
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent IGF-II analogues for FRET-based investigations into the binding of IGF-II to the IGF-1R†
J. M.
Cottam Jones
*a,
P. W. R.
Harris
bcd,
D. B.
Scanlon
a,
B. E.
Forbes
ef,
M. A.
Brimble
bcd and
A. D.
Abell
*ag
aDepartment of Chemistry, The University of Adelaide, Adelaide 5001, SA, Australia
bSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1010, New Zealand
dInstitute for Innovation in Biotechnology, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
eSchool of Molecular and Biomedical Sciences, The University of Adelaide, Adelaide 5005, SA, Australia
fSchool of Medicine, Flinders University of South Australia, Bedford Park, 5042, SA, Australia
gARC Centre of Excellence for Nanoscale BioPhotonics (CNBP), The University of Adelaide, South Australia, 5005, Australia
First published on 26th January 2016
Abstract
The interaction of IGF-II with the insulin receptor (IR) and type 1 insulin-like growth factor receptor (IGF-1R) has recently been identified as potential therapeutic target for the treatment of cancer. Understanding the interactions of IGF-II with these receptors is required for the development of potential anticancer therapeutics. This work describes an efficient convergent synthesis of native IGF-II and two non-native IGF-II analogues with coumarin fluorescent probes incorporated at residues 19 and 28. These fluorescent analogues bind with nanomolar affinities to the IGF-1R and are suitable for use in fluorescence resonance energy transfer (FRET) studies. From these studies the F19Cou IGF-II and F28Cou IGF-II proteins were identified as good probes for investigating the binding interactions of IGF-II with the IGF-1R and its other high affinity binding partners.
Introduction
Insulin-like growth factor II (IGF-II) is a 67-residue regulatory peptide that binds with high affinity to three receptors; the insulin receptor (IR), type 1 insulin-like growth factor receptor (IGF-1R), and type 2 insulin-like growth factor receptor (IGF-2R). The binding of IGF-II to these receptors promotes a range of responses, including cell growth, proliferation, differentiation and apoptosis.1–6Deregulation of the IGF system results in elevated levels of circulating IGF-II with an associated increase in binding to the IGF-1R and IR-A (insulin receptor isoform A).6–15 Activation of both receptors can then promote cancer cell growth and metastasis.16,17 While a structure of insulin bound to a fragment of the IR has been reported,18,19 detailed structural information on the interaction of IGF-II with either the IR-A or IGF-1R remains elusive.
A comparison of IGF-II and IGF-II analogues with the related ligands, IGF-I and insulin in competition binding assays on soluble receptors or cells expressing either the IR-A or the IGF-1R, does however, provide some information on the mechanisms of interaction of IGF-II with IGF-1R and IR-A.19–32 Alanine mutagenesis of IGF-II has identified two separate binding sites (defined as sites 1 and 2) that are critical for binding to the IGF-1R and IR and are analogous to the two insulin binding sites that bind the IR.33 However, precise molecular details of the contact residues within the receptors remain ill-defined. X-ray crystallographic analysis of insulin bound to a fragment of the IR18,19 suggests that the site 1 residues of IGF-II most likely contact the leucine-rich domain 1 and α-CT peptide.
Here we present the convergent synthesis of F19Cou IGF-II and F28Cou IGF-II and their use in a fluorescence resonance energy transfer (FRET) based study to define the interactions of IGF-II with the IGF-1R. A coumaryl glycine acceptor (see Scheme 1) was incorporated into IGF-II at Phe28 and at Phe19 to probe interactions with sites 1 and 2, respectively. The coumaryl glycine probe has advantages of a strong quantum yield and a large Stokes shift, while also exhibiting appropriate spectral overlap with tryptophan (Trp) as a FRET donor.34–41 Trp was deemed a suitable donor as these residues were identified as being positioned nearby likely IGF-II binding sites through analysis of the X-ray crystal structure of the Insulin:IR complex as well as IR and IGF-1R mutagenesis data.18,42,43
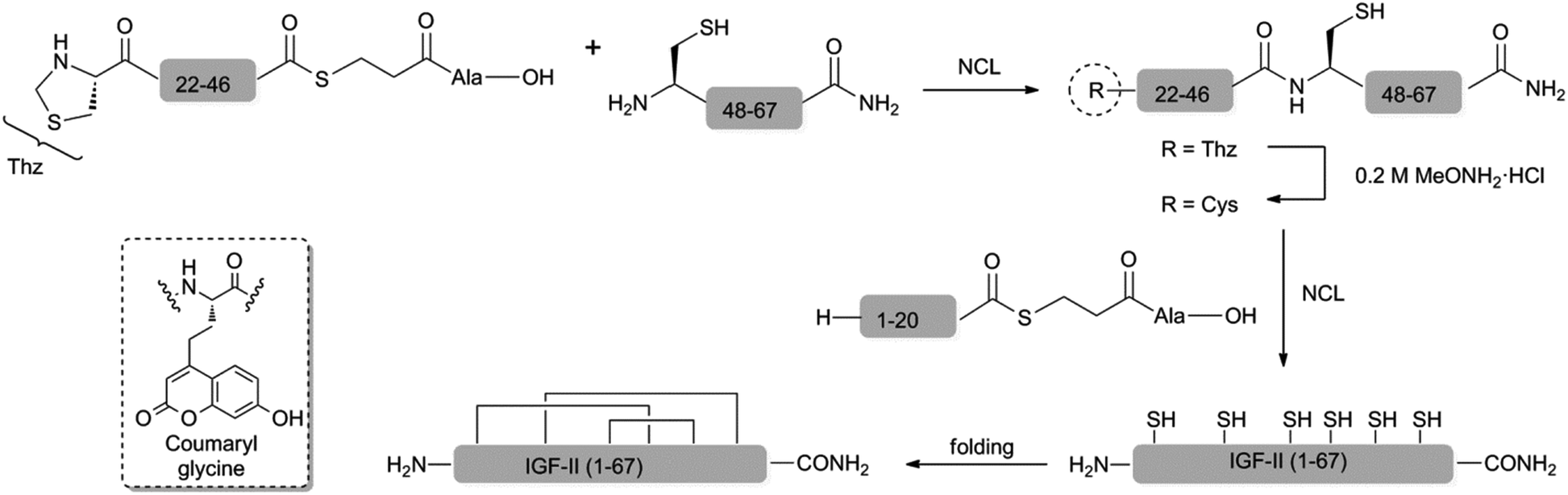 | ||
| Scheme 1 One-pot, three fragment ligation approach to the synthesis of IGF-II analogues. Inset: coumaryl glycine acceptor. NCL conditions: 6.0 M GnHCl, 200.0 mM Na2HPO4, 20.0 mM TCEP, 200.0 mM MPAA, pH of 6.7–7.0. Folding conditions: 2.50 M urea, 0.70 M Tris, 12.5 mM glycine, 2.0 mM EDTA, 0.5 mM DTT, 1.25 mM 2-hydroxyethyl disulphide at a pH of 9.1 and protein concentration of <0.10 mg mL−1. | ||
There are three reports on the chemical synthesis of IGF-II and all utilise a similar linear approach that requires multi-step post-cleavage deprotections and lengthy multi-step purification protocols.44–47 Furthermore, none are suitable for the incorporation of unnatural amino acids. The yield of IGF-II is presented in only two of these syntheses and this is somewhat problematically reported to be of the order of 2% (based on the starting resin). Thus a more modular approach to the coumaryl glycine-containing IGF-II analogues was deemed necessary. With this in mind we have developed an efficient convergent synthesis of IGF-II and its application to the preparation of two specific fluorescent IGF-II analogues; F19Cou IGF-II and F28Cou IGF-II (see Scheme 1).
Results and discussion
The six Cys residues (Cys9, Cys21, Cys46, Cys47, Cys51 and Cys60) of the IGF's provide suitable sites for a native chemical ligation (NCL) approach to the synthesis.48 The viability of such an approach to IGF-II and its analogues was first investigated with a two fragment-based synthesis of native IGF-II. This began with the synthesis of a C-terminal IGF-II (47–67) fragment by standard Fmoc-SPPS and the N-terminal IGF-II (1–46) thioester using an in situ neutralisation Boc-SPPS protocol (see ESI†).49 Ligation of the N-terminal IGF-II (1–46) thioester and the C-terminal IGF-II (47–67) fragment, in the presence of 6.0 M GnHCl, 200 mM Na2HPO4, 20 mM TCEP, 200 mM MPAA,50 at a pH of 6.8, gave the desired native IGF-II peptide (see ESI†). However, access to sufficient quantities of the required 46-residue fragment, or unnatural amino acid-containing derivatives thereof, proved impractical due to low yields associated with the assembly and purification of such a long peptide sequence. Therefore, a more convergent, three fragment-based approach to synthesis of the native IGF-II and fluorescent IGF-II analogues (F19Cou IGF-II and F28Cou IGF-II) was investigated as summarised in Scheme 1.The IGF-II peptides were assembled from three fragments of 20, 26 and 21 amino acids in length respectively, using iterative Val20 to Cys21 and Cys46 to Cys47 ligations as depicted in Scheme 1. Specific ligation sites were selected based on the size of the respective fragments and the predicted reactivity of the C-terminal residue bearing the thioester.51 Thus the Val20–Cys21 junction (rather than Leu8–Cys9) was selected as the N-terminal ligation site, where disconnection here gives rise to peptide fragments of similar size (approx. 20 residues)(see ESI† for IGF-II sequence).
The peptide thioesters (IGF-II (1–20) and IGF-II (Thz-46)) were prepared using an in situ neutralisation Boc-SPPS protocol,49 and the C-terminal IGF-II (47–67) fragment was synthesised using standard Fmoc-SPPS (see ESI†). The use of a manual in situ neutralisation Boc-SPPS protocol for the preparation of the N-terminal IGF-II (1–20) thioester prevented the formation of deletion by-products, which resulted from on-resin aggregation using an Fmoc-SPPS approach.47 The use of TFA in the deprotection of the Nα-Boc amino functionality was key in disrupting on-resin aggregation.52,53
The one-pot three fragment synthesis of the native IGF-II peptide (summarised in Fig. 1) began with ligation of the IGF-II (Thz-46) thioester with the C-terminal IGF-II (47–67) fragment under standard NCL conditions.50,54 LCMS analysis of the reaction mixture, after 60 min, revealed complete conversion to the IGF-II (Thz-67) fragment (see Fig. 1b). Treatment of this ligation mixture with methoxyamine hydrochloride (0.2 M) at pH 3,55 for 8 h gave complete conversion (as determined by LCMS) of the thiazolidone (Thz) to cysteine (refer to Fig. 1c). The ligation mixture was then returned to pH 6.8 and the N-terminal IGF-II (1–20) thioester was added. Complete conversion to the desired native IGF-II peptide was apparent after 36 h, based on LCMS analysis as shown in Fig. 1d, despite the expected slow reactivity of Val thioester.51 Native IGF-II peptide was isolated by solid phase extraction and purified by RP-HPLC. This material was then folded using the optimised folding conditions described by Delaine et al.56 (see ESI†) to give synthetic native IGF-II in an overall yield of 0.3% (from the IGF-II (Thz-67) thioester fragment) and importantly in improved purity (>98% based on RP-HPLC, see Fig. 2b) compared to previous reports.44,45
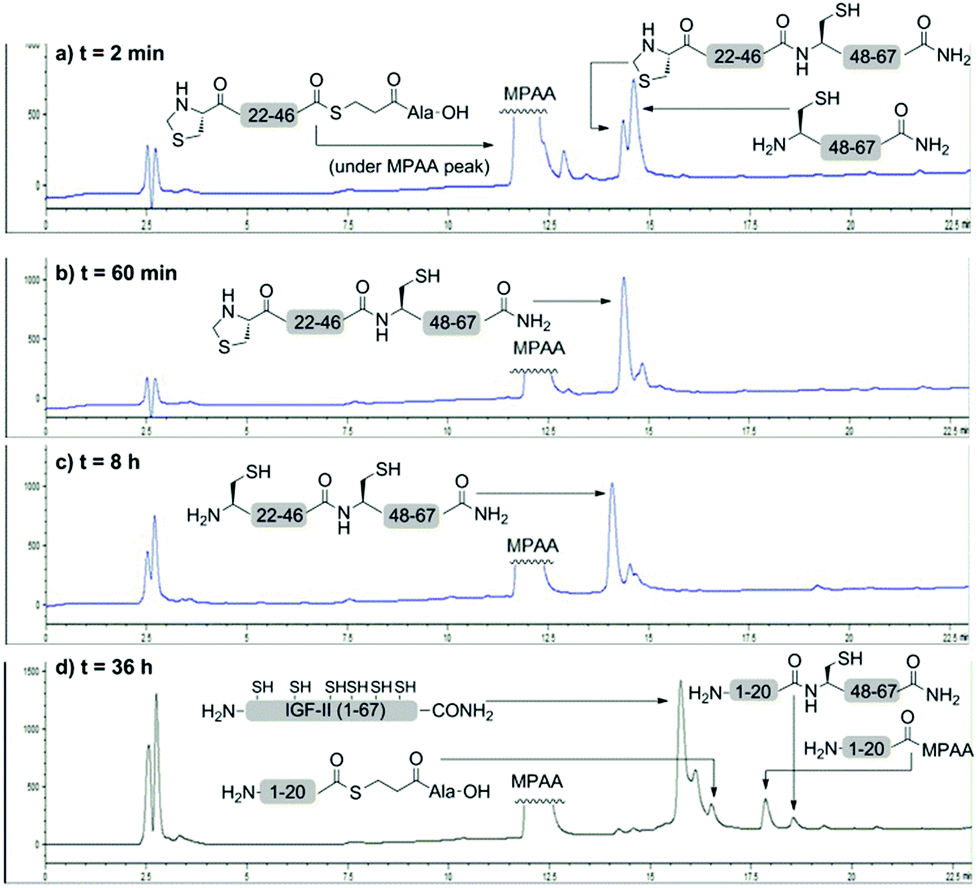 | ||
| Fig. 1 LCMS analysis of the one-pot three fragment synthesis of the native IGF-II peptide. Analysis of the cysteine-based ligation between the IGF-II (Thz-46) thioester and C-terminal IGF-II (47–67) fragment after (a) 2 min and (b) 60 min; (c) analysis of the thiazolidone deprotection of the IGF-II (Thz-67) fragment after 8 h; (d) analysis of the valine-based ligation between the N-terminal IGF-II (1–20) thioester and IGF-II (21–67) fragment after 36 h. | ||
 | ||
| Fig. 2 Purified synthetic IGF-II proteins synthesised using NCL approach. (a) Native IGF-II (2 fragment); (b) native IGF-II (3 fragment); (c) F19Cou IGF-II; (d) F28Cou IGF-II. | ||
The methodology was next applied to the synthesis of the two fluorescent IGF-II analogues, F19Cou IGF-II and F28Cou IGF-II. The F19Cou IGF-II peptide was synthesised by ligation of the N-terminal F19Cou IGF-II (1–20) thioester, IGF-II (Thz-46) thioester and C-terminal IGF-II (47–67) fragment. The F28Cou IGF-II peptide was similarly synthesised by ligation of the N-terminal IGF-II (1–20) thioester, F28Cou IGF-II (Thz-46) thioester and C-terminal IGF-II (47–67) fragment. Folding of the fluorescent IGF-II peptides was essentially as described above for the native IGF-II protein, and resulted in a single folded product (see ESI†).56 RP-HPLC was used to confirm the correct folding of the synthetic IGF-II analogues, which was consistent with folding profiles observed for other IGF-II analogues.33,57 Both the F19Cou IGF-II and F28Cou IGF-II proteins were isolated in excellent purity (>98% based on RP-HPLC, see Fig. 2c and d), and in moderate to low overall yields of 1% (from the IGF-II (Thz-67) thioester fragment) and 0.1% (from the F28Cou IGF-II (Thz-67) thioester fragment) respectively. A real advantage of the ligation methodology is that it is robust and highly reproducible. Sufficient quantities of peptide were obtained for the study, however the yield from protein folding could likely be improved if conducted on a larger scale.
Competition binding assays of the synthetic IGF-II analogues with the IGF-1R were conducted using solubilized immunocaptured IGF-1R58 and the resulting binding curves are depicted in Fig. 3. The IC50 values for the binding of the synthetic native IGF-II (two fragment synthesis) and synthetic native IGF-II (three fragment synthesis), to the IGF-1R were determined to be 2.1 ± 1.6 nM and 2.0 ± 1.2 nM respectively, see Table 1. These values are essentially identical to the IC50 values determined and those reported for the recombinant native IGF-II protein (2.1 ± 1.2 nM).56 The F19Cou IGF-II and the F28Cou IGF-II proteins gave IC50 values of 7.0 ± 1.3 nM and 6.5 ± 1.5 nM respectively. A small decrease in affinity compared to the native IGF-II protein was not unexpected since both Phe19 and Phe28 are reported to be important residues for IGF-II binding.33 Importantly, the observed three-fold decrease in binding affinity is consistent with reports for other IGF-II analogues with substitutions at Phe19 and Phe28.33,56
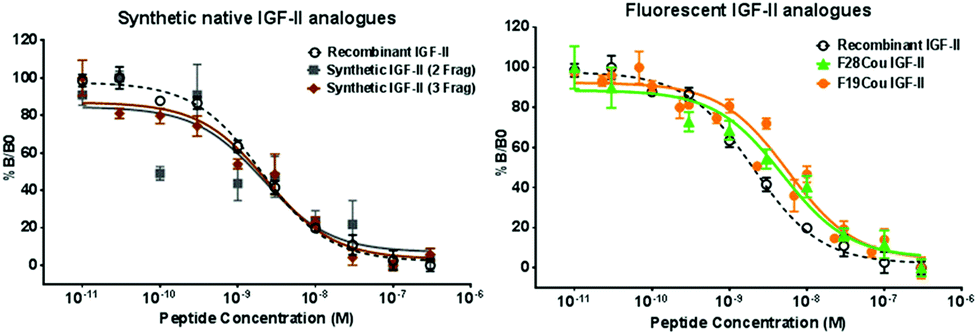 | ||
| Fig. 3 Competitive binding of the synthetic native IGF-II, F19Cou IGF-II, and F28Cou IGF-II proteins to immunocaptured IGF-IR. Immunocaptured IGF-1R was incubated with europium-labelled IGF-II (EuIGF-II) in the absence or presence of increasing concentrations of recombinant native IGF-II (black, dashed line), synthetic native IGF-II (two fragment: grey squares; three fragment: brown diamonds), F19Cou IGF-II (orange circles) or F28Cou IGF-II (green triangles). Results are expressed as a percentage of binding in the absence of competing ligand (Bo). Graphs show data pooled from three separate experiments and each data point is measured in triplicate per experiment, except data from the native IGF-II protein (two fragment) was derived from a single experiment performed in triplicate. Data is shown as the mean ± S.E. Error bars are shown when greater than the size of the symbols. | ||
| Protein | IC50 (nM) | Affinity relative to recombinant native IGF-II (%) |
|---|---|---|
| a Results derived from a single experiment performed in triplicate. | ||
| Recombinant native IGF-II | 2.1 ± 1.2 | 100 |
| Native IGF-II (2 fragment)a | 2.1 ± 1.6 | 100 |
| Native IGF-II (3 fragment) | 2.0 ± 1.2 | 105 |
| F19Cou IGF-II | 7.0 ± 1.3 | 30 |
| F28Cou IGF-II | 6.5 ± 1.5 | 32 |
A FRET analysis of the binding of both fluorescent IGF-II analogues to a soluble form of the IGF-1R (sIGF-1R)59 was next investigated. Native tryptophan fluorescence from the IGF-1R was used as the donor, with the coumarin contained within synthetic F19Cou IGF-II and F28Cou IGF-II proteins acting as an acceptor. Binding was investigated using a sensitized emission approach, where the presence of a FRET signal requires the tryptophan fluorescence of the IGF-1R to be quenched and the coumarin fluorescence of the IGF-II analogue is sensitized. The experiment involved titration of an IGF-II analogue (acceptor) into a solution of sIGF-1R (donor) until the protein and receptor were present in an equimolar ratio. After each addition of an IGF-II analogue, the sample was excited at 280 nm and 320 nm and the fluorescence emission spectrum for each wavelength was recorded (see ESI†). Control experiments for the IGF-II analogues (acceptor) and sIGF-1R (donor) were also analysed using the same methodology.
The resulting fluorescence emission spectra for the native IGF-II, F19Cou IGF-II, and F28Cou IGF-II, after excitation at 280 nm and 320 nm are shown in Fig. 4–6 respectively. Specifically fluorescence emission spectra of the IGF-II analogues (acceptor) in the absence and presence of the donor (sIGF-1R), the emission of the donor (sIGF-1R) and the summed emission spectrum resulting from the sum of the individual donor and acceptor spectra are displayed. The spectra reveal two key emissions, one at 332 nm resulting from excitation of the sIGF-1R (donor) and a second at 455 nm that arises from excitation of the coumarin within the synthetic F19Cou IGF-II and F28Cou IGF-II analogues (acceptor).
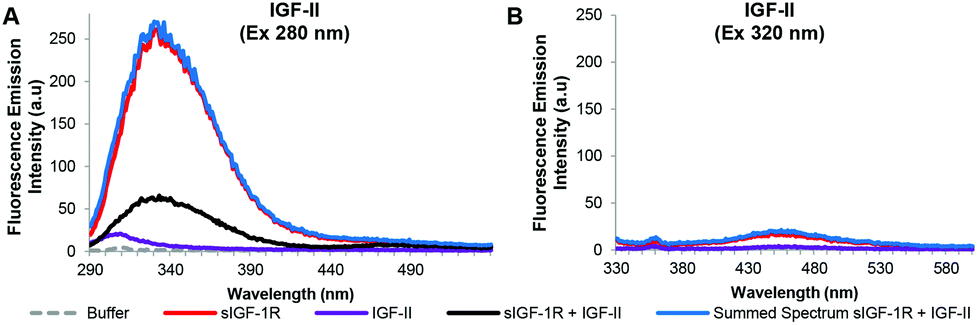 | ||
| Fig. 4 Summary of fluorescence emission spectra of the native IGF-II analogue and sIGF-1R after excitation at 280 nm (A) and 320 nm (B). Fluorescence emission spectra in the absence (purple solid line) and presence (black solid line) of the sIGF-1R, the emission of the sIGF-1R (red solid line) and the summed emission spectrum resulting from the sum of the individual donor (sIGF-1R) and acceptor (native IGF-II) spectra (blue solid lines) are displayed. Spectra were collected when the protein and receptor were present in an equimolar ratio at concentration of 0.20 μM, in 0.1 M sodium phosphate buffer at pH 7.2 after excitation at 280 nm or 320 nm. Spectra are derived from a single experiment, where each spectrum is averaged from three consecutive scans and have not been corrected for background fluorescence. | ||
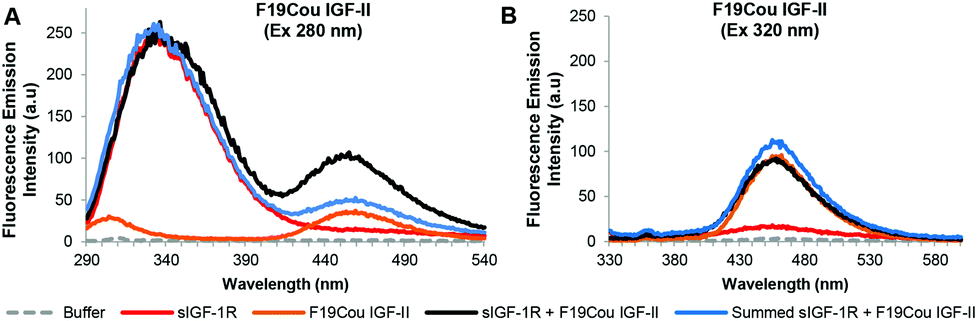 | ||
| Fig. 5 Summary of fluorescence emission spectra of the F19Cou IGF-II analogue after excitation at 280 nm (A) and 320 nm (B). Fluorescence emission spectra in the absence (orange solid line) and presence (black solid line) of the sIGF-1R, the emission of the sIGF-1R (red solid line) and the summed emission spectrum resulting from the sum of the individual donor (sIGF-1R) and acceptor (F19Cou IGF-II) spectra (blue solid lines) are displayed. Spectra were collected when the protein and receptor were present in an equimolar ratio at concentration of 0.20 μM, in 0.1 M sodium phosphate buffer at pH 7.2 after excitation at 280 nm or 320 nm. Spectra are derived from a single experiment, where each spectrum is averaged from three consecutive scans and have not been corrected for background fluorescence. | ||
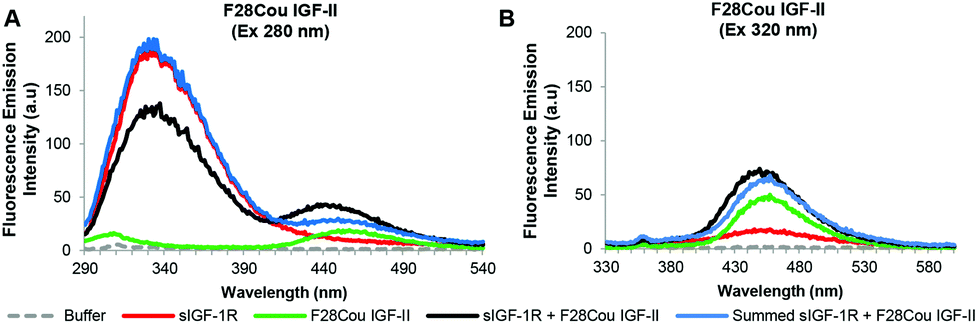 | ||
| Fig. 6 Summary of fluorescence emission spectra of the F28Cou IGF-II analogue after excitation at 280 nm (A) and 320 nm (B). Fluorescence emission spectra in the absence (green solid line) and presence (black solid line) of the sIGF-1R, the emission of the sIGF-1R (red solid line) and the summed emission spectrum resulting from the sum of the individual donor (sIGF-1R) and acceptor (F28Cou IGF-II) (blue solid lines) are displayed. Spectra were collected when the protein and receptor were present in an equimolar ratio at concentration of 0.19 μM, in 0.1 M sodium phosphate buffer at pH 7.2 after excitation at 280 nm or 320 nm. Spectra are derived from a single experiment, where each spectrum is averaged from three consecutive scans and have not been corrected for background fluorescence. | ||
The fluorescence emission spectra for the native IGF-II analogue are shown in Fig. 4. As expected, excitation at 280 nm of native IGF-II (control) alone (Fig. 4A: purple solid line) gave no fluorescence emission at 332 nm. This is because native IGF-II lacks endogenous Trp or a fluorescent probe. Similarly, excitation at 320 nm of native IGF-II alone (Fig. 4B: purple solid line) did not result in an increase in fluorescence emission at 455 nm as it lacks the coumarin fluorophore present in F19Cou IGF-II and F28Cou IGF-II. However, excitation at 280 nm of the sIGF-1R (donor) alone gave an intense emission at 332 nm (Fig. 4A: red solid line), which is due to the presence of endogenous Trp. Surprisingly, excitation at 280 nm of the sIGF-1R (donor) in the presence of native IGF-II resulted in fluorescence emission at 332 nm that was less intense than the fluorescence emission at 332 nm for the sIGF-1R alone (Fig. 4A: red solid line). This emission at 332 nm was also less intense than the sum of the individual emission spectra of native IGF-II and sIGF-1R (Fig. 4A: blue solid line). This large decrease in fluorescence at 332 nm indicates that the receptor (sIGF-1R) is extremely sensitive to ligand binding. The decrease is likely the result of extensive Trp quenching within the sIGF-1R, or perhaps a decrease in the quantum yield of the endogenous Trp within the sIGF-1R. This is an interesting result that suggests a change in the local environment of the IGF-1R Trp residues upon IGF-II binding, and supports the idea of a structural change in the receptor upon ligand binding.
An analysis of the fluorescence emission spectra for the F19Cou IGF-II analogue alone and in complex with the sIGF-1R is shown in Fig. 5. Excitation at 280 nm of the F19Cou IGF-II analogue alone (Fig. 5A: orange solid line) resulted in no fluorescence emission at 332 nm and a small intensity emission at 455 nm, and excitation of the sIGF-1R alone (donor) at 280 nm produced an intense emission peak at 332 nm (Fig. 5A: red solid line). Excitation at 280 nm of F19Cou IGF-II (acceptor) in the presence of the sIGF-1R (donor) (Fig. 5A: black solid line) resulted in fluorescence emissions at 332 nm and 455 nm. The fluorescence emission of the complex at 455 nm was increased compared to the fluorescence emission for the acceptor (F19Cou IGF-II) alone (Fig. 5A: orange solid line). Whereas the fluorescence emission of the complex at 332 nm was unchanged compared to the fluorescence emission spectrum for the donor (sIGF-IR) alone (Fig. 5A: red solid line), and was unchanged compared the sum of the individual emission spectra of the sIGF-1R and F19Cou IGF-II (Fig. 5A: blue solid line). This unaltered fluorescence emission at 332 nm suggests a lack of Trp quenching in the IGF-1R upon F19Cou IGF-II binding. Excitation at 320 nm of the F19Cou IGF-II (acceptor) alone (Fig. 5B: orange solid line) gave rise to fluorescence emission at 455 nm. The same fluorescence emission at 455 nm was also observed for the F19Cou IGF-II in complex with the sIGF-1R (acceptor & donor) (Fig. 5B: black solid line), after excitation at 320 nm. Importantly the fluorescence emission at 455 nm for F19Cou IGF-II in complex with the sIGF-1R (acceptor & donor) was identical in intensity to the emission for the F19Cou IGF-II alone (acceptor). These results demonstrate that fluorescence emission at 455 nm is enhanced upon F19Cou IGF-II binding to the receptor (sIGF-1R), and direct excitation (Ex 320 nm) of the coumarin residue is unaffected after ligand binding. Together these results confirm the F19Cou IGF-II coumarin fluorescence is sensitized.
Fig. 7 shows the acceptor emission data from the FRET interaction between F19Cou IGF-II and the sIGF-1R. Specifically, the extracted acceptor emission (Fig. 7: black dotted line) and the extracted FRET signal (Fig. 7: blue dotted line) are shown. Where the extracted acceptor emission (Fig. 7: black dotted line) was obtained by subtracting the donor (sIGF-1R) emission from the complex (sIGF-1R and F19Cou IGF-II) emission, and the FRET signal (Fig. 7: blue dotted line) was obtained by subtracting the donor (sIGF-1R) and acceptor (F19Cou IGF-II) spectra from the complex (sIGF-1R and F19Cou IGF-II) spectra. The extracted acceptor emission (Fig. 7: black dotted line) shows a fluorescence emission at 455 nm, which is consistent with coumarin emission. The emission was more intense than the fluorescence emission at 455 nm for the acceptor (F19Cou IGF-II) alone (Fig. 7: orange solid line). This increase in fluorescence emission at 455 nm is also visible in the extracted FRET signal shown in Fig. 7 (blue dotted line). Together these results suggest that the quantum yield of the Trp residues, within the IGF-1R, is enhanced by F19Cou IGF-II binding but is simultaneously and equally quenched by FRET. This results in the unaltered Trp emission seen in Fig. 5.
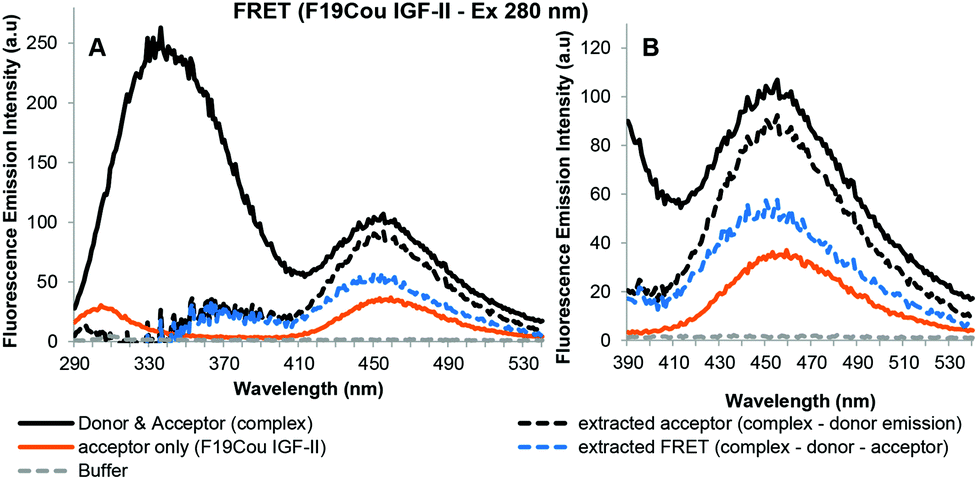 | ||
| Fig. 7 Sensitized fluorescence emission spectra for F19Cou IGF-II in complex with the sIGF-1R after excitation at 280 nm (A) with expansion between 390–540 nm (B). Fluorescence emission spectra of the complex (sIGF-1R and F19Cou IGF-II) uncorrected (black solid line), extracted acceptor emission (black dotted line) which was obtained by subtracting the donor (sIGF-1R) emission from the complex (sIGF-1R and F19Cou IGF-II) emission, acceptor (F19Cou IGF-II) emission (orange solid line), fluorescence emission spectrum resulting from FRET (blue dotted line), which was obtained by subtracting the donor (sIGF-1R) and acceptor (F19Cou IGF-II) spectra from the complex (sIGF-1R and F19Cou IGF-II) are displayed. Spectra were collected when the protein and receptor were present in an equimolar ratio at concentration of 0.2 μM, in 0.1 M sodium phosphate buffer at pH 7.2 after excitation at 280 nm. Spectra are derived from a single experiment, where each spectrum is averaged from three consecutive scans. | ||
Fig. 6 depicts the FRET analysis of the F28Cou IGF-II alone and in complex with the sIGF-IR. Excitation at 280 nm of F28Cou IGF-II alone (acceptor) (Fig. 6A: solid green line) gave a low intensity fluorescence emission at 455 nm, and excitation at 280 nm of the F28Cou IGF-II in complex with sIGF-IR (donor & acceptor) (Fig. 6A: black solid line), gave fluorescence emissions at 332 nm and 455 nm. The fluorescence emission at 332 nm was less intense than the emission at 332 nm for the sIGF-IR alone (Fig. 6A: red solid line). While the fluorescence emission at 455 nm for the complex was more intense than the emission at the same wavelength for the acceptor (F28Cou IGF-II) alone (Fig. 6A: green solid line).
The relative decrease in fluorescence at 332 nm between the complex and receptor alone is consistent with Trp quenching within the sIGF-1R upon F28Cou IGF-II binding, albeit to a lesser extent than with the native IGF-II (Fig. 4). While the increase in fluorescence emission at 455 nm for the complex compared to the acceptor (F28Cou IGF-II) alone, is consistent with a FRET interaction between the sIGF-1R and F28Cou IGF-II. Furthermore excitation at 320 nm gave fluorescence emission at 455 nm for both the F28Cou IGF-II in complex with the sIGF-1R (acceptor & donor) (Fig. 6B: black solid line) and F28Cou IGF-II alone (acceptor) (Fig. 6B: green solid line). This emission was only slightly more intense for the F28Cou IGF-II in complex with the sIGF-1R than for the F28Cou IGF-II alone, and suggests direct excitation (Ex 320 nm) of the coumarin residue is relatively unaffected after ligand binding.
These results are supported by the acceptor emission data shown in Fig. 8.
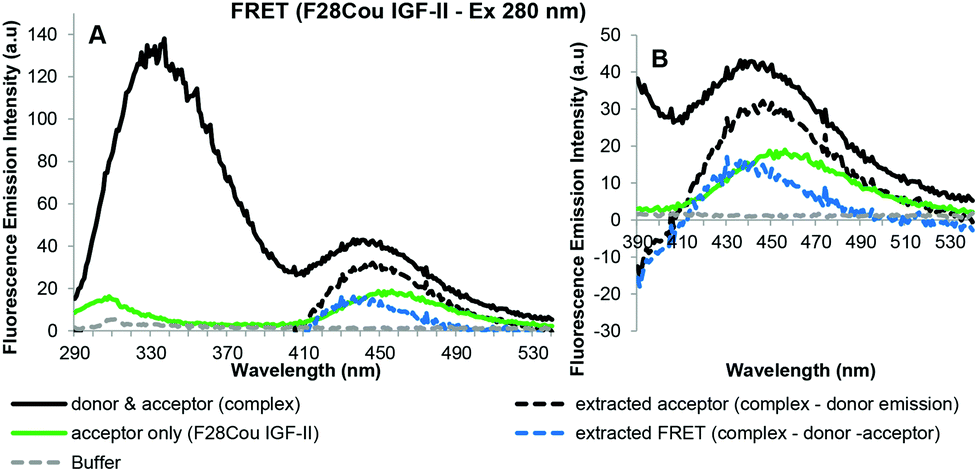 | ||
| Fig. 8 Sensitized fluorescence emission spectra for F28Cou IGF-II in complex with the sIGF-1R after excitation at 280 nm (A) with expansion between 390–540 nm (B). Fluorescence emission spectra of the complex (sIGF-1R and F28Cou IGF-II) uncorrected (black solid line), extracted acceptor emission (black dotted line) which was obtained by subtracting the donor (sIGF-1R) emission from the complex (sIGF-1R and F28Cou IGF-II) emission, acceptor (F28Cou IGF-II) emission (green solid line), fluorescence emission spectrum resulting from FRET (blue dotted line), which was obtained by subtracting the donor (sIGF-1R) and acceptor (F28Cou IGF-II) spectra from the complex (sIGF-1R and F28Cou IGF-II) are displayed. Spectra were collected when the protein and receptor were present in an equimolar ratio at concentration of 0.19 μM, in 0.1 M sodium phosphate buffer at pH 7.2 after excitation at 280 nm. Spectra are derived from a single experiment, where each spectrum is averaged from three consecutive scans. | ||
The extracted acceptor emission shown in Fig. 8 gave fluorescence emission at 455 nm, which is consistent with acceptor emission. This extracted emission was more intense than the fluorescence emission at 455 nm for the acceptor (F28Cou IGF-II) alone (Fig. 8: orange solid line). This emission is also shown in the extracted FRET signal (Fig. 8: blue dotted line). Together this data confirms the increase in the fluorescence emission at 455 nm is likely the result of a FRET interaction between the Trp residues of the IGF-1R and the coumarin of the F28Cou IGF-II analogue. A comparison of F19Cou IGF-II and F28Cou IGF-II acceptor emission spectra shown in Fig. 7 and 8 (blue dotted lines) shows the FRET signal for F19Cou IGF-II is stronger than the FRET signal for the F28Cou IGF-II analogue. This suggests that binding of F28Cou IGF-II to the sIGF-1R causes a decrease in the quantum yield of the receptor (sIGF-1R), leading to a decrease in fluorescence emission at 332 nm, which results in the reduced emission at 332 nm shown in Fig. 6.
A FRET interaction was expected for both site 1 (F28Cou IGF-II) and site 2 (F19Cou IGF-II) interactions as there are several naturally occurring Trp residues located adjacent to the putative IGF-II binding sites (including Trp residues 79, 127, 176, 244 for site 1 and 402, 404, 479, 519 601 and 618 for site 2) (see ESI†).19,27,28,43 Of these, the most likely donor candidates, identified from the X-ray crystallographic data of the insulin:IR interaction19,31 are Trp79, Trp519 and Trp544, as these residues are surface exposed and located approximately 18–25 Å from the proposed ligand binding site. As expected, an increase in fluorescence emission was not observed for the native IGF-II protein in the presence of the sIGF-1R, as it lacks a fluorescent probe (Fig. 4). However surprisingly binding of native IGF-II to the sIGF-1R causes a significant decrease in the Trp fluorescence of the sIGF-1R. The site 2, F19Cou IGF-II analogue (Fig. 5) displayed fluorescence emission that was sensitized, and Trp emission was unaltered. Finally, the F28Cou IGF-II analogue displayed Trp fluorescence which was lower in the presence of the sIGF-1R and the coumarin fluorescence of the F28Cou IGF-II analogue was sensitized. These results confirm that the coumarin probes of the F19Cou IGF-II and F28Cou IGF-II bind in close proximity (10–100 Å) to a Trp residue(s) within the IGF-1R and in turn Phe19 and Phe28 are appropriate sites within IGF-II for the incorporation of a fluorescent probe/FRET acceptor. The strong binding affinities and positive FRET results demonstrate these are good analogues for further FRET binding studies.
Conclusions
In conclusion, we report an efficient, modular synthesis of the native IGF-II protein and two fluorescent IGF-II analogues. A three fragment approach using a Val20–Cys21 ligation site, gave complete ligation in less than 47 h. The IGF-II proteins were isolated in comparable yields (0.1–1%) and importantly in higher purity compared to previously reported IGF-II synthesises (ca. 2%).44–46 The native IGF-II and two fluorescent IGF-II analogues bind with nanomolar affinity to the IGF-1R. Both the F19Cou IGF-II and F28Cou IGF-II proteins displayed a FRET interaction with the IGF-1R, with binding of the native IGF-II protein causing the largest quenching of IGF-IR fluorescence. These results demonstrate that the Trp residues within the sIGF-1R are extremely sensitive to ligand binding, and these coumarin probes bind in close proximity to Trp residues within the IGF-1R and as such are ideal analogues for investigating the interaction of IGF-II with its high affinity binding partners.Acknowledgements
The authors would like to acknowledge Ms Carlie Delaine for preparation of the sIGF-1R and Ms Clair Alvino for helpful discussions. The research was supported in part by the ARC Centre of Excellence in Nanoscale BioPhotonics (CNBP).Notes and references
- S. Kornfeld, Annu. Rev. Biochem., 1992, 61, 307–330 CrossRef CAS PubMed.
- E. Van Obberghen, Diabetologia, 1994, 37, S125–S134 CrossRef CAS PubMed.
- C. E. H. Stewart and P. Rotwein, Physiol. Rev., 1996, 76, 1005–1026 CAS.
- A. Denley, L. J. Cosgrove, G. W. Booker, J. C. Wallace and B. E. Forbes, Cytokine Growth Factor Rev., 2005, 16, 421–439 CrossRef CAS PubMed.
- A. Belfiore, F. Frasca, G. Pandini, L. Sciacca and R. Vigneri, Endocr. Rev., 2009, 30, 586–623 CrossRef CAS PubMed.
- L. K. Harris and M. Westwood, Growth Factors, 2012, 30, 1–12 CrossRef CAS PubMed.
- H. Yu and T. Rohan, J. Natl. Cancer Inst., 2000, 92, 1472–1489 CrossRef CAS PubMed.
- M. N. Pollak, E. S. Schernhammer and S. E. Hankinson, Nat. Rev. Cancer, 2004, 4, 505–518 CrossRef CAS PubMed.
- A. A. Samani, S. Yakar, D. LeRoith and P. Brodt, Endocr. Rev., 2007, 28, 20–47 CrossRef CAS PubMed.
- F. Frasca, G. Pandini, L. Sciacca, V. Pezzino, S. Squatrito, A. Belfiore and R. Vigneri, Arch. Physiol. Biochem., 2008, 114, 23–37 CrossRef CAS PubMed.
- M. Pollak, Nat. Rev. Cancer, 2008, 8, 915–928 CrossRef CAS PubMed.
- E. J. Gallagher and D. LeRoith, Endocrinology, 2011, 152, 2546–2551 CrossRef CAS PubMed.
- A. Belfiore and R. Malaguarnera, Endocr. – Relat. Cancer, 2011, 18, R125–R147 CrossRef CAS PubMed.
- J. Gao, Y. S. Chang, B. Jallal and J. Viner, Cancer Res., 2012, 72, 3–12 CrossRef CAS PubMed.
- M. Pollak, Clin. Cancer Res., 2012, 18, 40–50 CrossRef CAS PubMed.
- J. Brown, E. Y. Jones and B. E. Forbes, in Vitamins & Hormones, ed. L. Gerald, Academic Press, 2009, vol. 80, pp. 699–719 Search PubMed.
- H. M. El-Shewy and L. M. Luttrell, in Vitamins & Hormones, ed. L. Gerald, Academic Press, 2009, vol. 80, pp. 667–697 Search PubMed.
- J. G. Menting, Y. Yang, S. J. Chan, N. B. Phillips, B. J. Smith, J. Whittaker, N. P. Wickramasinghe, L. J. Whittaker, V. Pandyarajan, Z.-L. Wan, S. P. Yadav, J. M. Carroll, N. Strokes, C. T. Roberts, F. Ismail-Beigi, W. Milewski, D. F. Steiner, V. S. Chauhan, C. W. Ward, M. A. Weiss and M. C. Lawrence, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3395–E3404 CrossRef CAS PubMed.
- J. G. Menting, J. Whittaker, M. B. Margetts, L. J. Whittaker, G. K. W. Kong, B. J. Smith, C. J. Watson, L. Zakova, E. Kletvikova, J. Jiracek, S. J. Chan, D. F. Steiner, G. G. Dodson, A. M. Brzozowski, M. A. Weiss, C. W. Ward and M. C. Lawrence, Nature, 2013, 493, 241–245 CrossRef CAS PubMed.
- T. P. J. Garrett, N. M. McKern, M. Lou, M. J. Frenkel, J. D. Bentley, G. O. Lovrecz, T. C. Elleman, L. J. Cosgrove and C. W. Ward, Nature, 1998, 394, 395–399 CrossRef CAS PubMed.
- T. E. Adams and V. C. E. T. P. J. G. C. W. Ward, Cell. Mol. Life Sci., 2000, 57, 1050–1093 CrossRef CAS PubMed.
- C. W. Ward, T. P. J. Garrett, N. M. McKern, M. Lou, L. J. Cosgrove, L. G. Sparrow, M. J. Frenkel, P. A. Hoyne, T. C. Elleman, T. E. Adams, G. O. Lovrecz, L. J. Lawrence and P. A. Tulloch, Mol. Pathol., 2001, 54, 125–132 CrossRef CAS PubMed.
- P. De Meyts, Bioessays, 2004, 26, 1351–1362 CrossRef CAS PubMed.
- V. C. Epa and C. W. Ward, Protein Eng., Des. Sel., 2006, 19, 377–384 CrossRef CAS PubMed.
- M. Lou, T. P. J. Garrett, N. M. McKern, P. A. Hoyne, V. C. Epa, J. D. Bentley, G. O. Lovrecz, L. J. Cosgrove, M. J. Frenkel and C. W. Ward, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12429–12434 CrossRef CAS PubMed.
- N. M. McKern, M. C. Lawrence, V. A. Streltsov, M.-Z. Lou, T. E. Adams, G. O. Lovrecz, T. C. Elleman, K. M. Richards, J. D. Bentley, P. A. Pilling, P. A. Hoyne, K. A. Cartledge, T. M. Pham, J. L. Lewis, S. E. Sankovich, V. Stoichevska, E. Da Silva, C. P. Robinson, M. J. Frenkel, L. G. Sparrow, R. T. Fernley, V. C. Epa and C. W. Ward, Nature, 2006, 443, 218–221 CrossRef CAS PubMed.
- M. Keyhanfar, G. W. Booker, J. Whittaker, J. C. Wallace and B. E. Forbes, Biochem. J., 2007, 401, 269–277 CrossRef CAS PubMed.
- M. C. Lawrence, N. M. McKern and C. W. Ward, Curr. Opin. Struct. Biol., 2007, 17, 699–705 CrossRef CAS PubMed.
- C. Ward, M. Lawrence, V. Streltsov, T. Garrett, N. McKern, M. Z. Lou, G. Lovrecz and T. Adams, Acta Physiol., 2008, 192, 3–9 CrossRef PubMed.
- C. W. Ward and M. C. Lawrence, Bioessays, 2009, 31, 422–434 CrossRef CAS PubMed.
- B. J. Smith, K. Huang, G. Kong, S. J. Chan, S. Nakagawa, J. G. Menting, S.-Q. Hu, J. Whittaker, D. F. Steiner, P. G. Katsoyannis, C. W. Ward, M. A. Weiss and M. C. Lawrence, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6771–6776 CrossRef CAS PubMed.
- J. Whittaker, L. J. Whittaker, C. T. Roberts, N. B. Phillips, F. Ismail-Beigi, M. C. Lawrence and M. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11166–11171 CrossRef CAS PubMed.
- C. L. Alvino, K. A. McNeil, S. C. Ong, C. Delaine, G. W. Booker, J. C. Wallace, J. Whittaker and B. E. Forbes, J. Biol. Chem., 2009, 284, 7656–7664 CrossRef CAS PubMed.
- R. M. Christie and C.-H. Lui, Dyes Pigm., 1999, 42, 85–93 CrossRef CAS.
- R. M. Christie and C.-H. Lui, Dyes Pigm., 2000, 47, 79–89 CrossRef CAS.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, John Wiley & Sons, 2001 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- R. P. Haugland, The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, Life Technologies, 11th edn, 2010 Search PubMed.
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- J. Wang, J. Xie and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 8738–8739 CrossRef CAS PubMed.
- M.-P. Brun, L. Bischoff and C. Garbay, Angew. Chem., Int. Ed., 2004, 43, 3432–3436 CrossRef CAS PubMed.
- L. Whittaker, C. Hao, W. Fu and J. Whittaker, Biochemistry, 2008, 47, 12900–12909 CrossRef CAS PubMed.
- J. Whittaker, A. V. Groth, D. C. Mynarcik, L. Pluzek, V. L. Gadsboll and L. J. Whittaker, J. Biol. Chem., 2001, 276, 43980–43986 CrossRef CAS PubMed.
- C. H. Li, D. Yamashiro, R. Glenn Hammonds Jr. and M. Westphal, Biochem. Biophys. Res. Commun., 1985, 127, 420–424 CrossRef CAS PubMed.
- D. Yamashiro and C. H. Li, Int. J. Pept. Protein Res., 1985, 26, 299–304 CrossRef CAS PubMed.
- Y. M. Oh, H. L. Muller, H. P. Zhang, N. Ling and R. G. Rosenfeld, in Current Directions in Insulin-Like Growth Factor Research, ed. D. LeRoith and M. K. Raizada, Plenum Press Div Plenum Publishing Corp, New York, 1993, vol. 343, pp. 41–54 Search PubMed.
- J. Cottam, D. Scanlon, J. Karas, A. Calabrese, T. Pukala, B. Forbes, J. Wallace and A. Abell, Int. J. Pept. Res. Ther., 2013, 19, 61–69 CrossRef CAS.
- Y. Sohma, B. L. Pentelute, J. Whittaker, Q.-X. Hua, L. J. Whittaker, M. A. Weiss and S. B. H. Kent, Angew. Chem., Int. Ed., 2008, 47, 1102–1106 CrossRef CAS PubMed.
- M. Schnölzer, P. Alewood, A. Jones, D. Alewood and S. B. H. Kent, Int. J. Pept. Protein Res., 1992, 40, 180–193 CrossRef.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- R. C. d. L. Milton, S. C. F. Milton and P. A. Adams, J. Am. Chem. Soc., 1990, 112, 6039–6046 CrossRef CAS.
- M. Schnölzer, P. Alewood, A. Jones, D. Alewood and S. Kent, Int. J. Pept. Res. Ther., 2007, 13, 31–44 CrossRef.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CAS.
- M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS PubMed.
- C. Delaine, C. L. Alvino, K. A. McNeil, T. D. Mulhern, L. Gauguin, P. De Meyts, E. Y. Jones, J. Brown, J. C. Wallace and B. E. Forbes, J. Biol. Chem., 2007, 282, 18886–18894 CrossRef CAS PubMed.
- C. L. Alvino, S. C. Ong, K. A. McNeil, C. Delaine, G. W. Booker, J. C. Wallace and B. E. Forbes, PLoS One, 2011, 6 Search PubMed.
- A. Denley, E. R. Bonython, G. W. Booker, L. J. Cosgrove, B. E. Forbes, C. W. Ward and J. C. Wallace, Mol. Endocrinol., 2004, 18, 2502–2512 CrossRef CAS PubMed.
- K. H. Surinya, B. E. Forbes, F. Occhiodoro, G. W. Booker, G. L. Francis, K. Siddle, J. C. Wallace and L. J. Cosgrove, J. Biol. Chem., 2008, 283, 5355–5363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of peptide fragments, experimental conditions, analysis of ligations and fluorescent experiments. See DOI: 10.1039/c5ob02110c |
| This journal is © The Royal Society of Chemistry 2016 |