
A novel C,D-spirodioxene taxoid synthesized through an unexpected Pd-mediated ring cyclization†
Shao-Rong
Wang
a,
Pedro A.
Sánchez-Murcia
b,
Federico
Gago
b and
Wei-Shuo
Fang
*a
aState Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, 2A Nanwei Road, Beijing 100050, China. E-mail: wfang@imm.ac.cn; Tel: +86 10 63165229
bÁrea de Farmacología, Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28871 Alcalá de Henares, Unidad Asociada al Instituto de Química Médica del CSIC, E-28871 Madrid, Spain
First published on 25th November 2015
Abstract
A novel C,D-spirodioxene taxoid (6) was prepared from paclitaxel (1a), with the key steps including an unexpected Pd-mediated ring cyclization. The anti-tubulin activity of 6 was decreased relative to that of 1a and a previously reported C,D-spirolactone taxane (5). These observations could be rationalized on the basis of molecular modeling results. To the best of our knowledge, this is the first example indicating that 1,4-dioxenes can be synthesized from a mono-allyl vicinal diol through a Wacker-type cyclization. This strategy may be applicable to the synthesis of other C,D-spiro taxoids.
Introduction
Microtubule (MT) stabilizing agents such as paclitaxel (Taxol®, 1a), docetaxel (1b) and cabazitaxel (1c) are widely used for the treatment of ovarian cancer, breast cancer, and non-small-cell lung cancer etc., either as single agents or in combination with other drugs.

Although it was conceivable that an intact oxetane D-ring is crucial for bioactivity, more recent research studies challenged this belief, e.g. the cyclopropane analogue 2 showed significant MT stabilization activity,6 and D-seco taxane 3 also showed a potency on tubulin assembly promotion similar to that of 1a.7 This demonstrated that neither the intact oxetane ring nor an oxygen at C-5 might be necessary for binding of taxanes to MT/tubulin. However, none of the above two D-ring modified compounds possesses the potent cytotoxicity of their parent compounds 1a or 1b. Although it was believed that the divergence in cytotoxicity and MT/tubulin interaction for most D-seco taxanes may originate from other factors, such as their cell membrane permeability and chemical stability in cells, rather than interactions with tubulin/MTs, the experimental evidence is still lacking. It could be envisioned that the oxetane oxygen might play a critical role in cytotoxicity. In fact, Thoret et al. attempted to prove this by incorporating an oxygen atom at the C-5 position of 4-methyl paclitaxel to afford 4, but failed to restore the lost cytotoxicity.8 Thus, more effort should be made to demonstrate the exact function of the D-ring.
Recently, a C,D-spirolactone taxane (5) was reported to be able to stabilize MT (only 13 times less active than 1a) and to possess cytotoxic activity against U251 human glioma cells (IC50 values were in the range of 4–8 μM vs. 0.25–0.5 μM for 1a).9 In addition, it displayed a mechanistically distinct cytotoxic action in comparison with 1a, involving mTOR inhibition-dependent autophagy instead of the G2/M cell cycle block-associated apoptosis, which is the general mechanism of action for most classic MT-stabilizing agents. We hypothesize that the unique C,D-spiro ring in compound 5 would be responsible for its unusual mechanism.
To figure out the actual contribution of the D-ring for taxanes exerting their biological activities, some novel D-seco analogues were recently synthesized in our lab (unpublished work). During these efforts, a novel C,D-spirodioxene analogue 6 of paclitaxel was obtained through an unexpected Pd-mediated ring cyclization. Here we report its synthesis and bioassay results.
Results and discussion
The synthetic procedure is illustrated in Scheme 1. According to the reported protocol,5 intermediate 7 was obtained by multistep transformations starting from paclitaxel (1a). Since the double bond between C-5 and C-6, such as in 3, has been realized as an important element to “lock” the conformation of the C-ring, the 5-iodo has to be eliminated first. The 20-OH was protected by a triethylsilyl (TES) group; otherwise the oxetane ring would reclose under the elimination conditions. The elimination reaction was first attempted in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in toluene, but the yield of 9 was less than 50%. After some trials, it was found that treatment of 8 with LiCl accompanied by NaHCO3 in DMF furnished the product in high yield (91%). | ||
| Scheme 1 (a) Im, TESCl, DMF, 91%; (b) LiCl, NaHCO3, DMF, 90 °C, 91%; (c) HF/pyridine, MeCN, 95%; (d) IBX, EtOAc, 80 °C, 91%; (e) (PPh3)4Pd, ally-t-butylcarbonate, toluene, 95%; (f) NaBH4, MeOH, THF, 97%; (g) Im, TESCl, DMF, 92%; (h) PhLi, THF, 87%; (i) HF/pyridine, MeCN, 57%; (j) PdCl2, CuCl, H2O, DMF, O2, 83%; (k) HF/pyridine, MeCN, THF, 74%. | ||
At the very beginning, we attempted to replace the C-4 substituent to an allyl ether before the opening of the D-ring. However, it was found that the elimination of the 5-iodo could not be realized in the presence of any substituent larger than 4-OH. According to the 1HNMR data (not shown), it was suggested that, upon modification of the 4-OH, the conformation of the C-ring changed to such an extent that it prevented the elimination reaction.
Unexpectedly, the allylation of 4-OH in 9 was found to be extremely difficult, which could not be realized in the presence of various bases and active allyl reagents, although the 4-O-methylation worked smoothly under similar conditions (unpublished work). Treatment of 9 by (PPh3)4Pd and ally-t-butylcarbonate only afforded mainly the mono-allylated product at the C-3′N position. It was then found that the 4-O-allylation reaction could only be realized after the 20-OH was oxidized to an aldehyde. Thus, 20-O-TES was selectively removed in the presence of HF-pyridine, followed by oxidation of the primary hydroxyl by 2-iodoxybenzoic acid (IBX) to 20-aldehyde 11, on which the 4-OH allylation was realized (95% yield) without affording the C-3′N allylated by-product. Next, 20-aldehyde was reduced to hydroxyl with NaBH4, and the newly formed hydroxyl group was protected by a TES group to afford 14. After the opening of the 1,2-carbonate with PhLi and subsequent selective de-silylation of 20-O-TES by HF-pyridine, the intermediate 16 was obtained in 50% yield over two steps. It was noticed that the selective de-silylation of 20-O-TES 15 was much slower than that of 9. We take this as an indication of the decreased selectivity and yield due to steric hindrance by the neighbouring allyl group. When 16 was treated under Wacker oxidation conditions, the D-seco compound bearing a C,D-spiro ring 17 was unexpectedly obtained in 83% yield. The double bond between C-5 and C-6 was not affected under these conditions. Finally, desilylation of 17 afforded spiro-taxoid 6 in 74% yield.
Numerous deuterium-labelling studies have been done to elucidate the mechanism of the Wacker reaction, and those results indicated that an intramolecular hydrogen transfer is involved in the reaction procedure, i.e. no exchange between any alkyl hydrogen atoms and solvent protons occurs.10 On the basis of this rationale, the most plausible mechanism for the Wacker-type cyclization of 16 is proposed in Scheme 2. The palladium is first coordinated to C-4 allyl to form the π-complex 18, then the olefin is readily attacked by C-20 OH (an intramolecular proton transfer), leading to the formation of the six-membered ring, which is followed by a β-elimination to afford intermediate 20. After the hydrogen atom re-inserts into the molecule, a chloride-assisted deprotonation occurs to afford the final C,D-spirodioxene product 17. It seems that an unprotected C-20 OH is crucial to this reaction, as starting from the C-20 OTES compound 15, only the classic Wacker oxidation occurred to give ketone 22 under similar Wacker oxidation conditions (Scheme 3). To the best of our knowledge, this is the first example indicating that 1,4-dioxene analogues could be synthesized from mono-allyl vicinal diol through a Wacker-type cyclization, and this strategy may be applicable to the synthesis of other C,D-spiro taxoids in the future.
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Comparison of the 3J13,14α coupling constants (Table 1) indicates that the H13–C13–C14–H14α dihedral angle related to the puckering of the A-ring should be similar for 5 and 6 but strikingly different from that of 1a (Fig. 1). Indeed, in the ab initio optimized geometries this angle is smaller in 5 and 6 (∼121°) than it is in 1a (∼147°) and the computed 3J13,14α values, according to the Karplus Equation, are in consonance with the experimental measurements (Fig. 1 and Table 1). These modelled structures also account for the increment in ca. +0.5 ppm of the chemical shift of H14α due to the difference in orientation of the proximal electron lone pair of O4 in 5 and 6vs.1a (Table 1).
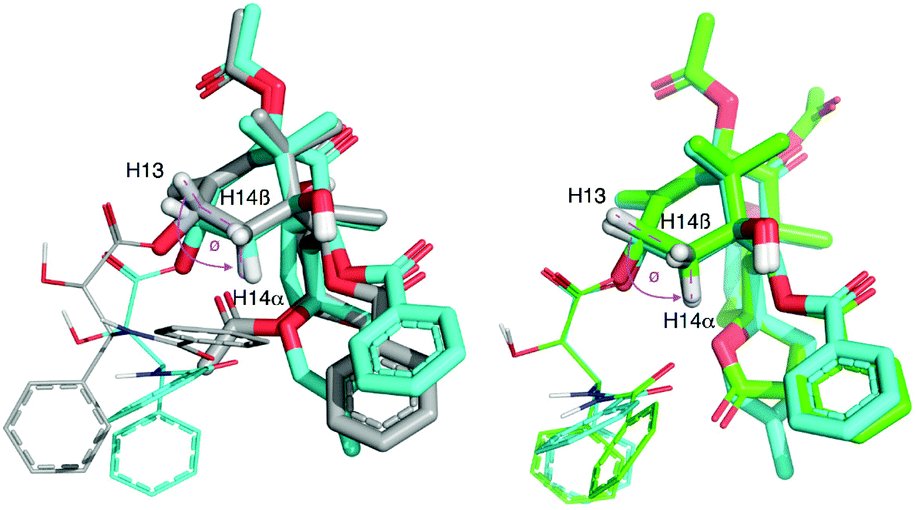 | ||
| Fig. 1 Comparison of the ab initio optimized geometry of 6 (C atoms in cyan) with those of (left) 1a (C atoms in grey) and (right) 5 (C atoms in green) using the common ring B for molecular superposition. The H13–C13–C4–H14α dihedral angle (Ø) is highlighted in pink and the distance separating H14a and O4 in 5 and 6 is shown as a two-headed arrow. In all cases, substituents on O13 are shown as lines for clarity. | ||
| Cmpd | H2 | H3 | H13 | H14α | H14β | 3 J 2,3 | 3 J 13,14β | 3 J 13,14α | Ø (°) | 3 J 13,14α (theoretical)d |
|---|---|---|---|---|---|---|---|---|---|---|
a From ref. 11.
b From ref. 9.
c Dihedral angle between H13–C13–C4–H14α based-on the optimized structures of 1a, 5 and 6.
d Calculated coupling constants based-on the optimized structures of 1a, 5 and 6, according to the Karplus equation 3JHH = Jo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2Ø, where Jo = 14 if Ø = 90–180° and Jo = 10 if Ø = 0–90°. cos2Ø, where Jo = 14 if Ø = 90–180° and Jo = 10 if Ø = 0–90°.
|
||||||||||
|
1aa |
5.67 | 3.79 | 6.23 | 2.35 | 2.28 | 7.0 | 9.0 | 9.0 | 147.5 | 10.0 |
|
5b |
5.89 | 3.16 | 6.03 | 2.81 | 2.45 | 5.5 | 10.5 | 4.0 | 122.0 | 3.9 |
| 6 | 5.44 | 3.44 | 6.03 | 2.94 | 2.29 | 6.0 | 10.8 | 4.2 | 120.0 | 3.5 |
The tubulin binding affinity and cytotoxicity of 6 turned out to be much weaker than those of the parent compound 1a (Table 2). Compared to compound 5, compound 6 is much less potent in the tubulin assay while its cytotoxicity is only slightly weaker (although these results may not be directly comparable because the measurements were performed in different cell lines).9 Inspection of the recent structure of microtubules with 1a bound to β-tubulin (PDB: 3J6G12) suggests that the bulkiness of the six-membered spiro ring in 6 most likely hampers proper lodging of this molecule into the taxane-binding site, thus accounting for the observed loss of affinity and cytotoxicity.
| Tubulin binding constant (Kb 35 °C, 107 M−1) | Cytotoxicity IC50 (nM) A2780 cells | |
|---|---|---|
| 1a | 1.43 ± 0.17 | 2.4 |
| 6 | ≤0.01 | 11830 |
Conclusions
In summary, we achieved the synthesis of a novel C,D-spiro taxane analogue 6 by a procedure in which a Wacker-type cyclization was involved as a key step to form the spirodioxene ring. Although the bioactivity of this novel taxane was not remarkable, a similar strategy used for its synthesis could be applied to enrich the structural diversity of other taxoids. For example, starting from the key intermediates 12 and 13, various cyclization methods13 could be used to prepare new analogues of 6 and other molecules bearing different spiro rings, to facilitate further structure–activity relationship studies and exploration of other unusual mechanisms of action.Experiment section
General
NMR spectra were recorded on Varian Mercury series spectrometers. The mass spectra (MS) were measured on an Agilent 1100 LC/MSD high performance ion trap mass spectrometer or LCQ ESI mass spectrometer. Commercially available reagents and solvents were used without further purification. All anhydrous reactions were performed under a positive pressure of argon and with solvents dried before use through a standard workflow. Column chromatography was carried out with silica gel as the stationary phase.:1) to give compound 8 (0.15 g, 91%) as a white solid. 1H NMR (CDCl3, 300 MHz): δ 7.77 (d, J = 7.8 Hz, 2H), 7.51–7.42 (m, 5H), 7.34–7.22 (m, 3H), 7.07 (d, J = 6.9 Hz, 1H), 6.58 (s, 1H), 6.13 (t, J = 7.5 Hz, 1H), 5.87 (d, J = 8.1 Hz, 1H), 5.02 (br s, 1H), 4.62 (d, J = 1.5 Hz, 1H), 4.55 (dd, J = 3.9, 10.5 Hz, 1H), 4.34 (s, 1H), 4.23 (d, J = 5.1 Hz, 1H), 4.05 (d, J = 9.3 Hz, 1H), 3.77 (d, J = 9.6 Hz, 1H), 3.66 (d, J = 4.5 Hz, 1H), 3.32 (dd, J = 6.0, 15.6 Hz, 1H), 2.50–2.34 (m, 2H), 2.46 (s, 3H), 2.22–2.10 (m, 1H), 2.18 (s, 3H), 1.24 (s, 6H), 1.13 (s, 3H), 0.97–0.91 (m, 18H), 0.79 (s, 9H), 0.70–0.52 (m, 12H), −0.13 (s, 3H), −0.36 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 201.6, 171.3, 169.1, 166.5, 152.4, 143.8, 139.1, 134.5, 132.8, 131.5, 128.7 (2C), 128.3 (2C), 127.4, 127.0 (4C), 89.5, 82.0, 75.6, 75.4, 73.1, 70.8, 70.7, 62.1, 61.7, 55.8, 46.0, 44.5, 41.2, 39.2, 32.4, 26.4, 25.5 (3C), 20.8, 19.8, 18.2, 17.7, 13.6, 6.6 (3C), 5.8 (3C), 5.1 (3C), 4.2 (3C), −5.5, −5.9; MS (ESI+) m/z 1226.7 (M + Na)+; HRMS (ESI): m/z calcd for C57H87INO13Si3+ (M + H)+: 1204.4524, found: 1204.4511.
:1) to give compound 9 (0.11 g, 91%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 7.79 (d, J = 7.6 Hz, 2H), 7.56–7.41 (m, 3H), 7.38–7.32 (m, 4H), 7.29–7.21 (m, 2H), 6.56 (s, 1H), 6.01 (d, J = 10.4 Hz, 1H), 5.88 (brd, J = 8.0 Hz, 1H), 5.81 (d, J = 8.0 Hz, 1H), 5.52 (d, J = 10.4 Hz, 1H), 4.77 (s, 1H), 4.47 (s, 1H), 4.41 (s, 1H), 4.25–4.16 (m, 2H), 3.72 (d, J = 9.8 Hz, 1H), 3.15 (dd, J = 15.6, 4.4 Hz, 1H), 3.04 (d, J = 6.0 Hz, 1H), 2.56 (dd, J = 15.8, 9.9 Hz, 1H), 2.19 (s, 3H), 2.09 (s, 3H), 1.20 (s, 6H), 1.19 (s, 3H), 0.90–0.96 (m, 18H), 0.86 (s, 9H), 0.60–0.65 (m, 12H), −0.07 (s, 3H), −0.27 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 203.1, 170.6, 169.2, 166.3, 152.5, 141.1, 139.9, 135.4, 134.3, 132.4, 131.5, 128.6, 128.6, 128.3 (2C), 127.5, 127.0, 127.0, 126.5 (2C), 125.7, 89.5, 81.3, 75.8, 75.4, 73.2, 72.3, 71.1, 66.2, 61.3, 55.5, 50.0, 40.7, 32.6, 27.2, 25.6 (3C), 20.8, 19.0, 18.2, 16.9, 13.4, 6.7 (3C), 6.6 (3C), 5.2 (3C), 4.2 (3C), −5.5, −5.9; MS (ESI+) m/z 1098.6 (M + Na)+; HRMS (ESI): m/z calcd for C57H85NNaO13Si3+ (M + Na)+: 1098.5221, found: 1098.5237.
:1) to give compound 10 (16 mg, 95%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 7.77 (d, J = 7.5 Hz, 2H), 7.56–7.41 (m, 4H), 7.38–7.23 (m, 6H), 6.56 (s, 1H), 5.97–5.85 (m, 3H), 5.57 (d, J = 10.5 Hz, 1H), 4.77 (s, 2H), 4.46 (s, 1H), 4.25 (d, J = 6.0 Hz, 1H), 4.12 (d, J = 12.0 Hz, 1H), 3.56 (d, J = 12.0 Hz, 1H), 3.02 (m, 2H), 2.81 (brs, 1H), 2.55 (dd, J = 15.6, 10.0 Hz, 1H), 2.18 (s, 3H), 2.12 (s, 3H), 1.19 (s, 3H), 1.18 (s, 3H), 1.14 (s, 3H), 0.93 (t, J = 7.9 Hz, 9H), 0.84 (s, 9H), 0.63 (q, J = 8.0 Hz, 6H), −0.10 (s, 3H), −0.29 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 202.9, 170.5, 169.2, 167.0, 152.4, 141.0, 139.6, 135.6, 134.0, 132.5, 131.8, 128.7 (2C), 128.4 (2C), 127.6, 127.0, 127.0, 126.5 (2C), 90.0, 81.6, 75.7, 75.4, 72.7, 72.1, 71.0, 65.5, 61.4, 55.3, 50.8, 40.7, 32.7, 27.1, 25.5 (3C), 20.8, 19.0, 18.2, 16.9, 13.2, 6.7 (3C), 5.2 (3C), −5.6, −5.9; MS (ESI+) m/z 984.3 (M + Na)+; HRMS (ESI): m/z calcd for C51H71NNaO13Si2+ (M + Na)+: 984.4356, found: 984.4368.
:1) to yield compound 11 (3.2 mg, 91%). 1H NMR (CDCl3, 300 MHz): δ 9.82 (s, 1H), 7.80 (d, J = 6.6 Hz, 2H), 7.55–7.44 (m, 3H), 7.40–7.19 (m, 6H), 6.59 (s, 1H), 5.98–5.79 (m, 3H), 5.48 (dd, J = 10.1, 2.1 Hz, 2H), 4.81 (brs, 1H), 4.50 (d, J = 1.7 Hz, 1H), 4.32 (d, J = 5.5 Hz, 1H), 3.26 (d, J = 5.5 Hz, 1H), 2.89 (dd, J = 15.7, 4.6 Hz, 1H), 2.54 (dd, J = 15.7, 9.9 Hz, 1H), 2.21 (s, 3H), 2.14 (s, 3H), 1.43 (s, 3H), 1.21 (s, 3H), 1.20 (s, 3H), 0.95 (t, J = 7.9 Hz, 9H), 0.84 (s, 9H), 0.65 (q, J = 7.9 Hz, 6H), −0.09 (s, 3H), −0.28 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 201.7, 197.0, 170.5, 169.2, 166.8, 151.4, 141.7, 139.5, 133.0, 133.0, 131.8, 129.8, 128.7 (2C), 128.4 (2C), 127.7, 127.1 (2C), 126.6 (2C), 89.2, 80.4, 78.1, 75.7, 75.3, 71.5, 70.8, 61.8, 55.4, 49.3, 41.1, 32.4, 26.5, 25.5 (3C), 20.8, 19.2, 18.2, 16.7, 13.5, 6.7 (3C), 5.1 (3C), −5.5, −5.9; MS (ESI+) m/z 982.5 (M + Na)+; HRMS (ESI): m/z calcd for C51H69NNaO13Si2+ (M + Na)+: 982.4200, found: 982.4199.
:1) to give compound 12 (63 mg, 95%) as a white solid. 1H NMR (CDCl3, 300 MHz): δ 10.03 (s, 1H), 7.82 (d, J = 7.2 Hz, 2H), 7.60–7.26 (m, 8H), 7.13 (d, J = 9.0 Hz, 1H), 6.51 (s, 1H), 6.21–6.01 (m, 2H), 6.02–5.80 (m, 2H), 5.63 (d, J = 9.4 Hz, 1H), 5.22 (d, J = 17.2 Hz, 1H), 5.12 (d, J = 10.5 Hz,1H), 4.68 (s, 1H), 4.56 (d, J = 2.0 Hz, 1H), 4.42 (d, J = 6.0 Hz, 1H), 4.22 (dd, J = 13.1, 5.3 Hz, 1H), 4.09 (dd, J = 13.0, 5.1 Hz, 1H), 3.22 (dd, J = 15.8, 6.4 Hz, 1H), 3.07 (d, J = 5.9 Hz, 1H), 2.62 (dd, J = 15.7, 10.0 Hz, 1H), 2.18 (s, 3H), 2.02 (s, 3H), 1.29 (s, 3H), 1.24 (s, 3H), 1.13 (s, 3H), 0.91 (t, J = 7.9 Hz, 9H), 0.84 (s, 9H), 0.60 (q, J = 7.9 Hz, 6H), −0.03 (s, 3H), −0.24 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 202.0, 200.8, 171.4, 169.2, 166.5, 152.0, 142.3, 138.5, 134.3, 134.2, 132.7, 131.8, 131.5, 128.8 (2C), 128.5 (2C), 127.8, 127.0 (2C), 126.9, 126.6 (2C), 117.8, 89.7, 82.0, 81.0, 75.5, 75.1, 71.9, 70.6, 66.1, 60.9, 55.6, 49.9, 41.3, 32.2, 26.2, 25.6 (3C), 20.8, 19.8, 18.1, 16.3, 12.8, 6.7 (3C), 5.2 (3C), −5.1, −5.7; MS (ESI+) m/z 1000.5 (M + H)+; HRMS (ESI): m/z calcd for C54H73NNaO13Si2+ (M + Na)+: 1022.4513, found: 1022.4527.
:1) to give compound 13 (82 mg, 97%) as a white solid. 1H NMR (CDCl3, 600 MHz): δ 7.82 (d, J = 7.2 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.48 (t, J = 7.2 Hz, 2H), 7.35–7.26 (m, 5H), 7.12 (d, J = 9.0 Hz, 1H), 6.49 (s, 1H), 6.17 (dd, J = 2.4, 10.8 Hz, 1H), 6.09 (t, J = 8.4 Hz, 1H), 5.99 (m, 1H), 5.71 (dd, J = 1.2, 10.8 Hz, 1H), 5.62 (dd, J = 1.8, 9.0 Hz,1H), 5.28 (dd, J = 1.2, 16.8 Hz), 5.11 (dd, J = 1.2, 10.2 Hz, 1H), 4.65 (brs, 1H), 4.56 (d, J = 2.4 Hz, 1H), 4.32–4.24 (m, 4H), 3.92 (d, J = 13.2 Hz, 1H), 3.14 (dd, J = 7.2, 15.6 Hz, 1H), 2.88 (d, J = 6.0 Hz, 1H), 2.55 (dd, J = 9.6, 15.6 Hz, 1H), 2.17 (s, 3H), 2.04 (s, 3H), 1.28 (s, 3H), 1.24 (s, 3H), 1.22 (s, 3H), 0.91 (t, J = 8.4 Hz, 9H), 0.84 (s, 9H), 0.60 (q, J = 8.4 Hz, 6H), −0.04 (s), −0.23 (s); 13C NMR (CDCl3, 150 MHz): δ 202.9, 171.6, 169.2, 166.6, 152.5, 142.1, 138.3, 135.5, 134.2, 131.7, 131.1, 130.1, 129.1, 128.7 (2C), 128.4 (2C), 127.8, 127.0 (2C), 126.7 (2C), 116.7, 89.9, 81.6, 80.9, 75.6, 75.0, 72.4, 70.9, 66.4, 64.6, 61.4, 55.6, 49.4, 41.3, 32.2, 26.1, 25.6 (3C), 20.8, 20.0, 18.1, 16.1, 12.7, 6.7 (3C), 5.2 (3C), −5.0, −5.6; MS (ESI+) m/z 1024.5 (M + Na)+; HRMS (ESI): m/z calcd for C54H75NNaO13Si2+ (M + Na)+: 1024.4669, found: 1024.4666.
:1) to give compound 14 (85 mg, 92%) as a white solid. 1H NMR (CDCl3, 600 MHz): δ 7.82 (d, J = 7.2 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.48 (t, J = 7.2 Hz, 2H), 7.38–7.26 (m, 5H), 7.17 (d, J = 9.0 Hz, 1H), 6.49 (s, 1H), 6.17 (dd, J = 2.4, 10.8 Hz, 1H), 6.03 (t, J = 7.8 Hz, 1H), 5.92 (m, 1H), 5.57 (dd, J = 1.2, 10.8 Hz, 1H), 5.60 (dd, J = 2.4, 9.0 Hz, 1H), 5.22 (dd, J = 1.8, 16.8 Hz, 1H), 5.06 (dd, J = 1.2, 10.2 Hz, 1H), 4.64 (brs, 1H), 4.57 (d, J = 2.4 Hz, 1H), 4.32–4.24 (m, 1H), 4.27 (d, J = 5.4 Hz, 1H), 4.21 (dd, J = 5.4, 13.8 Hz, 1H), 4.17 (d, J = 12.6 Hz, 1H), 3.97 (d, J = 12.6 Hz, 1H), 2.80 (d, J = 6.0 Hz, 1H), 3.22 (dd, J = 6.6, 15.6 Hz, 1H), 2.52 (dd, J = 9.6, 15.6 Hz, 1H), 2.17 (s, 3H), 2.01 (s, 3H), 1.26 (s, 3H), 1.25 (s, 3H), 1.22 (s, 3H), 0.97–0.88 (m, 18H), 0.84 (s, 9H), 0.64–0.56 (m, 12H), −0.02 (s, 3H), −0.22 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 203.4, 171.7, 169.2, 166.5, 152.7, 142.1, 138.5, 135.9, 134.2, 131.7, 131.1, 129.9, 128.7 (2C), 128.4 (2C), 127.7, 127.5, 127.0 (2C), 126.8 (2C), 116.0, 89.8, 81.6, 81.6, 75.7, 75.0, 72.5, 71.1, 66.5, 64.0, 61.4, 55.6, 49.9, 41.2, 32.3, 26.2, 25.6 (3C), 20.8, 19.9, 18.1, 16.3, 12.1, 6.7 (3C), 6.6 (3C), 5.3 (3C), 4.2 (3C), −4.9, −5.6; MS (ESI+) m/z 1133.6 (M + NH4)+; HRMS (ESI): m/z calcd for C60H89NNaO13Si3+ (M + Na)+: 1138.5534, found: 1138.5587.
:1) to afford compound 15 (35 mg, 87%, based on 54% substrate conversion). 1H NMR (CDCl3, 400 MHz): δ 8.12 (d, J = 7.6 Hz, 2H), 7.82 (d, J = 7.4 Hz, 2H), 7.64–7.26 (m, 11H), 7.17 (d, J = 8.6 Hz, 1H), 6.58 (s, 1H), 6.25–6.10 (m, 2H), 6.05 (d, J = 10.8 Hz, 1H), 5.68 (d, J = 8.5 Hz, 1H), 5.61–5.49 (m, 2H), 5.41 (d, J = 17.3 Hz, 1H), 5.22 (d, J = 10.6 Hz, 1H), 4.67 (s, 1H), 4.62 (s, 1H), 4.34–4.16 (m, 2H), 3.82 (d, J = 11.8 Hz, 1H), 3.70 (d, J = 11.9 Hz, 1H), 3.26–3.14 (m, 2H), 2.18 (s, 3H), 2.16–2.10 (m, 1H), 2.06 (s, 3H), 1.26 (s, 6H), 1.15 (s, 3H), 0.91 (t, J = 8.2 Hz, 9H), 0.81 (s, 9H), 0.68 (t, J = 8.0 Hz, 9H), 0.60 (q, J = 7.8 Hz, 6H), 0.24 (q, J = 7.9 Hz, 6H), −0.03 (s, 3H), −0.29 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 203.1, 171.6, 169.5, 167.1, 166.7, 139.1, 138.7, 136.6, 134.4, 133.3, 132.9, 131.6, 130.3 (2C), 129.9, 129.8, 128.7 (2C), 128.5 (2C), 128.3 (2C), 127.6, 127.0 (2C), 126.7 (2C), 126.5, 115.6, 82.2, 78.1, 75.2, 75.1, 75.1, 73.0, 71.9, 67.3, 63.6, 60.8, 55.8, 53.3, 43.2, 35.3, 26.9, 25.5 (3C), 20.9, 19.9, 18.1, 15.4, 11.2, 6.7 (3C), 6.5 (3C), 5.4 (3C), 3.9 (3C), −5.2, −5.8; MS (ESI+) m/z 1216.5 (M + Na)+; HRMS (ESI): m/z calcd for C66H95NNaO13Si3+ (M + Na)+: 1216.6003, found: 1216.6004.
:1) to give compound 16 (12 mg, 57%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 8.13 (d, J = 7.7 Hz, 2H), 7.81 (d, J = 7.6 Hz, 2H), 7.60–7.40 (m, 6H), 7.39–7.26 (m, 5H), 7.13 (d, J = 8.7 Hz, 1H), 6.58 (s, 1H), 6.28–6.15 (m, 2H), 6.03 (d, J = 10.1 Hz, 1H), 5.74–5.65 (m, 2H), 5.54 (d, J = 6.9 Hz, 1H), 5.44 (d, J = 17.2 Hz, 1H), 5.25 (d, J = 10.4 Hz, 1H), 4.67 (s, 1H), 4.62 (s, 1H), 4.36 (d, J = 11.7 Hz, 1H), 4.24 (dd, J = 13.1, 5.8 Hz, 1H), 3.83 (d, J = 12.3 Hz, 1H), 3.75 (d, J = 12.3 Hz, 1H), 3.25 (d, J = 6.8 Hz, 1H), 3.13 (dd, J = 15.3, 8.1 Hz, 1H), 2.19 (s, 3H), 2.17–2.09 (m, 1H), 2.07 (s, 3H), 1.27 (s, 3H), 1.26 (s, 3H), 1.15 (s, 3H), 0.93 (t, J = 7.9 Hz, 9H), 0.81 (s, 9H), 0.61 (q, J = 7.9 Hz, 6H), −0.04 (s, 3H), −0.29 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 202.5, 171.6, 169.5, 167.2, 166.7, 139.1, 138.6, 136.1, 134.4, 133.4, 132.9, 131.7, 130.3 (2C), 129.8, 129.3, 128.9, 128.7 (2C), 128.7 (2C), 128.4 (2C), 127.7, 127.0 (2C), 126.7 (2C), 116.6, 81.4, 78.6, 75.3, 75.2, 75.0, 72.8, 71.7, 67.2, 64.2, 60.7, 55.8, 52.3, 43.1, 35.4, 26.8, 25.5 (3C), 20.9, 20.1, 18.1, 15.3, 11.8, 6.7 (3C), 5.3 (3C), −5.2, −5.8; MS (ESI+) m/z 1102.5 (M + Na)+; HRMS (ESI): m/z calcd for C60H81NNaO13Si2+ (M + Na)+: 1102.5139, found: 1102.5141.
:1) to give compound 17 (5.8 mg, 83%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 8.21 (d, J = 7.8 Hz, 2H), 7.83 (d, J = 7.6 Hz, 2H), 7.57–7.19 (m, 12H), 6.67 (s, 1H), 6.50 (s, 1H), 6.19–6.10 (m, 2H), 5.74–5.64 (m, 2H), 5.47 (d, J = 6.4 Hz, 1H), 4.80 (s, 1H), 4.53 (s, 1H), 4.16 (d, J = 11.0 Hz, 1H), 3.97 (d, J = 11.0 Hz, 1H), 3.47 (d, J = 6.2 Hz, 1H), 3.06 (dd, J = 15.5, 5.6 Hz, 1H), 2.20 (s, 3H), 2.12 (dd, J = 15.5, 10.5 Hz, 1H), 2.04 (s, 3H), 1.42 (s, 3H), 1.32 (s, 3H), 1.25 (s, 3H), 1.07 (s, 3H), 0.92 (t, J = 7.9 Hz, 9H), 0.83 (s, 9H), 0.62 (q, J = 7.9 Hz, 6H), −0.06 (s, 3H), −0.28 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 202.2, 171.0, 169.5, 167.1, 166.5, 139.1, 138.8, 134.5, 133.7, 133.5, 132.2, 131.6, 130.9 (2C), 128.9, 128.7 (2C), 128.5 (2C), 128.4 (2C), 127.7, 127.6, 127.0 (2C), 126.6 (2C), 121.5, 77.7, 76.3, 75.5, 75.2, 74.7, 72.6, 70.9, 67.7, 60.5, 56.0, 53.0, 42.7, 35.0, 27.6, 25.5, 21.0 (3C), 18.8, 18.2, 16.0. 15.0, 14.0, 6.7 (3C), 5.3 (3C), −5.5, −5.9; MS (ESI+) m/z 1100.5 (M + Na)+; HRMS (ESI): m/z calcd for C60H79NNaO13Si2+ (M + Na)+: 1100.4982, found: 1100.4974.
:1) to give compound 6 (4.2 mg, 74%) as a white solid. 1H NMR (CDCl3, 600 MHz): δ 8.17 (d, J = 7.2 Hz, 2H), 7.84 (d, J = 7.2 Hz, 2H), 7.58–7.32 (m, 11H), 7.23 (d, J = 9.0 Hz, 1H), 6.54 (s, 1H), 6.16 (s, 1H), 6.08 (dd, J = 2.4, 10.8 Hz, 1H), 6.03 (dd, J = 3.6, 10.2 Hz, 1H), 5.84 (dd, J = 2.4, 9.0 Hz, 1H), 5.72 (dd, J = 1.8, 10.8 Hz, 1H), 5.44 (d, J = 6.0 Hz, 1H), 4.75 (d, J = 2.4 Hz, 1H), 4.74 (brs, 1H), 4.07, (d, J = 11.4 Hz, 1H), 4.03, (d, J = 11.4 Hz, 1H), 3.44 (d, J = 6.0 Hz, 1H), 2.94 (dd, J = 4.2, 15.6 Hz, 1H), 2.29 (dd, J = 10.8, 15.6 Hz, 1H), 2.24 (s, 3H), 1.72 (s, 3H), 1.49 (s, 3H), 1.30 (s, 3H), 1.19 (s, 3H), 1.10 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 202.9, 171.5, 170.7, 167.0, 166.4, 139.4, 138.6, 134.0, 133.7, 133.1, 132.1, 131.9, 130.6 (2C), 128.8, 128.7 (4C), 128.5, 128.5 (2C), 128.1, 127.4, 127.0 (2C), 127.0 (2C), 120.5, 77.6, 76.5. 75.5, 74.4, 74.0, 72.4, 72.3, 67.5, 60.3, 54.6, 52.6, 42.3, 35.1, 28.5, 20.9, 18.4, 16.4, 15.1, 13.1; MS (ESI+) m/z 872.4 (M + Na)+; HRMS (ESI): m/z calcd for C48H51NNaO13+ (M + Na)+: 872.3253, found: 872.3267.
:1) to give compound 22 (24 mg, 70%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 8.11 (d, J = 7.7 Hz, 2H), 7.82 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 7.5 Hz, 1H), 7.51–7.27 (m, 11H), 6.58 (s, 1H), 6.22 (t, J = 8.8 Hz, 1H), 5.66 (d, J = 10.9 Hz, 1H), 5.57 (m, 2H), 5.49 (d, J = 6.8 Hz, 1H), 4.71 (d, J = 2.2 Hz, 1H), 4.62 (s, 1H), 4.35 (s, 2H), 3.78 (d, J = 11.6 Hz, 1H), 3.70 (d, J = 11.6 Hz, 1H), 3.22 (d, J = 6.8 Hz, 1H), 2.92 (dd, J = 15.2, 8.0 Hz, 1H), 2.19 (s, 3H), 2.12 (s, 3H), 2.10 (s, 3H), 2.09–2.02 (m, 1H), 1.25 (s, 6H), 1.16 (s, 3H), 0.91 (t, J = 7.9 Hz, 9H), 0.77 (s, 9H), 0.67 (t, J = 8.0 Hz, 9H), 0.59 (d, J = 7.9 Hz, 6H), 0.23 (d, J = 8.0 Hz, 6H), −0.11 (s, 3H), −0.31 (s, 3H); 13C NMR (CDCl3, 150 MHz): δ 208.6, 202.7, 171.7, 169.4, 167.1, 166.9, 139.3, 138.6, 134.5, 133.4, 132.5, 131.6, 130.2 (2C), 130.0, 129.2, 128.6 (4C), 128.3 (2C), 128.1, 127.5, 127.1 (2C), 126.9 (2C), 83.2, 78.6, 75.4, 75.1, 75.0, 72.8, 71.5, 69.1, 68.2, 60.8, 55.7, 52.4, 43.1, 35.2, 26.6, 25.6, 25.5 (3C), 20.9, 20.1, 18.2, 15.4, 11.2, 6.7 (3C), 6.4 (3C), 5.3 (3C), 3.9 (3C), −5.5, −5.9; MS (ESI+) m/z 1232.5 (M + Na)+; HRMS (ESI): m/z calcd for C66H95NNaO14Si3+ (M + Na)+: 1232.5953, found: 1232.5962.
Tubulin and ligands
Calf brain tubulin was purified as described in the literature.14 All the compounds were diluted in dimethyl sulfoxide (DMSO) to a final concentration of 10 mM and stored at −20 °C. Commercially available paclitaxel and docetaxel were used as positive controls.Measurement of equilibrium constants for binding of taxoid ligands to microtubules
The equilibrium binding constants of the different ligands to the paclitaxel binding site of the assembled microtubules were measured as described.15 A mixture of 50 nM paclitaxel binding sites in glutaraldehyde-stabilized microtubules, and 50 nM Flutax-2 in a GAB (glycerol assembly buffer; 3.4 M glycerol, 10 mM NaPi, 1 mM EGTA, 6 mM MgCl2, 0.1 mM GTP, pH 6.5–6.7) was sampled in a 96-well plate for 200 μL per well. Increasing concentrations of the ligands were added to the wells. After being incubated at 35 °C for 20 min, the fluorescence anisotropies were measured using an Infinite F200® microplate reader (Tecan). The excitation and emission wavelengths were set up at 485 nm and 535 nm respectively. Binding constants (Kb) for compounds reversibly displacing Flutax-2 were calculated using a PC program (Equigra v5), which was kindly provided by Dr J. F. Díaz from Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas (Madrid, Spain). This program fits the experimental data by least squares to the equilibrium binding constant of the ligand investigated, employing the known values of the reference ligand Flutax-2.Cell lines and culture
A2780 and A2780/AD cells were cultured in RPMI 1640, supplemented with 10% fetal bovine serum. To maintain the cell drug resistance phenotype, doxorubicin was added to A2780/AD medium, maintaining a concentration of 100 nM. HeLa and HeLa/βIII cells were cultured in DMEM, supplemented with 10% fetal bovine serum. And the latter was additionally supplemented with 0.5 mg per mL G418 sulfate. All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.Cell cytotoxicity assay
Cell viability was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described.16 Briefly, 3000 cells per well for HeLa and HeLa/βIII were seeded in 96-well plates with Dulbecco's Modified Eagle's Medium. 4000 cells per well for A2780 and 15000 cells per well for A2780/AD were seeded in 96-well plates with the RPMI 1640 medium respectively, and then incubated under normal conditions overnight. Cells were treated with different concentrations of the ligands for 72 h, then 20 μL of 5 mg per mL MTT solution was added to the wells. The mixtures were incubated for 4 h at 37 °C until crystals were formed. After the medium was removed, 150 μL of DMSO was added to each well. The plates were gently agitated for 15 minutes until the color of the reaction was uniform and the absorbance was measured at 570 nm with an Infinite F200® microplate reader (Tecan). The vehicle-only treated cells served as the indicator of 100% cell viability. The 50% inhibitory concentration (IC50) was defined as the concentration that reduced the absorbance of the vehicle-only treated wells by 50% in the MTT assay.
Theoretical calculations
PyMOL molecular graphics software (v. 1.3, Schrödinger LLC) was employed for structure visualization, molecular editing and figure preparation. The initial geometry of 1a was obtained from PDB entry 3J6G12 and those of 5 and 6 were model-built in PyMOL. Geometry optimization was performed using the B3LYP (Becke, three-parameter, Lee-Yang-Parr)17 exchange–correlation functional together with a 3-21G basis set, as implemented in program Gaussian 09 (Revision D.01). Chloroform effects were taken into account by means of the standard IEF-SCRF continuum solvent method.Acknowledgements
S. R. W. and W. S. F. thank the National Natural Science Foundation of China (Grant no. 30930108 and no. 81202434) for financial support. F. G. is grateful to the Spanish Ministerio de Economía y Competitividad (SAF2012-39760-C02-02) and Comunidad Autómoma de Madrid (S2010-BMD-2457). We also thank Mr Ning Yan for his work on the cytotoxicity evaluation.Notes and references
- (a) R. Matesanz, I. Barasoain, C. G. Yang, L. Wang, X. Li, C. de Inés, C. Coderch, F. Gago, J. J. Barbero, J. M. Andreu, W. S. Fang and J. F. Díaz, Chem. Biol., 2008, 15, 573–585 CrossRef CAS PubMed; (b) C. Coderch, Y. Tang, J. Klett, S. E. Zhang, Y. T. Ma, S. R. Wang, R. Matesanz, B. Pera, A. Canales, J. Jiménez-Barbero, A. Morreale, J. F. Díaz, W. S. Fang and F. Gago, Org. Biomol. Chem., 2013, 11, 3046–3056 RSC.
- T. C. Boge, M. Hepperle, D. G. V. Velde, C. W. Gunn, G. L. Grunewald and G. I. Georg, Bioorg. Med. Chem. Lett., 1999, 9, 3041–3046 CrossRef CAS PubMed.
- L. Merckle, J. Dubois, E. Place, S. Thoret, F. Gúeritte, D. Guenard, C. Poupat, A. Ahond and P. Potier, J. Org. Chem., 2001, 66, 5058–5065 CrossRef CAS PubMed.
- R. Marder-Karsenti, J. Dubois, L. Bricard, D. Gúenard and F. Gúeritte-Voegelein, J. Org. Chem., 1997, 62, 6631–6637 CrossRef CAS.
- A. A. L. Gunatilaka, F. D. Ramdayal, M. H. Sarragiotto, D. G. I. Kingston, D. L. Sackett and E. Hamel, J. Org. Chem., 1999, 64, 2694–2703 CrossRef CAS PubMed.
- J. Dubois, S. Thoret, F. Gúeritte and D. Gúenard, Tetrahedron Lett., 2000, 41, 3331–3334 CrossRef CAS.
- L. Barboni, G. Giarlo, M. Ricciutelli, R. Ballini, G. I. Georg, D. G. VanderVelde, R. H. Himes, M. Wang, A. Lakdawala and J. P. Snyder, Org. Lett., 2004, 6, 461–464 CrossRef CAS PubMed.
- S. Thoret, F. Gúeritte, D. Gúenard and J. Dubois, Org. Lett., 2006, 8, 2301–2304 CrossRef CAS PubMed.
- M. V. Trmcic, R. V. Matovic, G. I. Tovilovic, B. Z. Ristic, V. S. Trajkovic, Z. B. Ferjancic and R. N. Saicic, Org. Biomol. Chem., 2012, 10, 4933–4942 CAS.
- J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef CAS PubMed.
- G. N. Chmurny, B. D. Hilton, S. Brobst, S. A. Look, K. M. Witherup and J. A. Beutler, J. Nat. Prod., 1992, 55, 414–423 CrossRef CAS.
- G. M. Alushin, G. C. Lander, E. H. Kellogg, R. Zhang, D. Baker and E. Nogales, Cell, 2014, 157, 1117–1129 CrossRef CAS PubMed.
- (a) N. H. Andersen, S. W. Hadley, J. D. Kelly and E. R. Bacon, J. Org. Chem., 1985, 50, 4144–4151 CrossRef CAS; (b) B. Wirz, R. Schmid and J. Foricher, Tetrahedron: Asymmetry, 1992, 3, 137–142 CrossRef CAS; (c) S. Ghorai and A. Bhattacharjya, Org. Lett., 2005, 7, 207–210 CrossRef CAS PubMed; (d) X. Chaminade, L. Coulombel, S. Olivero and E. Dunach, Eur. J. Org. Chem., 2006, 16, 3554–3557 CrossRef; (e) Z. Lu and S. S. Stahl, Org. Lett., 2012, 14, 1234–1237 CrossRef CAS PubMed; (f) A. B. Weinstein, D. P. Schuman, Z. X. Tan and S. S. Stahl, Angew. Chem., Int. Ed., 2013, 52, 11867–11870 CrossRef CAS PubMed.
- J. F. Díaz, M. Menéndez and J. M. Andreu, Biochemistry, 1993, 32, 10067–10077 CrossRef.
- R. M. Buey, I. Barasoain, E. Jackson, A. Meyer, P. Giannakakou, I. Paterson, S. Mooberry, J. M. Andreu and J. F. Díaz, Chem. Biol., 2005, 12, 1269–1279 CrossRef CAS PubMed.
- P. Cai, P. Lu, F. J. Sharom and W. S. Fang, Cancer Lett., 2013, 341, 214–223 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR spectra of compounds 8–17, 6, and 22, and geometry optimization Gaussian log files of compounds 1a, 5 and 6. See DOI: 10.1039/c5ob02131f |
| This journal is © The Royal Society of Chemistry 2016 |