
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemically re-bridging disulfide bonds and the discovery of a thiomaleimide mediated photodecarboxylation of C-terminal cysteines†
Daniel A.
Richards
a,
Sally A.
Fletcher
a,
Muriel
Nobles
b,
Hanno
Kossen
a,
Lauren
Tedaldi
a,
Vijay
Chudasama
a,
Andrew
Tinker
b and
James R.
Baker
*a
aDepartment of Chemistry, University College London, 20 Gordon St, London, UK. E-mail: j.r.baker@ucl.ac.uk; Tel: +44 (0)2076 792653
bBarts and The London, Queen Mary's School of Medicine and Dentistry, Charterhouse Square, London, UK
First published on 25th November 2015
Abstract
Described in this work is a novel method for photochemically manipulating peptides and proteins via the installation of cysteine-selective photoactive tags. Thiomaleimides, generated simply by the addition of bromomaleimides to reduced disulfide bonds, undergo [2 + 2] photocycloadditions to reconnect the crosslink between the two cysteine residues. This methodology is demonstrated to enable photoactivation of a peptide by macrocyclisation, and reconnection of the heavy and light chains in an antibody fragment to form thiol stable conjugates. Finally we report on an intriguing thiomaleimide mediated photochemical decarboxylation of C-terminal cysteines, discovered during this study.
The spatial and temporal control offered by photochemistry, along with its reagentless nature, presents powerful opportunities for the highly targeted manipulation of biomolecules. Research in this area has included the development of ‘photoclick’ reactions for bioconjugation1 and methods to enable the activation of proteins using light.2 Activators include photochromic switches (such as azobenzenes) which have been used as protein photoregulators and as switches for peptide activation.2b,3 Photolabile linkers, which upon release serve to ‘uncage’ to release the active species, have also been employed for this purpose.2a,4 This controlled activation has application in probing biological systems and in prodrugs, serving as a light mediated triggering mechanism.
We envisaged an alternative mechanism of photoactivation, which could be achieved by manipulating the disulfide bonds in proteins using photoactive chemical tags (Scheme 1). Essential disulfides present a target that is known to be susceptible to topological changes upon reduction, and thus deactivation. Developing a method to photochemically rebridge such a system would then trigger reactivation of the peptide, due to reformation of the covalent cross-link and thus the bioactive conformation. The following properties were proposed to be useful in the design of such a system; (a) efficient installation of tags; (b) rapid and high yielding photochemical reaction; and (c) minimal chemical perturbation upon irradiation, to give greatest prospect of reforming the pharmacophore.
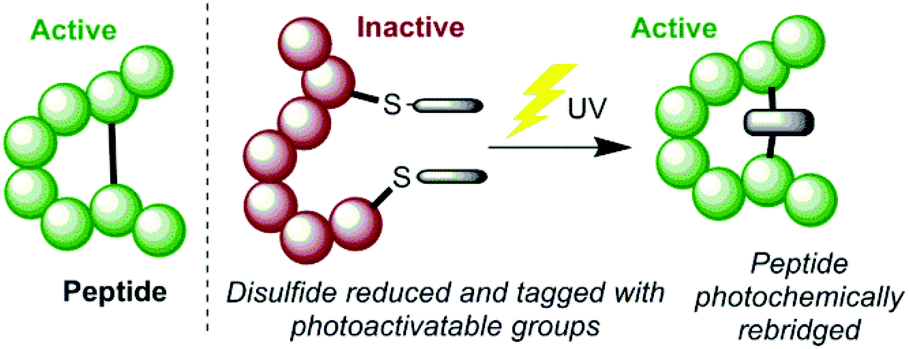 | ||
| Scheme 1 The proposed method for photochemically activating peptides by manipulating a disulfide bond. | ||
We have recently reported on the highly efficient [2 + 2] photodimerisation of thiomaleimides 1,5 generated readily by the addition of bromomaleimides to thiols,6 that appeared to meet these criteria well (Scheme 2). The reaction occurs rapidly (5 min) at low dilution in buffered aqueous conditions, quantitatively and both regio- and diastereo-selectively, to generate cyclobutanes 2 which contain a rigid two carbon bridge between the two thiols.
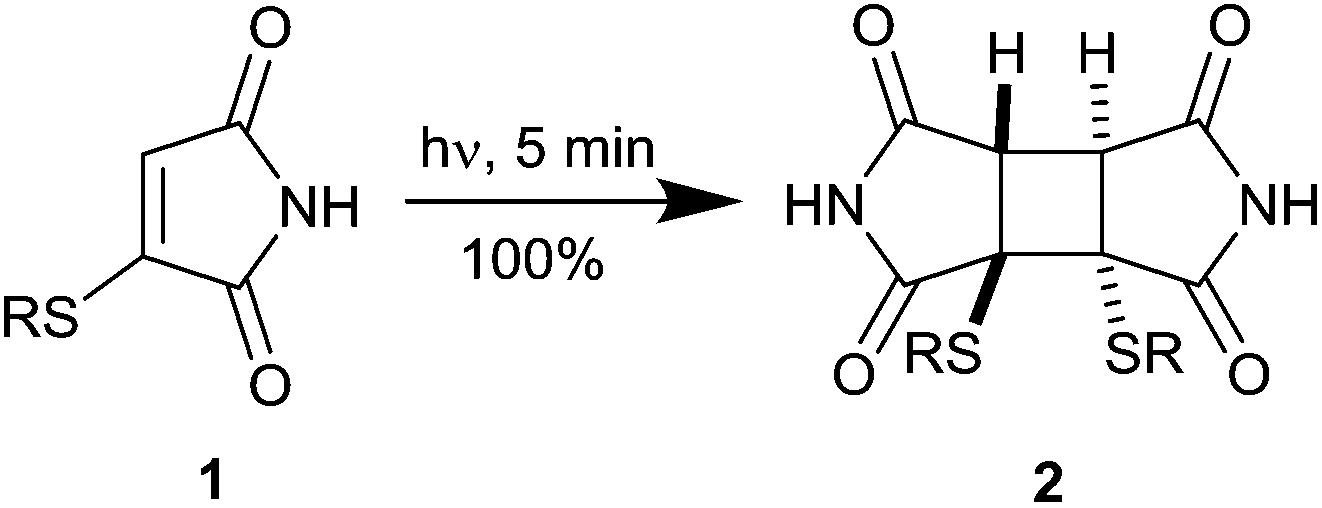 | ||
| Scheme 2 The photochemical dimerisation of thiomaleimides.5 | ||
It was thus hypothesised that by coupling two modes of reactivity of maleimides; selective bioconjugation to cysteine residues and [2 + 2] photocyloadditions, a novel method of photochemically manipulating disulfide bonds could be developed. Given the importance of disulfide bonds for the structure and function of a range of peptides and proteins, this could offer a method with diverse applications. A suitable model to test this hypothesis was found in the small cyclic peptide octreotide (3), a synthetic mimic of somatostatin used for the treatment of neuroendocrine tumours.7 Reduction of the single disulfide bond with TCEP was followed by modification of each liberated cysteine with bromomaleimide 4.8 A large excess of bromomaleimide 4 was employed to preclude the possibility of any competing succinimide bridging8i.e. one bromomaleimide bridging both cysteines. The resulting doubly modified bioconjugate was purified by HPLC and confirmed as 5 by MALDI mass spectrometry (ESI Fig. 1b and 2f†). After isolation 5 was irradiated using a 5 W LED with a λmax of 365 nm to effect photochemical re-bridging generating peptide 6 (Scheme 3). Though no mass change accompanies this reaction, changes in the HPLC trace suggest completion within 2 minutes (ESI Fig. 1c†). No further changes are observed upon prolonged irradiation. Notably the HPLC trace shows the formation of multiple peaks, with four major products; MALDI analysis revealed them to all have identical masses (m/z 1211, ESI Fig. 2†). This outcome is likely due to the formation of a mixture of the different diastereo- and regio-isomers of bridged conjugate 6; which is in contrast to the highly selective intermolecular reaction.5 NMRs obtained on the crude conjugates 5 and 6 confirmed the expected loss of olefenic peaks and appearance of cyclobutane C–H's (ESI Fig. 3†). Further evidence that photochemical cyclisation had occurred was gained through testing the thiol reactivity of the conjugate before and after irradiation. As expected, reaction of bis-thiomaleimide 5 with ethanedithiol resulted in conjugate addition;9 intriguingly this dithiol formed a bridge between the two maleimides to generate expanded macrocyle 7. In contrast, after irradiation of 5 the peptide was totally unreactive with ethanedithiol. This confirmed the loss of the maleimide conjugate acceptors and indicated complete conversion of open chain conjugate 5 to bridged conjugate 6.
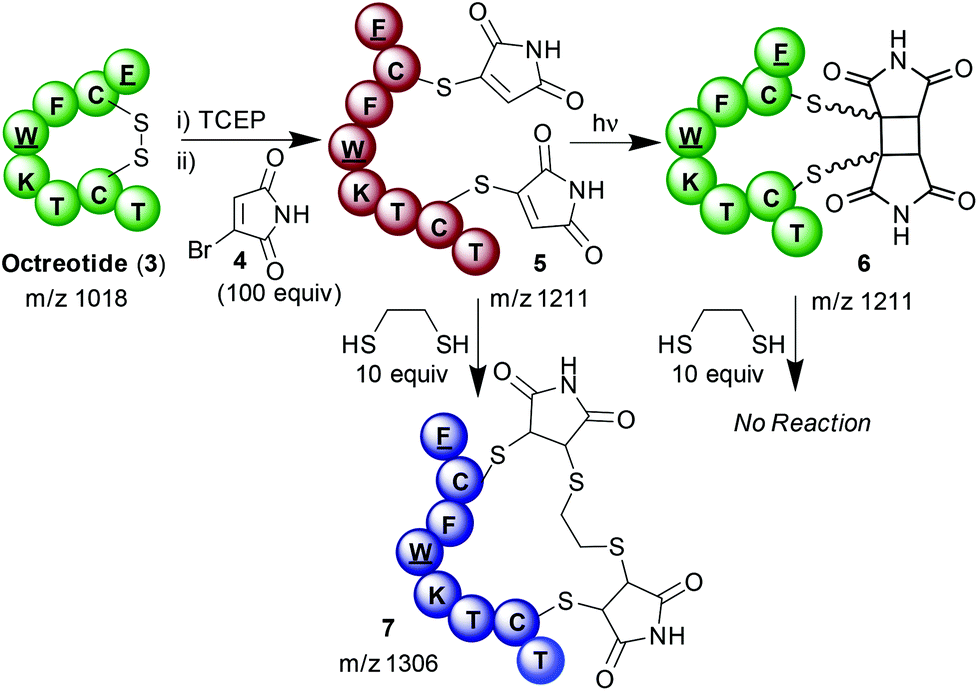 | ||
| Scheme 3 The tagging and photorebridging of Octreotide 3. | ||
To test the hypothesis that octreotide could be photochemically activated by disulfide re-bridging, we tested native octreotide 3 and conjugate 5 before and after irradiation in a signalling assay. We transiently transfected human somatostatin receptor 2 (SSTR2) into HEK293 cells and examined for current activation after agonist application using whole-cell patch clamping (ESI Fig. 6†).10 Concentrations were chosen to achieve maximum activation with each peptide construct. Octreotide 3 was found to be a potent agonist achieving significant current even at 10 nM concentration. The acyclic peptide conjugate 5 was essentially inactive even up to 1 μM, having lost its structure and thus pharmacophore. Pleasingly, by irradiating the sample to generate cyclised peptide 6 activity towards the SSTR2-G protein pathway is returned (Fig. 1). It is notable that this activity is significantly attenuated, even at 100 nM concentration. This is likely due to a combination of the deleterious effect of the 2-carbon bridge on the highly constrained structure of octreotide (as recently described)10 and the mixture of isomers obtained which will have varying activities. Despite this reduction in potency, this still represents a photochemically mediated activation and the first example of achieving this through rebridging of a disulfide bond.
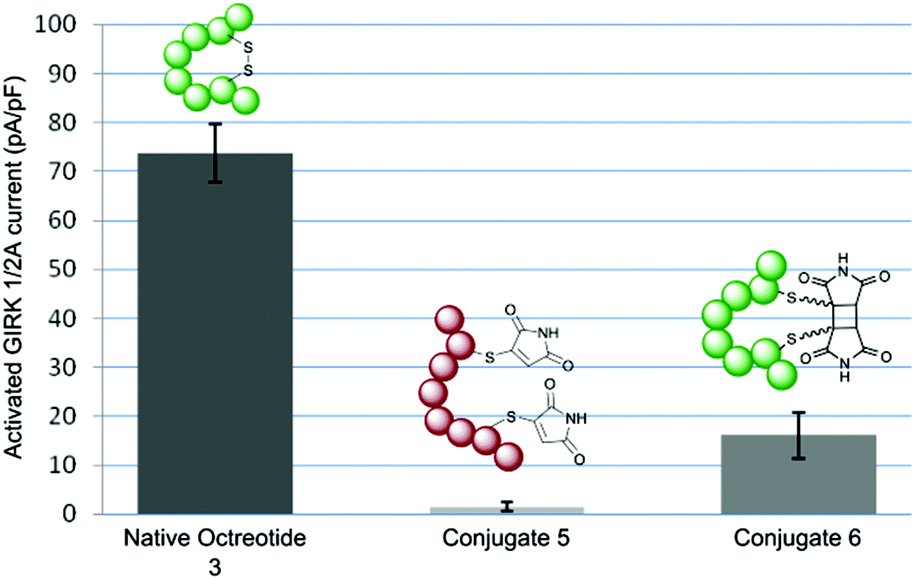 | ||
| Fig. 1 Column chart showing maximum GIRK current activation upon application of native octreotide 3 (10 nM), conjugate 5 (1 μM) and conjugate 6 (100 nM). | ||
Given the success of this [2 + 2] photocycloaddition on a small peptide, which confirmed that the reaction could be transferred from small molecule cysteine studies,5,6c we wanted to test the reaction on a larger protein. We have recently reported that bromomaleimides and other Next Generation Maleimides (NGMs) can be employed to site-selectively modify antibodies and antibody fragments, to generate conjugates with powerful prospective therapeutic and diagnostic applications.11 We thus decided to test the photochemical rebridging strategy on a Fab antibody fragment. Fab fragments contain one solvent accessible disulfide bond, cross-linking the heavy and light chains. In the case of such proteins, the disulfide bond is not crucial to activity as the structure is retained in its absence due to sufficient intermolecular forces. As such, photochemical rebridging in this context would not reactivate the antibody fragment; instead it would serve two purposes. Firstly, it would challenge the capability of this crosslinking reaction on a large biomolecule. Secondly it would provide a method of photochemically stabilising protein conjugates – by regenerating the interchain covalent bond whilst removing the thiol instability.11d
The Fab fragment 8 of the therapeutic monoclonal antibody Herceptin was chosen as the model protein.11e Reduction of the disulfide bond by incubation with TCEP was followed by bis-modification of the two liberated cysteines with an excess of bromomaleimide 4. Subsequent irradiation at 365 nm effected successful re-bridging of the chains (Scheme 4). SDS-PAGE was a highly effective analysis method for this reaction sequence (Fig. 2a). Lane 1 shows the ca. 50 kDa protein, which upon reduction is observed as separate heavy and light chains (lane 2) under these denaturing conditions. Bis-maleimide conjugate 10a retains these two bands as expected (lane 3), then upon irradiation the successful cross-linking of heavy and light chains is observed to afford bridged conjugate 11a (lane 4). By densitometry analysis the yield of the cycloaddition can be approximated at 85%. Retention of binding avidity of 10a and 11a to HER2 was confirmed using ELISA (Fig. 2c). Notably complete thiol stability was shown by incubation of 11a with DTT, BME and GSH, with no reactions observed. This is an important feature of this photochemical re-bridging, as thiol instability of antibody conjugates based on classical maleimides has recently been demonstrated as a problem for therapeutic applications.12
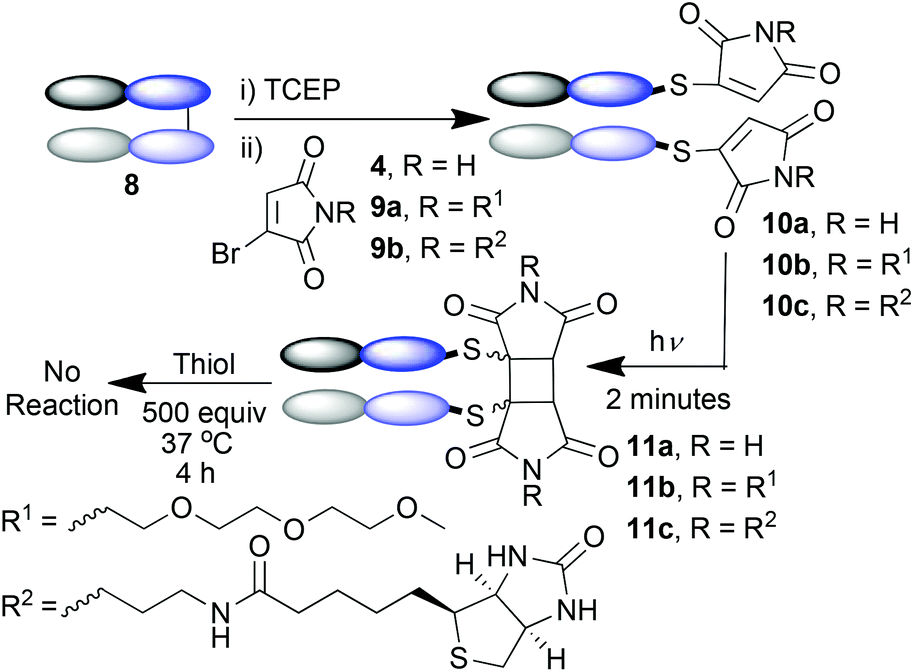 | ||
| Scheme 4 The photochemical re-bridging of a Fab fragment. | ||
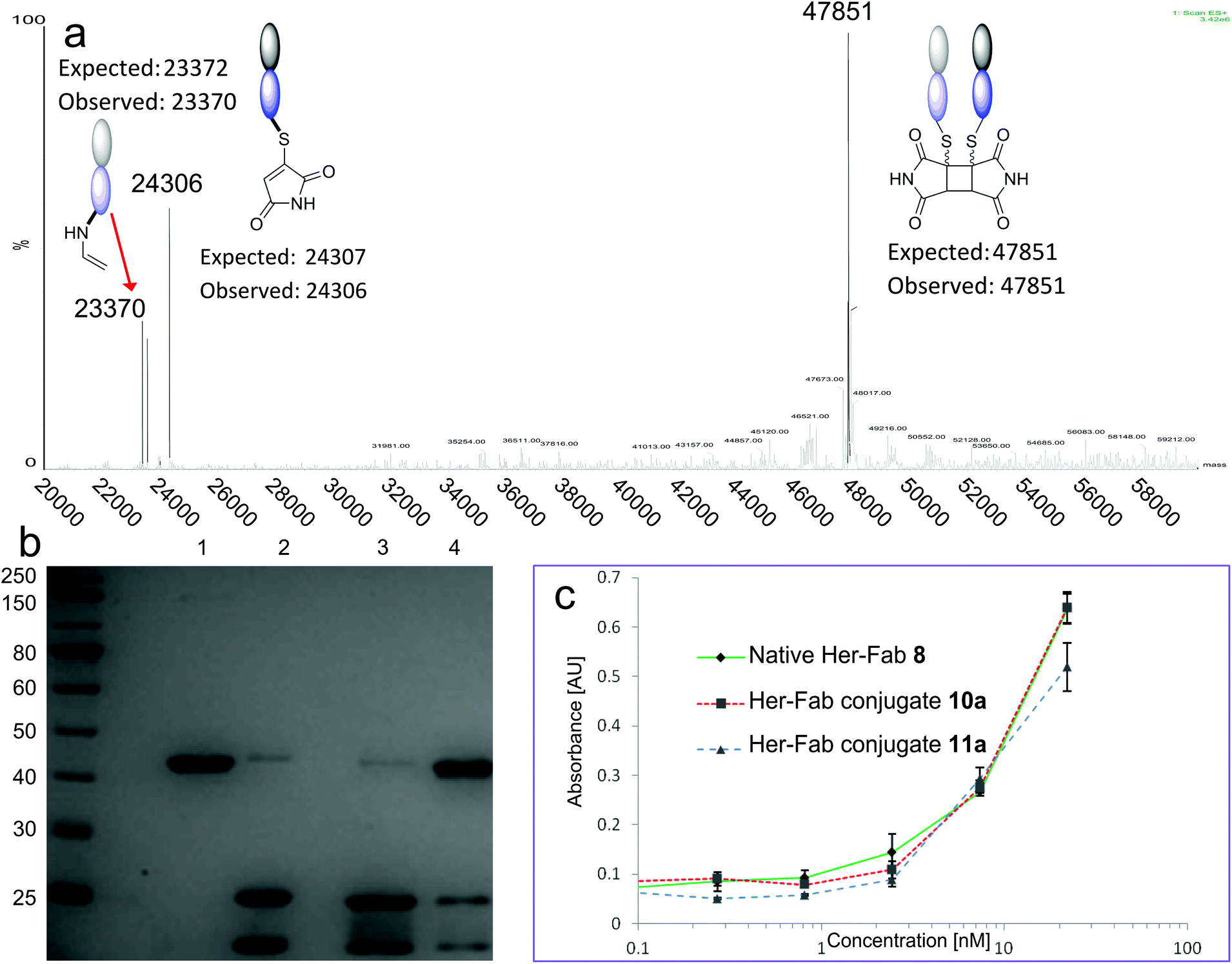 | ||
| Fig. 2 (a) Deconvoluted mass spectrum of conjugate 11a. Arrow denotes decarboxylative side reaction from Scheme 4. (b) SDS-PAGE analysis. Lane 1; native Her Fab 8. Lane 2; reduced Fab. Lane 3; conjugate 10a. Lane 4; conjugate 11a. For comparable SDS-PAGE and LCMS analysis of 10, 11b–c see ESI.† (c). ELISA against HER2 for Native Her Fab 8, conjugate 10a and conjugate 11a. | ||
To demonstrate that this photo-crosslinking could be carried out with protein conjugates towards functional applications, bromomaleimides 9a13 and 9b, with a short appended oligoethylene glycol motif and a biotin respectively, were employed. Successful re-bridging was observed in each case (see ESI†). LCMS data for all conjugates confirmed the reconnection of the heavy and light chains to afford 11a–c as the major outcome of the irradiation (e.g.Fig. 2a). Intriguingly a minor product observed by LCMS (marked by red arrow in Fig. 2a) hinted at an unprecedented reaction of the C-terminal cysteinyl-thiomaleimide on the light chain which was worthy of further investigation. The loss of 76 Da relative to the MWt of the unmodified light chain implied a decarboxylative release of the thiomaleimide, to afford enamides 12a–c (Scheme 5).
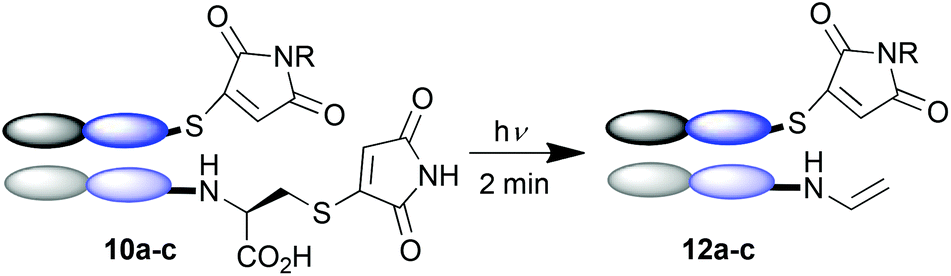 | ||
| Scheme 5 A proposed photochemical side reaction leading to trace formation of conjugates 12a–c. | ||
This reaction would be related to decarboxylative processes observed to occur on phthalimides containing pendent carboxylic acids tethered to the imide nitrogen.14 If demonstrated on thiomaleimides, this would represent a novel mode of photoreactivity of maleimides. It would also offer new opportunities in phototriggered release of cargo from proteins containing C-terminal cysteines.
To confirm this reaction a small molecule study was carried out. Irradiation of 13 (see ESI Scheme 2† for synthesis), in the presence of sodium acetate as a base, resulted in complete decarboxylation to produce ene-carbamate 16 in a 95% yield (Scheme 6). This confirmed the discovery of this new maleimide mediated decarboxylative photocleavage. Mechanistically, a similar sequence can be postulated to that suggested for the phthalimides;14b thus carboxylate 14 formation is followed by absorption of a photon by the maleimide and subsequent electron transfer to generate 15. Decarboxylation and C–S bond fragmentation generates enamide 16. The expected by-product would be a thiomaleimide anion; unfortunately any attempts to isolate this component were unsuccessful, suggesting oligomerisation or polymerisation was occurring. The knowledge gained from the discovery of this novel reaction is currently being used to inform the design of more general maleimide based photocleavable linkers.
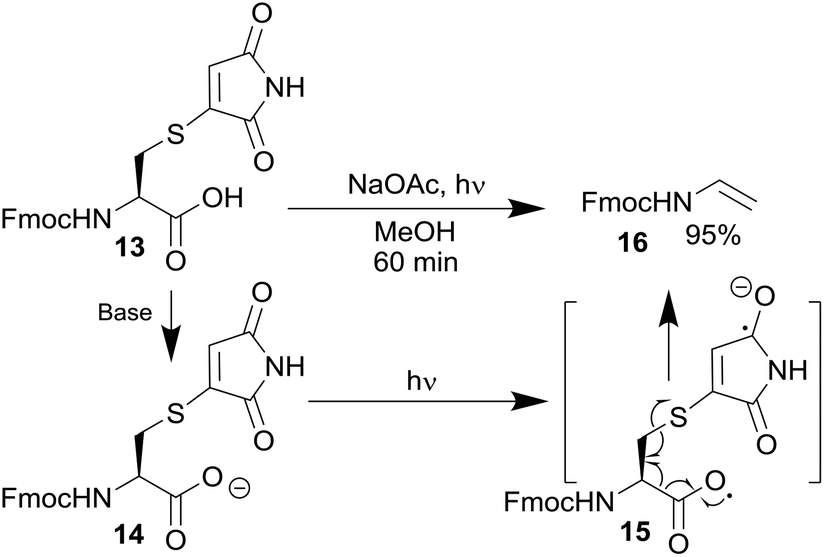 | ||
| Scheme 6 The photochemical decarboxylation of thio-maleimide 13 to produce enecarbamate 16. | ||
Conclusions
This work has described a strategy for the photochemically triggered re-bridging of reduced disulfide bonds. This is achieved by the installation of photoactive maleimide tags on the two cysteine residues, which then undergo [2 + 2] photocyloadditions to achieve the desired cross-linking. Disulfide bonds play a key role in the structure and function of many peptides and proteins, and thus it is envisaged that this methodology could have broad applicability in photochemically manipulating such biomolecules. It is anticipated that the effect of the incorporated bridge on the activity will vary, depending on the size and rigidity of the biomolecule, the role of the disulfide bond in its structure and its proximity to the pharmacophore.In this study we have demonstrated application to the photo-reactivation of Octreotide and the reconnection of heavy and light chains of an antibody fragment to generate thiol-stable antibody conjugates. During the course of the work an interesting side reaction was also observed, leading to the discovery of a novel decarboxylation reaction of C-terminal cysteines. This reaction could represent a new approach to photochemical uncaging, and is worthy of further exploration. The high yields, biocompatibility, and fast reaction times highlight the suitability of substituted maleimides reagents as tools for the photochemical manipulation of biomolecules.
Acknowledgements
The authors gratefully acknowledge Novartis, UCL and BBSRC for supporting this research, Dr Simon Watson for helpful discussions, Dr Abil Aliev for NMR technical support and Dr Kersti Karu for assistance with mass spectrometry. We would also like to acknowledge Dr Enrique Miranda Rota and Prof. Kerry Chester for support in the use of the ELISA assay.Notes and references
- (a) M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed; (b) C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- (a) A. Specht, F. Bolze, Z. Omran, J.-F. Nicoud and M. Goeldner, HFSP J., 2009, 3, 255–264 CrossRef CAS PubMed; (b) W. Szymanski, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef CAS PubMed.
- (a) C. Renner, U. Kusebauch, M. Loweneck, A. G. Milbradt and L. Moroder, J. Pept. Res., 2005, 65, 4–14 CrossRef CAS PubMed; (b) L. G. Ulysse and J. Chmielewski, Chem. Biol. Drug Des., 2006, 67, 127–136 CrossRef CAS PubMed; (c) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- L. M. Tedaldi, A. E. Aliev and J. R. Baker, Chem. Commun., 2012, 48, 4725–4727 RSC.
- (a) L. M. Tedaldi, M. E. B. Smith, R. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC; (b) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed; (c) J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2014, 12, 557–560 RSC; (d) M. E. B. Smith, M. B. Caspersen, E. Robinson, M. Morais, A. Maruani, J. P. M. Nunes, K. Nicholls, M. J. Saxton, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2015, 13, 7946–7949 RSC.
- L. Anthony and P. U. Freda, Curr. Med. Res. Opin., 2009, 25, 2989–2999 CrossRef CAS PubMed.
- C. Marculescu, H. Kossen, R. E. Morgan, P. Mayer, S. A. Fletcher, B. Tolner, K. A. Chester, L. H. Jones and J. R. Baker, Chem. Commun., 2014, 50, 7139–7142 RSC.
- R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 CAS.
- S. A. Fletcher, P. K. B. Sin, M. Nobles, E. Arstad, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2015, 13, 9559–9563 CAS.
- (a) F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 Search PubMed; (b) E. A. Hull, M. Livanos, E. Miranda, M. E. B. Smith, K. A. Chester and J. R. Baker, Bioconjugate Chem., 2014, 25, 1395–1401 CrossRef CAS PubMed; (c) F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC; (d) J. P. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC; (e) L. Castaneda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. B. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187–8189 RSC; (f) A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed; (g) M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016 10.1039/C5SC02666K.
- (a) S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed; (b) B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saadi, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, N. Trung, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawlil, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- L. Castaneda, Z. V. F. Wright, C. Marculescu, T. M. Tran, V. Chudasama, A. Maruani, E. A. Hull, J. P. M. Nunes, R. J. Fitzmaurice, M. E. B. Smith, L. H. Jones, S. Caddick and J. R. Baker, Tetrahedron Lett., 2013, 54, 3493–3495 CrossRef CAS PubMed.
- (a) M. Oelgemoller, P. Cygon, J. Lex and A. G. Griesbeck, Heterocycles, 2003, 59, 669–684 CrossRef CAS; (b) A. Soldevilla and A. G. Griesbeck, J. Am. Chem. Soc., 2006, 128, 16472–16473 CrossRef CAS PubMed; (c) A. Soldevilla, R. Perez-Ruiz, Y. D. Miara and A. Griesbeck, Chem. Commun., 2010, 46, 3747–3749 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C5OB02120K |
| This journal is © The Royal Society of Chemistry 2016 |