
Total synthesis and preliminary SAR study of (±)-merochlorins A and B†
Hongzhi
Yang
a,
Xue
Liu
b,
Qingong
Li
a,
Longbo
Li
a,
Jing-Ren
Zhang
*bc and
Yefeng
Tang
*ac
aThe Comprehensive AIDS Research Center, and Department of Pharmacology & Pharmaceutical Sciences, School of Medicine, Tsinghua University, Beijing 100084, P.R. China. E-mail: yefengtang@tsinghua.edu.cn; Tel: +86 10 62798236
bDepartment of Basic Medical Sciences, School of Medicine, Tsinghua University, Beijing 100084, China
cCollaborative Innovation Center for Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China
First published on 14th October 2015
Abstract
A modular synthesis of merochlorins A and B, two naturally occurring antibiotics, has been achieved concisely from readily available building blocks in 4–6 steps. The key steps include the bio-inspired tandem phenol oxidative dearomatization/[5 + 2] and [3 + 2] cycloadditions to construct the tricyclic cores of the targets, and the intermolecular Diels–Alder reaction followed by dehydrogenative aromatization to assemble the remaining aromatic units. The antibacterial activities of merochlorins A, B and some advanced synthetic intermediates were also evaluated, which provided valuable information on the structure–activity relationship (SAR) of this class of new antibiotics.
Introduction
Due to the ever-increasing threat of antibiotic resistance, there is an urgent need to develop new antibacterial agents with novel chemical structures and mechanisms of action.1 Natural products are arguably the most valuable resource for the discovery of new antibiotics.2 A recent paradigm appeared in 2012, when a pair of chlorinated meroterpenoids, merochlorins A (1) and B (2) (Fig. 1a), was identified from the marine bacterium Streptomyces sp. strain CNH-189 by Moore, Fenical and co-workers.3 Both 1 and 2 display broad-spectrum antibacterial activity against various Gram-positive organisms, such as clinically relevant methicillin-resistant Staphylococcus aureus strains (MIC = 2–4 μg mL−1) and vancomycin-resistant Enterococcus faecium (MIC = 2 μg mL−1),3,4 which renders them promising drug candidates for the development of new antibacterial agents. However, it was found that 1 was inactive (MIC > 64 μg mL−1) against the examined bacterial strains when tested in Mueller–Hinton broth (MHB) containing 10% human serum,4 which somewhat compromised its potential as a drug candidate. While further structural optimization may provide a solution to address this issue, the scarcity of 1 and 2 in natural resource makes it impractical to achieve such a goal. Thus, the development of an efficient and modular synthetic approach to access ample supply of 1 and 2 as well as their analogues is in great demand.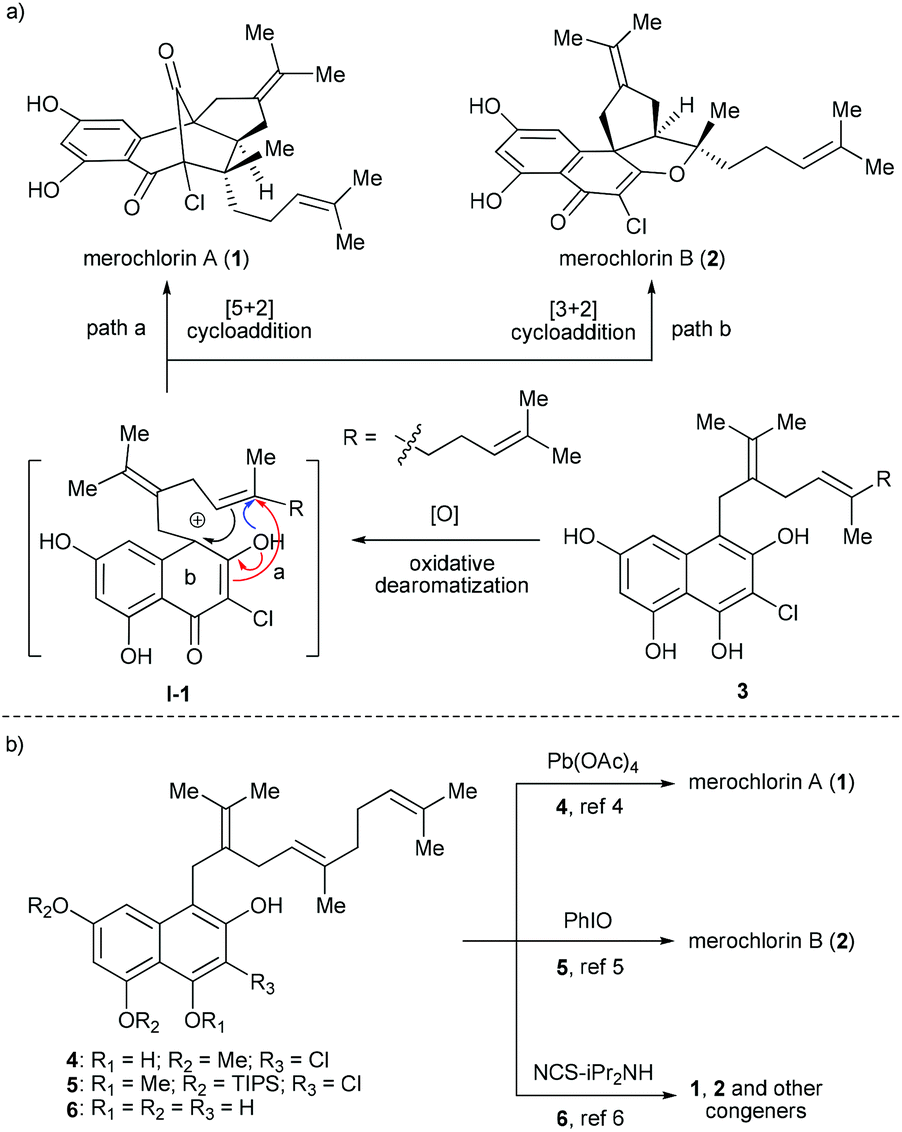 | ||
| Fig. 1 (a) Proposed biosynthetic pathway and (b) reported biomimetic synthesis of merochlorins A and B. | ||
While 1 and 2 bear different molecular skeletons, from the biosynthetic point of view they should be traced back to a common tetrahydroxynaphthalene (THN) precursor 3 (Fig. 1a). It was believed that 3 could first undergo phenol oxidative dearomatization to form a carbocation intermediate I-1, which then diverts into 1 and 2via [5 + 2] and [3 + 2] cycloadditions, respectively.3 Interestingly, this hypothesis was quickly validated by several elegant synthetic studies. In 2013, George and co-workers reported the first biomimetic synthesis of 1via tandem oxidative dearomatization/[5 + 2] cycloaddition (Fig. 1b).5 Subsequently, the first biomimetic synthesis of 2 was achieved by Trauner and co-workers via tandem oxidative dearomatization/[3 + 2] cycloaddition.6 Of note, only a single and different target was obtained in each of the above studies, probably due to the subtle difference between the substrates (4vs.5) and oxidants [Pd(OAc)4vs. PhIO] employed thereof. Another breakthrough appeared in 2014, when Moore and co-workers realized the enzymatic synthesis of 1 and 2 from pre-merochlorin 6 using a vanadium-dependent haloperoxidase.7a Meanwhile, they also accomplished the chemical synthesis of 1, 2 and several other related congeners with the same precursor using NCS/iPr2NH as both an oxidant and a chlorination agent.7b However, in this case both 1 and 2 generated only as minor components in relatively low yields. In this regard, a more efficient and practical approach enabling the diverted synthesis of merochlorins A and B remains to be established.
Results and discussion
Attracted by the prominent biological profiles of merochlorins A and B, we launched a program toward their synthesis. Our strategic plan is depicted in Scheme 1. As shown, we envisioned that both 1 and 2 could be obtained from the corresponding enone precursors 7 and 8via an intermolecular Diels–Alder reaction followed by dehydrogenative aromatization. Naturally, the key intermediates 7 and 8 could be accessed from the common precursor 9via the bio-inspired tandem oxidative dearomatization/[5 + 2] and [3 + 2] cycloadditions, respectively. In turn, 9 could be traced back to the readily accessible fragments 108 and 115,9via Friedel–Crafts alkylation. Of note, different from the previously reported strategies,5,6,7bwe projected to install the aromatic ring of the targets in the late stage of the synthesis, which not only avoids the tedious procedure for the preparation of fully functionalized THN derivatives (e.g. 5),6 but also enables access to other merochlorin analogues by using different diene components. The later feature is particularly important for further biomedical studies. Herein we report the successful implementation of the above strategy for the diverted total synthesis of merochlorins A and B. Moreover, we also evaluated the anti-bacterial activities of 1 and 2 as well as several advanced synthetic intermediates, which shed light on the preliminary structure–activity relationship (SAR) of this new class of antibiotics.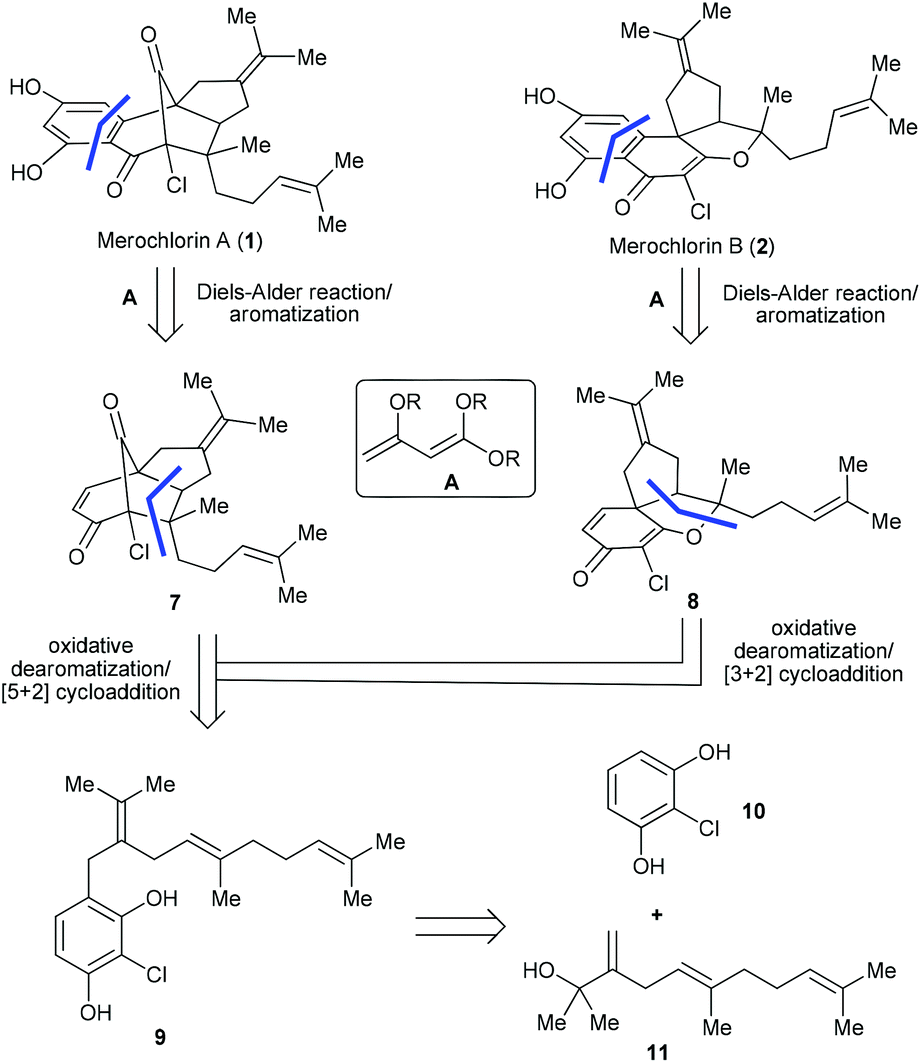 | ||
| Scheme 1 Retrosynthetic analysis of merochlorins A and B. | ||
Our studies commenced from the synthesis of the precursor 9. To this end, the fragments 108 and 115,9 were first prepared following the literature methods. Next, the assembly of 10 and 11 was achieved via a BF3·OEt2-promoted Friedel–Crafts alkylation,10 which led to the desired product 9 in 52% yield. With 9 secured in a scalable manner, we turned to explore the key step of our synthesis, the biomimetic oxidative dearomatization induced [5 + 2] and [3 + 2] cycloadditions.11 Since hypervalent iodine(III) reagent-mediated phenol dearomatization followed by other transformations have been well documented,12 we first evaluated some commonly used hypervalent iodine(III) reagents [e.g., PhI(OAc)2 and PhI(CF3CO2)2] in our scenario. To our delight, when the reaction was performed with PhI(OAc)2 (PIDA) in CH3CN at room temperature, the desired transformations did proceed to afford the tricyclic compounds 7 and 8 in 10% and 20% yields, respectively (condition 1, Scheme 2). A simple evaluation of the solvent effect revealed that comparable efficiency could be obtained with CF3CH2OH, while DCM and (CF3)2CHOH afforded inferior results (conditions 2–4). Interestingly, while a similar combined yield of 7 and 8 (30%) was obtained with PhI(CF3CO2)2 (PIFA) employed as an oxidant (condition 5), the selectivity of the products was notably improved (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 = 1:4). To further improve the efficiency of the transformations, we also evaluated some other reaction parameters including the temperature and additives (conditions 6–9), however, no significant improvement could be made. Eventually, we attempted some other oxidants (e.g., Pb(OAc)4, DDQ and FeCl3) in these reactions. It was found that while most of the reactions failed to give the desired products, Pb(OAc)4 proved to be the optimal choice (condition 10), which furnished 7 and 8 in 17% and 23% yields, respectively. It should be pointed out that no apparent side-products could be isolated in this reaction, implying that the moderate yields of the transformations mainly arouse from the severe decomposition of the starting material. Although the efficiency of the above reaction remains to be improved, it enables the rapid access of the two key intermediates en route to 1 and 2 in one pot, thus differentiating itself from the previous studies.5,6
:8 = 1:4). To further improve the efficiency of the transformations, we also evaluated some other reaction parameters including the temperature and additives (conditions 6–9), however, no significant improvement could be made. Eventually, we attempted some other oxidants (e.g., Pb(OAc)4, DDQ and FeCl3) in these reactions. It was found that while most of the reactions failed to give the desired products, Pb(OAc)4 proved to be the optimal choice (condition 10), which furnished 7 and 8 in 17% and 23% yields, respectively. It should be pointed out that no apparent side-products could be isolated in this reaction, implying that the moderate yields of the transformations mainly arouse from the severe decomposition of the starting material. Although the efficiency of the above reaction remains to be improved, it enables the rapid access of the two key intermediates en route to 1 and 2 in one pot, thus differentiating itself from the previous studies.5,6
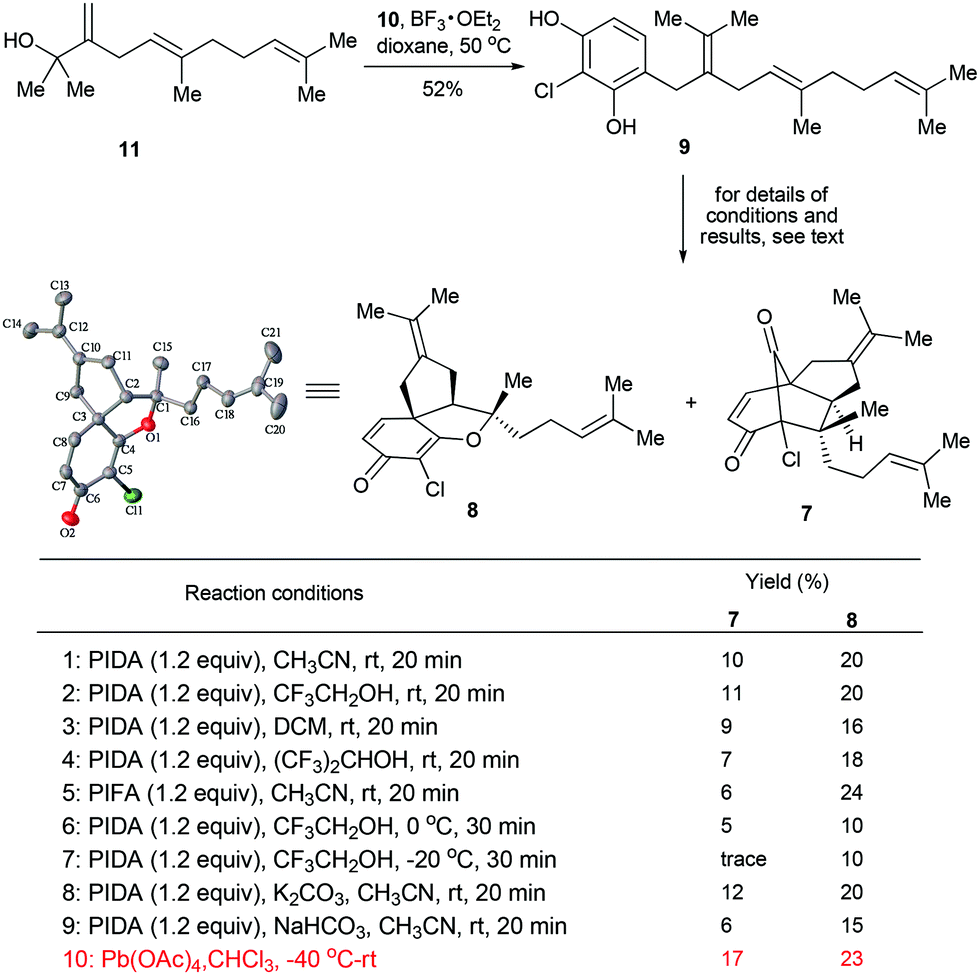 | ||
| Scheme 2 Synthesis of tricyclic intermediates 7 and 8. | ||
Having secured the synthesis of the tricyclic intermediates 7 and 8, we then moved to complete the total synthesis of 1 and 2. Based on the proposed strategic plan, the aromatic rings of 1 and 2 could be constructed via Diels–Alder reactions followed by aromatization. Of note, while a similar strategy has been put into practice in many precedents,13 most of them employed quinone derivatives as dienophiles, which are distinct from our case. To explore the feasibility of our design, we first attempted the Diels–Alder reaction between the dienophile 7 and diene 12a.14 To our delight, the reaction worked well under the thermal conditions (toluene, sealed tube, 160 °C, 4 h), providing 13a as a major product. It was found that 13a was unstable and readily converted into a mixture of 14a and 14b upon chromatography. Besides, a small amount of 1 was also detected at this stage, which might be generated from 14avia auto-oxidation with the action of air. Inspired by this observation, an operationally simple one-pot protocol was developed for the synthesis of 1. Thus, the resulting mixtures of Diels–Alder reactions were directly treated with 1 N HCl and then exposed to air for 12 h, which finally afforded 1 in 15% overall yield in 3 steps. Alternatively, the dehydrogenative aromatization could also be accelerated by using the Jones reagent as an extra oxidant,13b which gave a slightly improved overall yield (18%) in a shorter reaction time (Scheme 3).
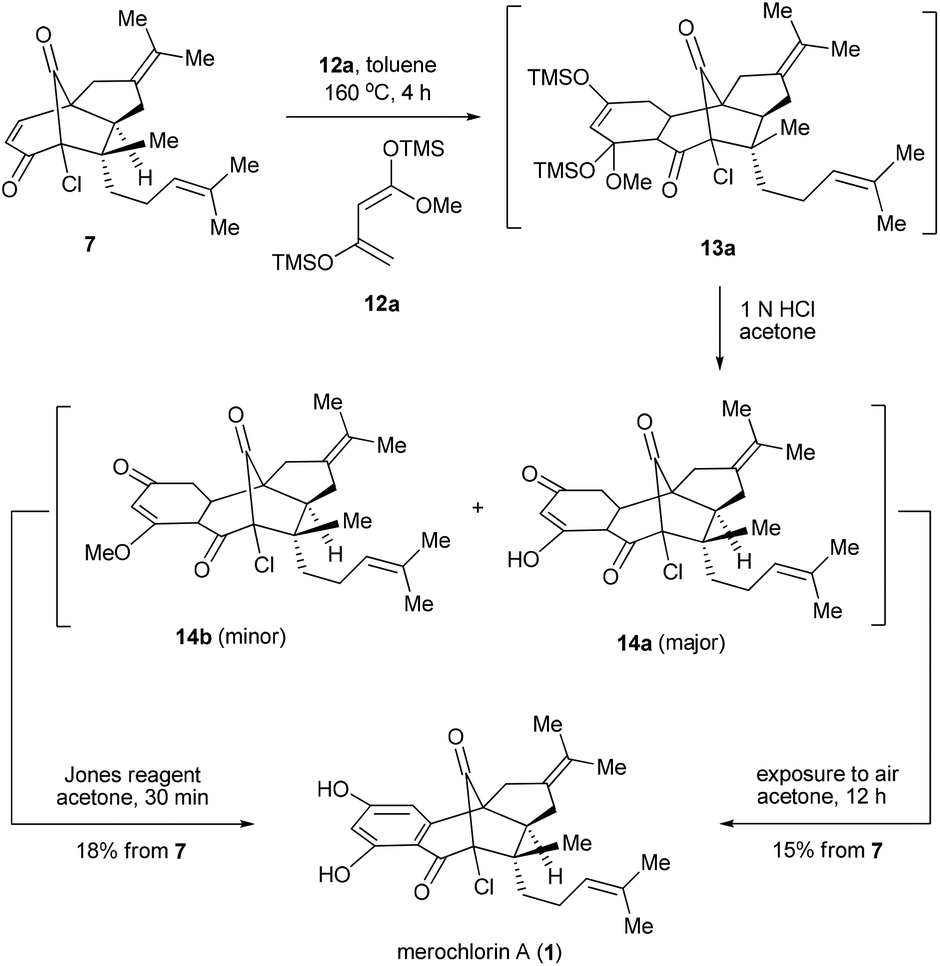 | ||
| Scheme 3 One-pot synthesis of merochlorin A. | ||
In parallel with the aforementioned work, an alternative and stepwise route towards 1 was also developed with 12b15 employed as the diene partner (Scheme 4). In this scenario, the Diels–Alder reaction could go to completion with a relatively low temperature and short reaction time (toluene, 100 °C, 1 h). After the acidic work-up, 14b was obtained as the predominant product,16 which, upon the further treatment of I2 in MeOH,17 provided the di-methylated merochlorin A (15) in 50% overall yield. The endgame of the total synthesis of 1 was then completed by adopting a modified two-step procedure developed by George and co-workers.5 Thus, upon treatment of 15 with LiCl in DMF at 135 °C (microwave irradiation) for 5 minutes led to the mono-methylated merochlorin A (16) as a major product, along with a small amount of 1. The resulting mixtures, after a simple work-up, were further subjected to the same conditions as above, which finally afforded 1 and 16 in 40% and 16% yields, respectively. It should be pointed out that, while the further conversion of 16 to 1 could be achieved with the elongation of reaction time, severe decomposition of the final product was observed, resulting in decreased yield of 1.
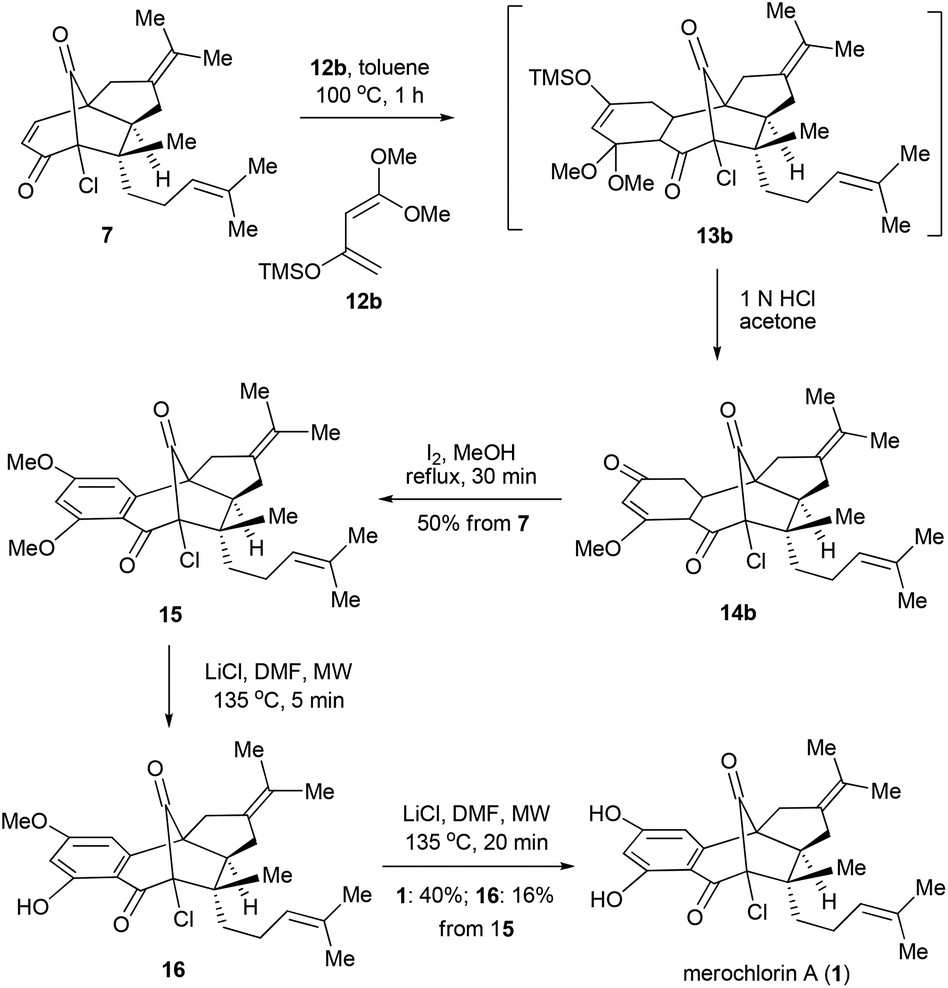 | ||
| Scheme 4 Alternative stepwise synthesis of merochlorin A. | ||
With the total synthesis of 1 achieved, we then moved to synthesize merochlorin B (2). Out of our expectation, the Diels–Alder reaction between 8 and 12a failed to proceed under the conditions employed above (toluene, sealed tube, 160 °C, 4 h), indicating that the dienophile 8 displayed relatively lower reactivity than 7. Gratifyingly, we quickly found that this problem could be resolved by using the less sterically hindered and more reactive diene 12b as a reaction partner. Indeed, the Diels–Alder reaction of 8 with 12b went to completion in refluxing toluene at 110 °C for 4 h, which, after a simple acidic work-up, afforded the desired product 18 in 78% isolated yield. The structure of 18 was confirmed by the X-ray crystallography.18 Next, the dehydrogenative aromatization of 18 was attempted with the same conditions (I2, MeOH) used for the synthesis of 1, however, only a low yield of the corresponding product (structure not shown) was obtained. Thus, an alternative approach via Saegusa oxidation was employed, wherein 18 was first converted into the corresponding silyl enol ether, which then advanced to the mono-methylated merochlorin B (19) in 50% overall yield.19 While the efficiency of this transformation appeared to be moderate, substantial amounts of 18 (ca. 40%) were recovered and could be recycled for the second time, thus improving the overall yield to ca. 70%. Finally, demethylation of 19 was achieved under the same conditions as mentioned above, which gave merochlorin B (2) in 60% yield (Scheme 5).
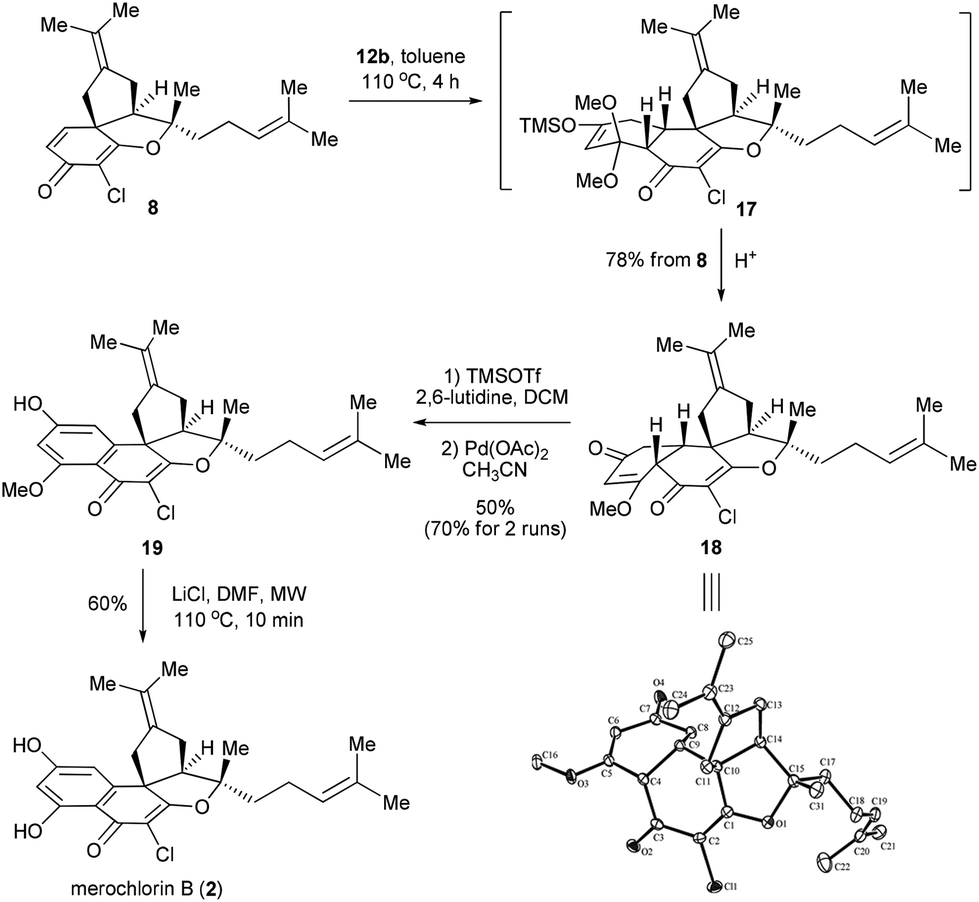 | ||
| Scheme 5 Total synthesis of merochlorin B. | ||
The accessibility of 1, 2 and several advanced synthetic intermediates (e.g., 7, 8, 15, 16, 18, and 19) in the current work enabled us to perform the preliminary SAR study on this class of antibiotics. It should be pointed out that, although extensive effort has been devoted to the total synthesis of 1 and 2,5–7 little has been devoted to explore their SAR. As shown in Table 1, both 1 and 2 exhibited potent inhibitory activity against the examined bacterial strains including E. faecalis, S. aureus, and S. pneumoniae which was in good agreement with the previous results.3,4 In comparison, all of the synthetic intermediates only showed weak or no inhibitory activity. These outcomes revealed that the aromatic domains of 1 and 2 played a very important role for their antibacterial activity (7vs.1; 8vs.2). Particularly, it proved that both the free hydroxyl groups on the aromatic ring were essential for their antibacterial activity, since either the mono- or di-methylated merochlorins (15, 16 and 19) turned out to be inactive in our test. The above information is valuable for the development of new merochlorin-derived antibiotics.
| Compound | MIC (μg mL−1) | ||
|---|---|---|---|
| E. faecalis | S. aureus | S. pneumoniae | |
| a Enterococcus faecalis ATCC29212. b Staphylococcus aureus ATCC29213. c Streptococcus pneumoniae NCTC7466. | |||
| 1 | 1–2 | 1–2 | 8–16 |
| 2 | 1–2 | 1–2 | 8–16 |
| 7 | 32 | 16–32 | 16–32 |
| 8 | >64 | >64 | >64 |
| 15 | >64 | >64 | >64 |
| 16 | >64 | >64 | >64 |
| 18 | >64 | >64 | >64 |
| 19 | >64 | >64 | >64 |
| Vancomycin | 1–2 | 0.5–1 | 0.25–0.5 |
| Ampicillin | 1–2 | 1–2 | 0.125–0.25 |
Conclusions
In summary, we have achieved a concise and modular synthesis of merochlorins A and B, two naturally occurring antibiotics, from the readily accessible fragments. The key elements of our synthesis include the bio-inspired tandem phenol oxidative dearomatization/[5 + 2] and [3 + 2] cycloadditions to access the tricyclic cores of the targets, and the intermolecular Diels–Alder reactions followed by aromatization to assemble the remaining aromatic units. Moreover, the antibacterial activities of 1, 2 and some advanced synthetic intermediates were also evaluated, and the outcomes shed light on the preliminary SAR of this class of new antibiotics. We are now working on the development of new merochlorin analogs with simplified structures and improved drug properties.Experimental section
General information
Unless otherwise mentioned, all reactions were carried out under a nitrogen atmosphere and anhydrous conditions and all reagents were purchased from commercial suppliers without further purification. Solvent purification was conducted according to ref. 20. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. Reactions were monitored by Thin Layer Chromatography on plates (GF254) supplied by Yantai Chemicals (China) using UV light as a visualizing agent and an ethanolic solution of phosphomolybdic acid and cerium sulfate, and heat as developing agents. If not specially mentioned, flash column chromatography uses silica gel (200–300 mesh) supplied by Tsingtao Haiyang Chemicals (China). NMR spectra were recorded on a Bruker AV400 instrument. TMS was used as an internal standard for 1H NMR (0 ppm), and a solvent signal was used as a reference for 13C NMR (CDCl3, 77.16 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, br = broad, td = triplet of doublets, qd = quartet of doublets, m = multiplet. Infrared (IR) spectra were recorded on a Thermo Nicolet Avatar 330 FT-IR spectrometer. High-resolution mass spectra (HRMS) were recorded on a Bruker ESI-Q/TOF MS, low-resolution mass spectral analyses were performed with a Waters AQUITY UPLCTM/MS. The microwave reactions were conducted with a CEM microwave reactor (model: DISCOVERY-SP, 300W).:1) afforded compound 9 (1.63 g, 52%) as a thick oil. Rf = 0.38 (silica gel, PE/EtOAc = 6:1); IR (film): 3460, 2966, 2913, 2856, 1612, 1493, 1440, 1165, 869, 802, 610; 1H NMR (400 MHz, CDCl3) δ 6.84 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 8.4 Hz, 1H), 5.80 (s, 1H), 5.34 (s, 1H), 5.08 (t, J = 6.0 Hz, 1H), 4.99 (t, J = 6.7 Hz, 1H), 3.37 (s, 2H), 2.68 (d, J = 6.8 Hz, 2H), 2.06–2.03 (m, 2H), 1.97–1.93 (m, 2H), 1.80 (s, 3H), 1.79 (s, 3H), 1.68 (s, 3H), 1.60 (s, 3H), 1.51 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 150.4, 150.1, 135.5, 131.3, 129.6, 128.4, 128.3, 124.4, 122.3, 119.0, 107.3, 107.2, 39.7, 32.3, 30.6, 26.6, 25.7, 20.8, 20.6, 17.7, 16.0 ppm; HRMS (ESI) m/z [M − H]− calcd for C21H29ClO2 347.1787, found 347.1787.
:1, 10:1, 6:1) to give compound 7 (84 mg, 17%) as a yellow oil and compound 8 (113 mg, 23%) as a yellow solid. 7: Rf = 0.45 (silica gel, PE/EtOAc = 6:1); IR (film): 2971, 1914, 1854, 1775, 1685, 1601, 1446, 1374, 810, 737; 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 9.2 Hz, 1H), 6.33 (d, J = 9.2 Hz, 1H), 4.97 (t, J = 6.9 Hz, 1H), 3.21 (d, J = 16.5 Hz, 1H), 2.53–2.35 (m, 3H), 2.21–2.15 (m, 1H), 2.08–1.99 (m, 2H), 1.90–1.83 (m, 1H), 1.67 (s, 3H), 1.63 (s, 3H), 1.62 (s, 3H), 1.57 (s, 3H), 1.17–1.10 (m, 1H), 0.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.8, 191.9, 159.6, 132.3, 131.6, 129.2, 124.1, 123.2, 93.9, 60.8, 57.8, 43.4, 40.6, 33.4, 32.6, 25.6, 22.6, 21.0, 20.9, 19.7, 17.6 ppm; HRMS (ESI) m/z [M + H]+ calcd for C21H27ClO2 347.1772, found 347.3043. 8: Rf = 0.24 (silica gel, PE/EtOAc = 6:1); IR (film): 2971, 2916, 2860, 1651, 1596, 1385, 1297, 1063, 820, 737; 1H NMR (400 MHz, CDCl3) δ 6.74 (d, J = 9.6 Hz, 1H), 6.16 (d, J = 9.6 Hz, 1H), 5.03 (t, J = 6.5 Hz, 1H), 2.85 (d, J = 17.2 Hz, 1H), 2.73–2.62 (m, 1H), 2.57–2.48 (m, 3H), 2.03–1.95 (m, 2H), 1.69–1.68 (m, 2H), 1.67 (s, 3H), 1.63 (s, 3H), 1.56 (s, 6H), 1.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 181.6, 174.5, 143.8, 132.5, 130.4, 127.0, 125.8, 122.8, 105.0, 97.1, 59.1, 51.3, 44.9, 44.6, 32.5, 25.6, 22.3, 22.1, 21.5, 21.3, 17.6 ppm; HRMS (ESI) m/z [M + H]+ calcd for C21H27ClO2 347.1772, found 347.3031.
:1) to give 15 (138 mg, 50%) as a white solid. Rf = 0.3 (silica gel, PE/EtOAc = 2:1); IR (film): 2954, 2920, 2850, 1595, 1462, 1259, 1092, 1013, 795, 733; 1H NMR (400 MHz, CDCl3) δ 6.51 (d, J = 1.7 Hz, 1H), 6.39 (d, J = 1.8 Hz, 1H), 5.00 (t, J = 6.7 Hz, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 3.15 (d, J = 15.2 Hz, 1H), 2.74 (d, J = 15.2 Hz, 1H), 2.46–2.31 (m, 3H), 2.16–2.07 (m, 1H), 1.85–1.80 (m, 1H), 1.77 (s, 3H), 1.74–1.67 (m, 1H), 1.64 (s, 3H), 1.62 (s, 3H), 1.56 (s, 3H), 1.33–1.24 (m, 1H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.6, 187.3, 165.2, 164.0, 151.6, 131.7, 131.6, 124.0, 123.7, 114.8, 100.0, 97.1, 91.1, 61.2, 58.9, 56.3, 55.7, 44.3, 38.4, 32.1, 29.7, 25.6, 22.8, 21.1, 20.8, 17.7, 16.0 ppm; HRMS (ESI) m/z [M + H]+ calcd for C27H33ClO4 457.2146, found 457.2141.
:1–4:1) as a white solid. Rf = 0.5 (silica gel, PE/EtOAc = 4:1); IR (film): 2954, 2920, 2849, 1623, 1462, 1377, 1264, 1202, 1021, 800; 1H NMR (400 MHz, CDCl3) δ 12.33 (s, 1H), 6.43 (d, J = 2.0 Hz, 1H), 6.36 (d, J = 2.1 Hz, 1H), 5.00 (t, J = 6.5 Hz, 1H), 3.86 (s, 3H), 3.14 (d, J = 15.4 Hz, 1H), 2.71 (d, J = 15.5 Hz, 1H), 2.46–2.30 (m, 3H), 2.14–2.10 (m, 1H), 1.86–1.81 (m, 1H), 1.77 (s, 3H), 1.72–1.67 (m, 1H), 1.65 (s, 6H), 1.58 (s, 3H), 1.23–1.19 (m, 1H), 0.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 199.3, 194.2, 166.8, 166.6, 149.6, 132.2, 131.4, 124.0, 123.4, 110.7, 102.5, 99.2, 90.6, 61.4, 58.6, 55.9, 45.3, 39.4, 31.9, 29.1, 25.6, 22.7, 21.1, 20.9, 17.7, 16.7 ppm; HRMS (ESI) m/z [M − H]− calcd for C26H31ClO4 443.1989, found 443.1976.
:1–4:1) to give merochlorin A (1) (18.8 mg, 40%) as a yellow solid, together with a substantial amount of 16 (7.8 mg, 16%). 1: Rf = 0.4 (silica gel, PE/EtOAc = 2:1); IR (film): 2960, 2923, 2853, 1625, 1583, 1453, 1255, 1168; 1H NMR (400 MHz, d6-DMSO) δ 11.99 (s, 1H), 11.36 (s, 1H), 6.46 (d, J = 2.0 Hz, 1H), 6.28 (d, J = 2.0 Hz, 1H), 5.01 (t, J = 7.1 Hz, 1H), 2.95 (d, J = 15.2 Hz, 1H), 2.73 (d, J = 15.2 Hz, 1H), 2.46–2.38 (m, 2H), 2.13–2.09 (m, 1H), 1.86–1.80 (m, 1H), 1.73 (s, 3H), 1.63 (s, 3H), 1.61 (s, 3H), 1.54 (s, 3H), 1.46 (dt, J = 13.4, 4.9 Hz, 1H), 1.20 (dt, J = 12.9, 4.1 Hz), 0.88 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 199.8, 193.0, 166.6, 165.8, 150.3, 132.4, 131.6, 124.3, 123.0, 109.6, 103.3, 101.8, 91.0, 61.3, 58.1, 45.1, 39.4, 31.7, 29.0, 25.8, 22.5, 21.3, 21.0, 17.9, 16.5 ppm; HRMS (ESI) m/z [M − H]− calcd for C25H29ClO4 427.1682, found 427.1679.
:1) to give 18 (78 mg, 78%) as a yellow solid. Rf = 0.2 (silica gel, PE/EtOAc = 2/1); IR (film): 2954, 2926, 2849, 1612, 1213, 1170, 1066, 981, 823, 730; 1H NMR (400 MHz, CDCl3) δ 5.41 (s, 1H), 5.05 (t, J = 6.9 Hz, 1H), 3.80 (s, 3H), 3.58 (d, J = 4.1 Hz, 2.89 (d, J = 17.3 Hz, 1H), 2.69 (t, J = 3.8, 3.9 Hz, 1H), 2.62–2.48 (m, 3H), 2.46–2.41 (m, 2H), 2.33–2.26 (m, 1H), 2.04–1.99 (m, 2H), 1.72–1.67 (m, 2H), 1.66 (s, 3H), 1.64 (s, 3H), 1.61 (s, 3H), 1.58 (s, 3H), 1.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.2, 184.9, 176.1, 174.6, 132.8, 130.3, 126.0, 122.6, 103.3, 102.8, 96.8, 59.3, 56.5, 49.9, 48.8, 44.2, 40.7, 40.5, 36.1, 31.4, 25.6, 22.0, 21.5, 21.3, 17.7 ppm; HRMS (ESI) m/z [M + H]+ calcd for C26H33ClO4 445.2140, found 445.1841.
:1) to give 19 (19.4 mg, 50%) as a white solid, together with recovered 18 (15.6 mg, 40%). The recovered 18 could be recycled for the second time, thus the overall yield could be improved to 70% after two runs. 19: Rf = 0.0.3 (PE/EtOAc = 1:2); IR (film): 2957, 2926, 2855, 1600, 1575, 1453, 1320, 1267, 1069, 730; 1H NMR (400 MHz, CDCl3) δ 9.36 (s, 1H), 6.59 (d, J = 1.4 Hz, 1H), 6.44 (d, J = 1.4 Hz, 1H), 5.04 (t, J = 6.9 Hz, 1H), 3.72 (s, 3H), 2.96–2.90 (m, 2H), 2.82 (d, J = 16.6 Hz, 1H), 2.64–2.60 (m, 1H), 2.52 (d, J = 16.6 Hz, 1H), 2.04–1.99 (m, 2H), 1.73–1.69 (m, 2H), 1.62 (s, 3H), 1.59 (s, 3H), 1.54 (s, 3H), 1.45 (s, 3H), 1.42 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 180.7, 173.0, 162.5, 162.3, 149.5, 132.5, 130.6, 126.1, 122.9, 111.1, 104.3, 103.1, 98.7, 96.5, 60.0, 55.7, 53.4, 49.2, 44.4, 34.9, 25.6, 22.4, 22.3, 21.4, 21.3, 17.6 ppm; HRMS (ESI) m/z [M + H]+ calcd for C26H31ClO4 443.1984, found 443.1981.
:1) to give merochlorin B (2) (23.2 mg, 60%) as a white solid. Rf = 0.17 (PE/EtOAc = 1:1); IR (film): 2960, 2926, 2855, 1600, 1450, 1261, 1089, 1058, 1021, 798; 1H NMR (400 MHz, d6-DMSO) δ 12.91 (s, 1H), 10.55 (s, 1H), 6.18 (d, J = 2.0 Hz, 1H), 6.16 (d, J = 2.0 Hz, 1H), 5.05 (t, J = 6.6 Hz, 1H), 2.99 (d, J = 7.6 Hz, 1H), 2.85 (d, J = 18.6 Hz, 1H), 2.81 (d, J = 18.4 Hz, 1H), 2.72 (d, J = 17.9 Hz, 1H), 2.47 (d, J = 18.3 Hz, 1H), 2.04–1.93 (m, 2H), 1.76–1.71 (m, 2H), 1.70 (s, 3H), 1.56 (s, 3H), 1.48 (s, 6H), 1.41 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 184.1, 176.0, 163.4, 163.3, 147.7, 131.3, 130.6, 125.5, 123.3, 105.9, 103.6, 101.0, 99.7, 97.9, 60.1, 52.0, 49.1, 43.2, 34.4, 25.4, 22.1, 21.9, 21.3, 21.2, 17.4 ppm; HRMS (ESI) m/z [M − H]− calcd for C25H29ClO4 427.1682, found 427.1680.
Materials for biological assays
E. faecalis ATCC 29212, S. aureus ATCC 29213, and S. pneumoniae NCTC7466 were included in this study. MHB broth was used for the MIC determination for all the bacteria. Dimethyl sulfoxide (DMSO) was used as the solvent for all the compounds, and the final concentration in the test solution is less than 1%. What's more, DMSO at the test concentration was used as vehicle control in the determination of MIC, and vancomycin and ampicillin were also used as quality control.Methods for biological assay
Minimum inhibitory concentration (MIC) represents the lowest concentration of an antimicrobial that inhibits the visible growth of a microorganism under a preset condition. In this study, MIC of each compound was determined using broth microdilution methods according to the guidelines described by the Clinical and Laboratory Standards Institute (CLSI). Bacteria were incubated to the mid-log phase and diluted to a final concentration 106 CFU per mL according to a previously determined conversion factor between optical density (OD) and colony forming unit (CFU) of the cultures for each bacterium. A suitable range of antimicrobial concentrations were chosen for this study (1 to 128 μg mL−1), and the antimicrobial stock solutions were diluted to 2 × concentrates relative to the final concentration in MHB (with 5% horse sheep for S. pneumoniae). The bacterial culture and antimicrobial solutions were mixed in a 1:1 volume ratio, and thus the final bacterial concentration was 5 × 105 CFU per mL, and then check. The growth of the bacterial cultures was checked by detecting change in culture turbidity or OD at the end of incubation, and MIC is between the lowest concentration without bacterial growth and the highest concentration with bacterial growth.
Acknowledgements
We acknowledge the financial support from the National Science Foundation of China (21272133) and the Beijing Natural Science Foundation (2132037).Notes and references
- (a) K. C. Nicolaou, J. C. Chen, D. J. Edmonds and A. A. Estrada, Angew. Chem., Int. Ed., 2009, 48, 660 CrossRef CAS PubMed; (b) E. D. Brown and G. D. Wright, Chem. Rev., 2005, 105, 759–774 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. F. Schäberle, F. Lohr, A. Schmitz and G. M. König, Nat. Prod. Rep., 2014, 31, 953 RSC; (b) B. Li, W. J. Wever, C. T. Walsh and A. A. Bowers, Nat. Prod. Rep., 2014, 31, 905 RSC; (c) M. Saleem, H. Hussain, I. Ahmed, T. Ree and K. Krohn, Nat. Prod. Rep., 2011, 28, 1534 RSC; (d) M. Saleem, M. Nazir, M. S. Ali, H. Hussain, Y. S. Lee, N. Riaz and A. Jabbar, Nat. Prod. Rep., 2010, 27, 238 RSC.
- L. Kaysser, P. Bernhardt, S.-J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988 CrossRef CAS PubMed.
- G. Sakoulas, S.-J. Nam, S. Loesgen, W. Fenical, P. R. Jensen, V. Nizet and M. Hensler, PLoS One, 2012, 7, e29439 CAS.
- H. P. Pepper and J. H. George, Angew. Chem., Int. Ed., 2013, 52, 12170 CrossRef CAS PubMed.
- R. Meier, S. Strych and D. Trauner, Org. Lett., 2014, 16, 2634 CrossRef CAS PubMed.
- (a) R. Teufel, L. Kaysser, M. T. Villaume, S. Diethelm, M. K. Carbullido, P. S. Baran and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11019 CrossRef CAS PubMed; (b) S. Diethelm, R. Teufel, L. Kaysser and B. S. Moore, Angew. Chem., Int. Ed., 2014, 53, 11023 CrossRef CAS PubMed.
- L. B. Yan, L. Wang, Y. J. Liang and S. Cheng, CN103172500A, 2013 Search PubMed-06-26.
- N. Biber, K. Mows and B. Plietker, Nat. Chem., 2011, 3, 938 CrossRef CAS PubMed.
- T. J. J. opczewski, M. P. Callahan, J. D. Neighbors and D. F. Wiemer, J. Am. Chem. Soc., 2009, 131, 14630 CrossRef PubMed.
- For relevant reviews on this topic, see: (a) S. Yamamura and S. Nishiyama, Synlett, 2002, 533 CrossRef CAS; (b) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed. For some inspiring examples, see: (c) Y. Shizuri, M. Ohkubo and S. Yamamura, Chem. Lett., 1989, 1113 Search PubMed; (d) S. Yamamura, Y. Shizuri, H. Shigenori, Y. Okuno and M. Ohkubo, Tetrahedron, 1991, 47, 635 CrossRef CAS; (e) H. Takakura, K. Toyoda and S. Yamamura, Tetrahedron Lett., 1996, 37, 4043 CrossRef CAS; (f) H. Takakura and S. Yamamura, Tetrahedron Lett., 1999, 40, 299 CrossRef CAS; (g) J. C. Green and T. R. Pettus, J. Am. Chem. Soc., 2011, 133, 1603 CrossRef CAS PubMed.
- For leading reviews on this topic, see: (a) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef; (b) L. F. Silva and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722 RSC.
- For selected examples of sequential Diels–Alder reaction–aromatization, see: (a) K. C. Nicolaou, C. R. H. Hale, C. Nilewski, H. A. Ioannidou, A. ElMarrouni, L. G. Nilewski, K. Beabout, T. T. Wang and Y. Shamoo, J. Am. Chem. Soc., 2014, 136, 12137 CrossRef CAS PubMed; (b) Z. T. Li, Y. X. Gao, Y. F. Tang, M. J. Dai, G. X. Wang, Z. G. Wang and Z. Yang, Org. Lett., 2008, 10, 3017 CrossRef CAS PubMed; (c) B. M. Trost, O. R. Thiel and H. C. Tsui, J. Am. Chem. Soc., 2003, 125, 13155 CrossRef CAS PubMed; (d) R. A. Tapia, C. Salas, A. Morello, J. D. Maya and A. Toro-Labbé, Bioorg. Med. Chem., 2004, 12, 2451 CrossRef CAS PubMed; (e) S. Jacques and B. Paul, Tetrahedron, 1984, 40, 3455 CrossRef.
- (a) M. Iqbal, N. Mistry and P. A. Clarke, Tetrahedron, 2011, 67, 4960 CrossRef CAS; (b) K. Takai, Y. Nawate, T. Okabayashi, H. Nakatsuji, A. Iida and Y. Tanabe, Tetrahedron, 2009, 65, 5596 CrossRef CAS; (c) A. Gerard, W. C. Donald, I. F. Geoffrey and W. R. Roger, Tetrahedron Lett., 1981, 22, 4347 CrossRef.
- (a) D. Barker, M. A. Brimble, P. Do and P. Turner, Tetrahedron, 2003, 59, 2441 CrossRef CAS; (b) D. Q. Chen, J. Q. Wang and N. I. Totah, J. Org. Chem., 1999, 64, 1776 CrossRef CAS PubMed.
- The stereochemistry of the newly generated chiral centers in the Diels–Alder adduct was not determined, since they transformed into achiral centers in the next step.
- M. D. ipakranjan, P. Pallab and R. D. Saroj, Tetrahedron, 2007, 63, 11781 CrossRef.
- X-ray CCDC 1412934 (8) and 1412881 (18) contain the supplementary crystallographic data for this paper.
- D. L. J. Clive and R. Sunasee, Org. Lett., 2007, 9, 2677 CrossRef CAS PubMed.
- D. D. Peerrin, W. L. Armarego and D. R. Perrins, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 1980 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The NMR spectra of compounds 1, 2, 7–9, 15, 16, 18 and 19, and the CIF file of compounds 8 and 18. CCDC 1412934 and 1412881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01946j |
| This journal is © The Royal Society of Chemistry 2016 |