
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective oxidative boron Heck reactions
A.-L.
Lee
*
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, Scotland, UK. E-mail: A.Lee@hw.ac.uk; Tel: +44 (0)131 4518030
First published on 29th October 2015
Abstract
This review highlights the use of the oxidative boron Heck reaction in enantioselective Heck-type couplings. The enantioselective oxidative boron Heck reaction overcomes several limitations of the traditional Pd(0)-catalysed Heck coupling and has subsequently allowed for intermolecular couplings of challenging systems such as cyclic enones, acyclic alkenes, and even site selectively on remote alkenes.
 A.-L. Lee | Ai-Lan Lee hails from Malaysia and obtained her MSci(Hons) (2000) and PhD (2004) from University of Cambridge, working under the supervision of Prof. Steven Ley. She was subsequently awarded a Lindemann Trust Fellowship (2004–2005) to work at Boston College with Prof. Amir Hoveyda on alkene metathesis. In 2006, Ai-Lan was appointed as a fixed-term Lecturer at the University of Edinburgh, carrying out research with Prof. David Leigh on rotaxane synthesis. She started as a Lecturer at Heriot-Watt University in 2007 and was promoted to Associate Professor, Reader in 2013. Her research interests include developing new gold- and palladium-catalysed reactions and catalysts. |
1. Introduction
Intramolecular asymmetric Pd(0) Mizoroki–Heck couplings1 were among the first catalytic enantioselective carbon–carbon bond forming reactions to be explored and have been successfully applied in natural product synthesis.2 In contrast, efficient intermolecular enantioselective Pd(0)-catalysed Heck reactions have proven more elusive, with the exception of couplings with specific “benchmark” cyclic olefins such as dihydrofuran and dihydropyrrole. Recently, however, much progress has been made in the field of enantioselective Heck-type couplings3,4 with the emergence of the Pd(II)-catalysed oxidative boron Heck variant (also called boron-Heck, or oxidative Heck) to overcome these limitations. The use of the oxidative Heck method has allowed for intermolecular couplings of more challenging systems, including desymmetrisation of quaternary centres, cyclic enones, acyclic alkenes and even site selectively on remote alkenes. This review will briefly outline the oxidative boron Heck reaction, followed by highlighting the major recent advances in enantioselective oxidative boron Heck couplings.2. Oxidative boron Heck reaction
Oxidative boron Heck (hereafter shortened to oxidative Heck) reactions are catalysed by Pd(II) instead of Pd(0) and differ from the traditional Pd(0)-catalysed Mizoroki–Heck reactions during the first step in the catalytic cycle (Schemes 1 and 2).5 The halide or triflate (R1X) in the Mizoroki–Heck reaction is replaced by the corresponding organoboronic acid [R1B(OH)2] in the oxidative Heck reaction. Therefore, the first step in the catalytic cycle is the transmetallation between the organoboronic acid and Pd(II) catalyst (Scheme 2), instead of an oxidative addition of Pd(0) into a halide or triflate. As such, the oxidative Heck reaction often does not require high temperatures or bases. However, an oxidant [usually O2, air, benzoquinone or Cu(OAc)2] is required to re-oxidise Pd(0) to Pd(II) at the end of the catalytic cycle.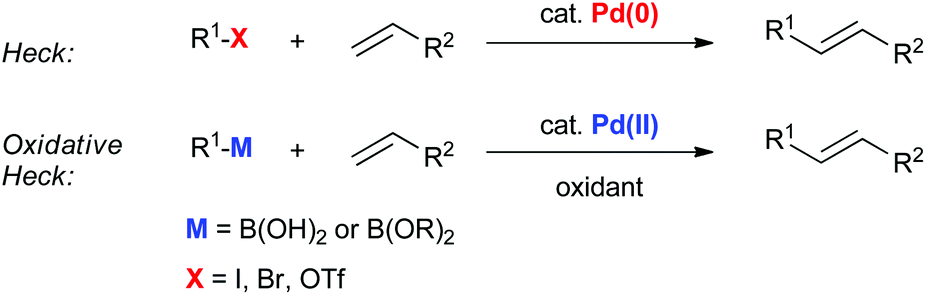 | ||
| Scheme 1 Difference between the Mizoroki–Heck and oxidative Heck reactions. | ||
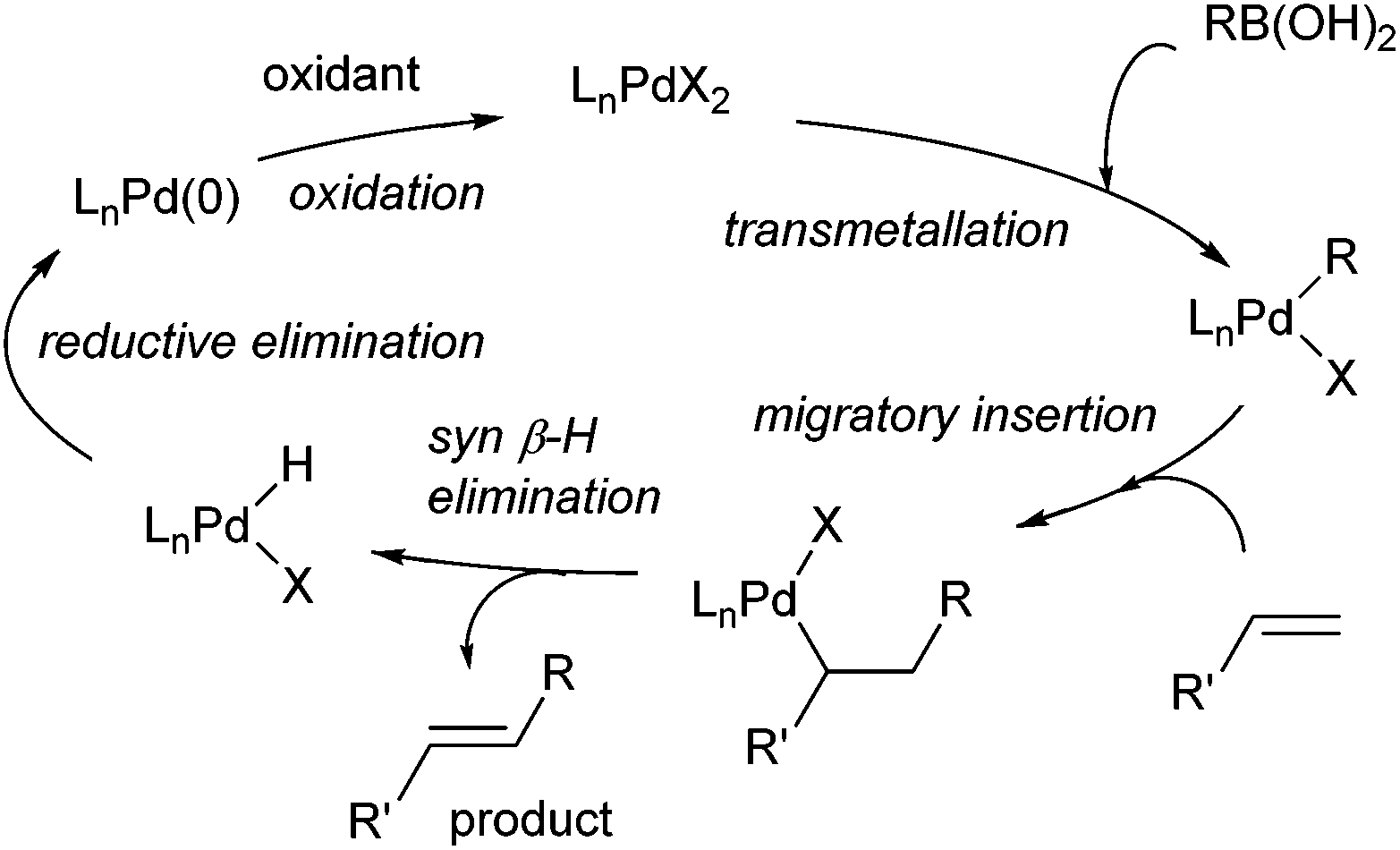 | ||
| Scheme 2 Proposed general mechanism for the oxidative boron Heck reaction. | ||
Oxidative Heck reactions are known to be efficient, mild (e.g. lower reaction temperatures, good functional group tolerance), tolerant of air and moisture and are capable of coupling challenging substrates such as highly substituted or cyclic olefins, many of which can be reluctant to undergo Pd(0)-catalysed Mizoroki–Heck couplings.6 Another advantage of changing from Pd(0) to Pd(II) catalysis is that both N-based as well as P-based ligands can now be used (vide infra). Furthermore, the absence of a halogen–Pd intermediate in the reaction means it is likely to proceed via the cationic rather than neutral route, which is thought to be important for enantioselectivity (Scheme 3).7
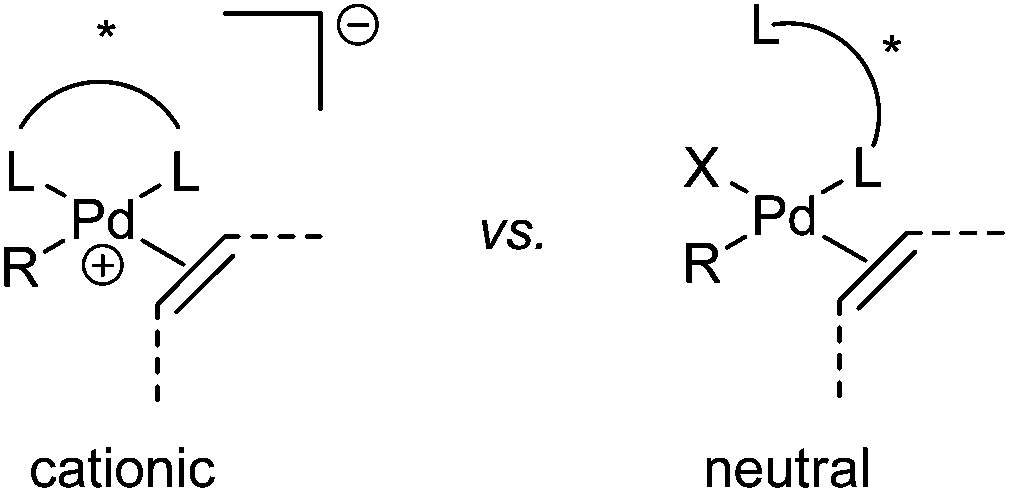 | ||
| Scheme 3 Proposed cationic vs. neutral pathway. | ||
The non-enantioselective and stoichiometric oxidative Heck coupling was first reported by Heck in 1975,8 while the catalytic version was developed by Uemura in 1994.9 However, it was not until the 2000's that the reaction was developed further, initially by Mori in 2001:10 reporting the use of Cu(OAc)2 as an oxidant, followed by the use of molecular oxygen as the oxidant by Jung in 2003.11 The first ligand modulated reaction (using dimethyl-1,10-phenanthroline) was reported by Larhed in 2004.12 Further investigations by Jung, Larhed and others led to the identification of bidentate N-ligands as optimal ligands, the discovery of base-free conditions, and air as the oxidant, which further improved the mildness and practicality of the reaction.6,13
The ability of the Pd(II)-oxidative Heck reaction to readily couple even cyclic substrates should be highlighted,14 as cyclic enones are notoriously reluctant to undergo intermolecular Pd(0)-catalysed Mizoroki–Heck couplings and often produce the conjugate addition products instead.15 This is usually attributed to the fact that cyclic systems such as 1 are stereochemically precluded from undergoing the final step in the traditional Pd(0) Heck cycle: the syn-β-H elimination.15a,16 It should be noted that Pd(II)-catalysed reactions with enones and boronic acids can, depending on the conditions, result in conjugate additions rather than Heck-type couplings,17 and the two reactions can sometimes also be competing pathways. Within this context, controlled switching between the Pd(II)-catalysed oxidative Heck and conjugate addition reactions have recently been reported (Scheme 4).18,19 A simple change of solvent from DCE (ClCH2CH2Cl) to DMSO switches the reaction from conjugate addition to oxidative Heck.
 | ||
| Scheme 4 Controlled switching between oxidative Heck and conjugate addition reactions. | ||
As shown in Scheme 5, the mechanism of the two reactions is thought to diverge after the transmetallation and migratory insertion steps. Intermediate I can either undergo epimerisation followed by syn-β-H elimination to produce the oxidative Heck product or protonolysis (possibly via Pd-enolate II) to produce the conjugate addition product. Conditions to promote the epimerisation of I to I′, and thereby allowing the syn-β-H elimination, are therefore required to favour the oxidative Heck reaction.
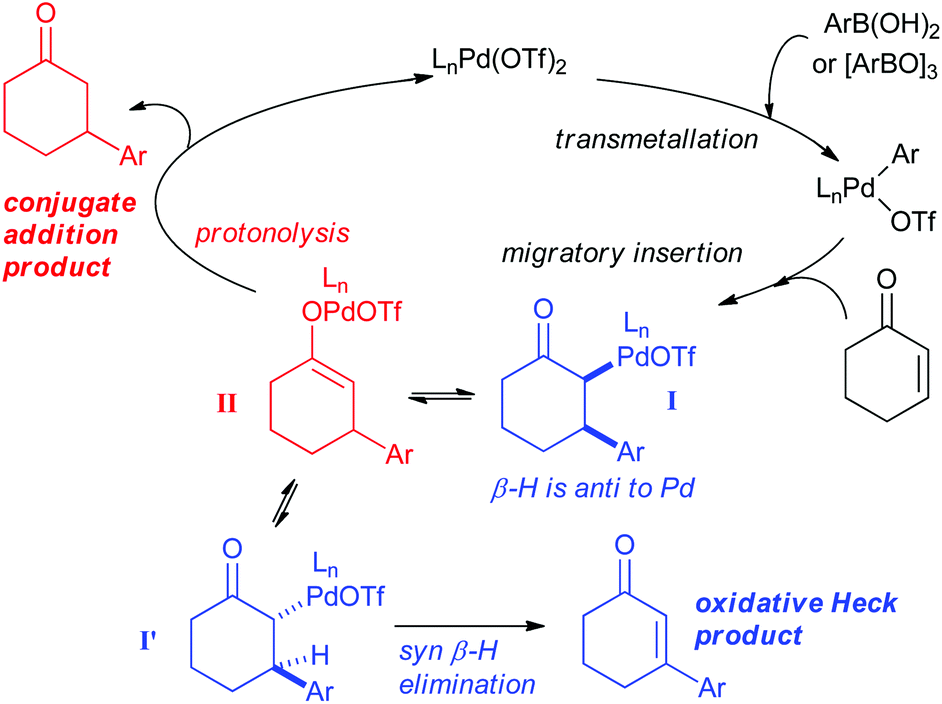 | ||
| Scheme 5 Proposed mechanisms for the Pd(II)-catalysed oxidative Heck and conjugate addition reactions. | ||
Although there are now opportunities to develop enantioselective Heck-type reactions on cyclic enone and related systems by adopting the oxidative Heck reaction protocol, sterically hindered alkene substrates are still a challenge18 and conditions must be optimised to avoid the competitive conjugate addition reaction. Indeed, while there are several reports on Pd(II)-catalysed asymmetric conjugate addition reactions on cyclic enones (and related structures),20 reports on the corresponding asymmetric oxidative Heck reaction have only begun to emerge (vide infra). Nevertheless, the ability to couple even highly substituted alkenes and cyclic enones (both challenging substrates under traditional Pd(0) catalysis) under mild conditions using a wide variety of ligands means that the oxidative Heck reaction has great potential to progress the field of asymmetric Heck-type reactions, especially in the area of the more challenging intermolecular couplings.
3. Enantioselective oxidative Heck reactions
3.1 Cyclic alkenes
The first reported attempt at enantioselective oxidative Heck coupling emerged in 2005 by Mikami and co-workers, who utilised the cyclic system 2.21 Following a screen of various chiral bidentate N as well as P ligands, the phosphine ligand (S,S)-chiraphos 4 was considered optimal, providing 5 with ees22 of up to 49% (Scheme 6). It is worth noting that enantioselective Heck-type coupling is possible with substrate 2, because the syn-β-H elimination occurs at the available position β′ instead of β, thus furnishing a chiral centre, and this also avoids any issues with having to epimerise at the β-position to form an available syn-β-H. Although this early work had a limited substrate scope (e.g. arylboronic acid 3a only) and modest enantioselectivities, it was nevertheless pioneering, and paved the way for further studies in the area. | ||
| Scheme 6 First example of enantioselective oxidative Heck reaction by Mikami. | ||
Since the main advantage of the oxidative Heck reaction is to enable asymmetric intermolecular Heck-type couplings, it is perhaps unsurprising that there appears to be only one report of an asymmetric intramolecular oxidative Heck reaction, again by Mikami and co-workers in 2007.23 A chiral quaternary carbon is installed in 80% ee (7) via ring closure of a sulphonamide species 6 (Scheme 7). However, the reduced alkene product 8 is always present as a side product.
 | ||
| Scheme 7 Intramolecular enantioselective oxidative Heck coupling by Mikami. | ||
As previously mentioned, there are a few privileged alkene substrates in enantioselective Heck-type couplings and 2,3-dihydrofuran 9 is one such alkene which is often used as a standard benchmark substrate for enantioselective intermolecular Heck reactions. In 2007, Gelman and co-workers demonstrated that good enantioselectivities of 10 (up to 86% ee) could be achieved using oxidative Heck coupling (Scheme 8).7 Chiral bidentate phosphine ligands (R)-BINAP or (R)-MeOBiphep were found to be optimal and enantioselectivities were generally moderate to good for a range of arylboronic acid coupling partners 3 with the exception of ortho substituted aryls, which gave poor ees. Once again, the enantioselective Heck-type reaction is possible on 2,3-dihydrofuran 9 because the syn-β-H elimination step occurs at the β′-position, where a syn-H is available. Isomerisation of the resulting 3,4-dihydropyran intermediate subsequently provides the enol ether product 10.
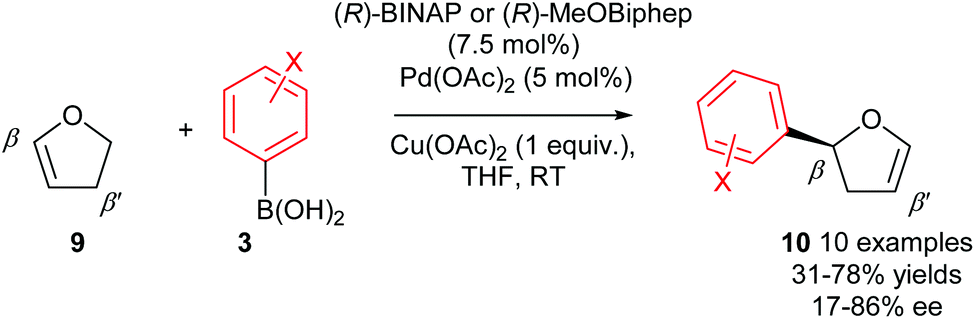 | ||
| Scheme 8 Enantioselective oxidative Heck couplings of 2,3-dihydrofuran by Gelman. | ||
Although the abovementioned early examples utilised chiral phosphine ligands, subsequent reports on enantioselective oxidative Heck couplings have tended to move away from phosphines in favour of N-ligands. For example, the use of the chiral dimeric tridentate NHC–amidate–alkoxide palladium(II) complex 12 allowed Jung and co-workers to significantly improve the enantioselectivities as well as substrate scope of the oxidative Heck reactions on substrates 11 (vs. Mikami's original report in 2005, Scheme 6). Not only are the ees now consistently 81–88%, but various substituents on the arylboronic acid 3 are now tolerated (Scheme 9). However, the current drawback to using catalyst 12 is that the yields are generally moderate (44–62%), due to a significant amount of deborylation in a side reaction to produce phenolic side-products.
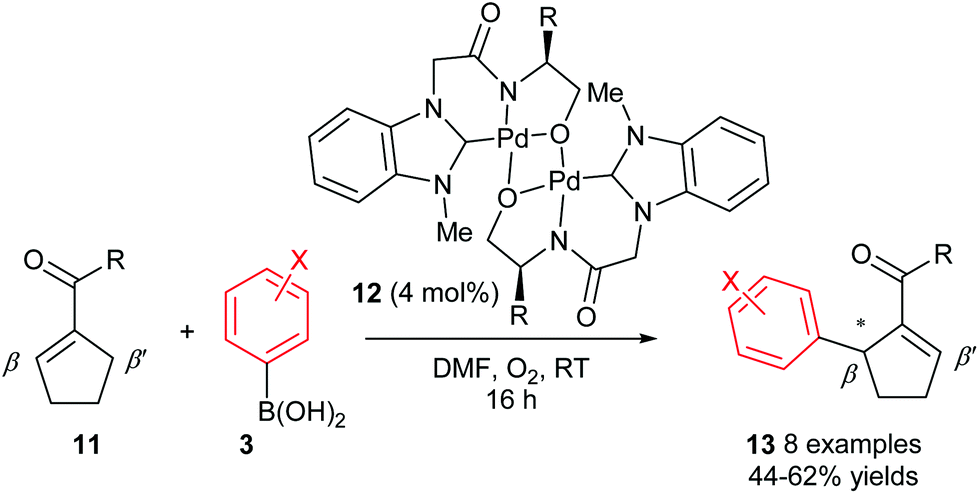 | ||
| Scheme 9 Higher enantioselectivities achieved by Jung using catalyst 12. | ||
So far, the enantioselective oxidative Heck reactions described form a new stereogenic centre in cyclic alkenes where conformational rigidity and hence restricted rotation around the C–C bond steers the β-H elimination away from the newly formed C–C bond (β′ instead of β, Schemes 6–9). Instead of the formation of a migrated cross-coupled product, our group was interested in exploiting the oxidative Heck coupling via a different approach: a direct coupling to form a stereogenic all-carbon quaternary centre via desymmerisation (Scheme 10).24 2,2-Disubstituted cyclopentene-1,3-diones 14 were chosen as substrates as this core is found in several biologically active natural products and metabolites.25 The use of chiral enantiopure PyOX ligands 16a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and 16b27 allowed for the oxidative Heck coupling of 14 with a range of arylboroxines 15 (dehydrated form of arylboronic acids) to yield 17 in up to 94:6 er (Scheme 10). A current limitation is that the er is modest when R is not an aryl substituent (e.g. R
26 and 16b27 allowed for the oxidative Heck coupling of 14 with a range of arylboroxines 15 (dehydrated form of arylboronic acids) to yield 17 in up to 94:6 er (Scheme 10). A current limitation is that the er is modest when R is not an aryl substituent (e.g. R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Bn in 14 with XOMe in 15 gives only 65:35 er of corresponding product 17). Nevertheless, the utility of the method was successfully demonstrated through the synthesis of (+)-preussidone 19 in one step from the enedione 14a, without the need for any protecting groups on the hydroxyls (Scheme 11).
Bn in 14 with XOMe in 15 gives only 65:35 er of corresponding product 17). Nevertheless, the utility of the method was successfully demonstrated through the synthesis of (+)-preussidone 19 in one step from the enedione 14a, without the need for any protecting groups on the hydroxyls (Scheme 11).
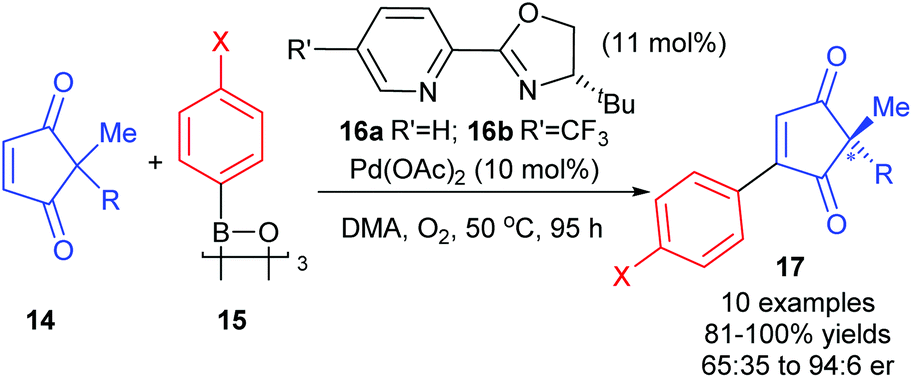 | ||
| Scheme 10 Desymmertisation of all-carbon quaternary centres by Lee. | ||
 | ||
| Scheme 11 Synthesis of (+)-preussidone using enantioselective oxidative Heck reaction. | ||
It should be noted that the solvent was switched from DMF for racemic studies to DMA (dimethylacetamide) for the enantioselective method, in order to avoid issues with competitive ligation from DMF.28 The temperature of the reaction was also important: at RT, the conjugate addition reaction became competitive, so higher temperatures were necessary for oxidative Heck couplings onto enediones 14.
In all of the examples highlighted in this section, the migratory insertion step is thought to be enantiodetermining, although mechanistic/modelling studies have yet to be carried out.
3.2 Acyclic alkenes
Intermolecular enantioselective Heck couplings of acyclic alkenes have traditionally been very challenging. For example, the first reported enantioselective intermolecular Heck coupling of a prochiral acyclic alkene by Uemura and co-workers occurred in a modest 17% ee under Pd(0) catalysis.29 Therefore, Jung and co-workers’ successful use of enantioselective Pd(II)-catalysed oxidative Heck reactions to achieve ees of up to 98% on challenging acyclic substrates are of substantial significance to the field.30In Jung's initial proof-of-concept studies, a chiral N-bidentate ligand (PyOX) was adopted as P-based ligands proved to be inefficient due to side reactions, including homocoupling and phenol formation.31 As shown in Scheme 12, 21 catalyses the oxidative Heck reaction of aryl boronic acids 3 and trisubstituted acyclic alkenes 20 at room temperature to furnish the migrated cross-coupled alkene 22 in moderate to good yields (67–79%) and enantioselectivities (62–75% ee). In contrast, the standard Pd(0)-catalysed Mizoroki–Heck reaction between iodobenzene and 20 using PPh3 as the ligand proceeded in <5% yield, even at high temperatures (140 °C).
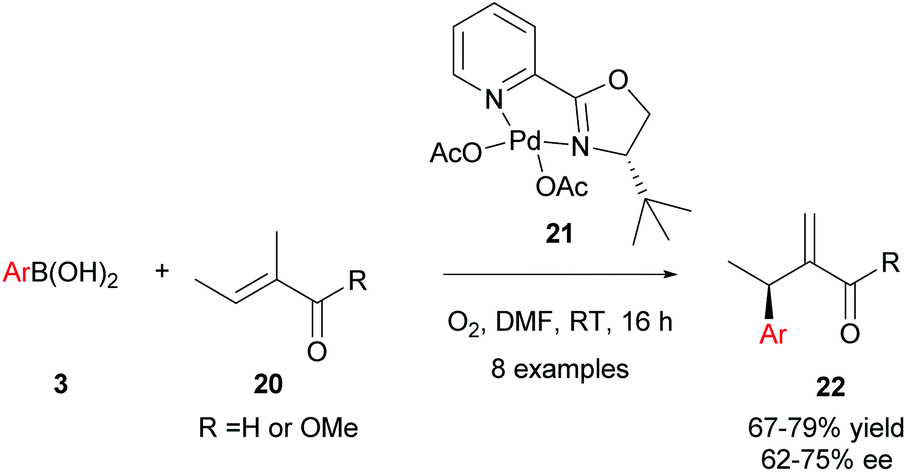 | ||
| Scheme 12 Jung's enantioselective oxidative Heck couplings on acyclic alkenes. | ||
It should be noted that the pre-formed catalyst 21 provided much higher enantioselectivities compared to commonly used in situ formation of 21via premixing Pd(OAc)2 with PyOX ligand 16a (Scheme 13). This observation was attributed to incomplete formation of 21 in the latter and/or relatively easy dissociation of the ligand 16a under the reaction conditions.
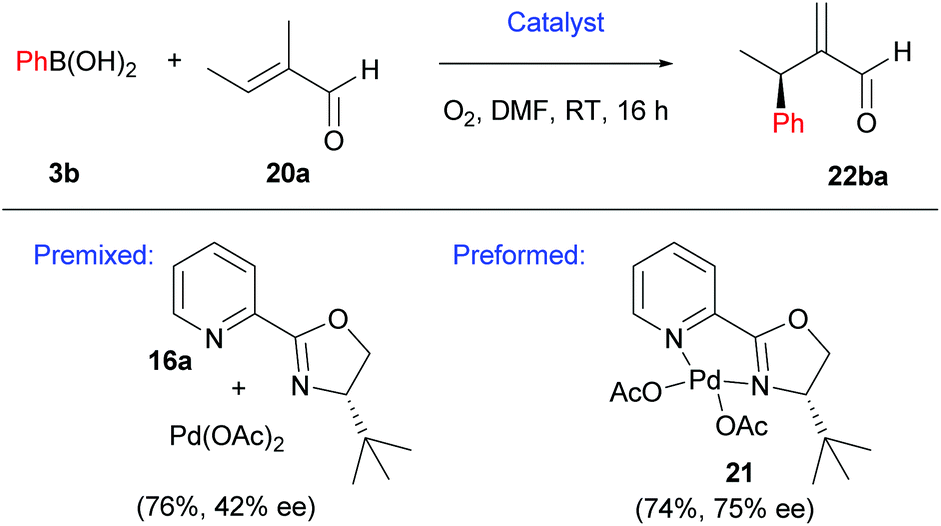 | ||
| Scheme 13 Premixed vs. preformed catalysts. | ||
As in Section 3.1, the migratory insertion step is proposed to be enantiodetermining. Fig. 1 shows the two plausible conformations for the alkene-coordinated structures during the migratory insertion step. The reaction is thought to proceed through coordination III instead of III′, because there is more steric repulsion between the alkene substituents (Me in III and Ac in III′) and the tert-butyl group on the oxazoline ring in III′ compared to III. The steric preference for conformation III thus leads to the observed enantioselectivity.
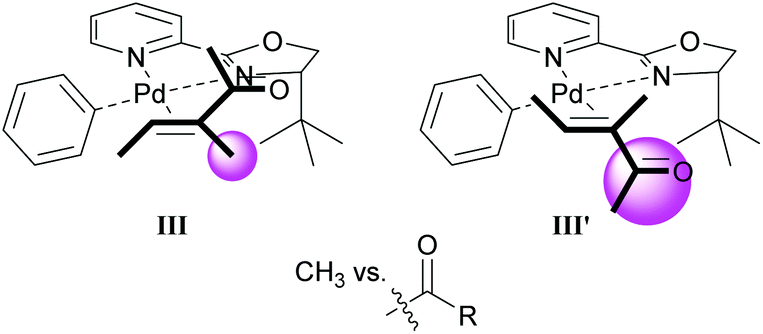 | ||
| Fig. 1 Proposed conformations for the enantiodetermining step in the oxidative Heck reaction with catalyst 21. | ||
Following this proof-of-concept work, Jung and co-workers decided to develop a tighter chiral Pd(II)-ligand complex by utilising a strongly coordinating NHC ligand, as well as a tridentate, rather than bidentate, ligand. To this end, novel air and water-stable chiral palladium(II) complexes with a tridentate N-heterocyclic carbene (NHC) amidate alkoxide ligand 12 were developed in 2008. Complexes 12 successfully catalysed the oxidative Heck couplings of 20 and 3 with excellent enantioselectivities (90–98% ee, Scheme 14).30 The high degree of asymmetric induction is proposed to stem from the tight binding of the tridentate ligands to Pd during the entire catalytic process. Although the yields of 22 were generally low to modest, the excellent enantioselectivities achieved using the oxidative Heck couplings (>90% ee) were unprecedented in intermolecular Heck-type reactions at the time.
 | ||
| Scheme 14 Asymmetric oxidative Heck couplings with excellent enantioselectivities. | ||
In their subsequent full paper, Jung and co-workers disclosed that both monomer 23 and dimer 12 were originally investigated as catalysts for the coupling between boronic acid 3c and alkene 20b, with each providing drastically different results (7% ee and 91% ee of coupled product 22cb respectively, Scheme 15). 1H NMR analysis reveals that the borate group is transferred to the alkoxide when the dimeric catalyst 12 is used (see borate intermediate IV, Scheme 16). In contrast, the borate group does not remain after transmetallation with the monomeric catalyst 23. The authors therefore suggest that the steric effect of the borate group in IV results in the higher enantioselectivities using dimeric catalyst 12.
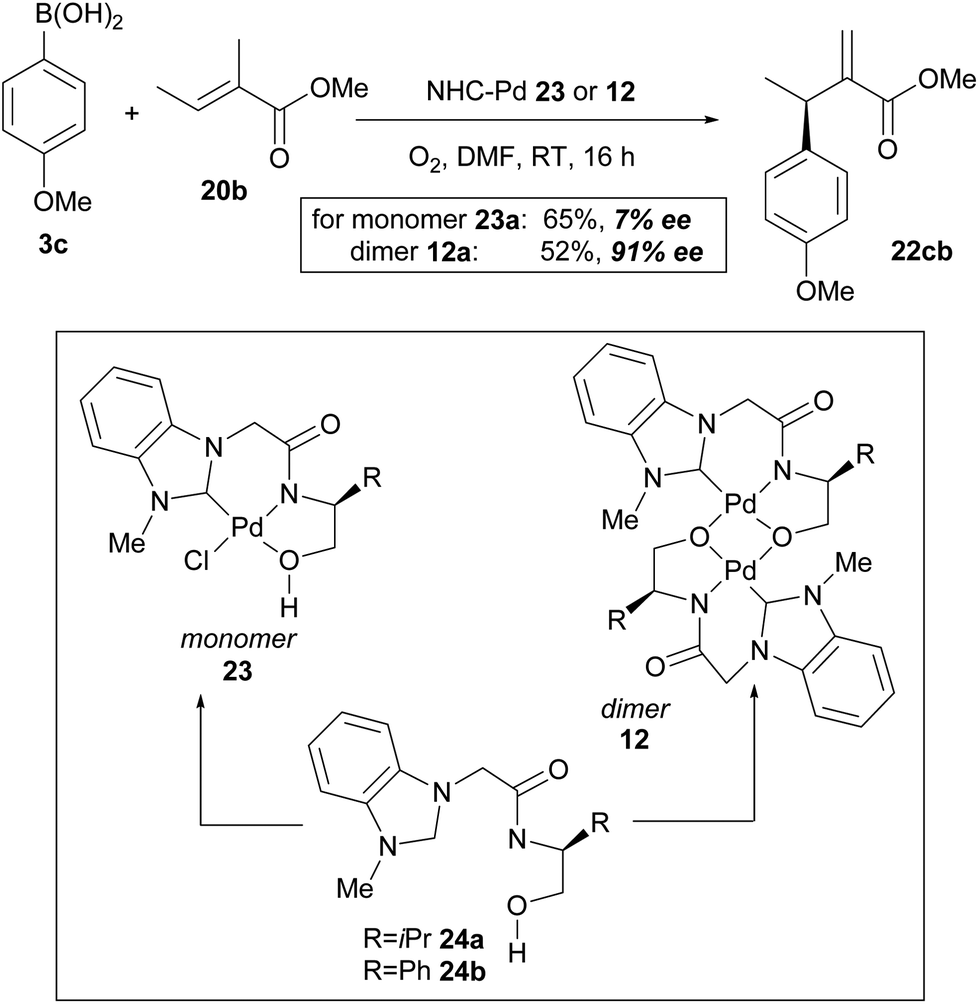 | ||
| Scheme 15 Significant enantioselectivity difference with monomeric and dimeric NHC–Pd catalysts. | ||
 | ||
| Scheme 16 Proposed mechanism of the enantioselective oxidative Heck reaction using dimeric catalyst 12a. | ||
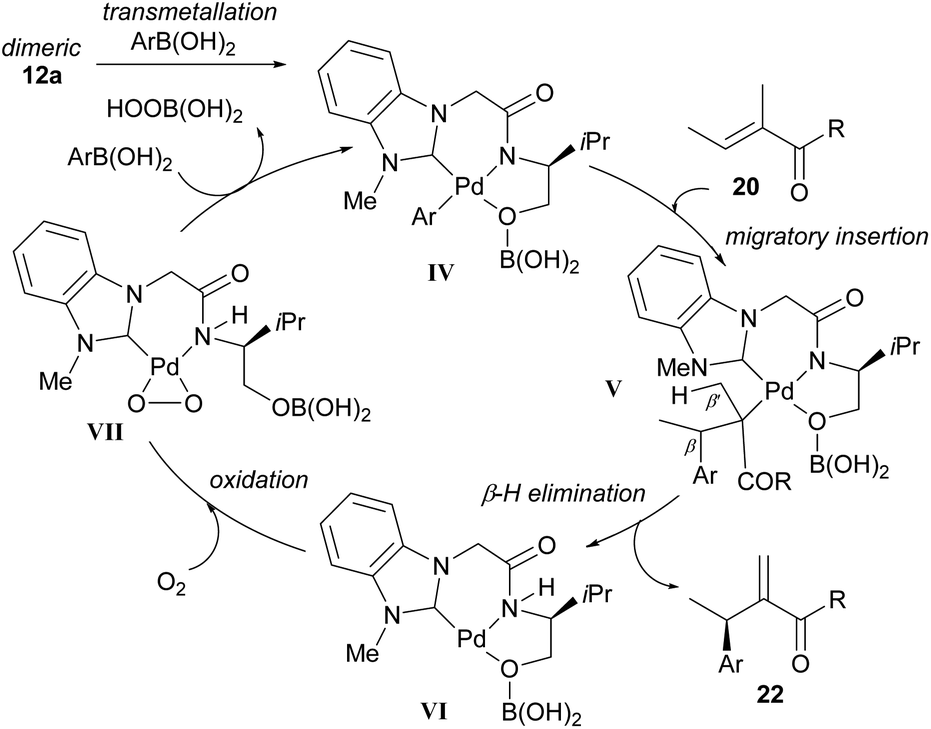
As before, the migratory insertion step is thought to be enantiodetermining. Fig. 2 shows the possible transition states for orientation of the alkene to the Pd complex IV. Approach by pathway A should be disfavoured by steric hindrance due to steric repulsion on the concave face. The alkene must therefore approach via pathway B. Of the two possible alkene coordination models E and F, coordination model E is disfavoured by steric hindrance between the alkenyl methyl substituent and the borate group. Therefore, coordination model F is favoured and results in the observed stereochemical (R)-configuration of product 22. The opposite alignment of the iso-propyl and borate group in IV was termed “counter axial groups” by the authors, and is thought to be the key factor for achieving high enantioselectivities. As such, the axial borate group governs the facial selectivity of the incoming alkene starting material when dimeric catalyst 12 is employed, and in contrast, this group is not present in monomeric catalyst 23, thereby resulting in lower enantioselectivities using the latter.
 | ||
| Fig. 2 Possible transition states for orientation of the alkene to Pd complex IV. Reprinted with permission from K. S. Yoo, J. O'Neill, S. Sakaguchi, R. Giles, J. H. Lee and K. W. Jung, J. Org. Chem., 2010, 75, 95. Copyright (2010) American Chemical Society.32 | ||
The proposed mechanism is shown in Scheme 16 and commences with transmetallation to produce the key borate species IV. Migratory insertion to form V and subsequent β-H elimination at position β′ produces the migrated cross-coupled product 22. Oxidation of the Pd(0) species VI by molecular oxygen produces a peroxo–palladium complex VII, which then reacts with arylboronic acid to regenerate the borate complex IV.
As described above, the enantioselectivities in this study are good to excellent (82%–94% ee, 11 examples), but the yields are poor to moderate (29%–61%). These poor yields are attributed to a significant amount of oxidative deborylation to produce phenolic side-products (e.g. in Scheme 15, 22cb is produced in 52% yield with side reactions: 43% deborylation, 2% homocoupling). Therefore, any further developments in the field will no doubt be centred upon achieving a highly enantioselective and high yielding catalytic system.
3.3 Redox–relay oxidative Heck Reaction
One of the most significant advances in asymmetric Heck-type chemistry in recent years is the redox–relay Heck-type reaction developed by Sigman and co-workers. The concept was first conceived using the Heck–Matsuda coupling27,33 (Heck-type variant using diazonium salts 25 as coupling partners, Scheme 17), but subsequent developments utilised the oxidative Heck reaction in order to improve the scope and practical application of the method. The work is exceptional because it not only allows for the formation of remote stereocentres, but also because of its excellent site selectivity and ability to distinguish between almost identical C–H bonds in the β-H elimination (see VIII→IX). The catalyst system imparts notable regioselectivity (from 80:20 for n = 2 to full selectivity for n = 0 in 27) during migratory insertion onto 26 and also promotes the migration of the alkene's unsaturation towards the alcohol in a redox isomerisation termed “redox–relay strategy” by the authors, to ultimately form the ketone product 27 (Scheme 17). While the racemic nature of the alkenol substrate does not bias enantioselection, the alkene configuration does (E → S and Z → R). Notably, the PyOX ligand 16b not only provides enantioinduction and regioselectivity, but also renders the Pd(II) hydride intermediates sufficiently electrophilic to reinsert into the alkene rather than dissociate from it.
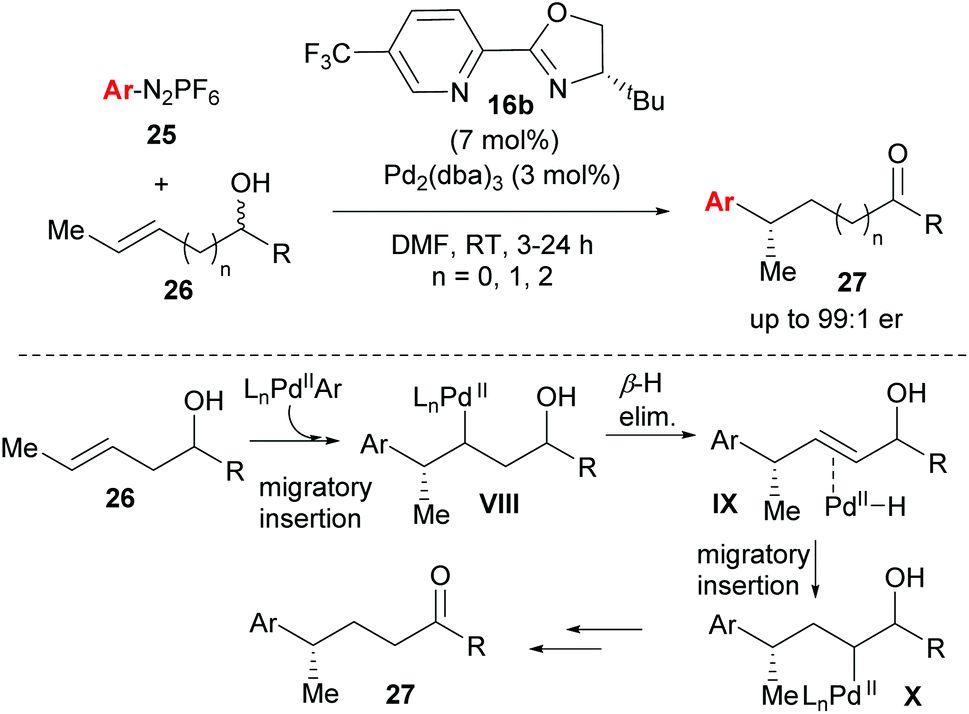 | ||
| Scheme 17 Enantioselective redox–relay Heck-Matsuda arylations of acyclic alkenyl alcohols by Sigman and co-workers. | ||
As described above, the redox–relay reaction was subsequently expanded to the oxidative Heck coupling (Scheme 18) and mechanistic investigations were carried out in order to determine what controls the regioselectivity of the reaction.34 Firstly, optimisation studies revealed that both Cu(OTf)2 and O2 oxidants as well as molecular sieves [to prevent retarded oxidation of Pd(0) to Pd(II) using just O2 or Cu(II)] were required for high conversions (Scheme 18). A thorough substrate scope investigation led to the conclusion that the (generally excellent) enantioselectivity is essentially independent of the nature of both reaction partners. In contrast, site selectivity is controlled by the nature of the arylboronic acid as well as substitution and chain length of the alkenyl alcohol substrate 27. A plot of site selectivity ratios vs. Hammett σ-values shows a clear correlation between the regioselectivity and the electronic nature of the arylboronic acid, with electron poor aryls providing high selectivities and electron rich aryls giving poor selectivity. A plot of site selectivity ratios vs.13C chemical shifts of the alkene (the most downfield-shifted C is distal from the alcohol) also reveals a clear trend (decreased selectivity as n increases), suggestive once again of electronic effects governing the site selectivity. The minor isomers also all exhibit high enantioselectivities. Using the evidence above, the authors suggest that the major and minor products arise from opposite faces of the alkene being presented to the catalyst during the migratory insertion step (Scheme 19). Additional support for the electronically influenced site selectivity was subsequently disclosed by DFT calculations on the reaction.35
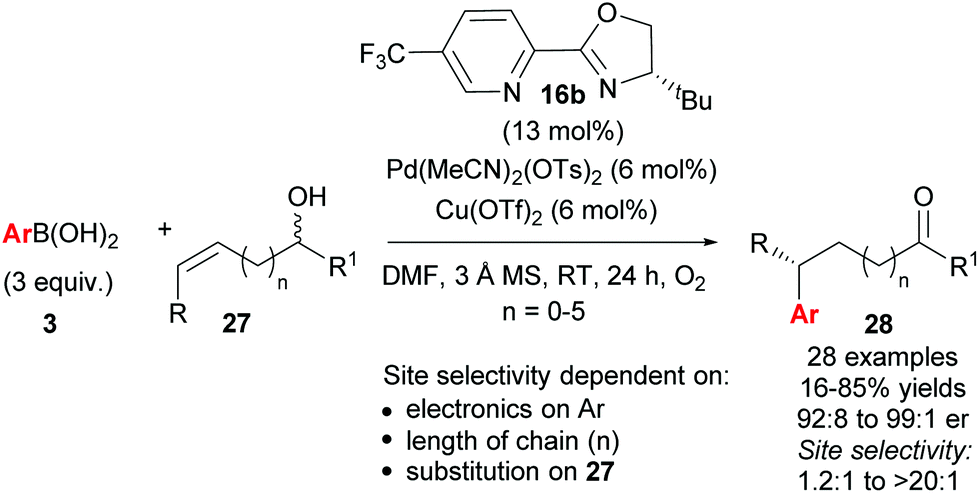 | ||
| Scheme 18 Enantioselective redox–relay oxidative Heck arylations of acyclic alkenyl alcohols. | ||
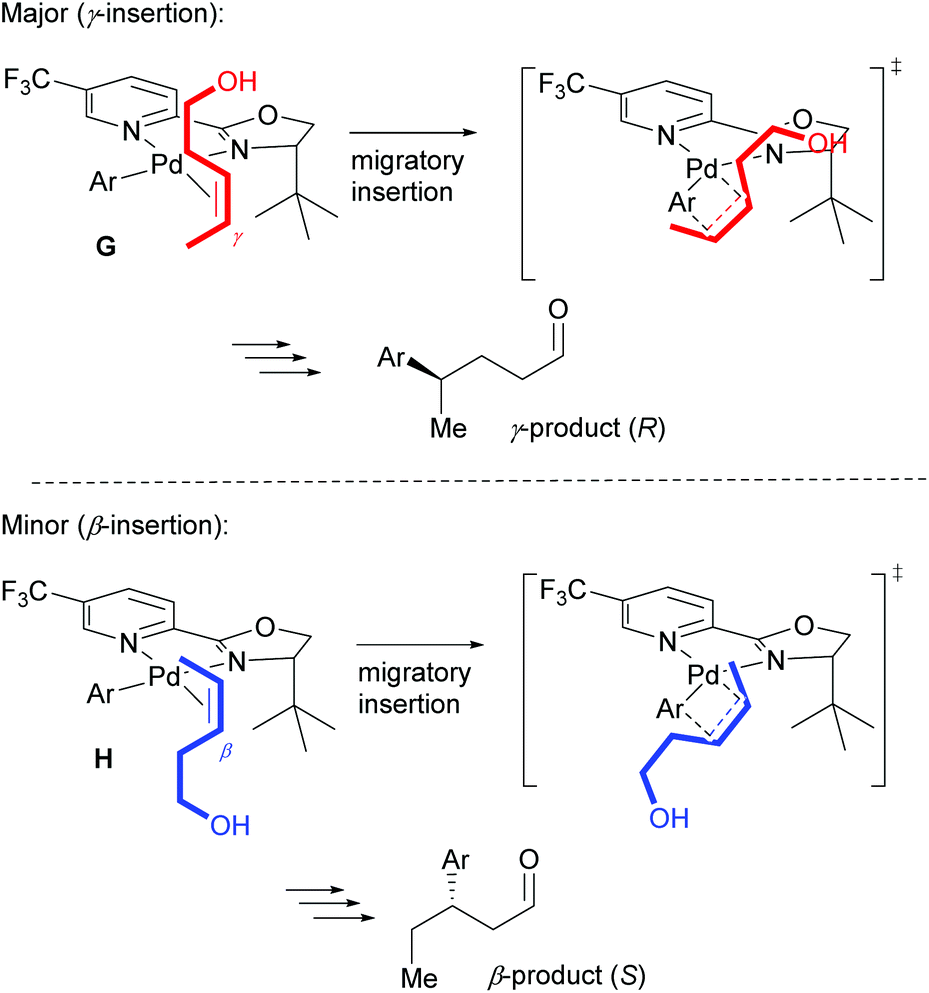 | ||
| Scheme 19 Proposed conformations towards the major and minor products. | ||
Following their initial study, the method was subsequently extended to enantioselective construction of remote quaternary centres (Scheme 20).36,37 Typically, quaternary stereocentres are prepared from substrates with pre-existing functional groups adjacent to the reaction site, with the location of the C–C bond formation strictly defined relative to the functional group in question. As such, the ability to install quaternary chiral centres which are remote from existing functional groups, using the oxidative Heck redox relay strategy, is truly exceptional. The reaction is highly site selective for the more hindered position of the alkene 29, which corroborates the authors’ earlier conclusion that the migratory insertion step is selective for the more downfield-shifted carbon, which is proposed to be controlled by remote dipole interactions of the alcohol functional group. In contrast to their results with disubstituted alkenes 27 (Scheme 18), the reaction with trisubstituted alkenes 29 is remarkably site selective irrespective of chain length as well as the electronics on the arylboronic acid 3, thereby providing an excellent substrate scope of various alkenyl alcohols 29 as well as aryls on 3. Enantioselectivities are once again excellent (92:8 to 99:1 er) and a highlight example is shown in Scheme 21: an alkene with sterically similar n-ethyl and n-butyl groups (29a) still provides product 30ba with a remarkable 97:3 er.
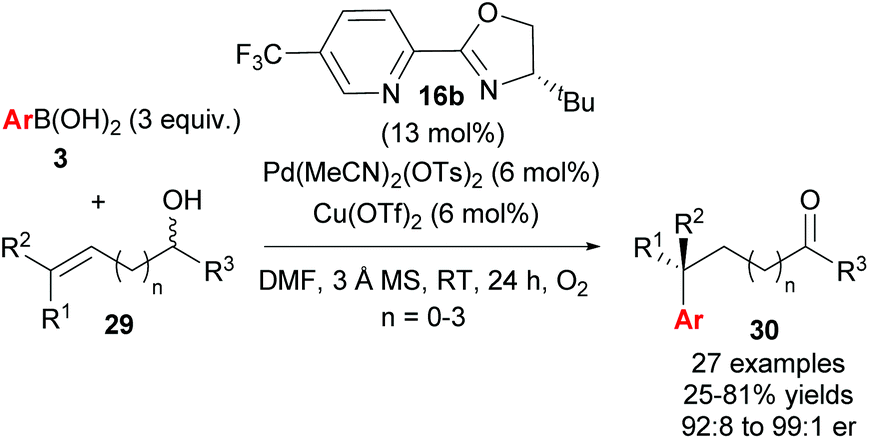 | ||
| Scheme 20 Enantioselective construction of remote quaternary centres by Sigman and co-workers. | ||
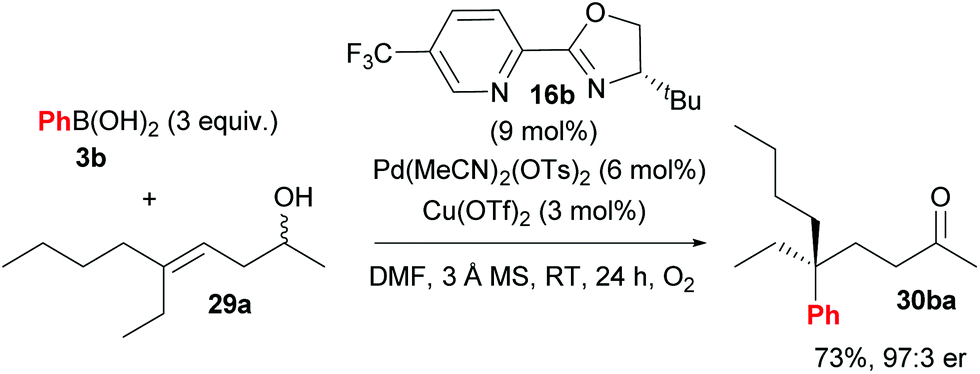 | ||
| Scheme 21 Remarkable enantioselectivity even for substrates with sterically similar substituents (n-ethyl vs. n-butyl). | ||
An exciting prospect for complex natural product synthesis is that the reaction has the ability to migrate through an existing chiral centre with preservation of enantiomeric composition, as well as catalyst controlled face-selection – two distinct diastereomers of 32 are formed by using different enantiomers of the catalyst (31→32, Scheme 22). This result implies that the catalyst remains ligated to the substrate and on the same face of the alkene throughout the relay process.
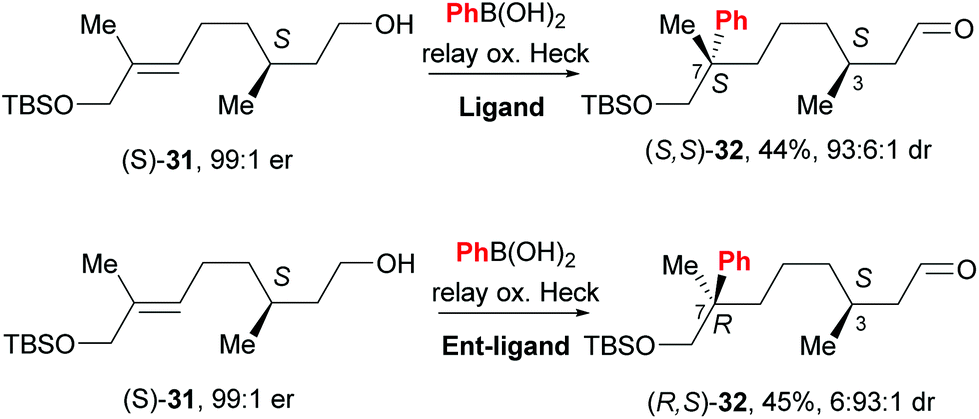 | ||
| Scheme 22 Configuration of pre-existing chiral centre is retained during chain walking process and face selectivity is catalyst controlled. | ||
Following this groundbreaking 2014 report, an area of further study was noted to be the expansion of the chemistry beyond the coupling of aryls to other saturated and unsaturated groups, and the ability of groups other than alcohol to intercept the chain-walking Pd-catalyst.38 Very recently, Sigman and co-workers have successfully demonstrated the ability of carbonyl groups to perform the latter chemistry (33→34, Scheme 23).39 There was a significant solvent effect on site selectivity and the use of DMA as solvent provided good to excellent regioselectivites for cases where n = 1. The advantage of using these alkenyl carbonyl substrates is that it now allows for iterative relay Heck reactions via a three-step approach (Scheme 24). An oxidative Heck redox–relay reaction on 33a followed by 1,2-reduction of 34a provides allylic alcohol 35, which can subsequently undergo an enantioselective Heck–Matsuda coupling to yield product 36, with two new stereocentres installed over the 3 steps.
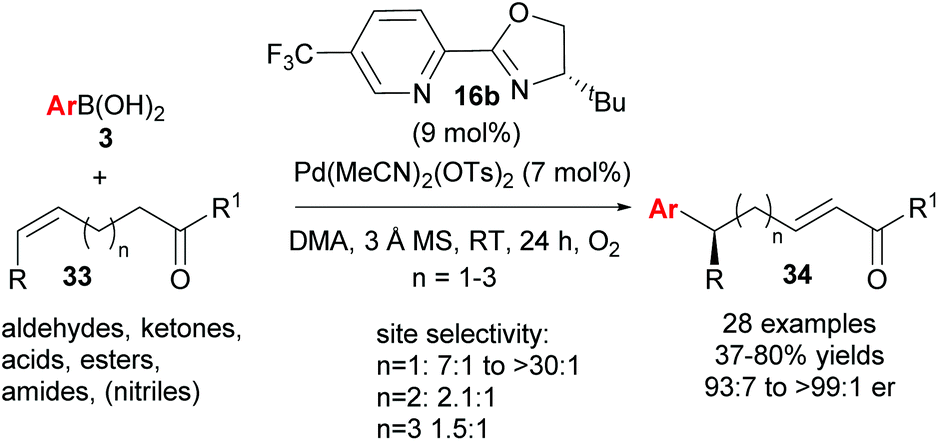 | ||
| Scheme 23 Enantioselective redox–relay oxidative Heck arylations of alkenyl carbonyl derivatives. | ||
 | ||
| Scheme 24 Iterative relay Heck reaction. | ||
4. Conclusions
Significant progress has been made in the field of enantioselective intermolecular Heck-type reactions in recent years, and in particular, enantioselective oxidative boron Heck couplings have contributed substantial advances. The field of enantioselective intermolecular Heck-type couplings has so far yet to achieve the same generality and application as the intramolecular counterpart; therefore, the advances highlighted in this review are of significance and bode well for the future of the field. Advances enabled by utilising the oxidative boron Heck reaction include enantioselective intermolecular couplings of more challenging systems such as desymmetrisation of quaternary centres, cyclic enones, acyclic alkenes and even site selectively on remote alkenes via a redox–relay coupling. Of note is the use of PyOX ligands, which appears to be instrumental in almost all of the most recent developments. Nevertheless, there is still plenty of room for improvement, including expansion of the chemistry beyond the coupling of aryls as well as to other challenging alkenes. Further developments in the field can therefore be anticipated.Acknowledgements
We thank the Leverhulme Trust (F/00 276/O) and EPSRC (EP/K00736X/1) for funding.Notes and references
- For recent reviews on asymmetric Heck reactions, see:
(a) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC
; (b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS
- Review: A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed
- For a recent highlight review, see: M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282 CrossRef CAS PubMed
- For selected recent advances in intermolecular asymmetric Heck couplings using Pd(0), see:
(a) S. Liu and J. Zhou, Chem. Commun., 2013, 49, 11758 RSC
- Review: B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 CrossRef CAS
- K. S. Yoo, C. H. Yoon and K. W. Jung, J. Am. Chem. Soc., 2006, 128, 16384 CrossRef CAS PubMed
- L. Penn, A. Shpruhman and D. Gelman, J. Org. Chem., 2007, 72, 3875 CrossRef CAS PubMed
- H. A. Dieck and R. F. Heck, J. Org. Chem., 1975, 40, 1083 CrossRef CAS
- C. S. Cho and S. Uemura, J. Organomet. Chem., 1994, 465, 85 CrossRef CAS
- X. Du, M. Suguro, K. Hirabayashi, A. Mori, T. Nishikata, N. Hagiwara, K. Kawata, T. Okeda, H. F. Wang, K. Fugami and M. Kosugi, Org. Lett., 2001, 3, 3313 CrossRef CAS PubMed
- Y. C. Jung, R. K. Mishra, C. H. Yoon and K. W. Jung, Org. Lett., 2003, 5, 2231 CrossRef CAS PubMed
- M. M. S. Andappan, P. Nilsson and M. Larhed, Chem. Commun., 2004, 218 RSC
- For other selected papers on oxidative Heck reaction, see:
(a) J. D. Crowley, K. D. Hanni, A.-L. Lee and D. A. Leigh, J. Am. Chem. Soc., 2007, 129, 12092 CrossRef CAS PubMed
- For example, see:
(a) A. Carrër, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef PubMed
- Conditions have recently been developed for Pd(0) Heck couplings onto cyclic enones but they are still fairly harsh (elevated temperatures) and the competing conjugate addition reaction is usually still observed. For example, see:
(a) Y. Fall, H. Doucet and M. Santelli, Tetrahedron, 2009, 65, 489 CrossRef CAS
- D. Tanaka and A. G. Myers, Org. Lett., 2004, 6, 433 CrossRef CAS PubMed
- Review: A. Gutnov, Eur. J. Org. Chem., 2008, 4547 CrossRef CAS
- S. E. Walker, J. Boehnke, P. E. Glen, S. Levey, L. Patrick, J. A. Jordan-Hore and A.-L. Lee, Org. Lett., 2013, 15, 1886 CrossRef CAS PubMed
- See also: Y. W. Kim and G. I. Georg, Org. Lett., 2014, 16, 1574 CrossRef CAS PubMed
- For selected examples, see:
(a) C. L. Boeser, J. C. Holder, B. L. H. Taylor, K. N. Houk, B. M. Stoltz and R. N. Zare, Chem. Sci., 2015, 6, 1917 RSC
- K. Akiyama, K. Wakabayashi and K. Mikami, Adv. Synth. Catal., 2005, 347, 1569 CrossRef CAS
- Note that while historically enantioselectivies have been quoted as enantiomeric excesses, the modern convention is to quote them as enantiomeric ratios. In this review, we have decided to quote the original numbers and conventions reported in the primary sources to avoid any misquoting of the primary results.
- K. Akiyama and K. Mikami, Heterocycles, 2007, 74, 827 CrossRef CAS
-
Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Blackwell Science Publ, Oxford, 2005 Search PubMed
-
(a) R. Antkowiak, W. Z. Antkowiak, I. Banczyk and L. Mikolajczyk, Can. J. Chem., 2003, 81, 118 CrossRef CAS
- H. Shimizu, J. C. Holder and B. M. Stoltz, Beilstein J. Org. Chem., 2013, 9, 1637 CrossRef PubMed
- E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed
- P. E. Glen, J. A. T. O'Neill and A.-L. Lee, Tetrahedron, 2013, 69, 57 CrossRef CAS
- K. Yonehara, K. Mori, T. Hashizume, K.-G. Chung, K. Ohe and S. Uemura, J. Organomet. Chem., 2000, 603, 40 CrossRef CAS
- S. Sakaguchi, K. S. Yoo, J. O'Neill, J. H. Lee, T. Stewart and K. W. Jung, Angew. Chem., Int. Ed., 2008, 47, 9326 CrossRef CAS PubMed
- K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neill and K. W. Jung, Org. Lett., 2007, 9, 3933 CrossRef CAS PubMed
- K. S. Yoo, J. O'Neill, S. Sakaguchi, R. Giles, J. H. Lee and K. W. Jung, J. Org. Chem., 2010, 75, 95 CrossRef CAS PubMed
- For other enantioselective Heck–Matsuda couplings, see:
(a) R. C. Carmona and C. R. D. Correia, Adv. Synth. Catal., 2015, 357, 2639 CrossRef CAS
- T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed
-
(a) Y. Dang, S. Qu, Z.-X. Wang and X. Wang, J. Am. Chem. Soc., 2014, 136, 986 CrossRef CAS PubMed
- T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed
- M. J. Hilton, B. Cheng, B. R. Buckley, L. Xu, O. Wiest and M. S. Sigman, Tetrahedron, 2015, 71, 6513 CrossRef CAS PubMed
- J. P. Morken, Nature, 2014, 508, 324 CrossRef CAS PubMed
- C. Zhang, C. B. Santiago, L. Kou and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 7290 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |