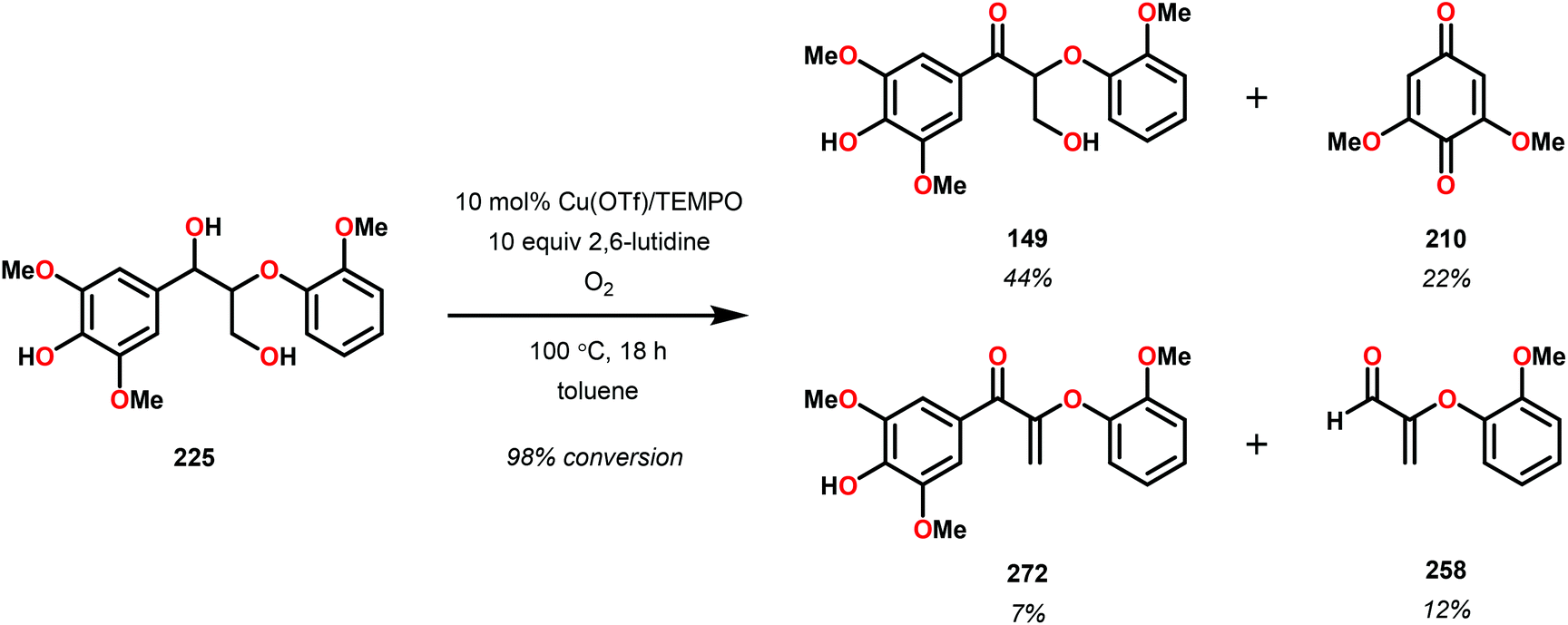 | ||
| Scheme 28 Catalytic oxidation of phenolic lignin model system β-O-4 lignin model 225 by the Cu(OTf)/TEMPO/2,6-lutidine system. | ||
Control reactions were run to highlight the importance of the TEMPO and 2,6-lutidine. Omitting both Cu(OTf) and TEMPO afforded only 2% conversion. In the absence of TEMPO, 60% conversion was observed after 18 h, indicating that oxidation can proceed without TEMPO, although it is evident that the TEMPO has a crucial role as a co-catalyst.181
Several catalytic systems have been developed for the aerobic oxidation of alcohols, lignin models, and diols, employing Cu and TEMPO.182–184 Although different mechanisms have been proposed for Cu/TEMPO based oxidations,185–189 Stahl and co-workers have proposed a mechanism relevant to the Cu(OTf)/TEMPO/2,6-lutidine reactions.190,191 Oxidation of alcohols by the oxoammonium salt is a well-understood process and this possibility has been probed by kinetic studies. However, Stahl and co-workers noticed a significant rate increase when switching from a CuII to a CuI source. For the mechanistic studies, Stahl and co-workers settled for a CuI(bpy)/TEMPO system where this system was compared to the TEMPO-catalyzed oxidation of alcohols. Monitoring of these reactions by in situ IR showed that the oxidation reactions employing stoichiometric oxoammonium (TEMPO) salts proceeded at a slower rate for both benzylic and aliphatic alcohols, implying that the oxoammonium cation is not the catalytically competent species in the TEMPO-catalyzed oxidation of alcohols, regardless of the 20-fold increase of the TEMPO reagent (Fig. 30).190
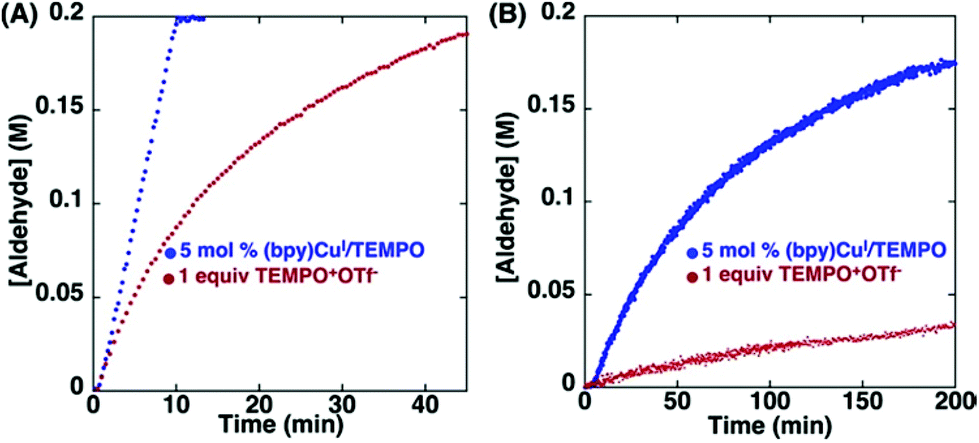 | ||
| Fig. 30 Monitoring of the formation of aldehyde by in situ IR. Oxidation of (A) benzyl alcohol and (B) cyclohexylmethanol by 5 mol% CuI(bpy)/TEMPO (blue) and stoichiometric TEMPO+OTf− (red). Reprinted with permission from ref. 190. Copyright 2013 American Chemical Society. | ||
From the spectroscopic studies, rate law calculations, kinetic isotope effect and Hammett studies, a mechanism was proposed and is depicted in Fig. 31. The authors proposed two separate half-reactions: (1) activation of O2 by the reduced Cu catalyst (CuI) (273 → 274), and (2) concerted oxidation of the alcohol where both CuII and TEMPO act as one-electron oxidants to achieve the overall two-electron conversion of the alcohol to the corresponding aldehyde (278 → 273).190
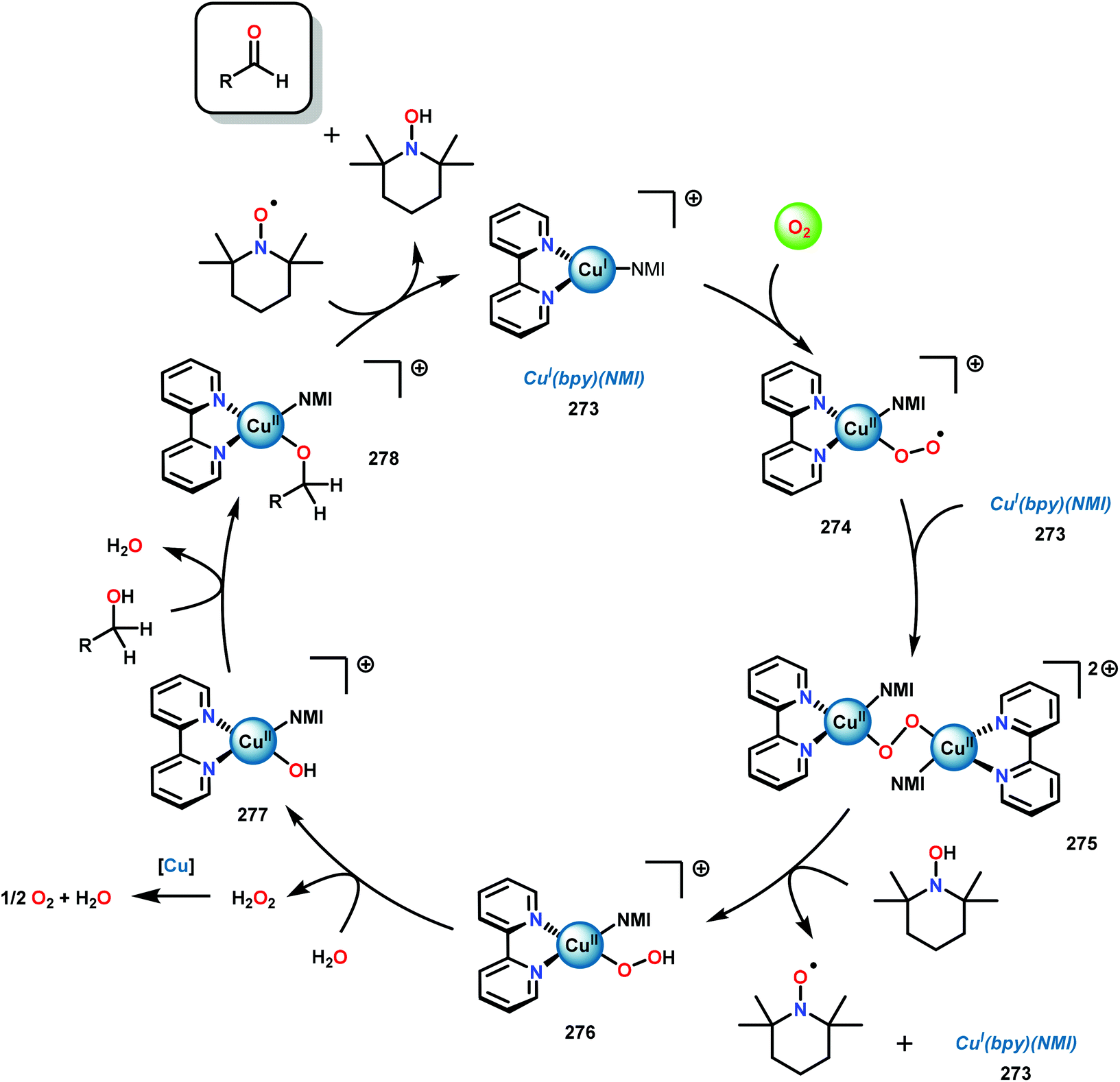 | ||
| Fig. 31 Proposed mechanism for catalytic CuI/TEMPO mediated oxidation of alcohols. NMI = N-methylimidazole. R = alkyl, aryl. | ||
Baker and co-workers noted that the mechanism outlined by Stahl and co-workers pertains to several key steps that may be occurring in the Cu(OTf)/TEMPO oxidation of lignin model systems. However, in contrast to the proposed mechanism in Fig. 31, which is proposed to be the fundamental mechanism operating under ambient temperatures, the Cu(OTf)/TEMPO experiments were conducted at elevated temperatures (100 °C). The increased temperatures most likely creates conditions where both CuI and CuII complexes with N-donor ligands are more labile, hence reducing the potential for achieving higher reactivity and selectivity.181 Additionally, Chiba and co-workers have also employed Cu for the aerobic oxidative cleavage of lignin model systems.192
In light of the potential for using oxoammonium salts as oxidants, Stahl and co-workers set out to develop a selective method for the aerobic oxidation of lignin model systems containing the β-O-4 linkage. Here, oxidation of the benzylic alcohol to produce the corresponding ketone was the desired transformation and their investigation started with β-O-4 lignin model system 61 (Fig. 32).180
 | ||
| Fig. 32 Possible product distribution for oxidation of lignin model system 61 by TEMPO-based catalytic protocols. | ||
An assortment of oxidation methods was investigated on model system 61 to identify suitable conditions for achieving high selectivity for the benzylic oxidation. In some of the applied catalytic systems, oxidation of the primary alcohol unit could also be observed and produced a “byproduct” (269), which is presumably the result of a retro-aldol reaction. In addition to these two oxidation products, the dehydrated ketone 280 was also isolated in certain cases. A CuBr/bpy/DABCO/TEMPO (10 mol%) system afforded 86% conversion but unfortunately afforded the dehydrated ketone 280 (23%) and aldehyde 269 (31%) as major products with the desired ketone 148 in merely 6% yield after 24 h at 60 °C using O2 as the terminal oxidant. Removal of DABCO increased conversion to 89% but afforded ketone 148 in only 10% yield and aldehyde 269 in 37% yield, together with other unidentifiable products. An aerobic system consisting of Pd(OAc)2/DMSO (5 mol% Pd(OAc)2) gave excellent selectivity, producing ketone 148 exclusively but in a modest yield of 42%. This could be slightly improved by employing Pd(OAc)2 (5 mol%) in a DMSO/H2O mixture (4:6) with added 2,9-dimethyl-1,10-phenanthroline (10 mol%) as a ligand. This combination produced 87% conversion of substrate 61 with the exclusive formation of ketone 148 in 56% yield. A TEMPO-based system using Fe(NO3)3 (5 mol%) and TEMPO/NaCl (10 mol%) in DCE afforded 100% conversion and produced the desired ketone 148 in 79% yield, but all metal-catalyzed oxidations paled in comparison to the final conditions, which consisted of AcNH–TEMPO (5 mol%), HNO3/HCl (10 mol% each), 1 atm O2 at 45 °C. These conditions were subsequently used on a variety of lignin model systems and generally afforded the desired ketones in high yields and excellent selectivity (Fig. 33).180 Even oxidized lignin, derived from aspen, could be depolymerized when treated with formic acid to afford >61 wt% of low-molecular-mass aromatics (Scheme 29).193
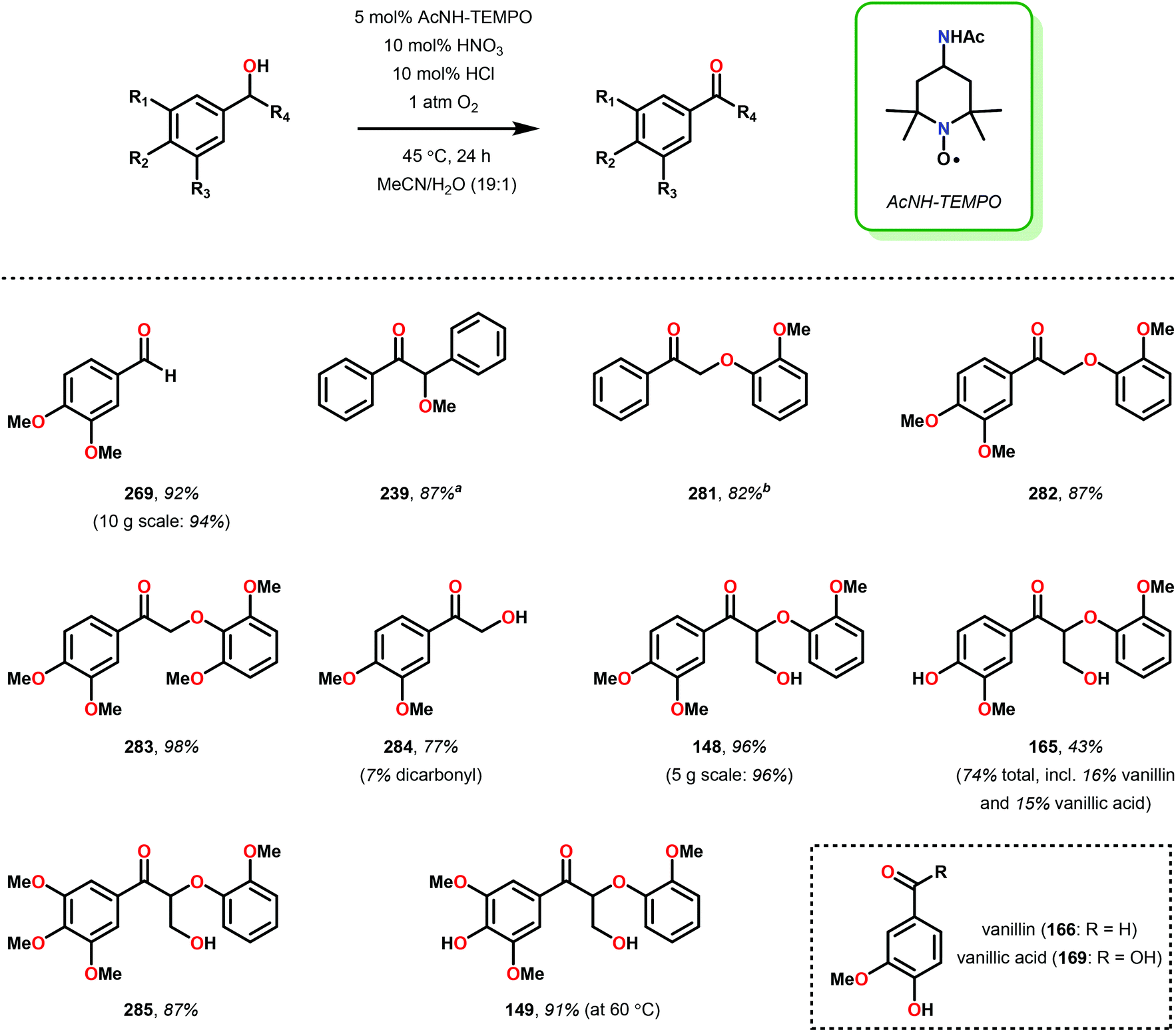 | ||
| Fig. 33 Metal-free aerobic oxidation of lignin models using the AcNH-TEMPO/HNO3/HCl catalyst system. Reaction conditions: 1 mmol of alcohol (0.2 M in MeCN/H2O 19:1). a 10 mol% AcNH-TEMPO, 36 h. b Yield after 36 h. | ||
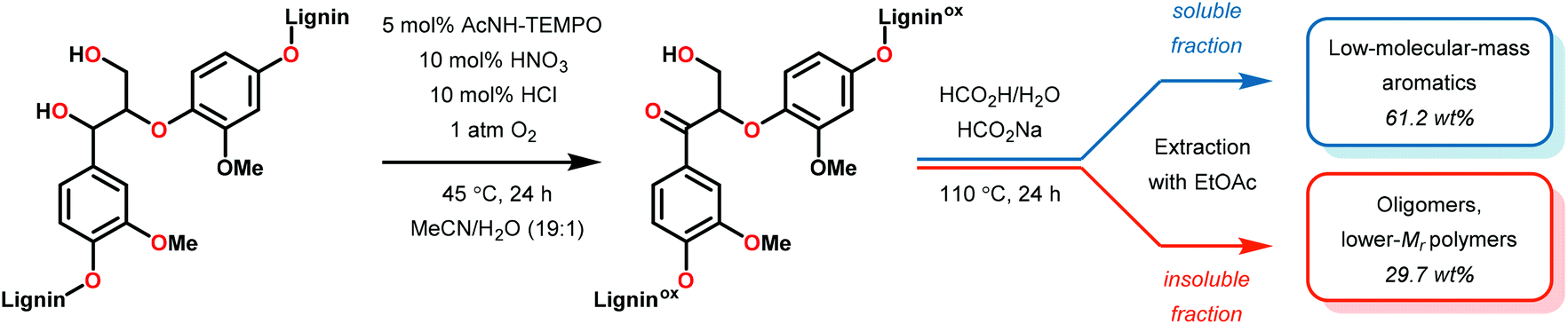 | ||
| Scheme 29 Two-step approach for depolymerization of aspen lignin. | ||
Stephenson and co-workers subsequently designed a two-step, overall redox-neutral approach to the degradation of lignin model compounds containing β-O-4 linkages. This method employed a recyclable oxidant, [4-AcNH-TEMPO]BF4, to carry out the oxidation of the benzylic alcohols with a subsequent photochemically-mediated reduction step to afford β-O-4 cleavage products (see also section 2). Stephenson and co-workers targeted the oxidation of the Cα–O in order to weaken the Cβ–O bond (see Fig. 35) for a subsequent photochemical reduction of the β-O-4 linkage using 1 mol% [Ir(ppy)2(dtbbpy)]PF6 (146) as the photocatalyst. 1 equivalent of Bobbitt salt in the presence of an equimolar amount of silica afforded the oxidized products in high yields for a variety of lignin model systems (Table 4).101 Additionally, a similar two-step approach has been reported by Westwood and co-workers where DDQ/t-BuONO was employed to affect the initial oxidation and stoichiometric amounts of Zn were used to carry out the subsequent reductive cleavage.194
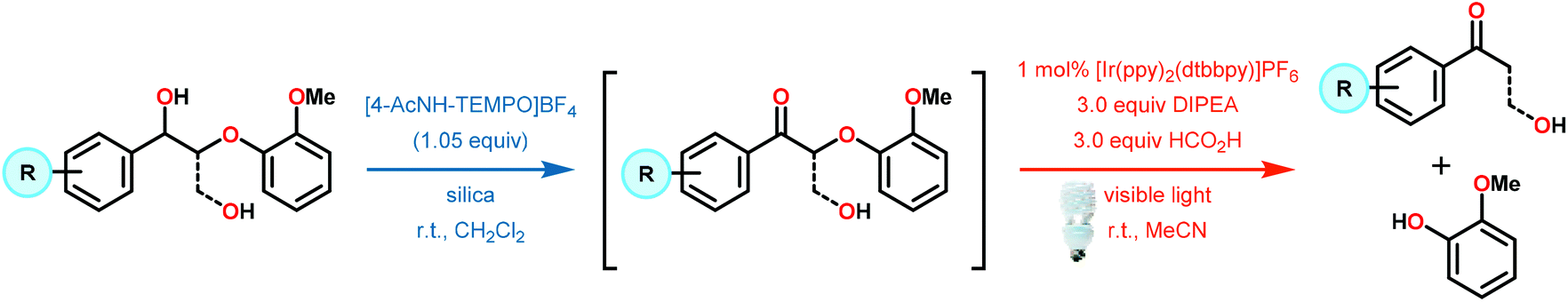
The extensive work with TEMPO-mediated oxidations of lignin model systems highlights that homogeneous catalysis with TEMPO reagents is indeed suitable for affecting the oxidation of lignin models. Selective oxidation of the benzylic alcohol as well as oxidative degradation of lignin models has shown how catalyst choice and catalyst amount can affect the efficiency and selectivity of the studied reaction. The knowledge acquired from this body of work is rapidly increasing as more advanced methods are being developed towards the depolymerization of more -sophisticated lignin model systems and native lignin itself.
4. Redox-neutral conversion of lignin
The ultimate atom-economical route for valorization of lignin would be to design a pathway where lignin is fragmented in a redox-neutral fashion. In this approach, the generated reductive equivalents stemming from the initial oxidation step would subsequently be used to reductively cleave the β-O-4 bond of the oxidized lignin fragment (see Fig. 34). This would allow carrying out the overall depolymerization of lignin through a one-pot tandem cleavage process without the use of stoichiometric additives or reagents. Although the two-step fragmentation process is formally redox-neutral, an initial oxidation step followed by a reduction step, the ideal redox-neutral transformation would utilize the same catalyst to perform both the oxidation and the reduction step and such catalytic systems are certainly not elementary to design. | ||
| Fig. 34 Principle for redox-neutral conversion of lignin and lignin model systems. | ||
Beckham and co-workers recently examined the bond dissociation energies (BDEs) of common bonds that are prevalent in native and oxidized lignin model dimers. By use of density functional theory (DFT) calculations the authors were able to quantitatively predict the relative BDEs for C–C and C–O bonds in an expanded range of lignin model compounds.102
It could be predicted that oxidation of the lignin model system (287) at either the γ-carbon (primary alcohol) or α-carbon (secondary alcohol) to an aldehyde or ketone, respectively, would significantly reduce the bond energy for C–O bond cleavage at the β-position (i.e. β-O-4 linkage). The BDEs for the ketone form (288) was shown to be on average 3 kcal mol−1 lower than the aldehyde form (289) (see Fig. 35).102 These results suggest that routes for depolymerization of lignin involving initial oxidation of either of the alcohol units in native lignin could lead to enhanced catalytic rates for the subsequent β-O-4 cleavage step.
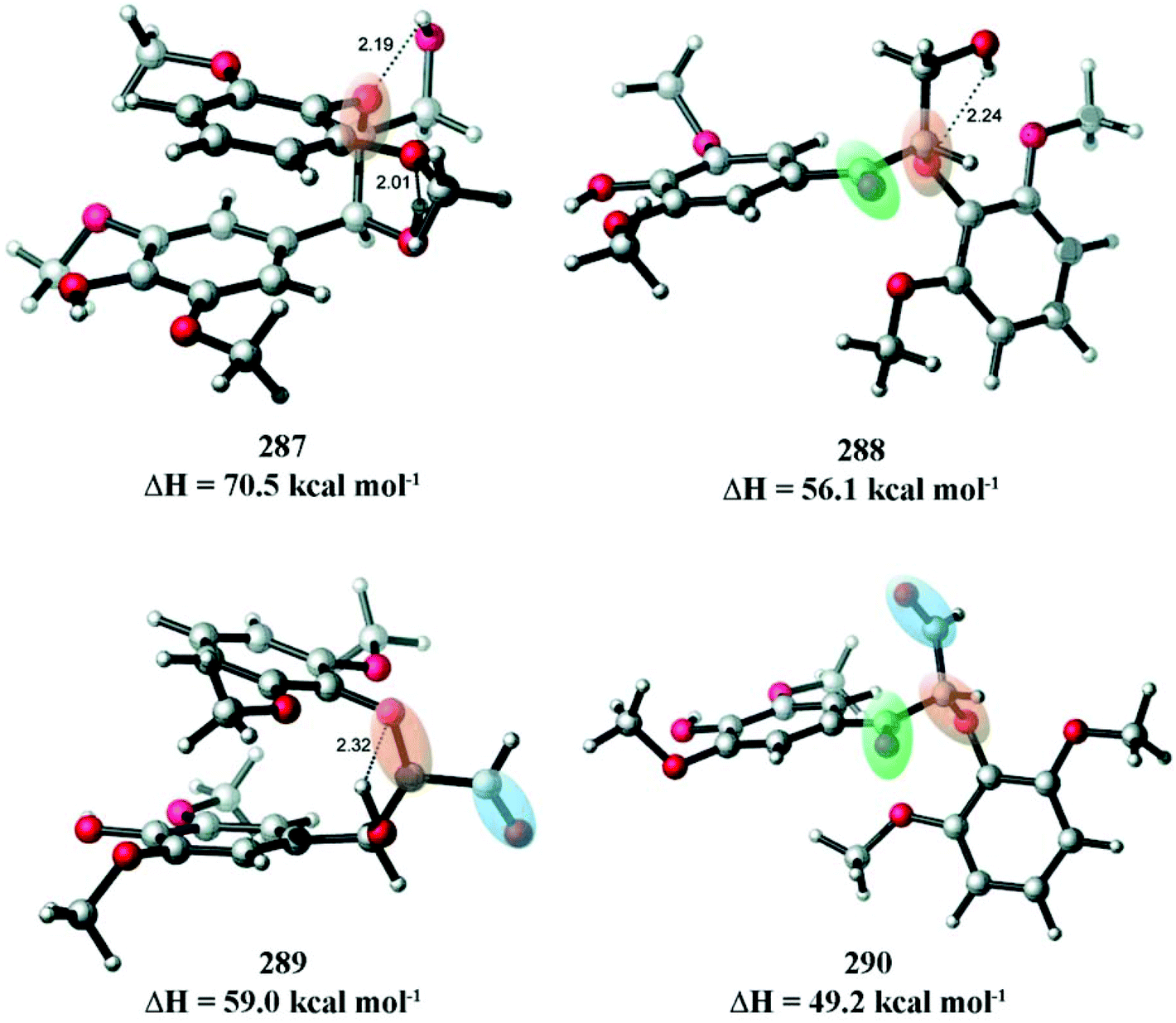 | ||
| Fig. 35 Comparison of β-O-4 in a native lignin model compound (287) and β-O-4 in various oxidized systems (288–290) for C–O bond cleavage at the β-position. Bond lengths are given in Å. The β-O-4 linkages (red) and the oxidized alcohol unit(s) (blue and green) have been highlighted for clarity. Adapted with permission from ref. 102. Copyright 2011 American Chemical Society. | ||
4.1 Ruthenium-promoted cleavage of lignin and β-O-4 model compounds
An elegant approach for the redox-neutral conversion of lignin model compounds was reported by Ellman, Bergman and co-workers.195 Ru(H)2(CO)(PPh3)3 and related complexes have previously been reported to be competent in affecting both dehydrogenation196,197 and C–O activation198,199 and thus seemed as a prominent starting point. Their approach utilized a Ru catalyst, produced in situ from Ru(H)2(CO)(PPh3)3 in combination with xantphos ((9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane)). The generated catalyst was able to affect the tandem catalytic dehydrogenation/C–O bond cleavage in a redox-neutral fashion without any additional reagents.195Initial experiments consisted of using 2-phenoxy-1-phenylethan-1-ol (116) as the model system to screen a variety of monodentate and bidentate ligands. In the absence of any ligand, catalytic amounts of Ru(H)2(CO)(PPh3)3 only resulted in minor amounts of cleaved products. Addition of monodentate phosphine ligands gave even lower conversion and use of bidentate ligands with a wide range of bite-angles were able to affect the dehydrogenation step to afford α-phenoxyacetophenone but were ineffective in promoting the subsequent β-O-4 reductive cleavage. However, quantitative yield of the cleaved products were obtained when using the wide bite-angle ligand xantphos, highlighting that a catalytic redox-neutral strategy is indeed achievable. A small set of lignin model systems were applied to the catalytic system and afforded in general high yields of the cleaved products (Table 5). However, a slight decrease in yield was observed when using a 2,6-dimethoxy substituted phenol (292).195
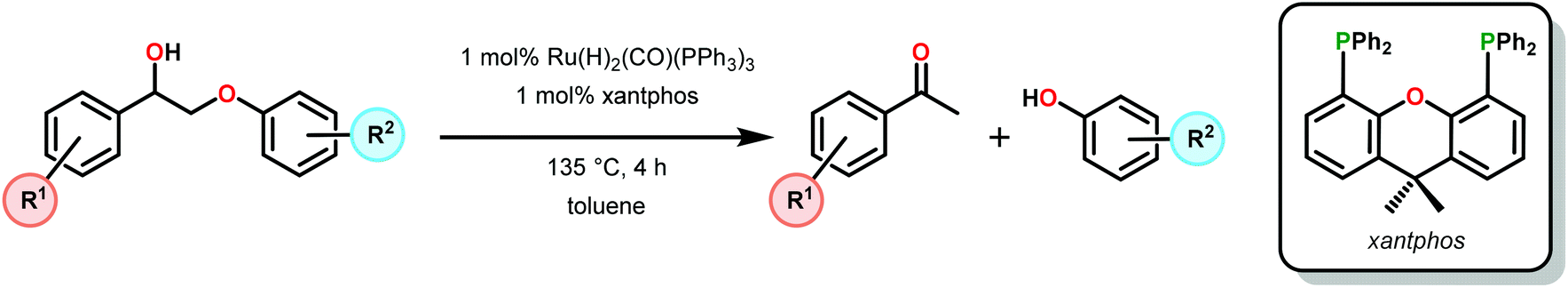
The authors subsequently applied the developed Ru-based catalytic system on an actual polymer of 2-aryloxy-1-arylethanol. Gratifyingly, the Ru-xantphos system was capable of providing quantitative depolymerization of poly(4′-hydroxy-1-phenethanol) to 4′-hydroxyacetophenone (107) by slightly modifying the reaction conditions (Scheme 30) in order to account for the poor solubility of the as-synthesized polymer. This result demonstrates the power of the designed catalytic system for redox-neutral cleavage of a lignin model polymer.195
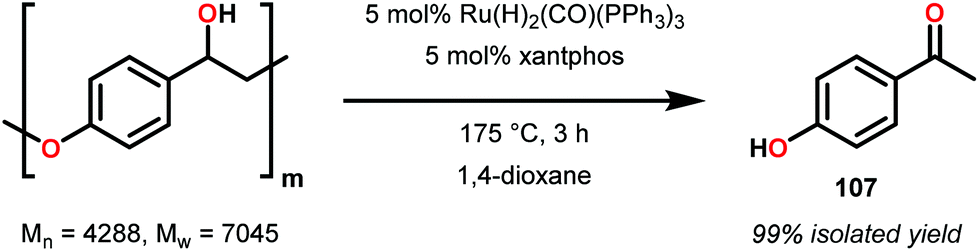 | ||
| Scheme 30 Redox-neutral depolymerization of lignin model polymer by the Ru(H)2(CO)(PPh3)3-xantphos system. | ||
Mechanistic studies revealed that blocking the dehydrogenation process inhibited the subsequent C–O bond cleavage (Scheme 31, top). In addition, the use of silane as a surrogate for molecular hydrogen successfully reduced the proposed intermediate 119 to afford the C–O bond cleaved products in 89% yield (Scheme 31, low). Lastly, deuterium labeling experiments using Et3SiD resulted in the formation of 55% deuterium incorporation at the α-keto position of the produced acetophenone. Collectively, these findings support the depicted mechanism in Fig. 36 where the Ru catalyst dehydrogenates the benzylic alcohol (116) to give the corresponding ketone (119). This is followed by C–O bond activation to produce a Ru-enolate (297), which upon hydrogenation results in a Ru-phenolate species (298), from which reductive elimination generates the phenol (62) to close the catalytic cycle.195 The group of Beckham subsequently performed DFT calculations on the Ru-xantphos system, which supported the suggested catalytic mechanism and further elucidated the kinetic and thermodynamic preferences of the Ru-based system.200
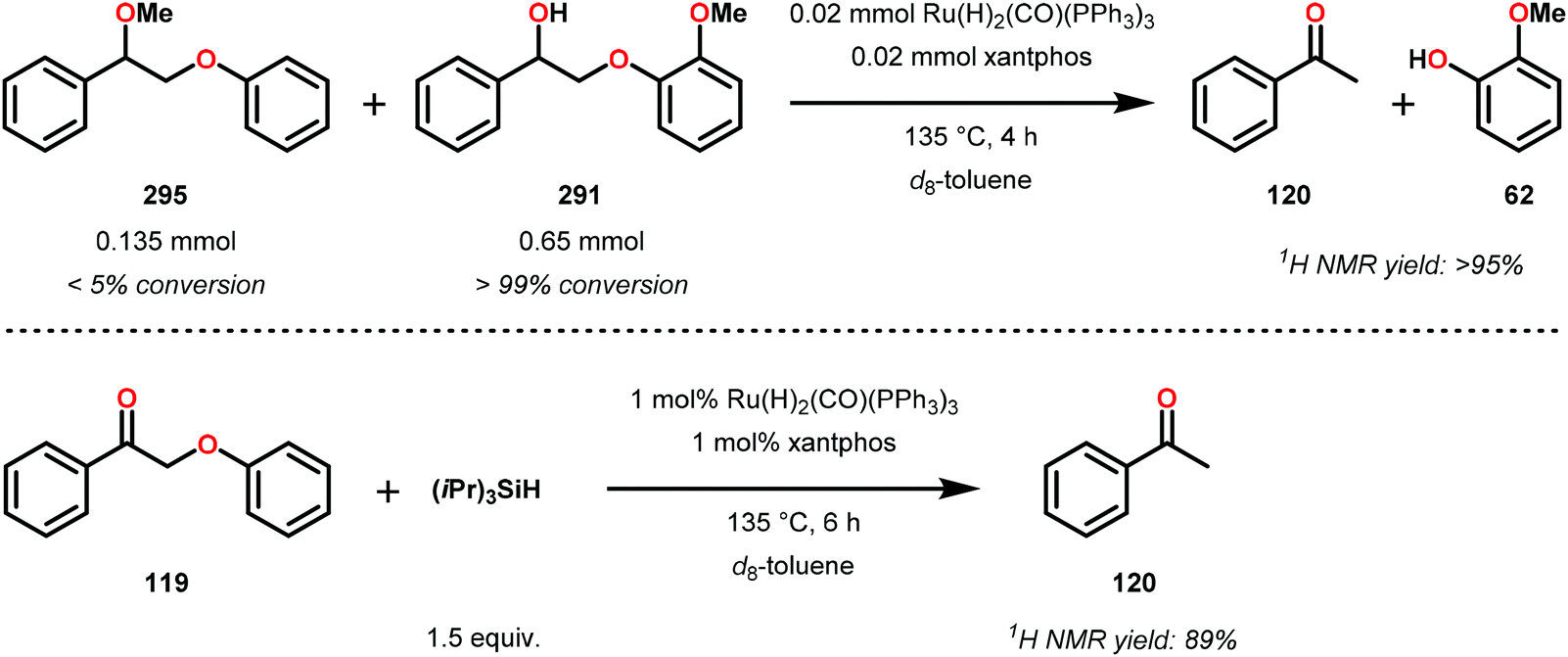 | ||
| Scheme 31 Mechanistic experiments conducted on the Ru(H)2(CO)(PPh3)3-xantphos system. | ||
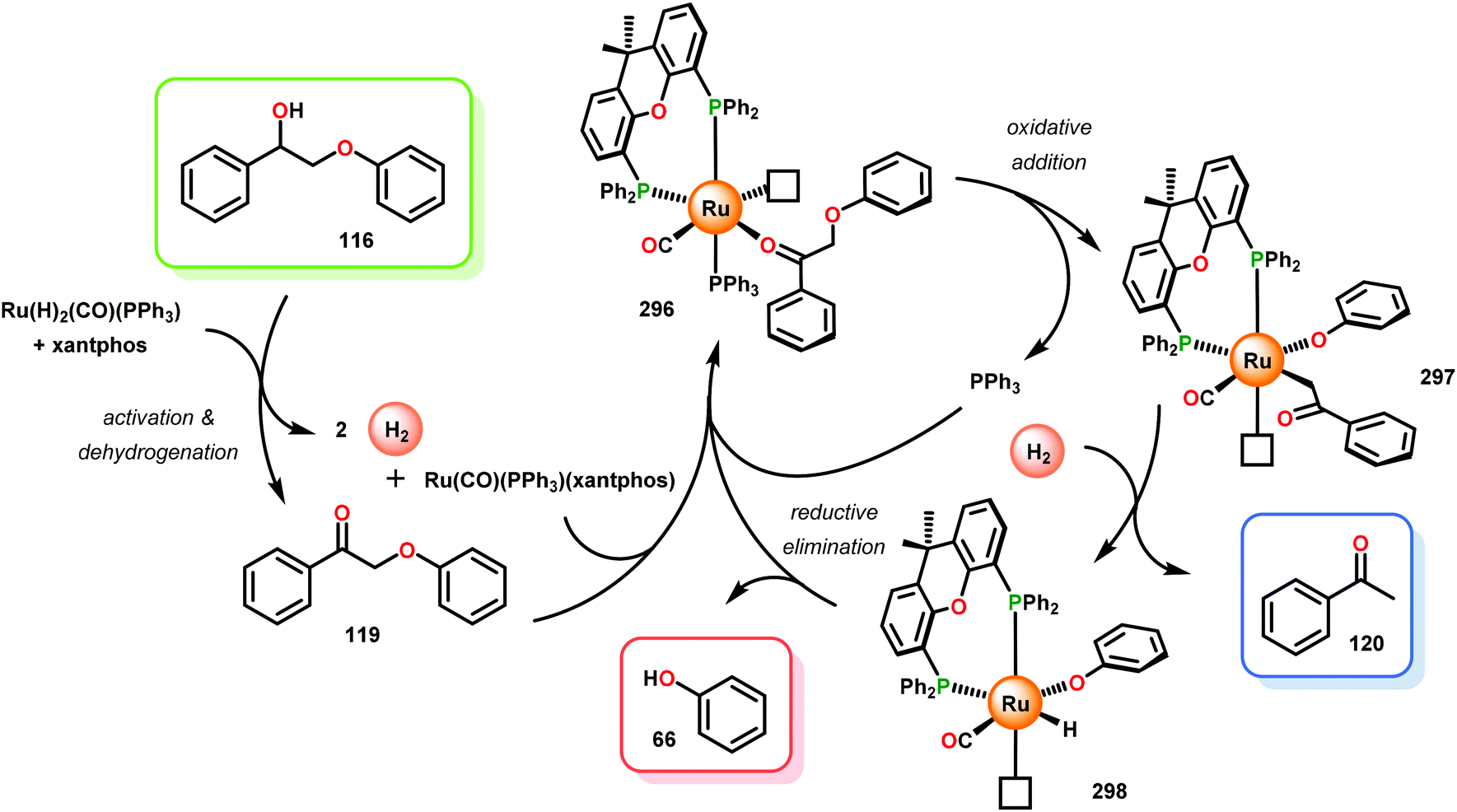 | ||
| Fig. 36 Proposed catalytic cycle for C–O bond cleavage by the Ru(H)2(CO)(PPh3)3-xantphos system. | ||
Although the discovery that the Ru-xantphos system was able to fragment lignin model compounds in a redox-neutral fashion, the necessity to implement the catalytic system on more complex and closely related native lignin systems should also be addressed. Inspired by Ellman, Bergman and co-workers, James and co-workers investigated the Ru-xantphos redox capability on more intricate model systems.201–203 Although the Ru(H)2(CO)(PPh3)3-xantphos system was applied to the efficient depolymerization of a synthetic polymer,195 James and co-workers found that the incorporation of a γ-OH functionality into the “simple” lignin model systems dramatically inhibited the formation of the hydrogenolysis products.201 When attempting to cleave the γ-OH containing compounds 299 and 300, only minor amounts of the hydrogenolysis product guaiacol were detected (Table 6). In addition to guaiacol, ketones 281 and 301 were observed, which most likely are formed by loss of formaldehyde. The conversions of compounds 299 and 300 under the studied conditions ranged from 10% up to 81%; however, the (by)products along with guaiacol and ketones 281 and 301 remain to be identified. Similar reactivity was also observed when using the reduced substrate 302, resulting in high conversion but with low yields of guaiacol and ketones 281 and 299 (Table 7).201 These findings clearly highlight that the inclusion of the γ-OH functionality severely restricts the catalytic hydrogenolysis reaction and limits the substrate scope of the Ru-xantphos system.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst (5 mol%) | Atmospherea | Conversionb (%) | Yieldb (%) | |
| 281/301 | 62 | |||||
| a 1 atm. b Yields and conversions were determined by 1H NMR based on two duplicate experiments. | ||||||
| 1 | 299 | Ru(H)2(CO)(PPh3)3-xantphos | Ar or H2 | 71–81 | 17–23 | 11–15 |
| 2 | 300 | Ru(H)2(CO)(PPh3)3-xantphos | Ar or H2 | 68–70 | 5–11 | 7–8 |
| 3 | 299 | — | Ar or H2 | 9–12 | 3–7 | 0 |
| 4 | 299 | Xantphos | Ar or H2 | 24–32 | 9–13 | 0 |
| 5 | 299 | Ru(H)2(CO)(PPh3)3 | Ar or H2 | 51–59 | 16–24 | 3–5 |
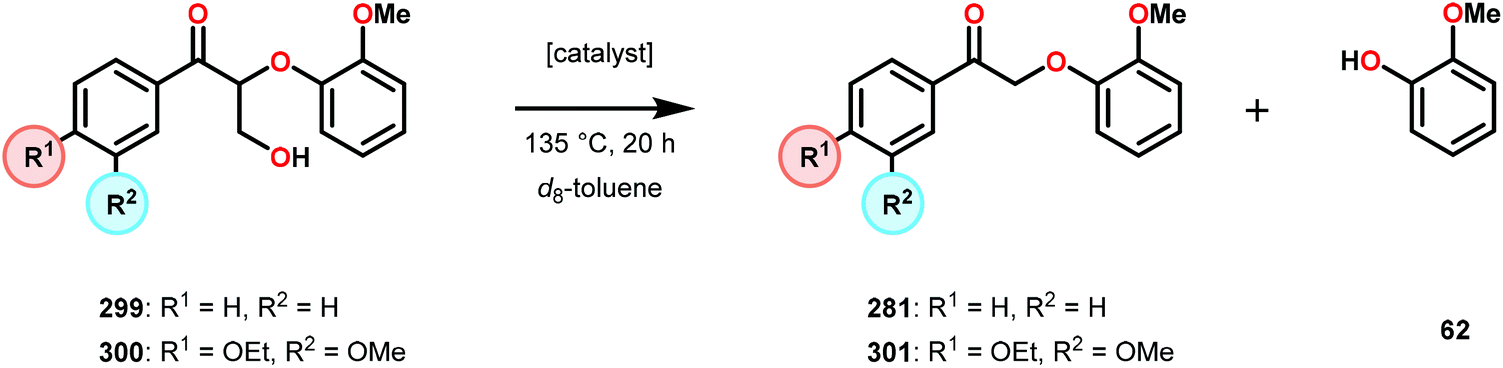
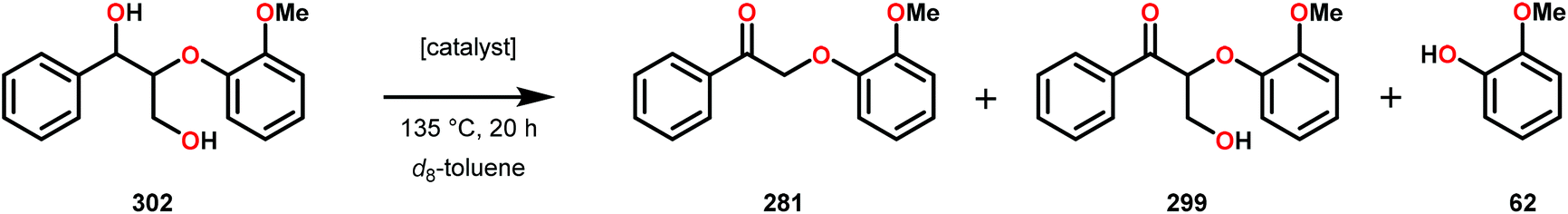
To probe the possible deactivation pathways, a scaled up reaction of ketone 299 and the Ru(H)2(CO)(PPh3)3-xantphos catalyst, resulted in the isolation of two white colored products, which were found to be Ru complexes 303 and 304 (Fig. 37). The structures of these complexes imply that substrate 299 has undergone a dehydrogenation accompanied by the formation of a stable Ru species where both α- and γ-carbons are coordinated to the metal center. Both of the isolated Ru complexes 303 and 304 were essentially inactive towards the β-O-4 cleavage of substrate 299. Interestingly, Ru complex 303 could not be interconverted to complex 304, implying that the two complexes originate from a common catalytic entity.201
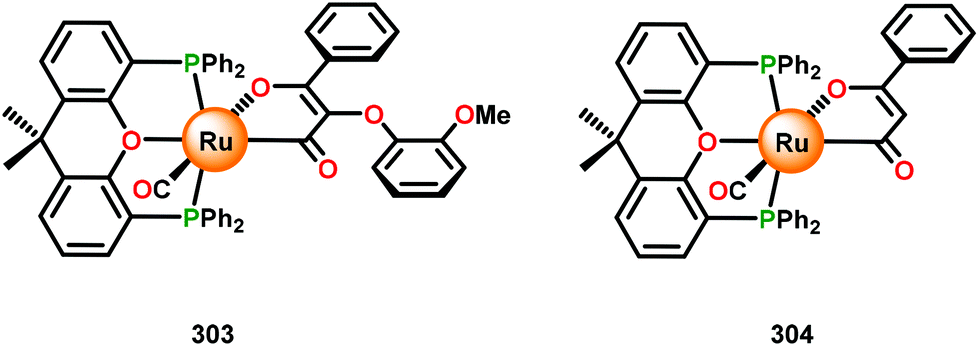 | ||
| Fig. 37 Structures of Ru complexes 303 and 304 generated after reaction with ketone 299. | ||
A tentative mechanism explaining the formation of Ru complexes 303 and 304 from reaction with ketone 299 is depicted in Scheme 32. It is assumed that the Ru(H)2(CO)(PPh3)3-xantphos catalyst dehydrogenates the primary alcohol moiety to form a Ru0-aldehyde species (307) with concomitant loss of H2. Such electron-rich Ru0-carbonyl intermediates have been documented to facilitate oxidative addition of the aldehyde unit to afford RuII-acyl-hydrido species.204 Oxidative addition of Ru0-aldehyde species 307 subsequently generates intermediate 308. The RuII-acyl-hydrido species 308 is proposed to be the common intermediate from which the two Ru complexes 303 and 304 are derived from. Loss of H2 from intermediate 308 would deliver Ru complex 303, or alternatively, if the H2 is used to hydrogenolyze the aryl ether moiety affords complex 304.201 This work highlights the deactivation pathways for the Ru(H)2(CO)(PPh3)3-xantphos system and that implementing redox-neutral fragmentation of simple lignin related compounds on more functionalized substrates housing the γ-OH unit is not straightforward and requires additional studies before redox-neutral conversion of native lignin can be carried out.
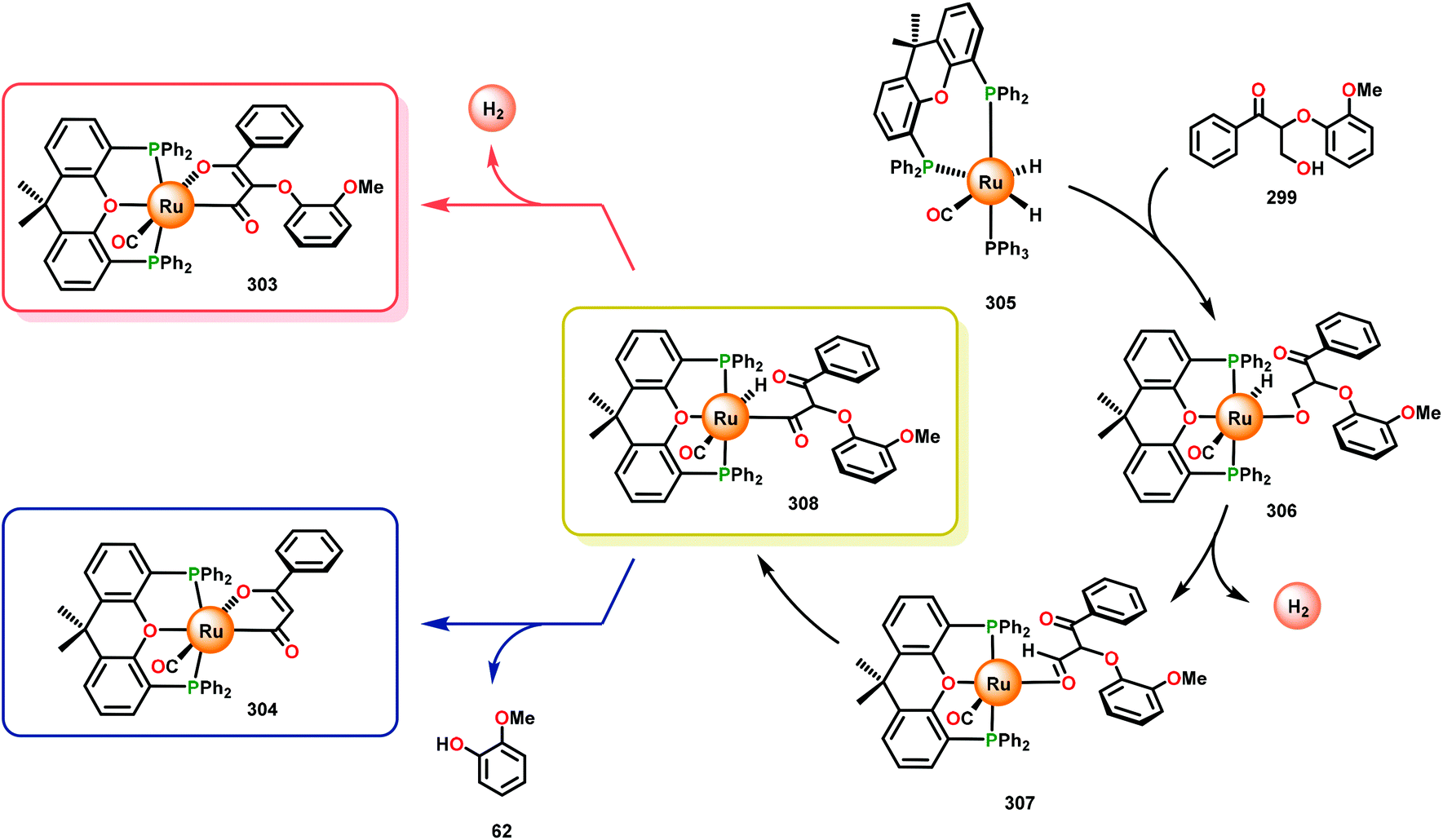 | ||
| Scheme 32 Proposed mechanism for formation of inactive Ru complexes 303 and 304 from reaction with lignin model compound 299. | ||
Subsequent studies by James and co-workers tried to address this lack of activity by modifying the γ-OH functionality to give model systems that would not irreversibly ligate to the Ru center.203 The γ-OH entity was therefore acetylated, a technique commonly employed to study and characterize lignin by NMR205 and mass spectrometry.206 Treating the acetylated compound 309 with the Ru(H)2(CO)(PPh3)3-xantphos catalyst under the previously established reaction conditions, resulted in complete conversion to hydrogenolysis products (see Table 8). However, attempts to transfer this methodology to acetylated native lignin were not successful.203 Although not being able to apply the strategy to native lignin, these findings are in sharp contrast to the studies with the non-acetylated substrates. Overall, the seminal studies on the Ru-xantphos system by Ellman, Bergman and James highlight the difficulties in identifying competent catalysts capable of mediating redox-neutral C–O bond activation of more complex systems that do not irreversibly ligate to the substrates.
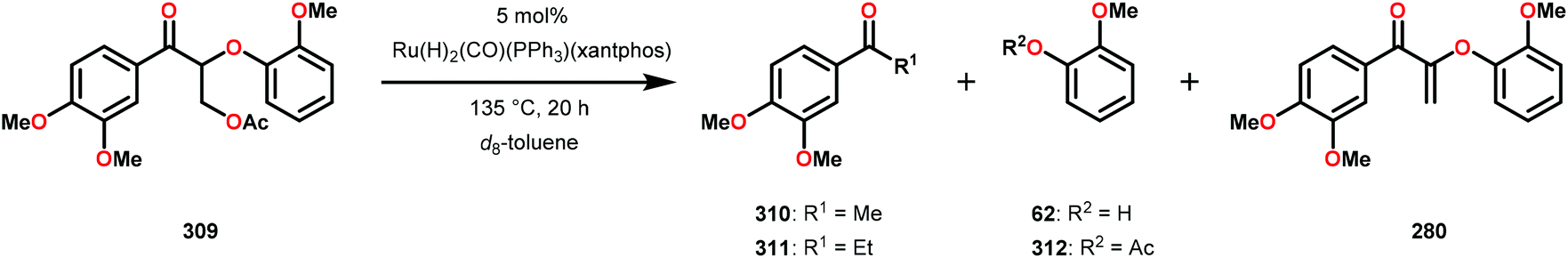
A subsequent study on the Ru(H)2(CO)(PPh3)3-xantphos system was carried out by Li and co-workers who demonstrated that RuHCl(CO)(PPh3)3 and Ru(H)2(CO)(PPh3)3 in the presence of inorganic bases, such as KOH, could be used in place of xantphos to affect the cleavage of β-O-4 lignin systems.207 In the absence of KOH, no product formation was observed when using pristine RuHCl(CO)(PPh3)3, which is in accordance with the earlier studies conducted by Ellman, Bergman and co-workers.195 Addition of KOH to either the RuHCl(CO)(PPh3)3 or Ru(H)2(CO)(PPh3)3 system had a dramatic impact on the catalytic activity. It is clear that KOH converts the inactive Ru precursors to active species in situ, which are responsible for the cleavage of the model substrates (Table 9). When applying the developed RuHCl(CO)(PPh3)3-base catalytic system on a lignin model compound housing a γ-OH unit, four products were observed (see Table 10). Relatively low yields of the corresponding veratryl-based products were obtained but with higher yields of guaiacol. A similar mechanism for the tandem dehydrogenation/hydrogenolysis was proposed as for the Ru-xantphos system (see Fig. 36).207
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Time (h) | Conversionb (%) | Yieldb (%) | |||
| R1 | R2 | Ketone | Phenol | ||||
| a Reactions were run with 0.8 mmol of the substrate in a Schlenk flask under an atmosphere of argon. b Yields and conversions were determined by GC relative to an internal standard. | |||||||
| 1 | 116 | H | H | 12 | 99 | 86 | 84 |
| 2 | 291 | H | 2-MeO | 36 | 93 | 80 | 75 |
| 3 | 292 | H | 2,6-(MeO)2 | 24 | 41 | 30 | 4 |
| 4 | 293 | 4-MeO | H | 12 | 91 | 80 | 77 |
| 5 | 294 | 3,4-(MeO)2 | 2-MeO | 24 | 95 | 66 | 45 |
| 6 | 313 | 3-OH-4-MeO | 2-MeO | 24 | 89 | — | 14 |
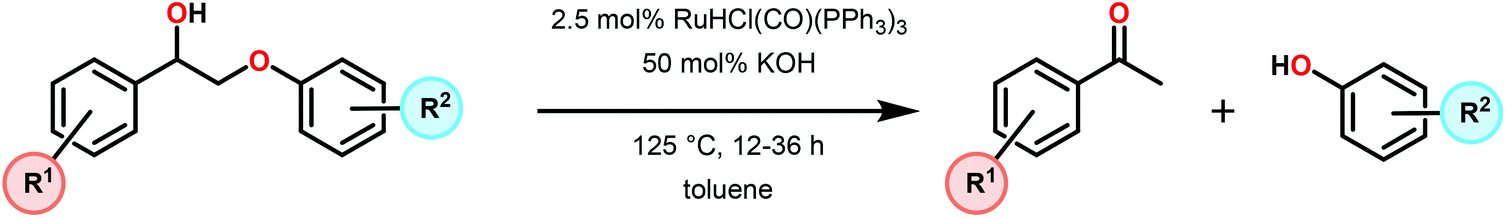
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Time (h) | Conversionb (%) | Yieldb (%) | |||
| 269 | 310 | 311 | 62 | |||
| a Reactions were run with 0.4 mmol of the substrate in a Schlenk flask under an atmosphere of argon. b Yields and conversions were determined by GC relative to an internal standard. c Minor product and not quantified. | ||||||
| 1 | 12 | 95 | 5 | 4 | —c | 18 |
| 2 | 24 | 94 | 7 | 6 | —c | 26 |
| 3 | 36 | 89 | 8 | 6 | —c | 26 |
| 4 | 48 | 91 | 11 | 4 | —c | 53 |
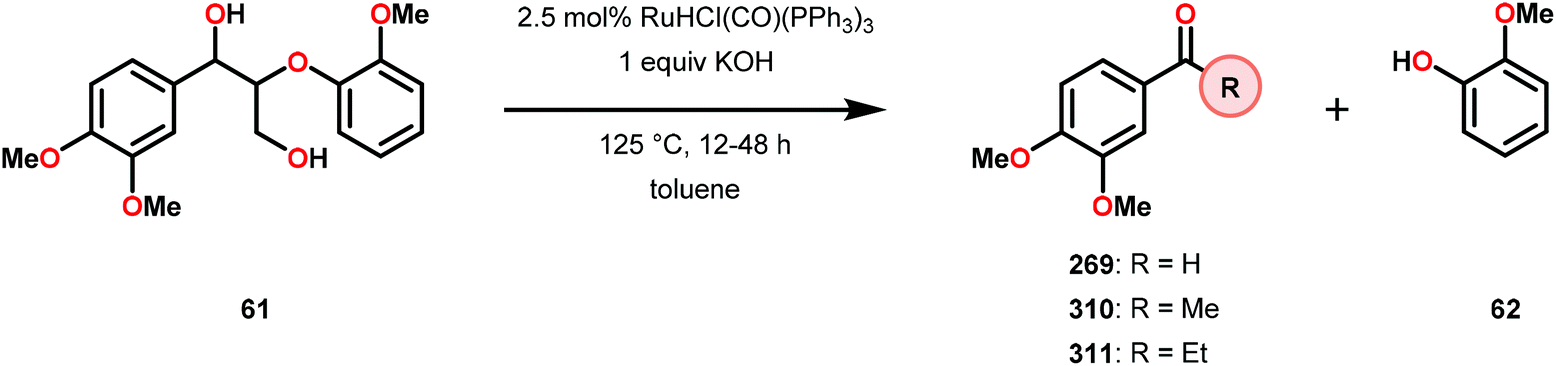
Since RuCl2(PPh3)3208 and related Ru complexes209,210 are known to mediate the direct alkylation of ketones with alcohols, via a so-called hydrogen borrowing strategy,211 Plietker and Weickmann recently coupled the redox-neutral β-O-4 cleavage to a sequential alkylation step where the derived acetophenone-based products could be alkylated with alcohols as the alkylating agent.212 The combination of the two processes allowed the formation of a variety of new structural motifs. Initial screening of the reaction conditions employed a variety of different Ru precursors (see Table 11). Simple Ru precursors such as RuCl2(PPh3)3 was found to efficiently cleave the β-O-4 linkage after activation with KOtamylate (Table 11, entry 2). It was revealed that a catalytic amount of EtOAc was necessary to obtain catalytic turnover (Table 11, entry 3). Thermal conditions, in the absence of any catalyst, did not yield any significant amounts of the cleaved products (Table 11, entry 1). Besides RuCl2(PPh3)3, several other common Ru precursors also gave the desired fragmented products in good to high yields (Table 11, entries 5–11). The influence of the monodentate phosphine ligands was also investigated and it could be concluded that more electron-donating phosphine ligands facilitated the cleavage (Table 11, entries 12 and 13), whereas more electron deficient and mixed alkyl–aryl phosphines were less effective (Table 11, entries 14 and 15).212
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | |
| 120 | 66 | ||
| a Reactions were carried out in a closed Schlenk tube under a N2 atmosphere with substrate 116 (0.125 mmol), Ru catalyst (5 mol%), KOtamylate (25 mol%) and EtOAc (25 mol%) in toluene (0.25 mL) at 140 °C for 18 h. b Determined by GC analysis using dodecane as internal standard. c Without EtOAc. d Reaction time of 4 h. | |||
| 1 | — | 2 | 13 |
| 2 | RuCl2(PPh3)3 | 94 | 94 |
| 3 | RuCl2(PPh3)3c | 23 | 27 |
| 4 | RuCl2(PPh3)3d | 68 | 74 |
| 5 | [Ru(p-cymene)Cl2]2 | 70 | 70 |
| 6 | [Ru(p-cymene)Cl2]2/PPh3 | 80 | 81 |
| 7 | Ru(OAc)2(PPh3)3 | 80 | 89 |
| 8 | Ru(OAc)2(PPh3)3c | 53 | 47 |
| 9 | Ru(dmso)4Cl2 | 52 | 58 |
| 10 | RuH2(PPh3)4 | 80 | 99 |
| 11 | RuHCl(CO)(PPh3)3 | 79 | 75 |
| 12 | RuCl2(P(p-MeC6H4)3)3d | 78 | 85 |
| 13 | RuCl2(P(p-MeOC6H4)3)3d | 72 | 84 |
| 14 | RuCl2(P(p-FC6H4)3)3d | 26 | 20 |
| 15 | RuCl2(PCyPh2)3d | 47 | 47 |
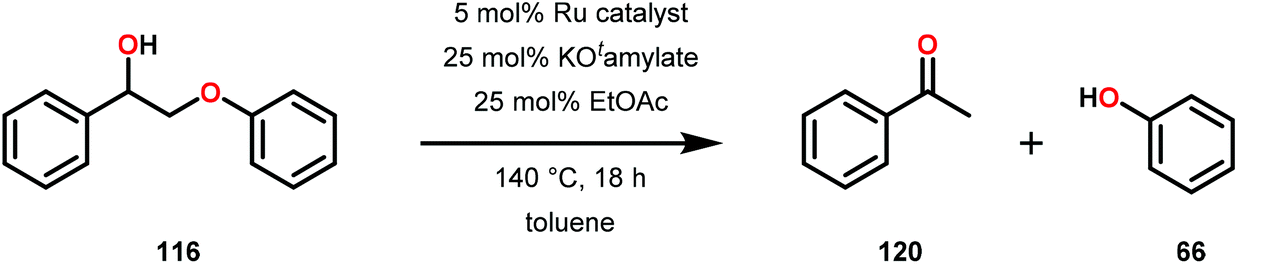
With the optimized reaction conditions in hand, a variety of lignin model systems were evaluated (Table 12). Model systems derived from the three different monolignols coumaryl-, coniferyl- and sinapyl alcohol were all successfully cleaved to the corresponding acetophenone and phenol products in good to excellent yields.212
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Yieldb (%) | |||
| R1 | R2 | Ketone | Phenol | ||
| a Reactions were carried out in a closed Schlenk tube under a N2 atmosphere with lignin model substrate (0.25 mmol), RuCl2(PPh3)3 (5 mol%), KOtamylate (25 mol%) and EtOAc (25 mol%) in toluene (0.5 mL) at 140 °C for 18 h. b Determined by GC analysis using dodecane as internal standard. c Isolated yield. | |||||
| 1 | 291 | H | 4-MeO | 59 | 53 |
| 2 | 293 | 4-MeO | H | 62 | 82 |
| 3 | 314 | 3,4-(MeO)2 | H | 98c | 98 |
| 4 | 294 | 3,4-(MeO)2 | 4-MeO | 81c | 62 |
| 5 | 315 | 3,4,5-(MeO)3 | H | 68c | 65 |
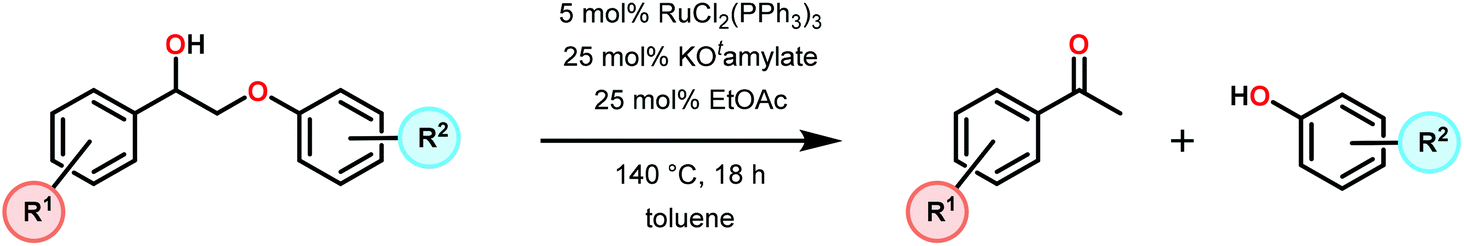
After completion of the β-O-4 cleavage of the lignin model systems, benzyl alcohol and a stoichiometric amount of KOH were added to the reaction mixture. Heating the resulting mixture for 16 h afforded the desired benzylated product in 71% yield (Table 13, entry 1). The Ru-catalyzed sequential β-O-4 cleavage/α-alkylation protocol was found to tolerate a wide variety of alcohols, such as benzylic-, heterobenzylic- and aliphatic alcohols (Table 13, entries 2–13). A tentative mechanism for the sequential β-O-4 cleavage/α-alkylation process is depicted in Scheme 33.212 Coupling the β-O-4 cleavage with a subsequent dehydrative α-alkylation process highlights that further in situ functionalization of the products stemming from cleavage of lignin model systems is viable for generation of a diverse array of lignin-derived value-added aromatic products.
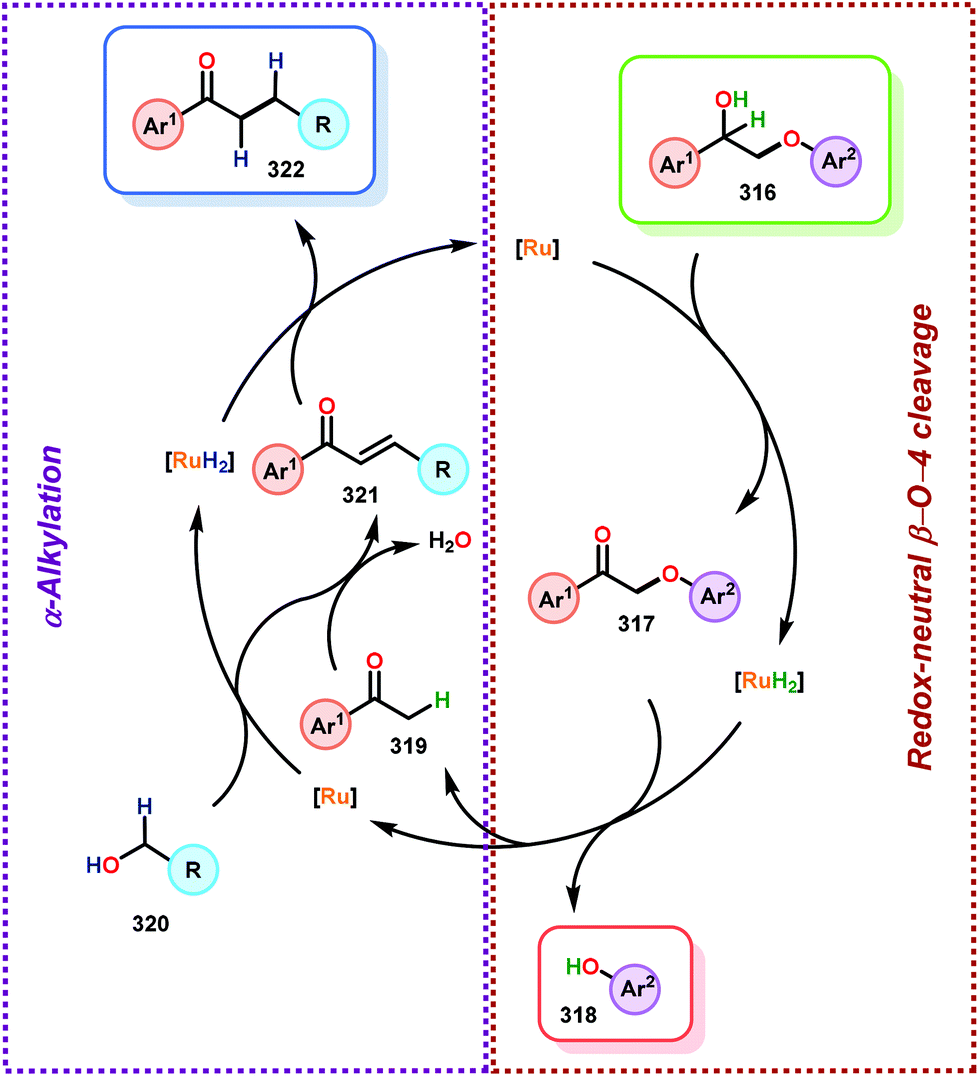 | ||
| Scheme 33 Plausible mechanism for the sequential β-O-4 cleavage/α-alkylation of lignin model compounds. | ||
|
|
|||
|---|---|---|---|
| Entry | Substrate | Alcohol | Yieldb (%) |
| a Reactions were carried out in a closed Schlenk tube under a N2 atmosphere with lignin model substrate (0.25 mmol), RuCl2(PPh3)3 (5 mol%), KOtamylate (25 mol%) and EtOAc (25 mol%) in toluene (0.5 mL) at 140 °C for 8 h. KOH (0.25 mmol) and alcohol (0.25 mmol) were then added and heated for additional 16 h. b Isolated yield. | |||
| 1 | R1 = H (116) | R2 = C6H5 | 71 |
| 2 | R1 = H (116) | R2 = 4-MeO-C6H4 | 43 |
| 3 | R1 = H (116) | R2 = 4-Cl-C6H4 | 51 |
| 4 | R1 = H (116) | R2 = 3,4-(MeO)2-C6H3 | 58 |
| 5 | R1 = H (116) | R2 = 2,4-Cl2-C6H3 | 47 |
| 6 | R1 = H (116) | R2 = 2-furanyl | 40 |
| 7 | R1 = H (116) | R2 = 2-thiophenyl | 40 |
| 8 | R1 = H (116) | R2 = CH2CH(CH3)2 | 47 |
| 9 | R1 = H (116) | R2 = C6H11 | 53 |
| 10 | R1 = 4-MeO (293) | R2 = C6H5 | 44 |
| 11 | R1 = 3,4-(MeO)2 (314) | R2 = C6H5 | 53 |
| 12 | R1 = 3,4-(MeO)2 (314) | R2 = C5H9 | 53 |
| 13 | R1 = 3,4,5-(MeO)3 (315) | R2 = C6H5 | 37 |

Leitner, Klankermayer and co-workers reported on an even more active Ru-based complex for the redox-neutral β-O-4 bond cleavage of lignin model system 116. By combining the Ru precursor [Ru(cod)(methallyl)2] (cod = 1,5-cyclooctadiene; methallyl = η3-C4H7) with tripodal phosphine based ligands, highly efficient Ru catalysts were obtained. Initial screening showed that monodentate and bidentate ligands such as PPh3 and dppp (dppp = 1,3-bis(diphenylphosphino)propane) resulted in low yields (Table 14, entries 1 and 2). However, when using the tripodal phosphine ligand 1,1,1-tris(diphenylphosphinomethyl)ethane (L1), a highly active catalytic system was obtained, affording the cleaved products 120 and 66 in 65% and 67% yield, respectively (Table 14, entry 4). Increased activity was observed with bis(diphenylphosphinoethyl)phenylphosphine (L2) where substrate 116 was selectively fragmented to produce acetophenone and phenol in 93% and 84% yield, respectively (Table 14, entry 5).213
|
|
|||
|---|---|---|---|
| Entry | Ligand | Yieldb (%) | |
| 120 | 66 | ||
| a Reactions were carried out with substrate 116 (0.2 mmol), Ru precursor (5 mol%) and ligand (5 mol%) in toluene (0.5 mL) at 135 °C for 2 h. b Determined by GC analysis using dodecane as internal standard. c 3 equiv. of PPh3 with respect to Ru precursor. dppp = 1,3-bis(diphenylphosphino)propane. | |||
| 1 | PPh3c | 5 | 5 |
| 2 | DPPP | 2 | 1 |
| 3 | Xantphos | 11 | 12 |
| 4 | L1 | 67 | 65 |
| 5 | L2 | 93 | 84 |
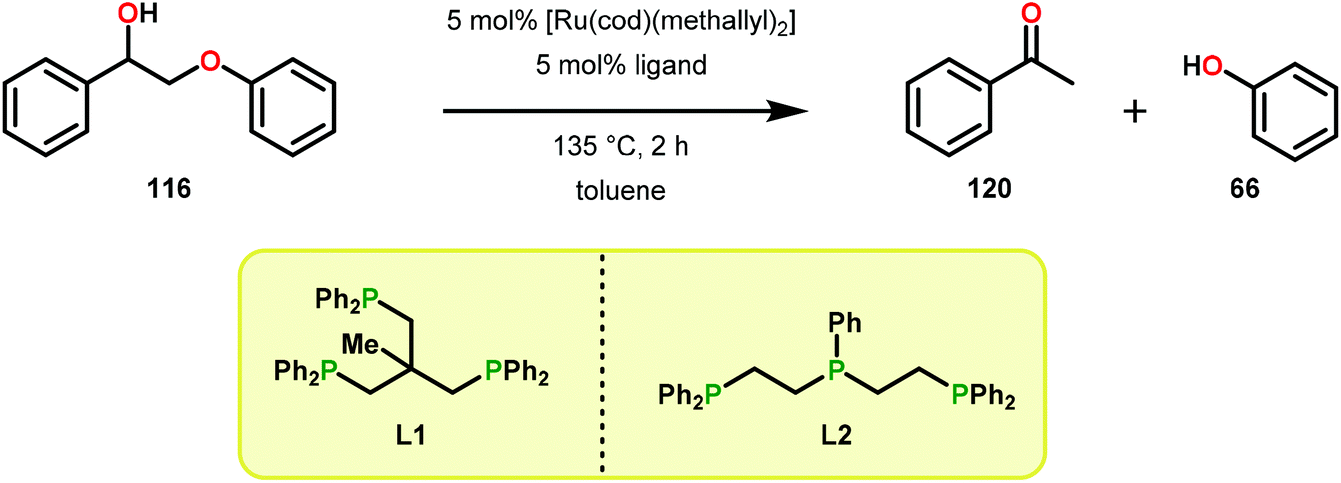
The unique activity prompted the authors to further study the catalytic system. A crystal structure of the Ru-tripodal phosphine complex showed that the structure was comprised of a doubly anionic [η3-C4H6]2− ligand where all three phosphorus atoms were coordinated to the RuII center to give complex 323 (Fig. 38). Conducting the catalytic experiments with isolated complex 323, cleavage was observed even at temperatures as low as 60 °C (Table 15).213 These results show that enhanced activity for the cleavage of lignin model substrates can be achieved by appropriate tailoring of the ligand scaffold.
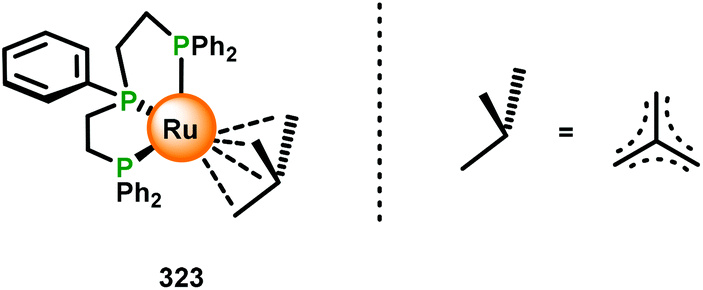 | ||
| Fig. 38 Molecular structure of Ru complex 323. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ru catalyst 323 (mol%) | T (°C) | Time (h) | Yieldb (%) | |
| 120 | 66 | ||||
| a Reactions were carried out with substrate 116 (0.2 mmol) and Ru complex 323 (5 mol%) in toluene (0.5 mL) at 60–135 °C for 2–20 h. b Determined by GC analysis using dodecane as internal standard. | |||||
| 1 | 5 | 60 | 20 | 26 | 25 |
| 2 | 5 | 80 | 20 | 75 | 75 |
| 3 | 5 | 100 | 20 | 94 | 92 |
| 4 | 5 | 135 | 2 | 96 | 92 |
| 5 | 1 | 135 | 2 | 91 | 90 |
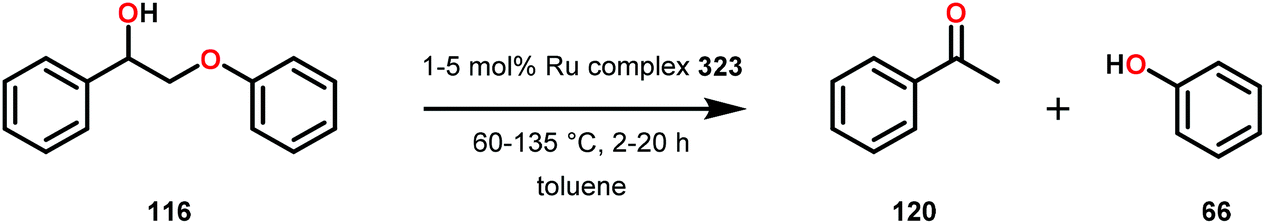
A follow-up study by Klankermayer and co-workers on these Ru-triphos systems revealed an unprecedented reactivity and selectivity of these complexes. The authors were able to carry out redox-neutral C–C bond cleavage on γ-OH containing lignin model substrates. Initial screening experiments using different tripodal-based phosphine systems showed that the use of Ru catalyst 329 afforded the C–C bond cleavage product 324 in 66% yield whereas the C–O bond cleavage product 62 was only detected in 4% yield (Table 16, entry 5). It could also be concluded that the catalyst had a strong influence on the catalytic activity (Table 16, entries 1–4).214
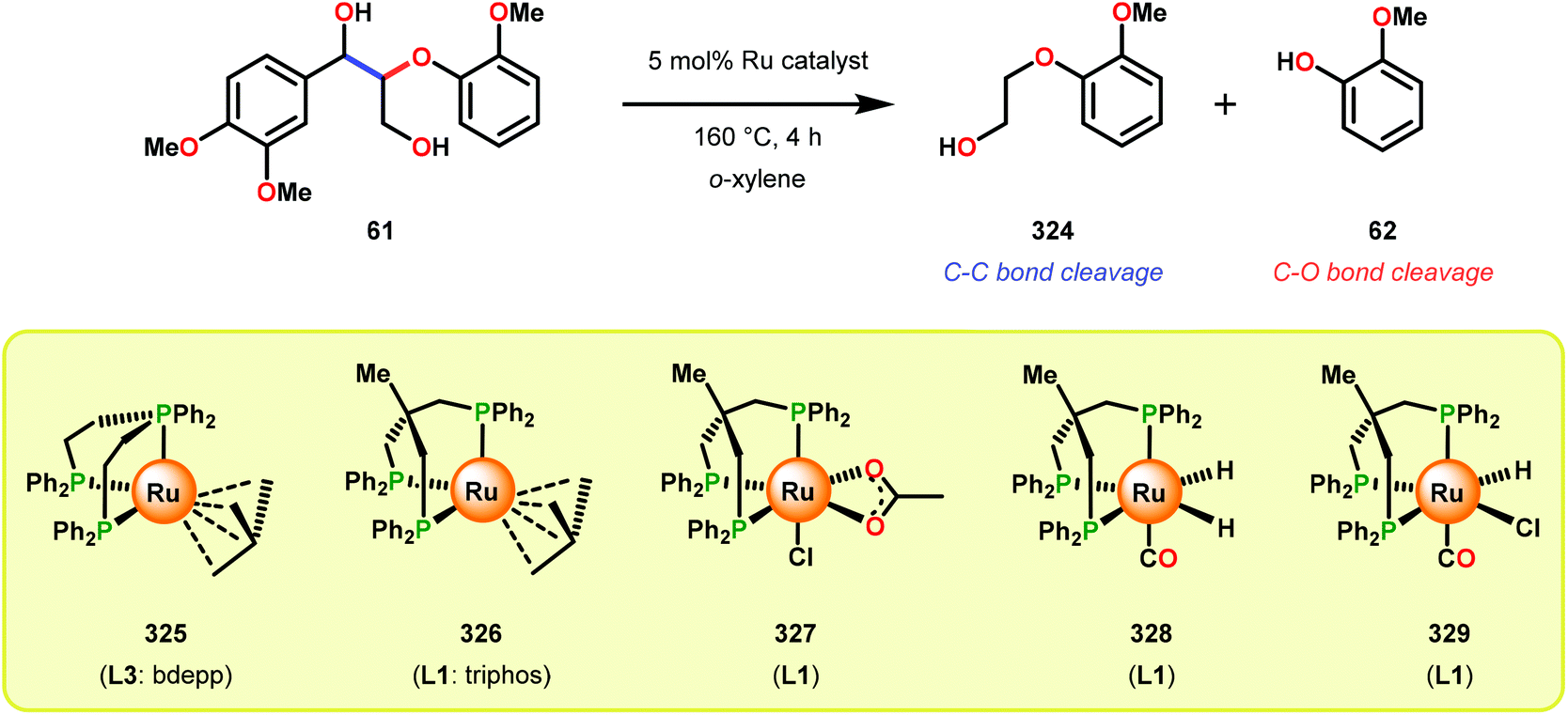
Since Ru complex 329 afforded the desired C–C bond cleavage product in good yield, the RuHCl(PPh3)3 precursor was used in combination with selected phosphine ligands (Table 17). When carrying out the catalytic reactions with the bidentate ligands xantphos or dppp, only minor amounts of the C–C bond cleavage products were detected (Table 17, entries 2 and 5); however, use of the tridentate triphos ligand L1 afforded the C–C cleavage products in high yields (∼76%) after 4 h (Table 17, entry 6). Comparable yields (70%) were observed after merely 1 h (Table 17, entry 7) when performing the reactions at 160 °C. These findings highlight the potential of the Ru-triphos complexes for mediating redox-neutral hydrogen transfer reactions. Different phenyl and phenol substituted lignin model systems were subsequently evaluated with the RuHCl(PPh3)3/triphos ligand L1 system. It was shown that the substitution pattern of the phenol ring had a moderate effect on the yield of the C–C bond cleavage (Table 18, entries 1–3 and 5). However, the sterically encumbered ortho functionalized substrates 332 only produced aldehyde 269 in 26% yield (Table 18, entry 4). This trend could also be observed with the highly substituted 2,4,5-trimethoxy model system 334, which yielded its corresponding C–C cleavage product in 14% yield (Table 18, entry 6).214
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | T (°C) | Time (h) | Yieldb (%) | |
| 269 | 324 | ||||
| a Reactions were carried out with substrate 61 (0.1 mmol) and RuHCl(PPh3)3 (5 mol%) in toluene (0.5 mL) at 140 °C for 1–4 h. The catalyst precursor/ligand solution was preheated at 140 °C for 2 h. b Determined by GC analysis using dodecane as internal standard. c Reaction was performed with ortho-xylene (0.5 mL) as the solvent. | |||||
| 1 | None | 140 | 4 | 8 | 3 |
| 2 | Xantphos | 140 | 4 | 2 | 2 |
| 3 | L1 | 140 | 4 | 64 | 64 |
| 4 | L2 | 140 | 4 | 26 | 31 |
| 5 | dppp | 140 | 4 | 8 | 6 |
| 6 | L1 | 160c | 4 | 75 | 77 |
| 7 | L1 | 160c | 1 | 69 | 70 |
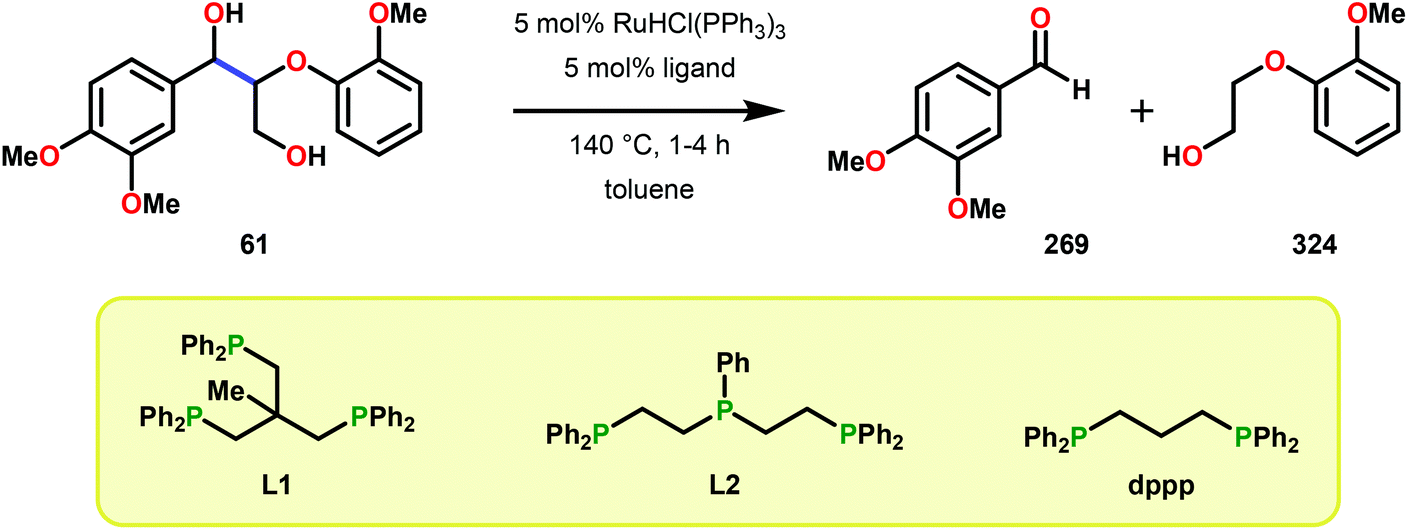
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Yieldb (%) | |
| Aldehyde | Alcohol | ||||
| a Reactions were carried out with substrate (0.1 mmol), RuHCl(PPh3)3 (5 mol%) and ligand L1 (5 mol%) in ortho-xylene (0.5 mL) at 160 °C for 4 h. The catalyst precursor/ligand solution was preheated at 140 °C for 2 h. b Determined by GC analysis using dodecane as internal standard. | |||||
| 1 | 61 | 3,4-(MeO)2 | 2-MeO | 75 | 77 |
| 2 | 330 | 3,4-(MeO)2 | 3,5-(MeO)2 | 86 | — |
| 3 | 331 | 3,4-(MeO)2 | H | 63 | — |
| 4 | 332 | 3,4-(MeO)2 | 2,6-(MeO)2 | 26 | — |
| 5 | 333 | 3-MeO-4-EtO | 2-MeO | — | 75 |
| 6 | 334 | 2,4,6-(MeO)3 | 2-MeO | — | 14 |
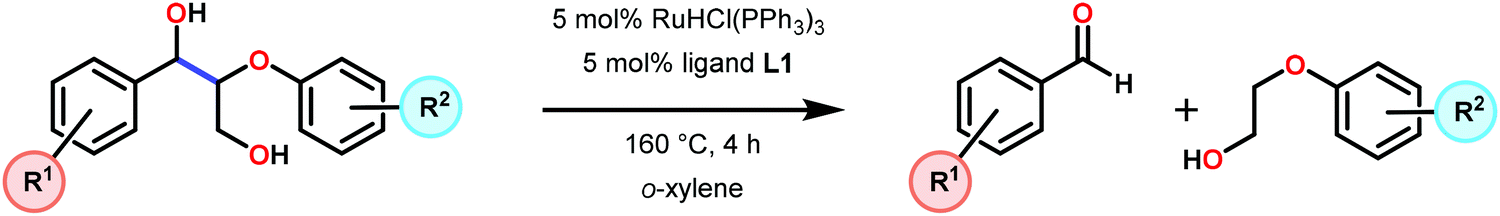
Insight into the catalytic mechanism for these hydrogen transfer reactions was obtained by studying a variety of substituted lignin model systems (Scheme 34). Bis(methylether) substrate 335 and the benzylic ethyl ether compound 336 were practically inactive toward C–C and C–O bond cleavage, giving the fragmented products in <2%. However, substrate 337 containing the benzylic alcohol and a primary methyl ether unit yielded the C–O bond cleavage product in 39% yield. This reactivity pattern suggests that both alcohol moieties need to be present in order for efficient C–C bond cleavage to occur, whereas C–O bond fragmentation only requires the benzylic alcohol. Additional support for this hypothesis was given from the catalytic activity of lignin model systems 338 and 339 containing tertiary alcohol units where exclusive formation of the C–O cleavage product was observed with substrate 339. Furthermore, species 344 (Scheme 35) was successfully characterized by NMR spectroscopy and was also formed upon reacting the substrate with RuHCl(CO)(L1), implying that this species is a potential resting state of the Ru-triphos system.214 In contrast to the catalytically inactive Ru species 303 (Fig. 37),201 isolated by James and co-workers, the pentacoordinated intermediate 344 maintains a vacant coordination site, which may in part account for this catalytic improvement.
 | ||
| Scheme 34 Reactivity pattern of the RuHCl(PPh3)3/triphos ligand L1 system. | ||
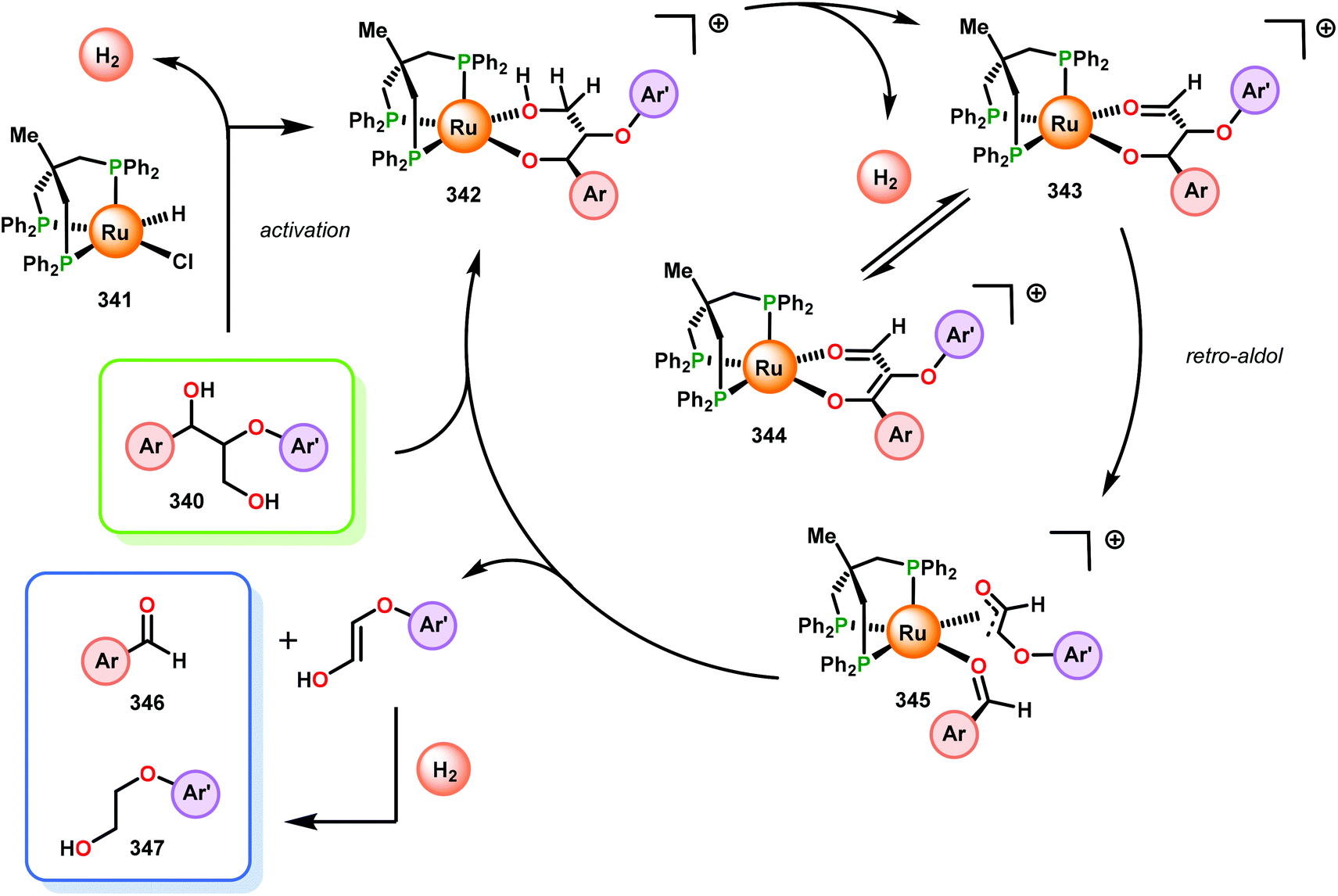 | ||
| Scheme 35 Proposed redox-neutral hydrogen transfer/retro-aldol mechanism for C–C bond cleavage of lignin model compounds by the Ru-triphos catalytic system. | ||
Based on these observations, a mechanistic proposal for the sequential redox-neutral hydrogen transfer/retro-aldol C–C bond cleavage was presented and is outlined in Scheme 35.214 The initial step in the proposed mechanistic pathway involves coordination of the substrate to the Ru catalyst, with concomitant liberation of H2 to generate 342. Subsequent dehydrogenation of intermediate 342 results in species 343, which undergoes a retro-aldol type C–C bond cleavage to form the product aldehyde 346 and Ru species 345, where the latter can react with the substrate, closing the catalytic cycle. The generated enol is subsequently hydrogenated under the reaction conditions to yield the observed saturated alcohol moiety.214 This work emphasizes that simple ligand tuning of homogeneous Ru-based catalytic systems can provide access to systems exhibiting unprecedented catalytic activity and selectivity towards the redox-neutral conversion of lignin model systems.
4.2 Other transition-metal based systems for redox-neutral conversion of lignin and lignin-related substrates
A novel reactivity pattern for carrying out redox-neutral degradation of lignin model systems was reported by Toste and co-workers.166 This catalytic system utilized a V-based catalyst where the redox-neutral fragmentation was proposed to proceed through a ketyl radical pathway. Initial experiments explored various V-based catalysts to selectively convert the lignin model system. A majority of the tested catalysts gave the oxidized ketone 349 as the major product (Table 19, entries 2–7). However, the use of V complexes with Schiff base ligands was shown to favor C–O bond cleavage over the benzylic oxidation, where ligands with larger bite angles displayed the highest selectivity (Table 19, entries 8–11). As with the Ru-based catalytic systems, subtle modifications in the ligand scaffold were able to change the reactivity of the V system from benzylic oxidation toward promoting C–O bond cleavage.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conversiona (%) | Yielda (%) | ||
| 348 | 62 | 349 | |||
| a Determined by 1H NMR spectroscopy versus an internal standard. | |||||
| 1 | None | 0 | — | — | — |
| 2 | VOSO4·xH2O | 34 | 2 | 2 | 6 |
| 3 | VO(acac)3 | 79 | 13 | 22 | 31 |
| 4 | VO(OiPr)3 | 82 | 5 | 11 | 45 |
| 5 | 350 | 86 | 6 | 6 | 59 |
| 6 | 351 | 66 | 13 | 14 | 41 |
| 7 | 352 | 55 | 3 | — | 37 |
| 8 | 353 | >95 | 61 | 45 | 27 |
| 9 | 354 | 86 | 70 | 62 | 8 |
| 10 | 355 | 95 | 65 | 50 | 18 |
| 11 | 256 | >95 | 82 | 57 | 7 |
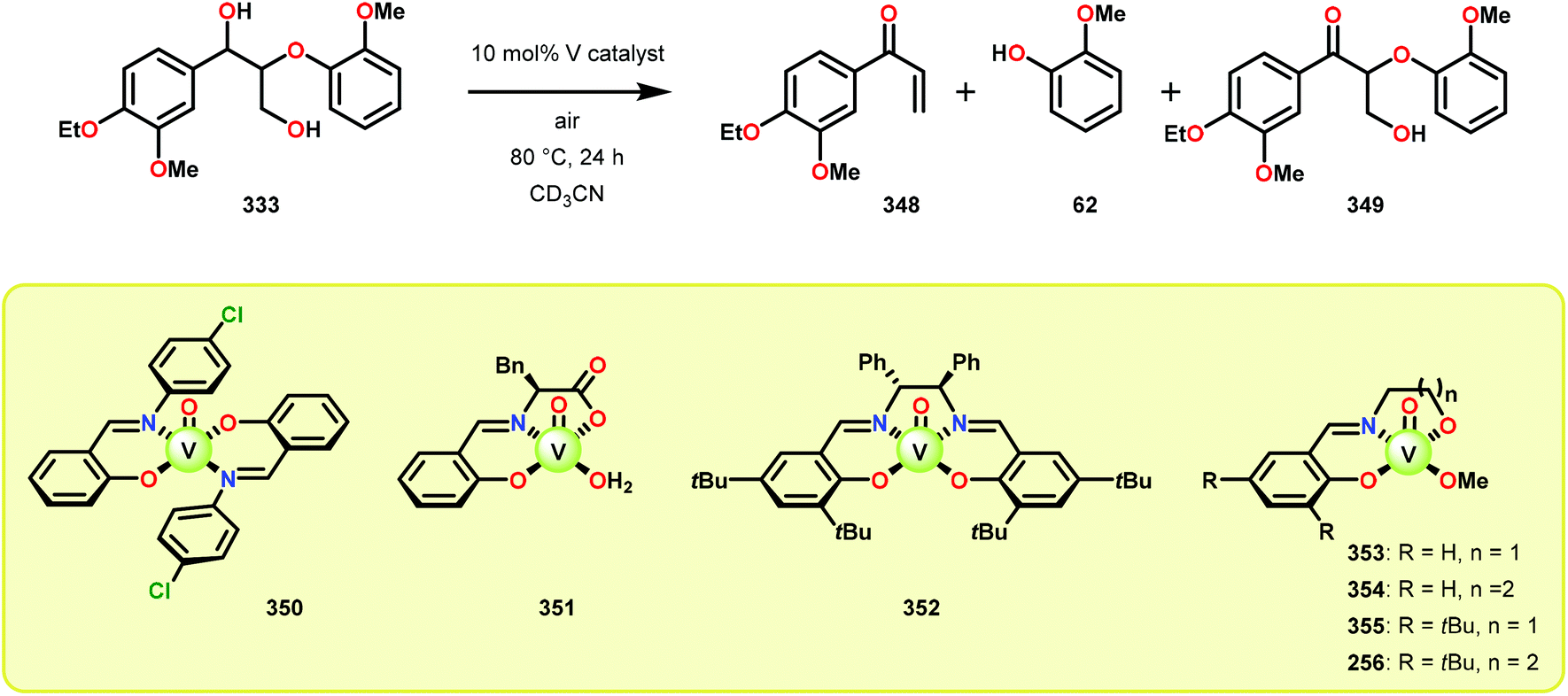
It was shown that molecular oxygen was not essential to achieve catalytic turnover. When performing the catalytic experiments under anaerobic conditions with V complex 256, a pale purple precipitate was isolated, which was revealed to be the VIV complex. These results suggest that the initial VV catalyst is reduced to a VIV species during the fragmentation. Fundamental insight into the mechanism of the non-oxidative C–O bond cleavage was obtained from the reactivity of different analogues of the lignin model substrate (Scheme 36, top). Using a substrate where the benzylic alcohol was methylated (356), afforded the conjugated aldehyde 357 in low yield, suggesting that ligand exchange with the benzylic hydroxyl moiety is essential for the catalytic reactivity. However, employing the benzyl methyl ether substrate 358, high conversion was observed to the desired C–O bond cleavage products 348 and 62 (see Scheme 36, lower).166
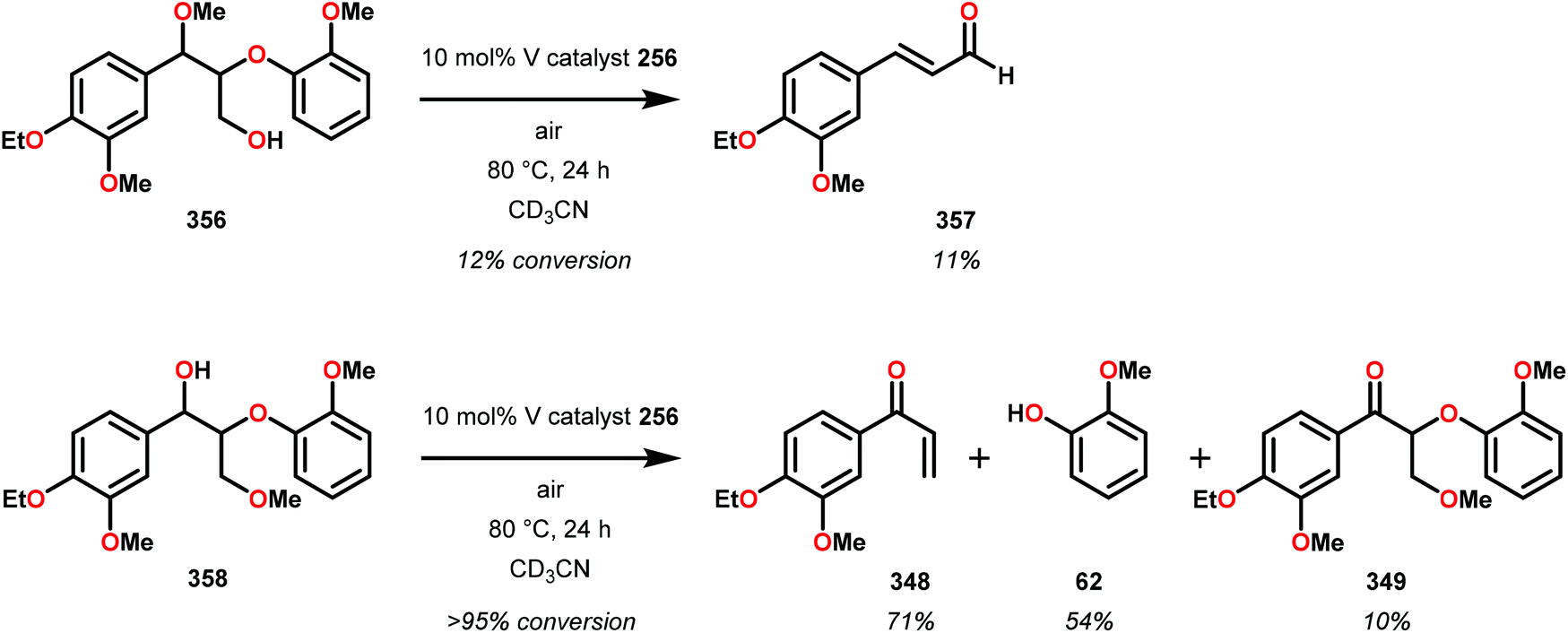 | ||
| Scheme 36 Reactivity study of V complex 256. | ||
Additional experiments consisted of performing the reaction in another solvent than MeCN. When conducting the cleavage in EtOAc, >95% conversion was observed, affording the cleaved products 348 and 62 in even higher yields (93% and 70%, respectively, cf. Table 19, entry 11). The trimeric lignin model compound 359 also underwent clean cleavage to furnish three monomeric species, showing that the V system can be applied on more intricate lignin systems (Scheme 37).166
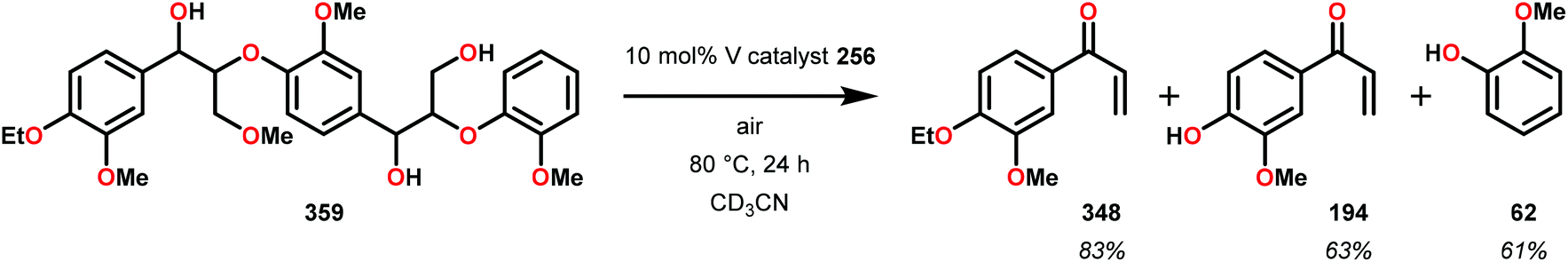 | ||
| Scheme 37 Redox-neutral cleavage of trimeric lignin model system 359 by V catalyst 256. | ||
A one-electron catalytic process was invoked and is depicted in Scheme 38. After the initial ligand exchange with the benzylic alcohol unit, the benzylic Hα is abstracted, producing a ketyl radical (361) from which subsequent fragmentation occurs to produce an aryloxy radical (362) and enolate 363. Elimination of the hydroxy moiety from the resulting enolate 363 generates the enone product 364 and VIV species 365, which can be reoxidized to VV by the produced aryloxy radical (362).166 This work demonstrates that the use of V catalysis may dramatically alter the reactivity by which lignin and related compounds can be cleaved. By subtle changes in the ancillary ligands, a novel fragmentation scenario was realized, affording functionalized aryl enones in high yields and good selectivities.
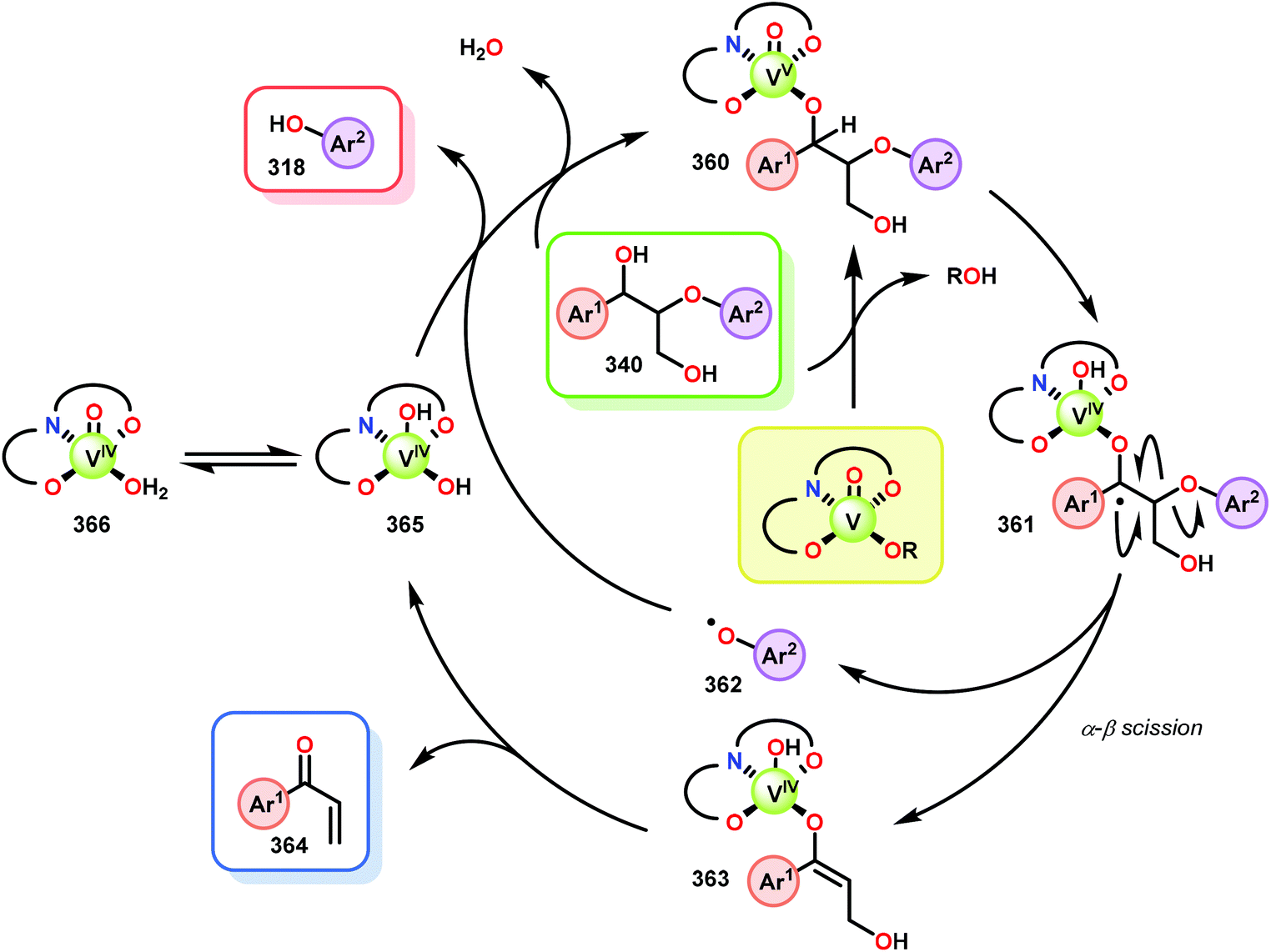 | ||
| Scheme 38 Proposed mechanism for the V-catalyzed redox-neutral cleavage of lignin model systems. | ||
In a subsequent study Toste and co-workers assessed the reactivity of the V-catalyzed redox-neutral depolymerization toward lignin extracted from Miscanthus giganteus with various solvents.215 The plant-derived lignin was extracted using dioxane, acetone and ethanol, affording dioxasolv, acetosolv and ethanosolv lignin, respectively. The degradation studies were carried out at 80 °C for 24 h in MeCN/THF or EtOAc/THF mixtures. For dioxasolv lignin, 24 h treatment with V catalyst 256 lowered the overall molecular weight distribution of the lignin sample, as determined by gel permeation chromatography (GPC) analysis. Similar results were obtained with acetosolv lignin, which produced a shift toward lower molecular weight species after catalysis. However, when using ethanosolv derived lignin, GPC analysis revealed a lesser degree of depolymerization. The lower reactivity of ethanosolv lignin was attributed to the prevalence of ethylated benzylic hydroxyl moieties (cf. Scheme 36), which is a result of the acidic pretreatment, thus highlighting that pretreatment and isolation significantly alter the structure of native lignin. The product mixture obtained from V-treated dioxasolv lignin was also analyzed and quantified by GC/MS. The primary phenolic products generated from this reaction are depicted in Fig. 39, where vanillin, syringaldehyde and syringic acid were found to be the most prevalent degradation products.
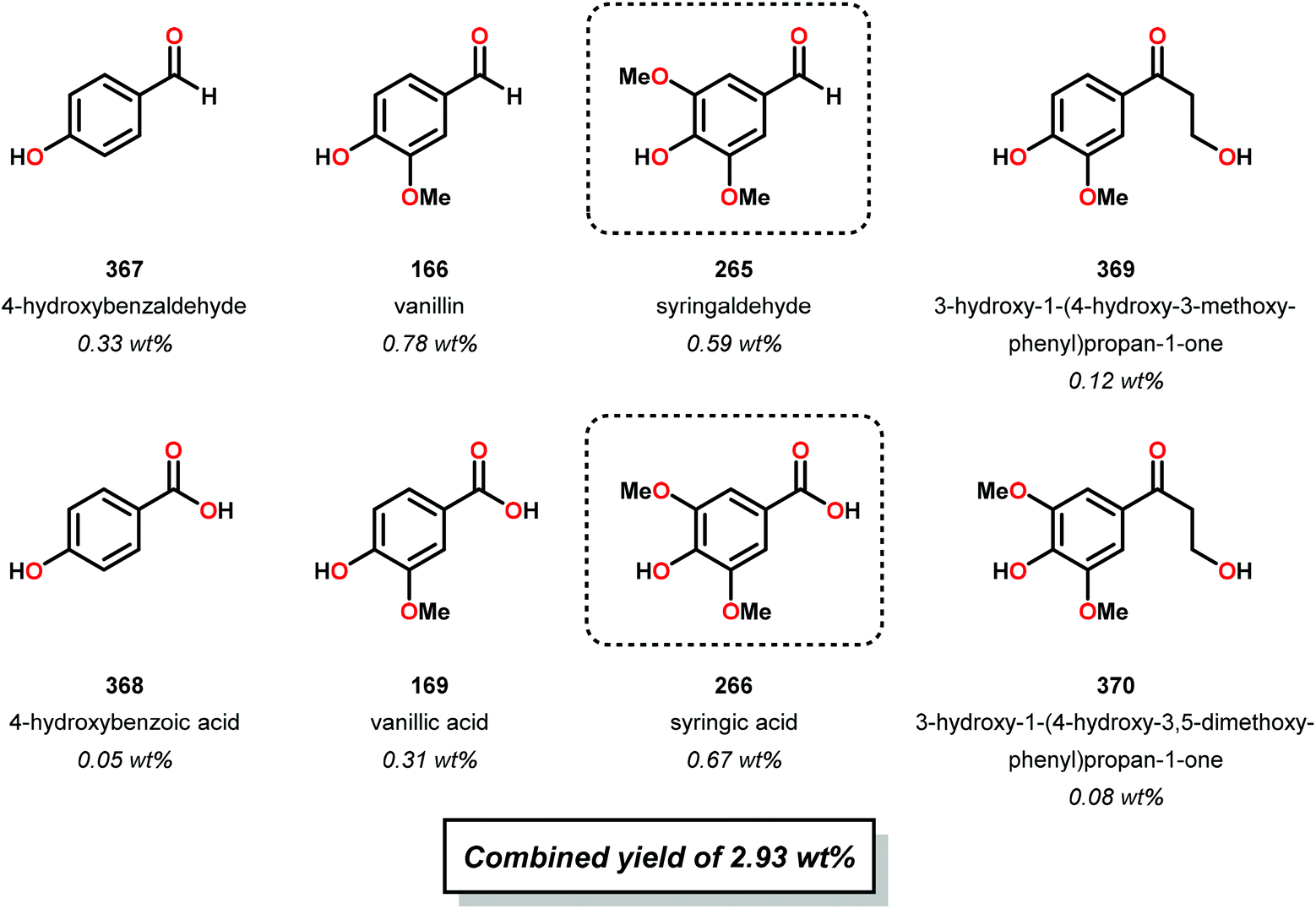 | ||
| Fig. 39 Identity and yield of products from V-catalyzed degradation of dioxasolv lignin derived from Miscanthus giganteus. | ||
Methyltrioxorhenium(VII) (MTO) has also been reported to mediate the cleavage of C–O bonds in lignin model systems to yield phenolic- and aldehyde-based products.216 In these reactions, methyldioxorhenium(V) (MDO), generated in situ by reduction from MTO, was proposed to be the catalytically active species. Heating a reaction mixture of MTO and lignin model system 291 at 155 °C afforded phenylacetaldehyde 371 and guaiacol (62) in high yields (Scheme 39). Recycling experiments showed that the catalyst could be reused at least five times without any significant loss of conversion or yield of guaiacol. However, attempts to carry out the cleavage on γ-OH functionalized lignin model system 61 proved to be unsuccessful. Although the reaction proceeded with a shorter reaction time and at lower temperature, only 9% yield of aldehyde 373 and 65% yield of guaiacol (62) were obtained (Scheme 40). In addition to the formation of compounds 373 and 62, several other products were detected. The decrease in selectivity for the γ-OH functionalized substrate 61 was not clear and further experiments are needed to explain the unsuccessful cleavage of the γ-OH containing substrate.
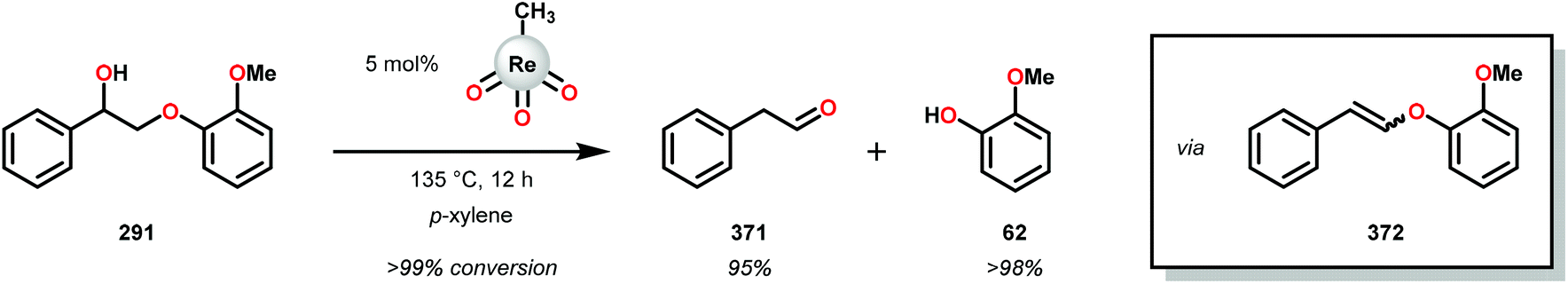 | ||
| Scheme 39 Methyltrioxorhenium (MTO) catalyzed C–O cleavage of lignin model system 291. | ||
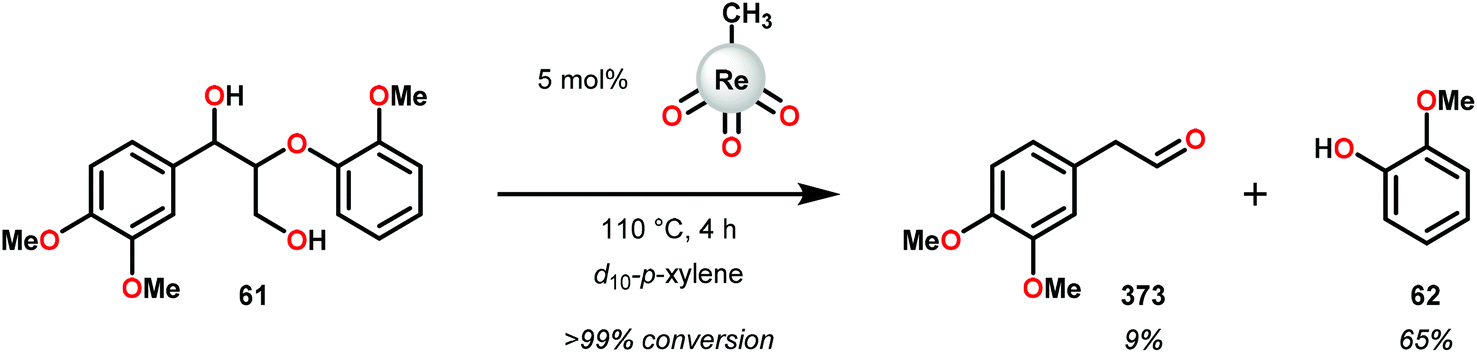 | ||
| Scheme 40 Re-catalyzed C–O cleavage of γ-OH functionalized lignin model system 61. | ||
Compared to the Ru-catalyzed C–O bond cleavage, the Re-catalyzed cleavage does not afford acetophenone but rather produces phenylacetaldehyde, presumably through a 1,2 shift of the O atom. In order to shed some light on the mechanistic pathway, isotopic labeling experiments were performed with 17O and deuterium labeled 291 (see Scheme 41). When using 291-17O (17O labeled at the benzylic hydroxyl group), complete incorporation of 17O was observed in the phenylacetaldehyde product (Scheme 41, top). For the deuterium labeled compound 291-D, labeled at the α-carbon, the deuterium labeled product 371-D was obtained, showing that the deuterium does not undergo migration. Based on the isotopic labeling studies and the detection of intermediate 372, a mechanism was proposed and is depicted in Scheme 42. In the proposed catalytic cycle MTO is initially reduced to MDO, which subsequently reacts with a second molecule of the substrate to give the Re-alkoxide species 374. Elimination from intermediate 374 generates enol ether 375 and ReV bis(hydroxo) intermediate 376, which formally adds an O- and H atom to enol ether 375 to produce an acetal-like Re-alkoxide (378). From species 378, MDO is liberated together with the observed cleavage products.216 The Re-catalyzed redox-neutral system constitutes a novel route for carrying out C–O cleavage in lignin model systems, however, additional understanding of the mechanistic pathway is vital in order to design improved Re-based systems capable of mediating C–O bond cleavage of γ-OH containing model systems.
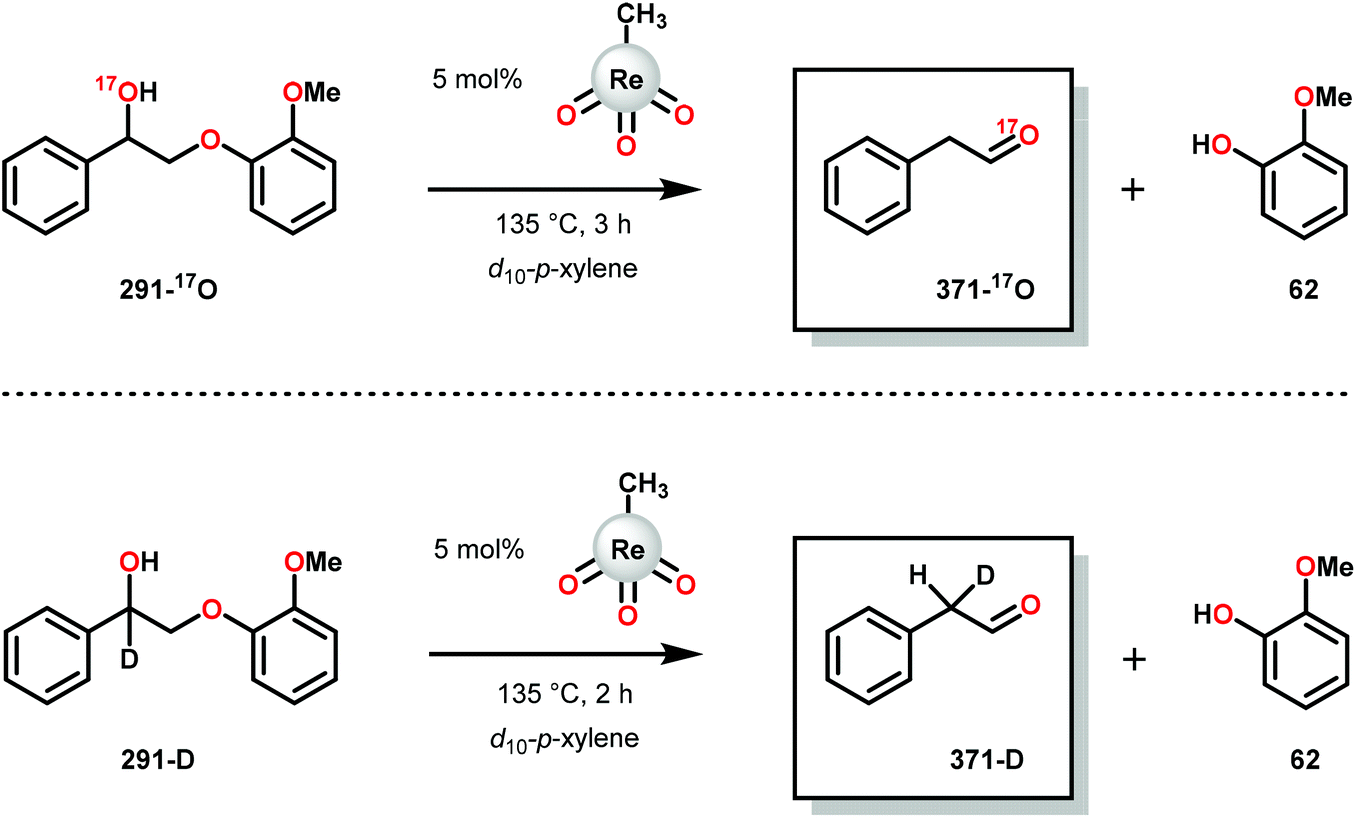 | ||
| Scheme 41 Isotopic labeling studies of the Re-catalyzed C–O cleavage reaction. | ||
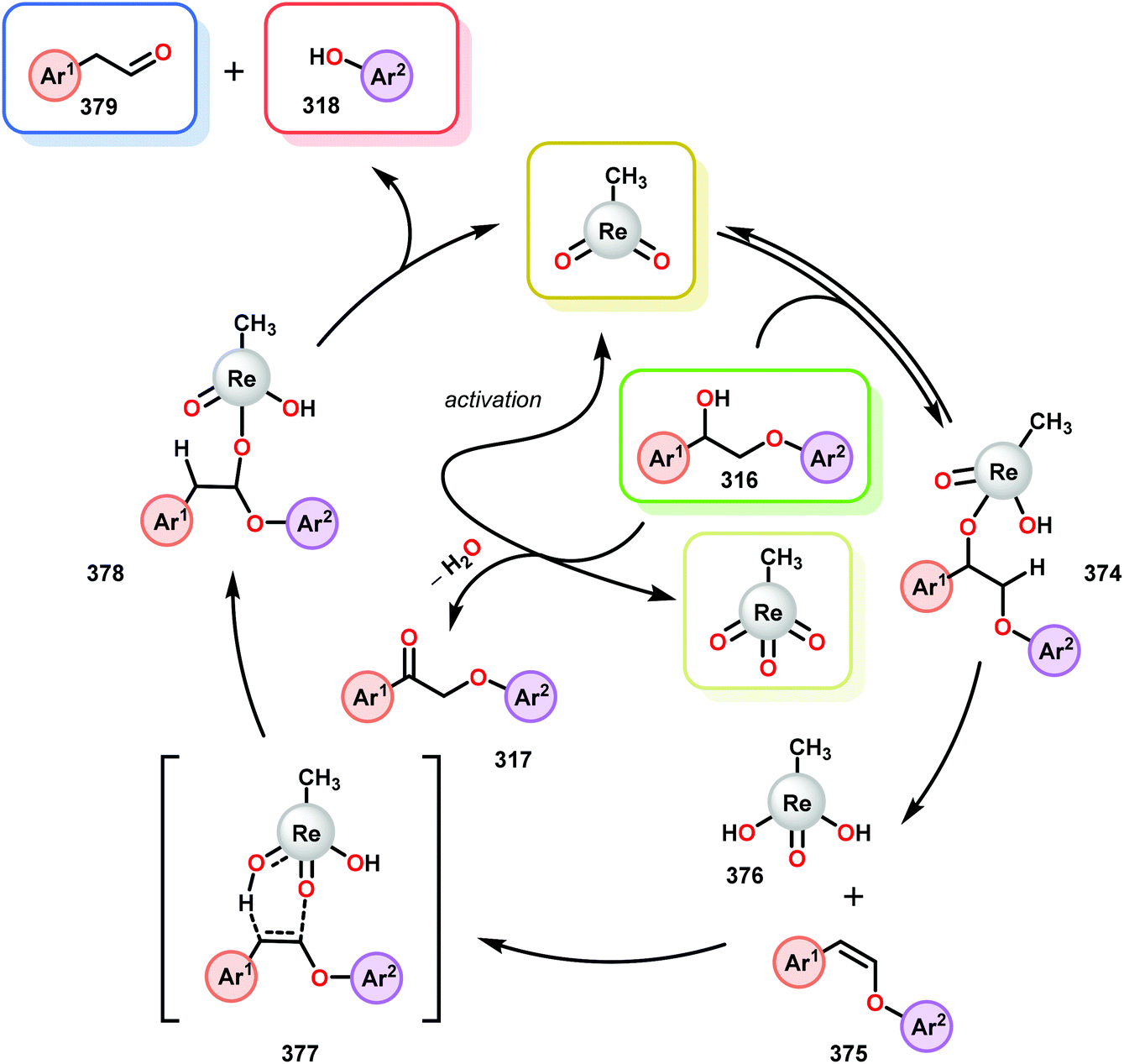 | ||
| Scheme 42 Proposed mechanism for Re-mediated redox-neutral C–O cleavage of lignin model systems. | ||
Samec and co-workers recently reported on the use of Pd/C as a heterogeneous catalyst for the redox-neutral C–O bond cleavage of various 2-aryloxy-1-arylethanols.217 The authors reasoned that commercially available Pd/C could indeed be a viable catalyst for mediating the redox-neutral conversion of lignin model systems (Fig. 40) inspired by their previous work on reductive cleavage of lignin model systems84,85 and because Pd on solid supports is known to be air- and moisture stable in a wide pH range. After initial screening, it could be established that the addition of a substoichiometric amount of NaBH4 in the presence of air resulted in efficient transformation of model system 116 (Table 20). It was reasoned that the required use of NaBH4 was to transform the initial thin Pd oxide layer to produce a reactive Pd surface. The use of 0.1 equivalents of NaBH4 was found to be optimal to efficiently mediate the redox-neutral conversion of model system 116. Employing <0.1 equivalents of NaBH4 yielded the benzylic oxidized product 119 and the addition of >0.1 equivalents NaBH4 resulted in reduction of the generated ketone 119 to afford 1-phenylethanol (124).217
 | ||
| Fig. 40 Pd/C catalyzed redox-neutral vs. reductive cleavage of β-O-4 lignin model compounds. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Yielda (%) | |
| Ketone | Phenol | ||||
| a Isolated yield based on two duplicate experiments. b Reaction was run for 4 h. | |||||
| 1 | 116 | H | H | 95 | 92 |
| 2 | 293 | 4-MeO | H | 98 | — |
| 3 | 291 | H | 2-MeO | 94 | — |
| 4b | 314 | 3,4-(MeO)2 | H | 95 | — |
| 5 | 99 | 4-MeO | 2-MeO | 99 | 95 |
| 6b | 294 | 3,4-(MeO)2 | 2-MeO | 98 | — |
| 7 | 292 | H | 2,6-(MeO)2 | 97 | 95 |
| 8b | 380 | 3,4-(MeO)2 | 2,6-(MeO)2 | 92 | — |
| 9b | 381 | 4-OH | 2-MeO | 97 | — |
| 10b | 382 | 3-MeO-4-OH | 2-MeO | 96 | — |
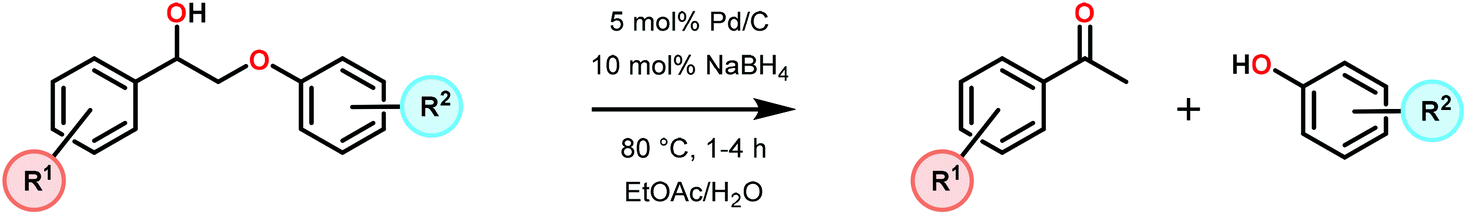
The scope of the Pd/C catalyzed redox-neutral reaction was subsequently investigated and it was found that a variety of substituted lignin model systems could be efficiently cleaved, generating the corresponding acetophenone and phenolic products in excellent yields (Table 20). Phenolic substrates were also tolerated and could be converted to the corresponding products in high yields (Table 20, entries 9 and 10). The Pd/C catalytic system was subsequently applied on a polymeric model substrate to determine the compatibility with more native lignin resembling substrates. The model polymer had a molecular weight of ∼6 kDa, similar to that found for native lignin. The model polymer underwent efficient cleavage and ketone 107 was isolated in near quantitative yield (Scheme 43).217
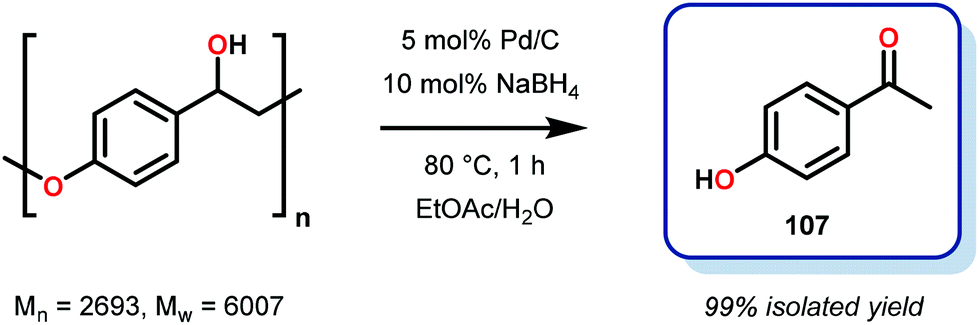 | ||
| Scheme 43 Pd/C catalyzed redox-neutral depolymerization of lignin model polymer. | ||
To gain insight into the mechanism of the redox-neutral transformation, experiments were performed with methylated substrate 295 and substrate 117. However, neither of these substrates gave any reaction (Scheme 44). In light of these results the authors proposed a mechanism (Scheme 45) where an initial activation step of the Pd surface occurs to generate the “activated” Pd/C, which subsequently reversibly dehydrogenates the benzylic alcohol unit in the lignin model substrate. The enolate form of the generated ketone is adsorbed to Pd to yield intermediate 383.217 The hydride can then be inserted either at the α or β-position, according to a Horiuti–Polanyi-type218,219 pathway, to give intermediates 385 or 387, respectively. C–O bond cleavage of intermediate 385 produces 386 where the enol moiety and aryloxide are still adsorbed, from which the desired products are subsequently liberated. Alternatively, C–O bond insertion of 387 forms carbene-type intermediate 388, which can undergo tautomerization to release the acetophenone product (389) and phenolic product (318).217
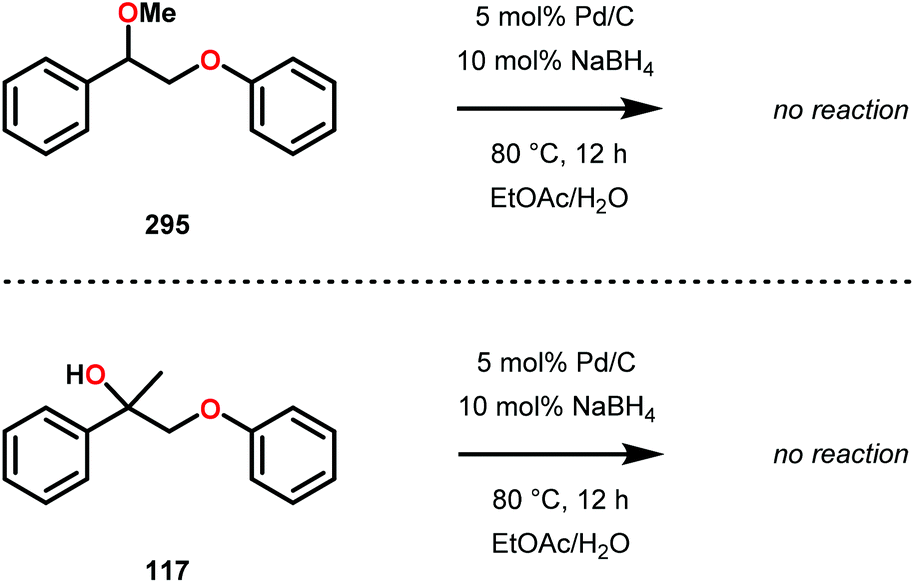 | ||
| Scheme 44 Redox-neutral C–O bond cleavage attempts of substrates 295 and 117. | ||
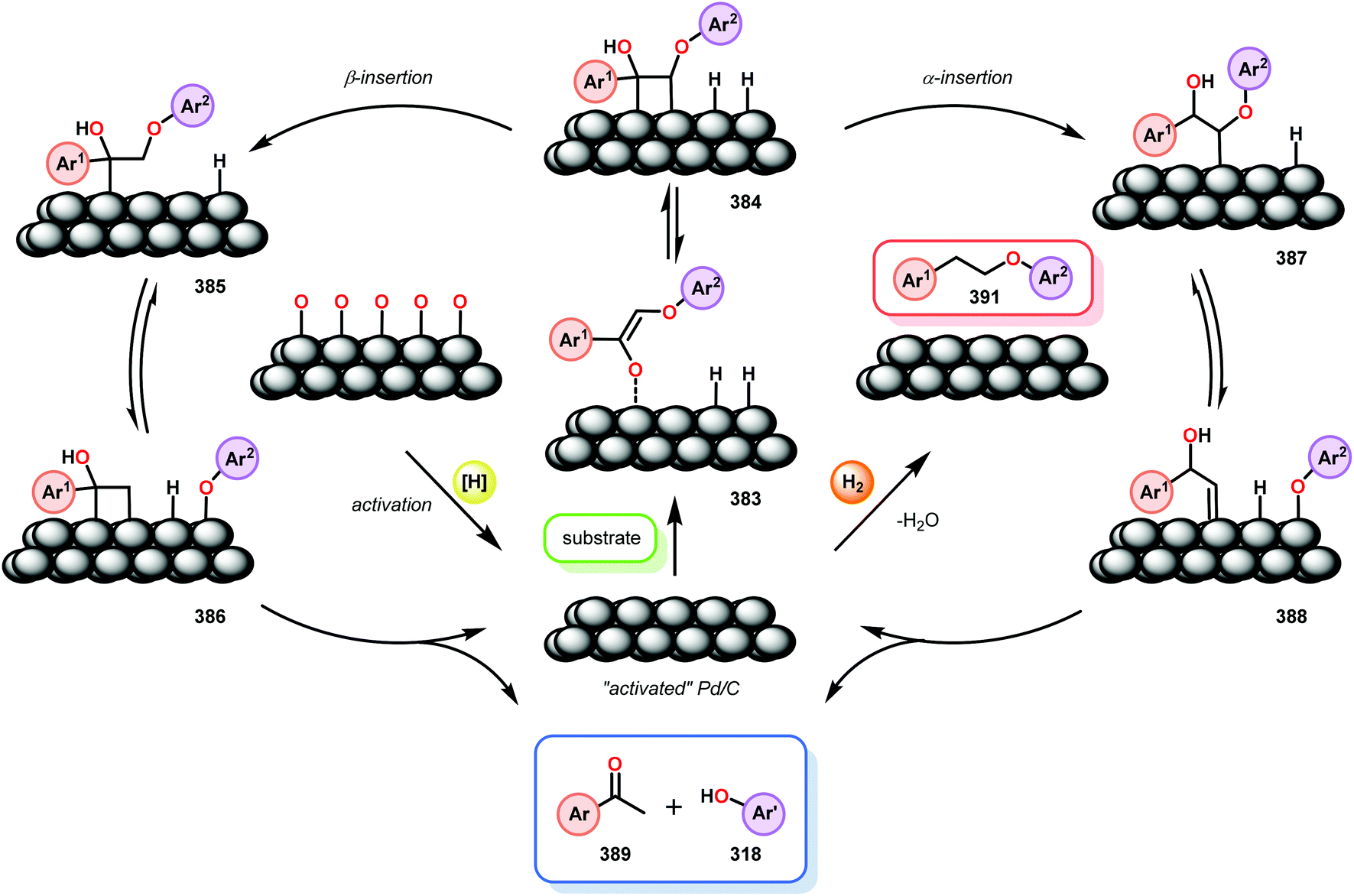 | ||
| Scheme 45 Proposed mechanism for the Pd/C catalyzed redox-neutral C–O bond cleavage of lignin model systems. | ||
The ability of Pd/C to mediate redox-neutral cleavage of lignin model systems highlights the implementation and scalability of the reaction. The switch in reactivity is intriguing and may be useful for designing novel catalytic protocols or for improving already existing systems.
5. Conclusions and outlook
The development of a sustainable and carbon-neutral biorefinery has emerged as a prominent scientific and engineering goal of modern society. As petroleum becomes less accessible, biomass-based carbon sources have emerged as potential feedstocks for fuel production and commodity chemical manufacturing. Lignin is the largest source of a renewable material which contains an aromatic skeleton and is the second most abundant component in biomass. While technologies to process cellulose and hemicellulose are well developed, lignin processing constitutes a considerably more challenging task. Lignin is currently regarded as a disposal liability and only used in low-value applications. The fact that lignin is currently an unexploited resource makes the valorization of lignin an ideal solution for the production of defined aromatic commodity chemicals.The difficulty in catalytically depolymerizing lignin originates from its highly irregular and complex structure. In this regard, transition metal catalysis constitutes an excellent option for studying the valorization of lignin owing to the relative ease by which these complexes can be accessed in combination with their tunability. Studying lignin valorization constitutes an opportunity for evaluating transition metal catalysis and for method development. From the work described in this review it is clear that three fundamentally distinguishable strategies have emerged for the conversion of lignin and lignin model systems; reductive, oxidative and redox-neutral cleavage.
It is obvious that several intriguing approaches have been devised for the cleavage of lignin model systems, which have had a crucial impact on the field. However, a majority of these methods are not sufficiently selective and low yielding. Several of the prominent pathways developed for fragmenting lignin model systems utilize ketyl radicals as key intermediates. Successfully controlling how these radicals are generated and fragmented could afford novel routes for selectively producing value-added products.
Redox-neutral lignin depolymerization represents an ideal approach to biomass valorization because it does not require a terminal oxidant/reductant. This is a particularly important consideration in the context of commodity chemical/fine chemical synthesis where the cost of stoichiometric reagents becomes prohibitively expensive on scale. Although the redox-neutral approach is certainly appealing, there remains a lack of fundamental knowledge for how to control and govern these catalytic systems to selectively generate aromatic products. To achieve this, it is essential that the scientific field shifts away from employing simple lignin models to more elaborate systems, or even samples of native lignin, to begin to address the compatibility issues encountered already in the elementary stages of catalyst design. Creative solutions based on these advances must be devised to ultimately furnish methods that are industrially applicable for the sustainable production of high-value commodity chemicals. It is clear that a final solution for lignin valorization will most likely utilize a combination of the different approaches highlighted in this review.
Acknowledgements
Financial support from the NSF (CHE-1440118), the Alfred P. Sloan Foundation, the Camille and Henry Dreyfus Foundation and University of Michigan is gratefully acknowledged. M.D.K. gratefully acknowledges financial support from the Swedish Research Council (637-2013-7314) and the Royal Swedish Academy of Agriculture and Forestry (Kungliga Skogs- och Lantbruksakademien) for a postdoctoral fellowship.References
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- S. R. Collinson and W. Thielemans, Coord. Chem. Rev., 2010, 254, 1854–1870 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- S. Dutta, K. C.-W. Wu and B. Saha, Catal. Sci. Technol., 2014, 4, 3785–3799 CAS.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- P. J. Deuss and K. Barta, Coord. Chem. Rev., 2016, 306, 510–532 CrossRef CAS.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1850–1860 CrossRef CAS PubMed.
- G. Brunow, in Biorefineries - Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH Verlag, Weinheim, Germany, 2006, vol. 2, pp. 151–163 Search PubMed.
- J. Ralph, J. Peng, F. Lu, R. D. Hatfield and R. F. Helm, J. Agric. Food Chem., 1999, 47, 2991–2996 CrossRef CAS PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- J. J. Bozell, Alternative, Renewable, and Novel Feedstocks for Producing Chemicals, US Dep. Energy Oak Ridge Natl. Lab., Oak Ridge TN, 2006, pp. 204–209 Search PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- P. Gallezot, ChemSusChem, 2008, 1, 734–737 CrossRef PubMed.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. B. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167–171 CrossRef PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef PubMed.
- J. H. Clark, R. Luque and A. S. Matharu, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 183–207 CrossRef PubMed.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- J. Zhu, X. Pan and R. S. Zalesny, Jr., Appl. Microbiol. Biotechnol., 2010, 87, 847–857 CrossRef CAS PubMed.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef CAS.
- J. B. Lindsey and B. Tollens, Liebigs Ann. Chem., 1892, 267, 341–366 CrossRef.
- J. Y. Zhu, X. J. Pan, G. S. Wang and R. Gleisner, Bioresour. Technol., 2009, 100, 2411–2418 CrossRef CAS PubMed.
- CycleWood solutions, http://cyclewood.com/technology/, (accessed October 2015).
- C. Fargues, Á. Mathias and A. Rodrigues, Ind. Eng. Chem. Res., 1996, 35, 28–36 CrossRef CAS.
- A. Lindner and G. Wegener, J. Wood Chem. Technol., 1988, 8, 323–340 CrossRef CAS.
- X. Pan, C. Arato, N. Gilkes, D. Gregg, W. Mabee, K. Pye, Z. Xiao, X. Zhang and J. Saddler, Biotechnol. Bioeng., 2005, 90, 473–481 CrossRef CAS PubMed.
- T. Ikeda, K. Holtman, J. F. Kadla, H.-m. Chang and H. Jameel, J. Agric. Food Chem., 2002, 50, 129–135 CrossRef CAS PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- J. Sauer and H. Adkins, J. Am. Chem. Soc., 1937, 59, 1–3 CrossRef CAS.
- J. V. Vaughen and W. A. Lazier, J. Am. Chem. Soc., 1931, 53, 3719–3728 CrossRef CAS.
- F. Krafft, Ber. Dtsch. Chem. Ges., 1877, 10, 2034–2036 CrossRef.
- E. E. Harris, J. D'Ianni and H. Adkins, J. Am. Chem. Soc., 1938, 60, 1467–1470 CrossRef CAS.
- C. P. Brewer, L. M. Cooke and H. Hibbert, J. Am. Chem. Soc., 1948, 70, 57–59 CrossRef CAS PubMed.
- L. W. Covert and H. Adkins, J. Am. Chem. Soc., 1932, 54, 4116–4117 CrossRef CAS.
- J. M. Pepper and H. Hibbert, J. Am. Chem. Soc., 1948, 70, 67–71 CrossRef CAS PubMed.
- For representative publications demonstrating NMR analysis of lignin, see: (a) G. Gellerstedt and D. Robert, Acta Chem. Scand., 1987, B41, 541–546 CrossRef CAS; (b) C.-L. Chen and D. Robert, Methods Enzymol., 1988, 161, 137–174 CAS; (c) N. Fukagawa, G. Meshitsuka and A. Ishizu, J. Wood Chem. Technol., 1991, 11, 373–396 CrossRef CAS; (d) R. M. Ede and I. Kilpeläinen, Res. Chem. Intermed., 1995, 21, 313–328 CrossRef CAS; (e) I. Kilpeläinen, J. Sipilä, G. Brunow and K. Lundquist, J. Agric. Food Chem., 1994, 42, 2790–2794 CrossRef; (f) E. Ammalahti, G. Brunow, M. Bardet, D. Robert and I. Kilpeläinen, J. Agric. Food Chem., 1998, 46, 5113–5117 CrossRef; (g) D. V. Evtuguin, C. P. Neto, A. M. S. Silva, P. M. Domingues, F. M. L. Amado, D. Robert and O. Faix, J. Agric. Food Chem., 2001, 49, 4252–4261 CrossRef CAS PubMed.
- NMR Database of Lignin and Cell Wall Model Compounds, http://www.ars.usda.gov/SP2UserFiles/Place/36553000/software/NMR/NMR_DataBase_Intro_&_Index.pdf. (accessed October 2015).
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed.
- A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed.
- A. C. Hillier, G. A. Grasa, M. S. Viciu, H. M. Lee, C. Yang and S. P. Nolan, J. Organomet. Chem., 2002, 653, 69–82 CrossRef CAS.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC.
- M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed.
- E. Geist, A. Kirschning and T. Schmidt, Nat. Prod. Rep., 2014, 31, 441–448 RSC.
- E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246–2247 CrossRef CAS.
- J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428–2432 CrossRef PubMed.
- D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed.
- B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed.
- P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- Sigma-Aldrich. http://www.sigmaaldrich.com/catalog/search?term=MFCD07369796&interface=MDL%20No.&N=0&mode=mode%20matchall&lang=en®ion=US&focus=product (accessed October 2015).
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480–5483 CrossRef CAS PubMed.
- J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- For examples of β-hydride elimination from Pd and Pt complexes, see: (a) F. Ozawa, T. Ito and A. Yamamoto, J. Am. Chem. Soc., 1980, 102, 6457–6463 CrossRef CAS; (b) S. Komiya, T. Morimoto, A. Yamamoto and T. Yamamoto, Organometallics, 1982, 1, 1528–1536 CrossRef CAS; (c) H. Bryndza, J. C. Calabresse, M. Marsi, C. Roe, W. Tam and J. Bercaw, J. Am. Chem. Soc., 1986, 108, 4805–4813 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, ChemSusChem, 2009, 2, 215–217 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412–414 RSC.
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS PubMed.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 CAS.
- S. Sawadjoon, A. Lundstedt and J. S. M. Samec, ACS Catal., 2013, 3, 635–642 CrossRef CAS.
- M. V. Galkin, S. Sawadjoon, V. Rohde, M. Dawange and J. S. M. Samec, ChemCatChem, 2014, 6, 179–184 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- X. Zhou, J. Mitra and T. B. Rauchfuss, ChemSusChem, 2014, 7, 1623–1626 CrossRef CAS PubMed.
- P. Rousu, P. Rousu and J. Anttila, Resour., Conserv. Recycl., 2002, 35, 85–103 CrossRef.
- A. N. Desnoyer, B. Fartel, K. C. MacLeod, B. O. Patrick and K. M. Smith, Organometallics, 2012, 31, 7625–7628 CrossRef CAS.
- Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674–12678 CrossRef CAS PubMed.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. J. Douglas, J. D. Nguyen, K. P. Cole and C. R. J. Stephenson, Aldrichimica Acta, 2014, 47, 15–25 CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, N. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef CAS.
- M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463–4466 CrossRef CAS PubMed.
- M. A. Mercadante, C. B. Kelly, J. M. Bobbitt, L. J. Tilley and N. E. Leadbeater, Nat. Protoc., 2013, 8, 666–676 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 1135–1138 CrossRef CAS.
- E. Hasegawa and D. P. Curran, J. Org. Chem., 1993, 58, 5008–5010 CrossRef CAS.
- P. R. Chopade, E. Prasad and R. A. Flowers, II, J. Am. Chem. Soc., 2004, 126, 44–45 CrossRef CAS PubMed.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 2596–2599 CrossRef CAS.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS.
- J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985–4987 RSC.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef CAS PubMed.
- F. R. Bou-Hamdan and P. H. Seeberger, Chem. Sci., 2012, 3, 1612–1616 RSC.
- M. Tien and T. K. Kirk, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 2280–2284 CrossRef CAS.
- M. Kuwahara, J. K. Glenn, M. A. Morgan and M. H. Gold, FEBS Lett., 1984, 169, 247–250 CrossRef CAS.
- H. E. Schoemaker, P. J. Harvey, R. M. Bowen and J. M. Palmer, FEBS Lett., 1985, 183, 7–12 CrossRef CAS.
- M. Tien and T. K. Kirk, Science, 1983, 221, 661–663 Search PubMed.
- S. H. Lim, K. Nahm, C. S. Ra, D. W. Cho, U. C. Yoon, J. A. Latham, D. Dunaway-Mariano and P. S. Mariano, J. Org. Chem., 2013, 78, 9431–9443 CrossRef CAS PubMed.
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- M. Shimada, T. Habe, T. Umezawa, T. Higuchi and T. Okamoto, Biochem. Biophys. Res. Commun., 1984, 122, 1247–1252 CrossRef CAS PubMed.
- T. Habe, M. Shimada, T. Okamoto, B. Panijpan and T. Higuchi, J. Chem. Soc., Chem. Commun., 1985, 1323–1324 RSC.
- M. Shimada, T. Habe, T. Higuchi, T. Okamoto and B. Panijpan, Holzforschung, 1987, 41, 277–285 CrossRef CAS.
- E. A. Mayeda, J. Am. Chem. Soc., 1975, 97, 4012–4015 CrossRef CAS.
- P. J. Kersten, M. Tien, B. Kalyanaraman and T. K. Kirk, J. Biol. Chem., 1985, 260, 2609–2612 CAS.
- A. Paszczynski, R. L. Crawford and R. A. Blanchette, Appl. Environ. Microbiol., 1988, 54, 62–68 CAS.
- G. Labat and B. Meunier, J. Org. Chem., 1989, 54, 5008–5011 CrossRef CAS.
- C. Crestini, R. Saladino, P. Tagliatesta and T. Boschi, Bioorg. Med. Chem., 1999, 7, 1897–1905 CrossRef CAS PubMed.
- F. Cui, T. Wijesekera, D. Dolphin, R. Farrell and P. Skerker, J. Biotechnol., 1993, 30, 15–26 CrossRef CAS.
- F. Cui and D. Dolphin, Bioorg. Med. Chem., 1994, 2, 735–742 CrossRef CAS PubMed.
- F. Cui and D. Dolphin, Can. J. Chem., 1995, 73, 2153–2157 CrossRef CAS.
- P. Zucca, G. Mocci, A. Rescigno and E. Sanjust, J. Mol. Catal. A: Chem., 2007, 278, 220–227 CrossRef CAS.
- P. Zucca, F. Sollai, A. Garau, A. Rescigno and E. Sanjust, J. Mol. Catal. A: Chem., 2009, 306, 89–96 CrossRef CAS.
- C. Crestini, A. Pastorini and P. Tagliatesta, J. Mol. Catal. A: Chem., 2004, 208, 195–202 CrossRef CAS.
- C. Crestini, A. Pastorini and P. Tagliatesta, Eur. J. Inorg. Chem., 2004, 4477–4483 CrossRef CAS.
- C. Zhu, W. Ding, T. Shen, C. Tang, C. Sun, S. Xu, Y. Chen, J. Wu and H. Ying, ChemSusChem, 2015, 8, 1768–1778 CrossRef CAS PubMed.
- W. Zhu and W. T. Ford, J. Mol. Catal., 1993, 78, 367–378 CrossRef CAS.
- F. Cui and D. Dolphin, Bioorg. Med. Chem., 1995, 3, 471–477 CrossRef CAS PubMed.
- R. D. Jones, D. A. Summerville and F. Basolo, Chem. Rev., 1979, 79, 139–179 CrossRef CAS.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- For reviews on metal Schiff-base complexes, see for example: (a) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC; (b) K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS; (c) S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- For reactivity studies of Co-Schiff base complexes with O2, see for example: (a) C. Floriani and F. Calderazzo, J. Chem. Soc. A, 1969, 946–953 RSC; (b) D. Chen and A. E. Martell, Inorg. Chem., 1987, 26, 1026–1030 CrossRef CAS; (c) D. Chen, A. E. Martell and Y. Sun, Inorg. Chem., 1989, 28, 2647–2652 CrossRef CAS.
- F. Basolo, B. M. Hoffman and J. A. Ibers, Acc. Chem. Res., 1975, 8, 384–392 CrossRef CAS.
- R. S. Drago, B. B. Corden and C. W. Barnes, J. Am. Chem. Soc., 1986, 108, 2453–2454 CrossRef CAS PubMed.
- J. J. Bozell, B. R. Hames and D. R. Dimmel, J. Org. Chem., 1995, 60, 2398–2404 CrossRef CAS.
- C. Canevali, M. Orlandi, L. Pardi, B. Rindone, R. Scotti, J. Sipilä and F. Morazzoni, J. Chem. Soc., Dalton Trans., 2002, 3007–3014 RSC.
- V. Sippola, O. Krause and T. Vuorinen, J. Wood Chem. Technol., 2004, 24, 323–340 CrossRef CAS.
- K. Kervinen, H. Korpi, M. Leskelä and T. Repo, J. Mol. Catal. A: Chem., 2003, 203, 9–19 CrossRef CAS.
- K. Kervinen, M. Allmendinger, M. Leskelä, T. Repo and B. Rieger, Phys. Chem. Chem. Phys., 2003, 5, 4450–4454 RSC.
- K. Kervinen, H. Korpi, J. G. Mesu, F. Soulimani, T. Repo, B. Rieger, M. Leskelä and B. M. Weckhuysen, Eur. J. Inorg. Chem., 2005, 2591–2599 CrossRef CAS.
- S. K. Badamali, R. Luque, J. H. Clark and S. W. Breeden, Catal. Commun., 2009, 10, 1010–1013 CrossRef CAS.
- S. K. Badamali, R. Luque, J. H. Clark and S. W. Breeden, Catal. Commun., 2011, 12, 993–995 CrossRef CAS.
- D. Cedeno and J. J. Bozell, Tetrahedron Lett., 2012, 53, 2380–2383 CrossRef CAS.
- B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed.
- B. Biannic, J. J. Bozell and T. Elder, Green Chem., 2014, 16, 3635–3642 RSC.
- A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2008, 130, 14474–14476 CrossRef CAS PubMed.
- E. Takezawa, S. Sakaguchi and Y. Ishii, Org. Lett., 1999, 1, 713–715 CrossRef CAS PubMed.
- T. R. Felthouse, J. Am. Chem. Soc., 1987, 109, 7566–7568 CrossRef CAS.
- S. Barroso, G. Blay, I. Fernández, J. R. Pedro, R. Ruiz-García, E. Pardo, F. Lloret and M. C. Muñoz, J. Mol. Catal. A: Chem., 2006, 243, 214–220 CrossRef CAS.
- S. Riaño, D. Fernández and L. Fadini, Catal. Commun., 2008, 9, 1282–1285 CrossRef.
- J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 2232–2248 CrossRef CAS.
- T. Hirao, Coord. Chem. Rev., 2003, 237, 271–279 CrossRef CAS.
- S. Gazi, W. K. H. Ng, R. Ganguly, A. M. P. Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, A. D. Sutton and D. L. Thorn, J. Am. Chem. Soc., 2009, 131, 428–429 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. Silks, Org. Lett., 2011, 13, 1908–1911 CrossRef CAS PubMed.
- For mechanistic investigations, see: (a) B. N. Wigington, M. L. Drummond, T. R. Cundari, D. L. Thorn, S. K. Hanson and S. L. Scott, Chem. – Eur. J., 2012, 18, 14981–14988 CrossRef CAS PubMed; (b) S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, L. A. Silks and D. L. Thorn, J. Am. Chem. Soc., 2010, 132, 17804–17816 CrossRef CAS PubMed.
- G. Zhang, B. L. Scott, R. Wu, L. A. Silks and S. K. Hanson, Inorg. Chem., 2012, 51, 7354–7361 CrossRef CAS PubMed.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2013, 3, 3111–3122 CrossRef CAS.
- V. L. Pardini, C. Z. Smith, J. H. P. Utley, R. R. Vargas and H. Viertler, J. Org. Chem., 1991, 56, 7305–7313 CrossRef CAS.
- V. L. Pardini, R. R. Vargas, H. Viertler and J. H. P. Utley, Tetrahedron, 1992, 48, 1221–1228 CrossRef.
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC.
- J. M. Bobbitt, C. Brückner and N. Merbouh, Org. React., 2009, 74, 103–424 CrossRef CAS.
- T. Vogler and A. Studer, Synthesis, 2008, 1979–1993 CAS.
- R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS.
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414–2415 RSC.
- P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805–811 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- B. Sedai and R. T. Baker, Adv. Synth. Catal., 2014, 356, 3563–3574 CrossRef CAS.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- B. L. Ryland, S. D. McCann, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 12166–12173 CrossRef CAS PubMed.
- M. F. Semmelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef CAS.
- E. T. T. Kumpulainen and A. M. P. Koskinen, Chem. – Eur. J., 2009, 15, 10901–10911 CrossRef CAS PubMed.
- T. A. Hamlin, C. B. Kelly, J. M. Ovian, R. J. Wiles, L. J. Tilley and N. E. Leadbeater, J. Org. Chem., 2015, 80, 8150–8167 CrossRef CAS PubMed.
- A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 CAS.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, ACS Catal., 2013, 3, 2599–2605 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, S. Chiba and T.-P. Loh, Chem. Commun., 2013, 49, 11439–11441 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Chem. Commun., 2008, 624–625 RSC.
- R. L. Chowdhury and J.-E. Bäckvall, J. Chem. Soc., Chem. Commun., 1991, 1063–1064 RSC.
- F. Kakiuchi, M. Usui, S. Ueno, N. Chatani and S. Murai, J. Am. Chem. Soc., 2004, 126, 2706–2707 CrossRef CAS.
- Y. Zhao and V. Snieckus, J. Am. Chem. Soc., 2014, 136, 11224–11227 CrossRef CAS PubMed.
- S. C. Chmely, S. Kim, P. N. Ciesielski, G. Jimenez-Oses, R. S. Paton and G. T. Beckham, ACS Catal., 2013, 3, 963–974 CrossRef CAS.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- A. Wu, B. O. Patrick and B. R. James, Inorg. Chem. Commun., 2012, 24, 11–15 CrossRef CAS.
- A. Wu, J. M. Lauzon and B. R. James, Catal. Lett., 2015, 145, 511–518 CrossRef CAS.
- L. Benhamou, V. César, N. Lugan and G. Lavigne, Organometallics, 2007, 26, 4673–4676 CrossRef CAS.
- C. Qu, T. Kishimoto, M. Kishino, M. Hamada and N. Nakajima, J. Agric. Food Chem., 2011, 59, 5382–5389 CrossRef CAS PubMed.
- T. Sonoda, T. Ona, H. Yokoi, Y. Ishida, H. Ohtani and S. Tsuge, Anal. Chem., 2001, 73, 5429–5435 CrossRef CAS PubMed.
- W. Huo, W. Li, M. Zhang, W. Fan, H.-m. Chang and H. Jameel, Catal. Lett., 2014, 144, 1159–1163 CrossRef CAS.
- C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, Tetrahedron Lett., 2002, 43, 7987–7989 CrossRef CAS.
- R. Martínez, D. J. Ramón and M. Yus, Tetrahedron, 2006, 62, 8988–9001 CrossRef.
- T. Kuwahara, T. Fukuyama and I. Ryu, Org. Lett., 2012, 14, 4703–4705 CrossRef CAS PubMed.
- For selected reviews on hydrogen borrowing strategies, see: (a) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed; (c) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed; (d) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- D. Weickmann and B. Plietker, ChemCatChem, 2013, 5, 2170–2173 CrossRef CAS.
- T. vom Stein, T. Weigand, C. Merkens, J. Klankermayer and W. Leitner, ChemCatChem, 2013, 5, 439–441 CrossRef CAS.
- T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS PubMed.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS.
- R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed.
- M. V. Galkin, C. Dahlstrand and J. S. M. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS PubMed.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
- F. Zaera, Phys. Chem. Chem. Phys., 2013, 15, 11988–12003 RSC.
| This journal is © The Royal Society of Chemistry 2016 |