
Electronically modified amine substituted alkynols for regio-selective synthesis of dihydrofuran derivatives†
Vijay
V
ad,
Manjusha V.
Karkhelikar
b,
B.
Sridhar
c,
Nedaossadat
Mirzadeh
d,
Suresh
Bhargava
d and
Pravin R.
Likhar
*ab
aIICT-RMIT Joint Research Center, CSIR-IICT, Hyderabad 500007, India
bOrganometallic Gp., I & PC Division, CSIR-IICT, Hyderabad 500007, India. E-mail: plikhar.iict@gov.in
cX-ray Crystallography Center, CSIR-IICT, Hyderabad-500007, India
dCenter of Advanced Materials and Industrial Chemistry, School of Applied Sciences, RMIT University, Melbourne, Australia
First published on 13th November 2015
Abstract
An efficient and simple approach has been developed for the regio-selective synthesis of iodo-substituted dihydrofurans from amine substituted alkynols. The resulting iodo-substituted dihydrofurans have been further diversified by C–C couplings and C–N coupling reactions to afford a diverse range of substituted dihydrofuran derivatives.
Introduction
Electrophilic cyclization is one of the power tools of contemporary organic synthesis. The protocol exemplifies the compatibility of a variety of functional groups and efficient synthesis of a large number of diverse cyclic compounds with interesting biological activities as well as physical properties.1 Likewise, the synthesis of various heterocycles by electrophilic cyclization, especially with I+ as an electrophile, has become the subject of considerable attention recently.2 Notable features of these reactions include ease of performance, short reaction time, ease of product isolation and mainly metal free conditions. More importantly, the presence of iodine enables further functionalization of the molecule. However, in electrophilic cyclization reaction of alkynes bearing two competitive nucleophiles, several factors govern the reaction pathway. The attack of the nucleophile mainly depends on the nucleophilicity of the competing functional groups, polarization of the alkyne triple bond and the cationic nature of the intermediates; these eventually could offer simultaneous ring annulation and lead to the formation of a diverse range of fused heterocycles in few steps.3 Although electrophilic cyclization of alkynes bearing two competing nucleophiles offers strategies for designing various heterocycles, the concept has not been the subject of extensive investigation.We have reported recently the selective formation of a pyrrole nucleus over indole by taking advantage of two competing amine nucleophiles in the electrophilic cyclization of alkynes; this was achieved by modifying the electronic properties of one of the amine groups and hence manipulating its nucleophilic attack.4 The present study highlights an efficient and simple approach for the regio-selective synthesis of iodocyclized dihydrofurans from alkynes bearing two different competing nucleophiles i.e. amine and alcohol. Iodo-dihydrofurans have further been diversified by palladium/copper catalyzed C–C coupling and C–N coupling reactions to afford a diverse range of substituted dihydrofuran derivatives. The preparation of dihydrofuran moieties using this route represents a simple and efficient procedure, addressing the synthesis challenges in the previously known methodology.
In the past few years there have been extensive reports on the synthesis of various kinds of furan substrates.5 However, the synthesis of dihydrofurans by electrophilic cyclization mediated by molecular iodine has been seldom reported.6 Dihydrofuran moiety has often been found in a diverse range of molecules which includes natural products, reaction intermediates, and biologically active compounds and materials.7 These five membered oxygenated heterocycles are usually prepared from oxidative coupling/annulation of 1,3 dicarbonyl compounds with olefins in the presence of excess transition metal salts as oxidants.8 Recently, hypervalent iodine, iodobenzenediacetate, was reported to promote the oxidative coupling/annulation of 1,3-cyclohexanedione with alkenes.9 Later, Lei et al. developed a synthetic approach for dihydrofurans and indolizines by iodine catalysed radical oxidative annulation of β-ketoester with alkenes in the presence of tert-butyl peroxybenzene as an oxidant.10
Results and discussion
The required alkynes were prepared in high yields following our previously reported procedure, by palladium/copper catalysed Sonogashira reaction under mild reaction conditions.4 The electrophilic cyclization of alkyne 1a (Scheme 1) was initiated with K2CO3 as the base and I2 as the electrophilic source. However, the reaction failed to give the hoped-for products 2a and/or 3a (Scheme 1), changing the reaction conditions and using different bases and solvents led to the same results. (Table 1) Attempts to use analogous substrates 1b & 1c (Scheme 1), for obtaining 2b–2c and 3b–3c respectively, were not successful either. The reactions gave no traces of the desired products. (Table 1) However, protection of the amine group with ester led to the formation of 5a, which was isolated in a pure state in 47% yield (Table 1, entry 4). Inspired by the observation, a variety of solvents, bases, additives and iodine sources were used to improve the yield of the reaction. Eventually, when a strong base such as KOtBu was used in acetonitrile in the presence of molecular iodine, a very good yield of 5a was obtained (entry 12). The use of additive (ceric ammonium nitrate, CAN) in combination with other iodine sources could not help in this iodocyclization reaction. The product obtained from the cyclization was fully characterized by 1H NMR, 13C NMR and HRMS. The formation of dihydrofuran 5a was visibly observed in the 1H NMR analysis when the protons of CH2 groups close to the triple bond and the hydroxyl group were shifted downfield; 2.68 ppm (CHa2, t) near to the triple bond shifted to 3.07 ppm (CHa2, t) and 3.78 ppm (CHb2, t) near to the oxygen shifted to 4.53 ppm (CHb2, t). This shift was more pronounced in the CHb2 group near to the oxygen than the CHa2 group near to the iodine. This effect can be attributed to the higher electronegative effect of the oxygen than that of iodine.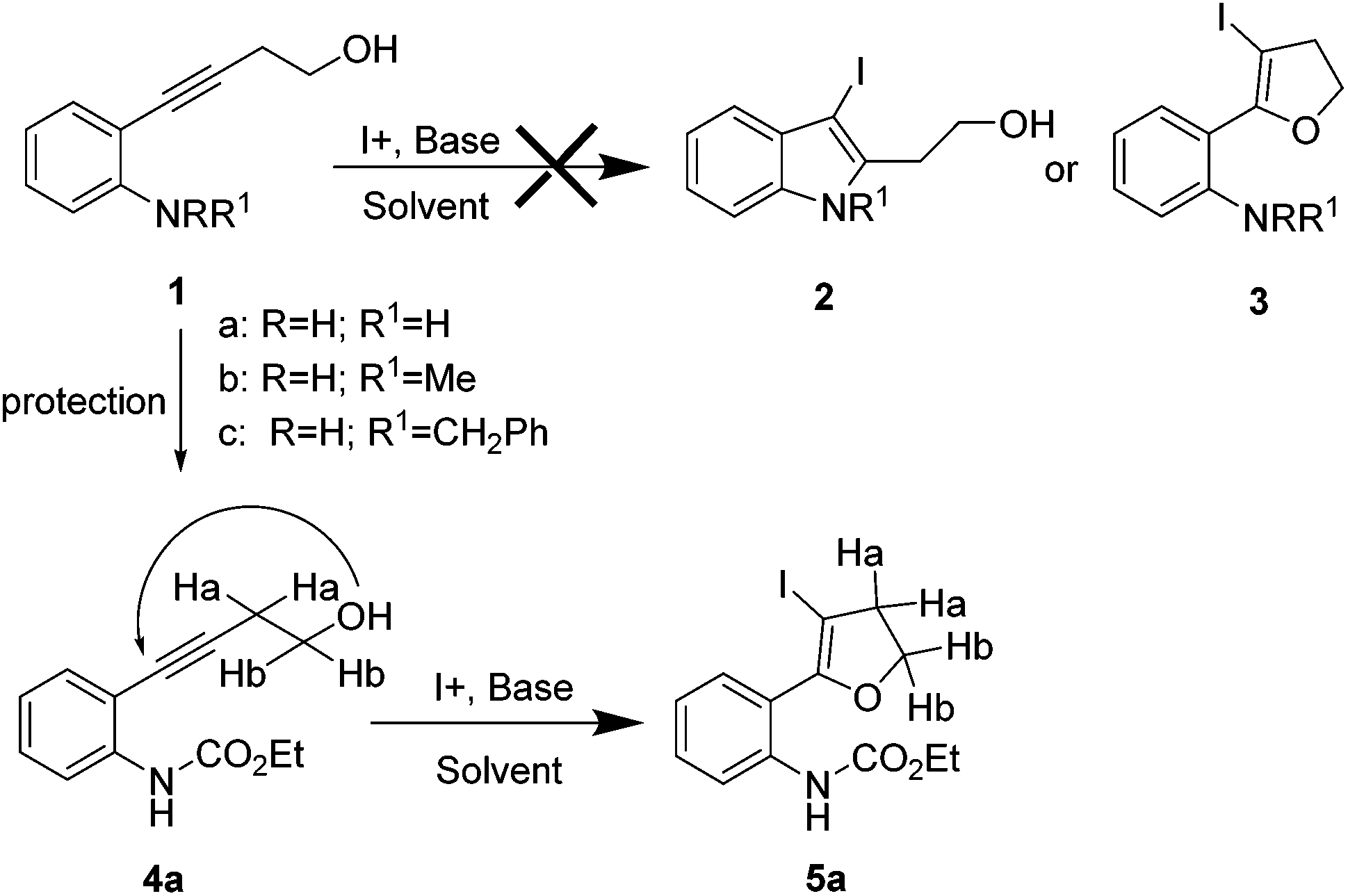 | ||
| Scheme 1 | ||
| Entry | 1a/4a | Solvent | Base (equiv.) | I+ (equiv.) | Yieldb (%) 2a/3a/5a |
|---|---|---|---|---|---|
| a Reaction conditions: alkynol 1a/4a (0.5 mmol), base, I+ source, solvent (2 mL) at room temperature for 2 h. b Yields of isolated products. c No desired product formed. d Inseparable mixture including 5a. | |||||
| 1 | 1a | MeCN | K2CO3 (3) | I2 (3) | — |
| 2 | 1a | DCM | K2CO3 (3) | I2 (3) | — |
| 3 | 1a | THF | NaHCO3 (3) | I2 (3) | — |
| 4 | 4a | MeCN | K2CO3 (3) | I2 (3) | 47 |
| 5 | 4a | MeCN | Cs2CO3 (3) | I2 (3) | 20 |
| 6 | 4a | MeCN | NaHCO3 (3) | I2 (3) | —c |
| 7 | 4a | MeCN | K2CO3 (3) | CAN(1.1) + I2 (3) | —d |
| 8 | 4a | MeCN | K2CO3 (3) | ICl (2) | —c |
| 9 | 4a | DCM | — | NIS (1.1) | —c |
| 10 | 4a | THF | K2CO3 (3) | I2 (3) | 17 |
| 11 | 4a | MeOH | K2CO3 (3) | I2 (3) | Trace |
| 12 | 4a | MeCN | KOt-Bu (3) | I 2 (3) | 78 |
Using optimized reaction conditions, the scope of the iodocyclization reaction was further extended to various amino alkynols (shown in Scheme 2). The effects of changing the substituents on the aromatic ring, the position of substituents on the aromatic ring and the use of various groups to protect the amine functionality were explored. The methodology successfully afforded the preparation of a range of iodo-substituted dihydrofurans 5a–5m in moderate to good yields. Though 5l could not be isolated in the pure state, we were able to detect it from the HRMS analysis (Table 2).
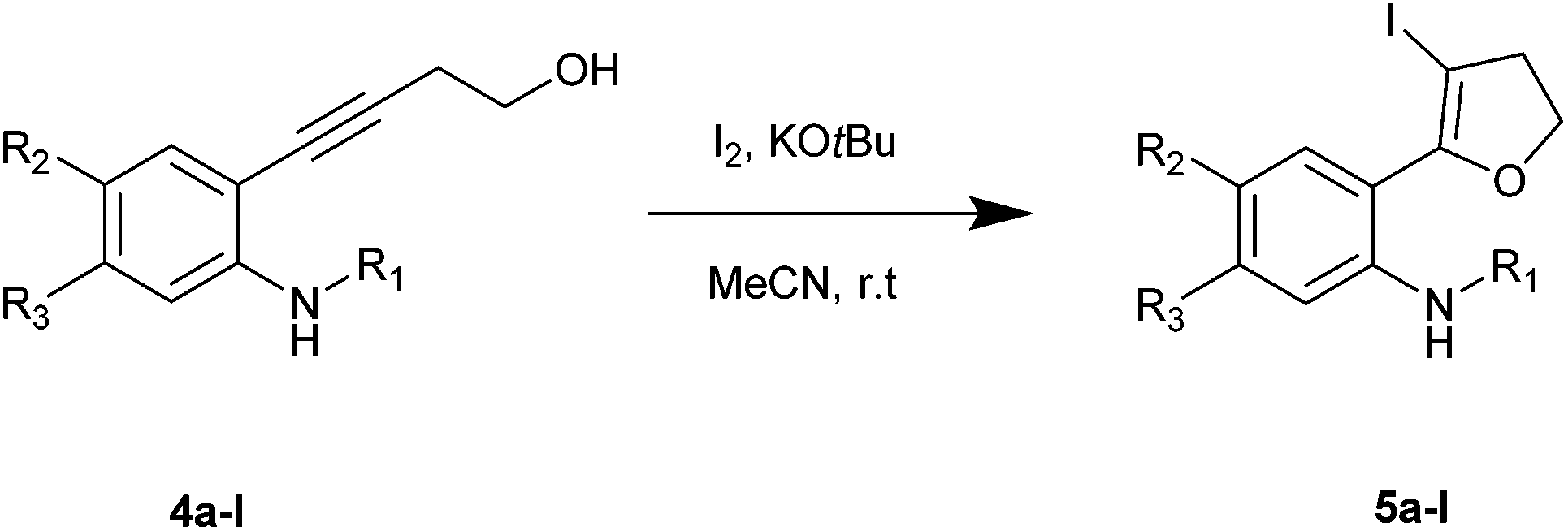 | ||
| Scheme 2 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
The molecular structure of 5e was confirmed by X-ray crystallography, and is shown in Fig. 1. Selected bond distances and angles for 5e are summarized in the caption to Fig. 1.
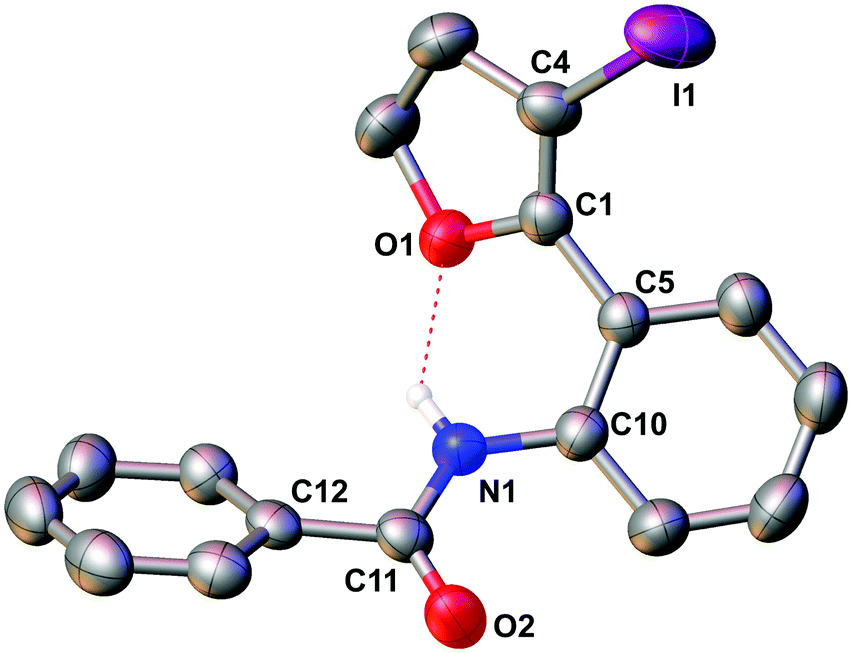 | ||
| Fig. 1 Molecular structure of 5e. Ellipsoids show 50% probability levels. Hydrogen atoms have been omitted for clarity.Selected bond distances (Å) and angles (°) in 5e: N(1)–C(10) 1.414(4), N(1)–C(11) 1.350(4), C(11)–O(2) 1.221(4), C(4)–I(1) 2.061(3), C(10)–N(1)–C(11) 127.9(3), N(1)–C(11)–O(2) 123.7(3). | ||
The iodocyclization reaction proceeded well with the reactants containing different protecting groups (5a, 5b, 5d, 5e) except for the methylated amine (5c). The slight decrease in the product yield was observed with alkynes bearing electron withdrawing groups on the aromatic ring (5f to 5i) but in contrast to the above observation, the reaction could not proceed with an electron donating substituent (5j). However, when the amine group was acetylated/benzoylated (4k and 4l), we were able to get the products 5k and 5l (compound 5l not isolated but confirmed by HRMS).
C–C and C–N coupling of iodo-dihydrofuran
With the set protocol investigated for iodocyclization of electronically modified amine substituted alkynols to various iodo-dihydrofuran substrates, we were curious to know the reactivity of iodo-dihydrofuran towards C–C and C–N bond forming reactions. In C–C coupling reactions, palladium catalysed Suzuki, Heck and Sonagashira coupling reactions were examined using the corresponding coupling partners, boronic acid, methyl acrylate and propargyl alcohol, respectively (Scheme 3). All the reactions were performed in the presence of a catalytic amount of bis(triphenylphosphine)palladium(II)dichloride and quantitative yields of the corresponding coupled product were observed. The products obtained in coupling reactions were analysed by 1H NMR, 13C NMR and HRMS. Interestingly, when substrate 4a was reacted with methyl acrylate in the presence of a palladium catalyst the product obtained was identical to the product 8a obtained in the coupling reaction of 5a with methyl acrylate. The structure of the product 8a was further confirmed by X-ray diffraction analysis and is shown in Fig. 2. Selected bond distances and angles for 8a are summarized in the caption to Fig. 2.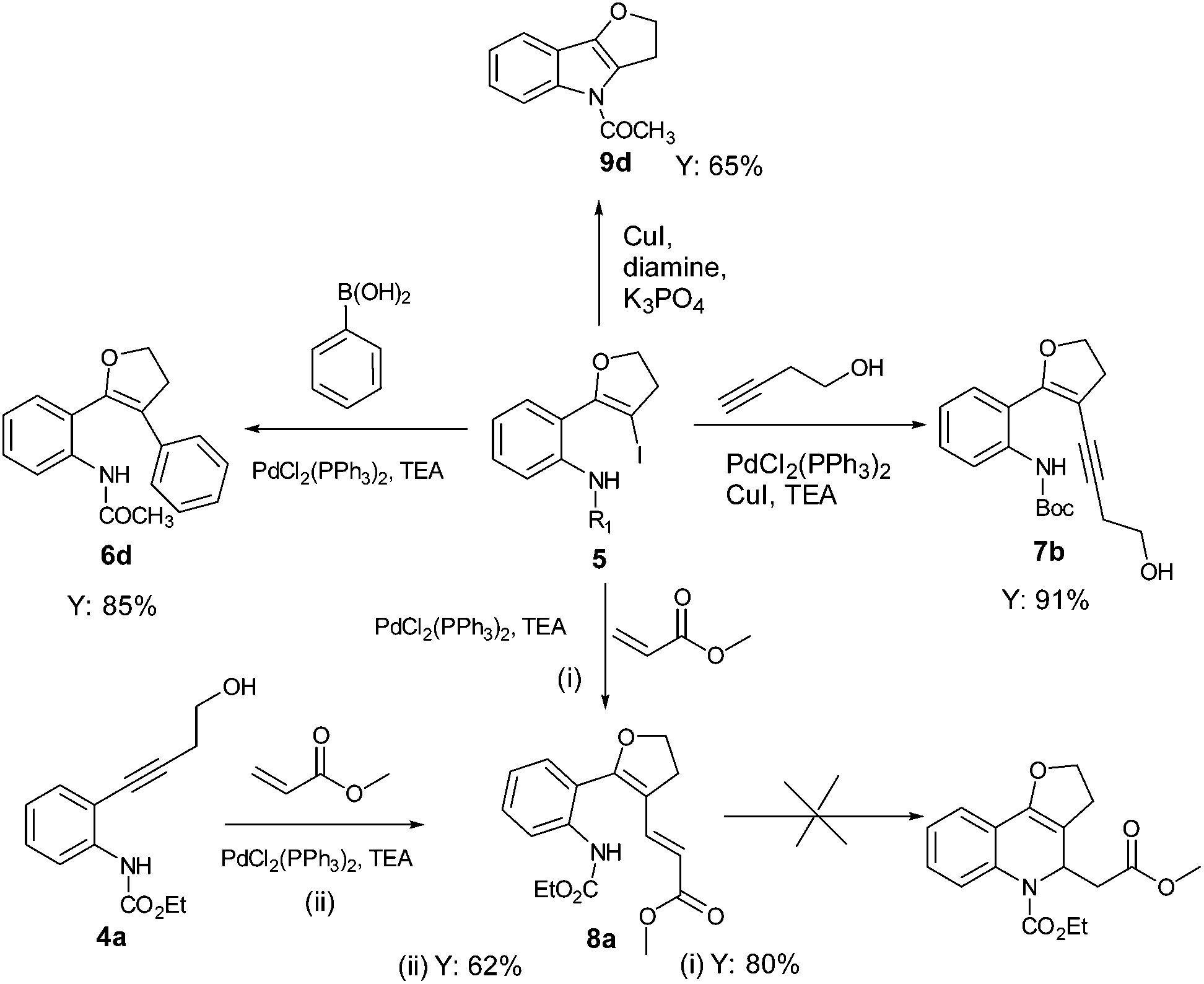 | ||
| Scheme 3 | ||
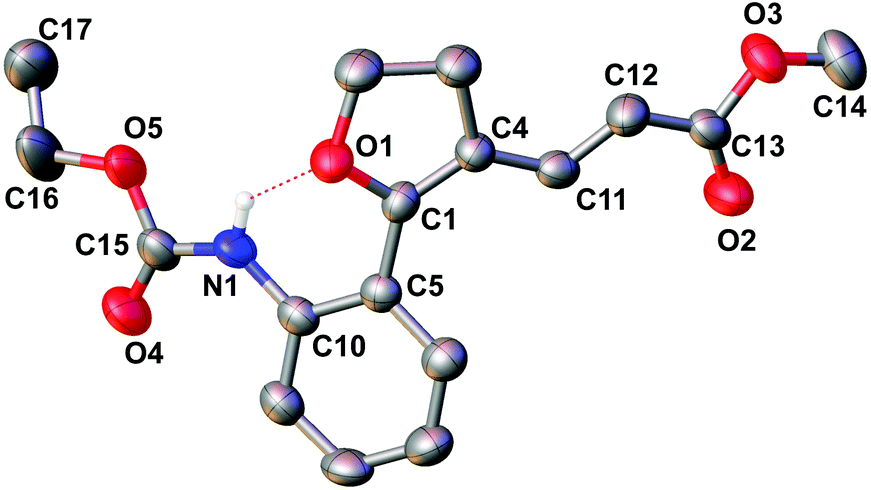 | ||
| Fig. 2 Molecular structure of 8a. Ellipsoids show 50% probability levels. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) in 8a: N(1)–C(10) 1.400(5), N(1)–C(15) 1.356(5), C(15)–O(4) 1.194(5), C(15)–O(5) 1.338(5), C(10)–N(1)–C(15) 129.6(4), N(1)–C(15)–O(4) 127.3(4), N(1)–C(15)–O(5) 107.6(3). | ||
Our effort to synthesize tricyclic fused heterocycles which are generally found in the major class of pharmaceutical products, biologically active compounds and materials11 from iodo-dihydrofuran by intramolecular C–N coupling in the presence of copper iodide, diamine and potassium phosphate was fruitful and furo-indole 9d was obtained in good yield. Unfortunately we were not successful in synthesizing a dihydrofuroquinolone system by intramolecular Michael addition under various reaction conditions, which might be because of the delocalization of the double bond with the double bond of the furan ring and lone pair electrons on the oxygen atom.
Conclusions
In summary, a regio-selective iodocyclization of amino alkynols to dihydrofurans is achieved by modifying the electronic properties of the amino group. The yield of regio-selective iodo-substituted dihydrofuran is optimized by the use of an appropriate iodine source, base and solvent. Further, the application of regioselectively synthesized dihydrofuran embedded with an iodo handle has been exploited for the synthesis of diversely substituted dihydrofurans using the well-known catalytic C–C coupling and C–N coupling. To the best of our knowledge, this is the first report on the preparation of a furo-indole moiety from amine substituted alkynol. Considering the easy availability of the starting materials, broad substrate scope, operationally simple methodology and the importance of the dihydrofuran scaffold in various ranges of substances, this method should find ample applications in various fields.Experimental
All reactions were carried out in oven dried glassware under an atmosphere of dry nitrogen. Chemicals were purchased from Aldrich and used as received unless mentioned otherwise. All solvents used were dried before use. Product purification by column chromatography was accomplished using a silica gel 60–120 mesh. Technical grade solvents were used for chromatography and distilled prior to use. NMR spectra were recorded in Fourier transform mode. The 1H NMR, 13C NMR and 19F NMR spectra were recorded on a 300 MHz, 400 MHz, and 500 MHz spectrophotometer using CDCl3 and TMS as the internal standard. Multiplicities in the 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, q = quartet, qt = quintet, m = multiplet and bs = broad singlet; coupling constants are reported in Hz. Low (MS) and high (HRMS) resolution mass spectra were recorded by an ion trap method and mass/charge (m/z) ratios are reported as values in atomic mass units. All the melting points are uncorrected.General procedure for the synthesis of ethyl (2-(4-hydroxybut-1-yn-1-yl)phenyl)carbamate (4a–4l) and characterization data
To a solution of protected 2-iodoaniline (1 mmol) in N,N-dimethyformamide (3 mL) were added 3-butyn-1-ol (1.2 mmol) and triethylamine (3 mmol), PdCl2(PPh3)2 (2 mol%) and CuI (1 mol%) were added to the above solution and stirred well under nitrogen atmosphere at room temperature until the reaction was complete (approx. 6 h). The reaction mixture was diluted with ethyl acetate and washed with brine solution. The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product obtained was purified by column chromatography over silica gel using a hexane–ethyl acetate mixture (70:30).
:20).
General procedure for the synthesis of N-(2-(3-phenyl-4,5-dihydrofuran-2-yl)phenyl)acetamide (6d) from N-(2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)acetamide (5d) and characterization data
To a solution of N-(2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)acetamide (1 mmol) and phenyl boronic acid (1.2 mmol) in DMF:H2O (4:1) (3 mL) were added Et3N (3 mmol) and PdCl2(PPh3)2 (5 mol%) under a nitrogen atmosphere and stirred at 80 °C until the reaction was complete. The reaction mixture was diluted with ethyl acetate and washed with brine solution. The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product obtained was purified by column chromatography over silica gel using a hexane–ethyl acetate mixture (60:40).
General procedure for the synthesis of tert-butyl (2-(3-(4-hydroxybut-1-yn-1-yl)-4,5-dihydrofuran-2-yl)phenyl)carbamate (7b) from tert-butyl (2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)carbamate (5b) and characterization data
To a solution of tert-butyl (2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)carbamate (1 mmol) in DMF (3 mL) were added 3-butyn-1-ol (1.2 mmol) and triethylamine (3 mmol). PdCl2(PPh3)2 (2 mol%) and CuI (1 mol%) were added to the above solution and stirred well under a nitrogen atmosphere at room temperature until the reaction was complete (approx. 6 h). The reaction mixture was diluted with ethyl acetate and washed with brine solution. The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product obtained was purified by column chromatography over silica gel using a hexane–ethyl acetate mixture (50:50).
:30).
General procedure for the synthesis of (E)-ethyl (2-(3-(3-oxobut-1-en-1-yl)-4,5-dihydrofuran-2-yl)phenyl)carbamate (8a) from ethyl (2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)carbamate (5a)
To a solution of ethyl (2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)carbamate (1 mmol) and methyl acrylate (2 mmol) in DMF (3 mL) were added Et3N (3 mmol) and PdCl2(PPh3)2 (5 mol%) under a nitrogen atmosphere and stirred at 80 °C until the reaction was complete (approx. 4 h). The reaction mixture was diluted with ethyl acetate and washed with brine solution, and then with water. The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product obtained was purified by column chromatography over silica gel using a hexane–ethyl acetate mixture (80:20).
General procedure for the synthesis of 1-(2H-furo[3,2-b]indol-4(3H)-yl)ethanone from N-(2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)acetamide (9d) and characterization data
To a solution of N-(2-(3-iodo-4,5-dihydrofuran-2-yl)phenyl)acetamide (1 mmol) in toluene (2 mL) were added CuI (10 mol%), K3PO4 (2 mmol), N,N-dimethylethylenediamine (20 mol%) and stirred under a nitrogen atmosphere at 80 °C until the reaction was complete (approx. 6 h). The reaction mixture was diluted with ethyl acetate and washed with brine solution, and then with water. The organic extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product obtained was purified by column chromatography over silica gel using a hexane–ethyl acetate mixture (50:50).
Acknowledgements
VV thanks RMIT University, Australia and RMIT-IICT Joint Research Center, Hyderabad for his fellowships. Authors are grateful to DIICT for providing all the facilities.Notes and references
- (a) A. Arcadi, S. Cacchi, G. Fabrizi, F. Marinelli and L. Moro, Synlett, 1999, 1432–1434 CrossRef CAS; (b) G. M. M. El-Taeb, A. B. Evans, S. Jones and D. W. Knight, Tetrahedron Lett., 2001, 42, 5945–5948 CrossRef CAS; (c) S. P. Bewand and D. W. Knight, Chem. Commun., 1996, 1007–1008 Search PubMed; (d) C. J. Nichols, J. J. J. Parks and J. Undergrad, Chem. Res., 2002, 1, 2732 Search PubMed; (e) D. W. Knight, Electrophile-Induced 5-EndoCyclizations, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and T. L. Gilchrist, Pergamon Press, Oxford, 2002, vol. 14, pp. 19 Search PubMed; (f) J. P. Waldo and R. C. Larock, Org. Lett., 2005, 7, 5203–5205 CrossRef CAS PubMed.
- (a) A. Arcadi, S. Cacchi, S. Di Giuseppe, G. Fabrizi and F. Marinelli, Org. Lett., 2002, 4, 2409–2412 CrossRef CAS PubMed; (b) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed; (c) C. Schultz-Fademrecht, M. Zimmermann, R. Fröhlich and D. Hoppe, Synlett, 2003, 1969–1972 CAS; (d) B. Singh, A. Chandra, S. Singh and R. M. Singh, Tetrahedron, 2011, 67, 505–511 CrossRef CAS.
- (a) S. Mehta, J. P. Waldo and R. C. Larock, J. Org. Chem., 2009, 74, 1141–1147 CrossRef CAS PubMed; (b) B. V. S. Reddy, M. Swain, S. M. Reddy, J. S. Yadav and B. Sridhar, J. Org. Chem., 2012, 77, 11355–11361 CrossRef PubMed.
- M. V. Karkhelikar, R. R. Jha, B. Sridhar, P. R. Likhar and A. K. Verma, Chem. Commun., 2014, 50, 8526–8528 RSC.
- (a) C.-H. Cho, F. Shi, D.-I. Jung, B. Neuenswander, G. H. Lushington and R. C. Larock, ACS Comb. Sci., 2012, 14, 403–414 CrossRef CAS PubMed; (b) S. Samanta, R. Jana and J. K. Ray, Tetrahedron Lett., 2009, 50, 6751–6754 CrossRef CAS; (c) X. Zhao, L. Zhang, X. Lu, T. Li and K. Lu, J. Org. Chem., 2015, 80, 2918–2924 CrossRef CAS PubMed; (d) Z. Chen, G. Huang, H. Jiang, H. Huang and X. Pan, J. Org. Chem., 2011, 76, 1134–1139 CrossRef CAS PubMed; (e) X. Wang, L. Liu, W. Chang and J. Li, Sci. China, Ser. B: Chem., 2009, 52, 1314–1320 CrossRef CAS; (f) D. Yue, T. Yao and R. C. Larock, J. Org. Chem., 2005, 70, 10292–10296 CrossRef CAS PubMed; (g) L. Y. Chen, J. R. Chen, H. G. Cheng, L. Q. Lu and W. J. Xiao, Eur. J. Org. Chem., 2014, 4714–4719 CrossRef CAS; (h) B. Schmidt and D. Geißler, Eur. J. Org. Chem., 2011, 4814–4822 CAS; (i) T. Okitsu, K. Nakata, K. Nishigaki, N. Michioka, M. Karatani and A. Wada, J. Org. Chem., 2014, 79, 5914–5920 CrossRef CAS PubMed.
- (a) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Chem. – Eur. J., 2012, 18, 5460–5489 CrossRef CAS PubMed; (b) R. Dembinski, Y. Li, D. Gundapuneni and A. Decker, Top. Heterocycl. Chem., 2012, 27, 65–100 CAS; (c) B. Wan, X. Jiang, G. Jia and S. Ma, Eur. J. Org. Chem., 2012, 4373–4379 CrossRef CAS; (d) R. Antonioletti, S. Malancona, F. Cattaruzza and P. Bovicelli, Tetrahedron, 2003, 59, 1673–1678 CrossRef CAS; (e) Y. Liu and S. Zhou, Org. Lett., 2005, 7, 4609–4611 CrossRef CAS PubMed.
- (a) Y. Liu, S. Zhang, J. Jung and T. Xu, in Bioact. Heterocycles V 2007, pp. 231–258 Search PubMed; (b) D. H. R. Barton, H. T. Cheung, A. D. Cross, L. M. Jackman and M. Martin-Smith, J. Chem. Soc., 1961, 5061–5073 RSC; (c) D. L. Wright, Chapter 1 Furans as versatile synthons for target-oriented and diversity-oriented synthesis, Elsevier Masson SAS, 2005, vol. 17 Search PubMed; (d) M. J. Palframan and G. Pattenden, Chem. Commun., 2014, 50, 7223–7242 RSC; (e) B. A. Keay, J. M. Hopkins and P. W. Dibble, Comprehensive Heterocyclic Chemistry III, Elsevier, 2008 Search PubMed; (f) P. Ghosh, M. Das, A. P. Rameshbabu, D. Das, S. Datta, S. Pal, A. B. Panda and S. Dhara, ACS Appl. Mater. Interfaces, 2014, 6, 17926–17936 CrossRef CAS PubMed; (g) D. Zabiegaj, E. Santini, M. Ferrari, L. Liggieri and F. Ravera, Colloids Surf., A, 2015, 1–8 Search PubMed.
- (a) E. Baciocchi and R. Ruzziconi, J. Org. Chem., 1991, 56, 4772–4778 CrossRef CAS; (b) M. G. Vinogradov, A. E. Kondorsky and G. I. Nikishin, Synthesis, 1988, 1988, 60–62 CrossRef; (c) E. I. Heiba, R. M. Dessau and P. G. Rodewald, J. Am. Chem. Soc., 1974, 96, 7977–7981 CrossRef CAS; (d) E.-A. I. Heiba and R. M. Dessau, J. Org. Chem., 1974, 39, 3456–3457 CrossRef CAS; (e) W. E. Fristad and J. R. Peterson, J. Org. Chem., 1985, 50, 10–18 CrossRef CAS; (f) B. B. Snider, R. Mohan and S. A. Kates, J. Org. Chem., 1985, 50, 3659–3661 CrossRef CAS; (g) Q. Zhang, R. M. Mohan, L. Cook, S. Kazanis, D. Peisach, B. M. Foxman and B. B. Snider, J. Org. Chem., 1993, 58, 7640–7651 CrossRef CAS.
- D. Kalpogiannaki, C.-I. Martini, A. Nikopoulou, J. A. Nyxas, V. Pantazi and L. P. Hadjiarapoglou, Tetrahedron, 2013, 69, 1566–1575 CrossRef CAS.
- S. Tang, K. Liu, Y. Long, X. Gao, M. Gao and A. Lei, Org. Lett., 2015, 17, 2404 CrossRef CAS PubMed.
- (a) J. Qin, P. Dhondi, X. Huang, M. Mandal, Z. Zhao, D. Pissarnitski, W. Zhou, R. Aslanian, Z. Zhu, W. Greenlee, J. Clader, L. Zhang, M. Cohen-Williams, N. Jones, L. Hyde and A. Palani, Bioorg. Med. Chem. Lett., 2011, 21, 664–669 CrossRef CAS PubMed; (b) Y. Xiong, B. Ullman, J.-S. K. Choi, M. Cherrier, S. Strah-Pleynet, M. Decaire, K. Feichtinger, J. M. Frazer, W. H. Yoon, K. Whelan, E. K. Sanabria, A. J. Grottick, H. Al-Shamma and G. Semple, Bioorg. Med. Chem. Lett., 2012, 22, 1870–1873 CrossRef CAS PubMed; (c) H. Gao, Q. L. Xu, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2014, 53, 2701–2705 CrossRef CAS PubMed; (d) H. J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4427 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1405059–1051824. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02033f |
| This journal is © The Royal Society of Chemistry 2016 |