
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concise synthesis of rare pyrido[1,2-a]pyrimidin-2-ones and related nitrogen-rich bicyclic scaffolds with a ring-junction nitrogen†
T. A.
Alanine
ab,
W. R. J. D.
Galloway
a,
S.
Bartlett
a,
J. J.
Ciardiello
a,
T. M.
McGuire
b and
D. R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk; Fax: +44 (0)1223-336362
bAstraZeneca UK Ltd., Alderly Park, Macclesfield, Cheshire SK10, UK
First published on 24th November 2015
Abstract
Pyrido[1,2-a]pyrimidin-2-ones represent a pharmaceutically interesting class of heterocycles. The structurally related pyrido[1,2-a]pyrimidin-4-ones are associated with a broad range of useful biological properties. Furthermore, quinolizinone-type scaffolds of these sorts with a bridgehead nitrogen are expected to display interesting physico–chemical properties. However, pyrido[1,2-a]pyrimidin-2-ones are largely under-represented in current small molecule screening libraries and the physical and biological properties of the pyrido[1,2-a]pyrimidin-2-one scaffold have been poorly explored (indeed, the same can be said for unsaturated bicyclic compounds with a bridgehead nitrogen in general). Herein, we report the development of a new strategy for the concise synthesis of substituted pyrido[1,2-a]pyrimidin-2-ones from readily available starting materials. The synthetic route involved the acylation of the lithium amide bases of 2-aminopyridines with alkynoate esters to form alkynamides, which were then cyclised under thermal conditions. The use of lithium amide anions ensured excellent regioselectivity for the 2-oxo-isomer over the undesired 4-oxo-isomer, which offers a distinct advantage over some existing methods for the synthesis of pyrido[1,2-a]pyrimidin-2-ones. Notably, different aminoazines could also be employed in this approach, which enabled access to several very unusual bicyclic systems with higher nitrogen contents. This methodology thus represents an important contribution towards the synthesis of pyrido[1,2-a]pyrimidin-2-ones and other rare azabicycles with a ring-junction nitrogen. These heterocycles represent attractive structural templates for drug discovery.
Introduction
Heterocyclic ring systems are the core scaffolds of a large proportion of all pharmaceuticals and agrochemicals.1–3 However, heterocyclic chemical space has, to date, been explored in an uneven and unsystematic fashion.2,4 Chemists have largely focused upon only a small subset of heterocyclic scaffolds, generally those that are both synthetically facile and have proven biological relevance.4 Unsurprisingly, there remains intense interest in the synthesis of both novel heterocyclic scaffolds and also unusual ring systems that have been largely underexploited in drug and agrochemical discovery.2,5–8 One class of uncommon heterocycles are the pyrido[1,2-a]pyrimidin-2-ones (compounds based around scaffold 1, Fig. 1). Quinolizinone-type scaffolds of these sorts have elicited interest within the pharmaceutical industry due to reports of encouraging biological activities and attractive predicted physico–chemical properties (such as high polarity and aqueous solubility compared to their non-nitrogenated analogues) as a result of their polar zwitterionic character (Fig. 1).1,9–12 The regioisomeric pyrido[1,2-a]pyrimidin-4-ones are associated with a broad range of useful biological properties and numerous synthetic routes towards substituted derivatives of this scaffold are available.9,12,13 In contrast, pyrido[1,2-a]pyrimidin-2-ones have been considerably less well studied.9,12,14 The relative scarcity of molecules based around the pyrido[1,2-a]pyrimidin-2-one scaffold can be attributed to a comparative lack of general and flexible methods for their construction. Existing synthetic strategies suffer from various drawbacks (vide infra). Thus, there is a need for new strategies to generate different pyrido[1,2-a]pyrimidin-2-ones so that the biological and physico–chemical properties of this heterocyclic system can be further investigated.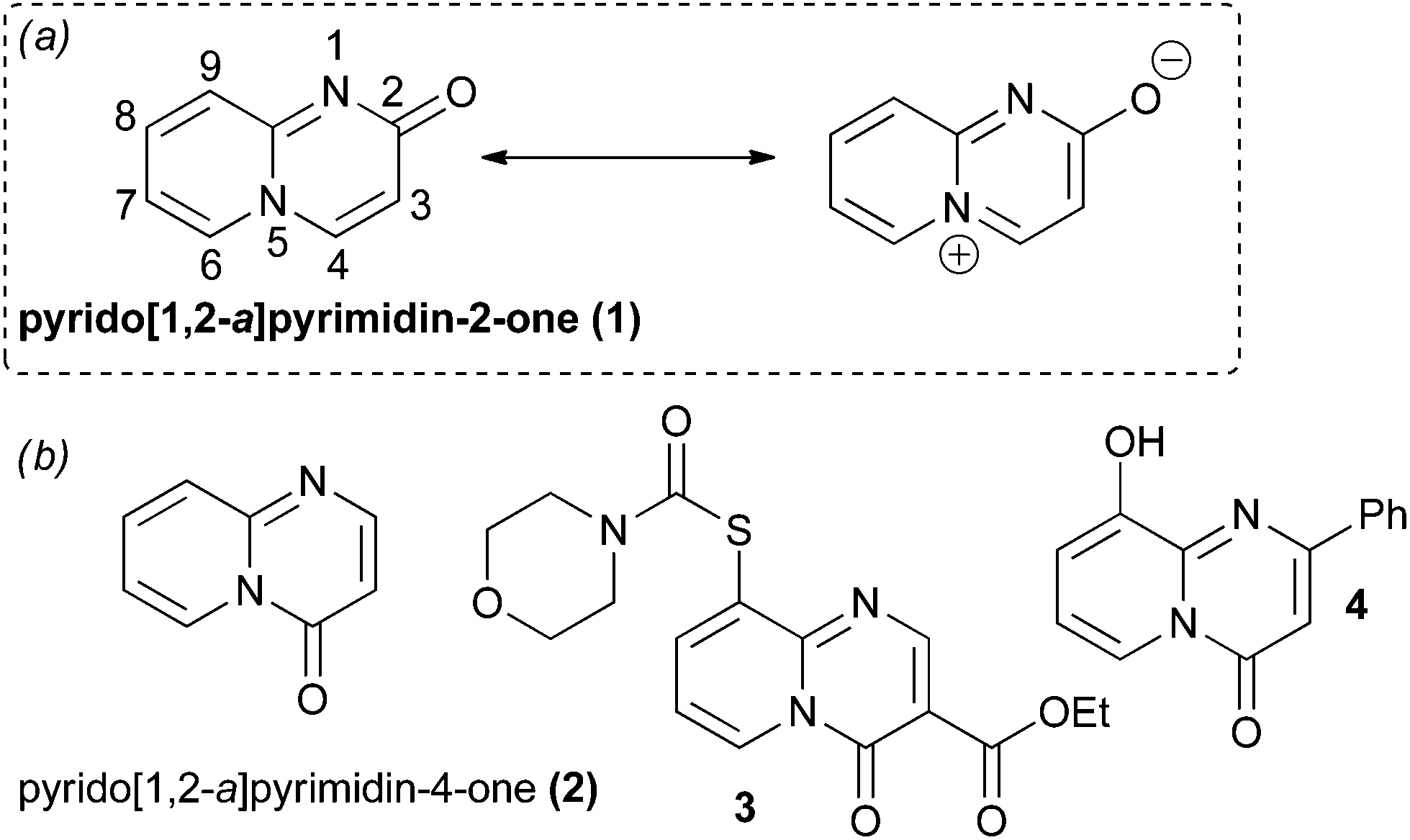 | ||
| Fig. 1 (a) The pyrido[1,2-a]pyrimidin-2-one core (1) and its dipolar canonical form. (b) The pyrido[1,2-a]pyrimidin-4-one core (2) and some examples of biologically active compounds that are based around this ring system. Compound 3 is an antiulcerative agent15 and compound 4 is an inhibitor of bacterial quorum sensing.16 | ||
We targeted the development of a more robust and general method for the synthesis of pyrido[1,2-a]pyrimidin-2-one derivatives. It was also hoped that the new methodology would enable access to other related quinolizinone-type systems with higher nitrogen contents; such very rare derivatives would be expected to be even more polar than pyrido[1,2-a]pyrimidin-2-ones and thus may have valuable properties such as enhanced aqueous solubility.
Reported methods for the synthesis of pyrido[1,2-a]pyrimidin-2-ones include:9,12 (i) the cyclisation of 2-aminopyridine with ethyl cyanoacetate at high temperature and pressure;17 (ii) the cyclisation of 2-aminopyridine with the Vilsmeier-Haack reagent;18 (iii) the reaction of 2-aminopyridines with hex-2-en-4-yne-1,6-dioate or allene-1,3-dicarboxylic esters;19 (iv) the acid-catalysed cyclisation of N-acetoacetylated 2-amino pyridines/picolines/quinolones19 and; (v) the addition of 2-aminopyridines to Baylis-Hillman acetates.20 These strategies all suffer from various drawbacks, including a restricted substrate scope, the need for harsh reaction conditions and poor regiocontrol (generation of mixtures of the isomeric 2-oxo and 4-oxo-derivatives 1 and 2).9,12 The most commonly employed method for the synthesis of pyrido[1,2-a]pyrimidin-2-one derivatives (of the general form 5) is the one-pot thermal cyclisative condensation of 2-aminopyridine derivatives 6 with activated alkynoates 7, typically carried out under neutral conditions (Scheme 1).9,12,21–24 In principle, this approach has several attractive features. It is conceptually straightforward, step-efficient, and proceeds from readily-available starting materials. Furthermore, since the approach is inherently modular in nature it would be expected that a range of novel analogues could be accessed through variation in the building blocks used. Unfortunately, the current substrate scope is limited to 2-aminopyridine derivatives and aminoquinoline and undesired side product formation has been reported.9,12,25 In addition, there are potential regioselectivity issues that could arise as a consequence of the fact that the 2-aminopyridine component has two different nucleophilic nitrogen centres and there are two possible modes of addition to the alkynoate component (direct or conjugate); in principle, both of the isomeric 2-oxo and 4-oxo-derivatives can be generated (vide infra).25 Herein, we describe the development of a new strategy for the synthesis of pyrido[1,2-a]pyrimidin-2-ones and related structures of the general form 8 from aminoazines 9 and alkynoate esters 7 which is based around this general approach (Scheme 1). The novelty of the new strategy centres on the use of a “defined” two-step procedure that involves acylation of the lithium amide bases of aminoazines with alkynoate esters to form alkynamides 10, which can then be cyclised under thermal conditions. Compared to one-pot thermal cyclisative condensation methods involving the combination of aminoazines and alkynoate esters, this new procedure offers a broader substrate scope; several previously unreported substituted pyrido[1,2-a]pyrimidin-2-one derivatives could be accessed using this new methodology, together with several extremely rare scaffolds with higher nitrogen contents (of the general forms 11–15) through the use of the corresponding aminoazine substrates with higher nitrogen contents (see Scheme 1 for data). Excellent regioselectivity for the desired 2-oxo-isomers of the bicyclic scaffolds over the undesired 4-oxo-isomers was observed in all cases. This new methodology thus represents an important contribution towards the synthesis of pyrido[1,2-a]pyrimidin-2-ones and other rare azabicycles with a ring-junction nitrogen. These heterocycles represent attractive structural templates for drug discovery, offering access to underexplored, yet biologically interesting chemical space and thus the potential to secure novel intellectual property.
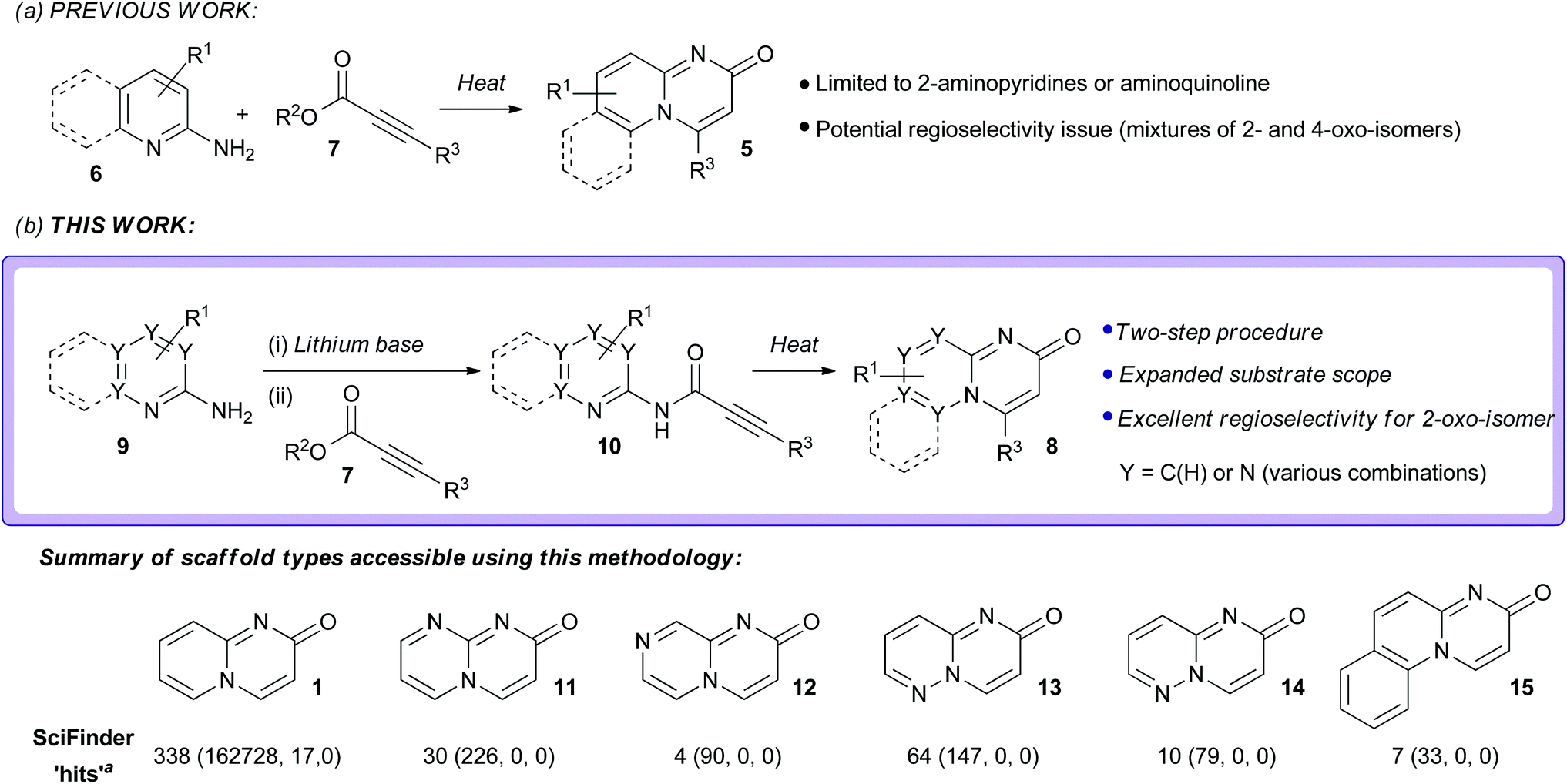 | ||
| Scheme 1 Synthesis of pyrido[1,2-a]pyrimidin-2-ones and related derivatives from aminoazines and alkynoate esters. aNumber ‘substances’ retrieved from a ‘Chemical Structure Substructure’ search with ‘Lock ring fusion or formation’ selected (August 2015). Data for the number of substances found are reported in the form: conventional substructure (closely associated tautomers and zwitterions, loosely associated tautomers and zwitterions, other). | ||
Results and discussion
Outline of the synthetic strategy
The exact mechanistic pathway of the cyclisative condensation of 2-aminopyridines 16 with activated alkynoates 7 to form pyrido[1,2-a]pyrimidin-2-ones 17 under typically employed neutral reaction conditions is not known. 2-Aminopyridines 16 are potentially nucleophilic at both the endocyclic pyridine nitrogen and the exocyclic amine nitrogen. In principle, either nitrogen centre could react first with the alkynoate, with direct and conjugate modes of addition possible in each case (Scheme 2, illustrated for 2-aminopyridines 16).26 In the case of initial reaction at the endocyclic pyridine nitrogen, conjugate addition would lead to intermediate 18 (Scheme 2, path A). Subsequent intramolecular lactam formation involving the 2-amino group would then furnish the pyrido[1,2-a]pyrimidin-2-one system 17. In the case of initial reaction at the exocyclic amine nitrogen, the pyrido[1,2-a]pyrimidin-2-one scaffold 17 would result from direct addition to the alkynoate to form 19 (Scheme 2, path B) followed by intramolecular conjugate addition of the endocyclic pyridine nitrogen. Under typically employed neutral/mildly basic conditions, it might be anticipated that reaction at the pyridine nitrogen would occur first since 2-aminopyridines 16 are known to typically react with electrophiles preferentially at the endocyclic pyridine nitrogen centre27–30 (Scheme 2, paths A and C). Indeed, there is some evidence that the first step of the reaction sequence is the conjugate addition of the pyridine nitrogen of 16 to alkynoate 7 (Scheme 2, path A).9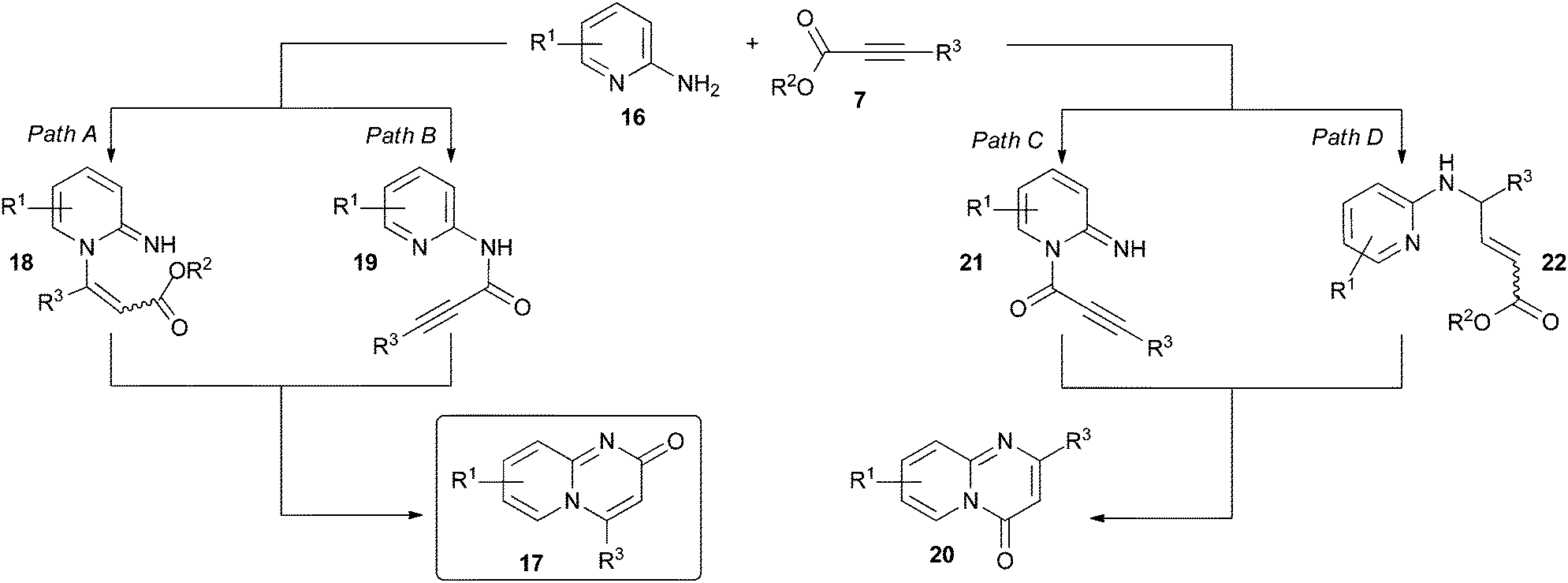 | ||
| Scheme 2 Potential mechanistic pathways leading to pyrido[1,2-a]pyrimidin-2-ones 17 and pyrido[1,2-a]pyrimidin-4-ones 20 from 2-aminopyridines 16 and alkynoates 7. An analogous set of pathways can be envisaged for the reaction of other aminoazine derivatives with 7 (not shown). | ||
However, initial reaction at either amine centre could also lead to the formation of the isomeric pyrido[1,2-a]pyrimidin-4-one scaffold 20. Direct addition of the pyridine nitrogen of 16 to the alkynoate 7 would furnish intermediate 21 (Scheme 2, path C); subsequent intramolecular conjugate addition of the exocyclic amine would then yield the 4-oxo scaffold 20. The 4-oxo scaffold 20 would also be the predicted product if the exocyclic amine adds initially to the alkynoate in a conjugate fashion to form 22 (Scheme 2, path D). Thus, in principle, both of the isomeric 2-oxo and 4-oxo-derivatives can be accessed by combination of aminopyridine derivatives and alkynoates 7.
Aminoazine substrates with higher nitrogen contents (such as diazines and triazines) would be expected to be inherently less nucleophilic than 2-aminopyridines 16. Thus, it would be anticipated that they would be expected to be considerably less reactive with alkynoates 7 under the neutral conditions typically employed in reactions of this sort, which may explain why they have been largely unexplored in this context. We hypothesised that this issue could be addressed through the use of basic reaction conditions, specifically the addition of alkynoate 7 to a pre-formed solution of the aminoazine substrate 9. The use of basic conditions would provide access to the corresponding amide anion, which would be expected to be substantially more reactive than the parent amine. Subsequent electrophilic attack would be expected to occur preferentially at the exocyclic amino group nitrogen over the endocyclic pyridine nitrogen (deprotonation of the exocyclic amino group is one method for preferentially directing electrophilic attack towards the exocyclic nitrogen in heterocyclic amidines such as 2-aminopyridine27,30). Furthermore, the deprotonated amide anion would be expected to be a harder nucleophile than the amino group (present under neutral or mildly basic conditions) and thus more regioselective for the acyl position of the alkynoate; that is, analogous to preferential reaction via path B in Scheme 2 rather than other competing modes of addition with 7. Isolation of the resulting amide intermediate 10 and subsequent thermal-induced cyclization should then furnish the azabicyclic framework 8. Thus, we anticipated that the adoption of this “defined” two-step protocol involving basic reaction conditions would effectively “bias” reaction progress towards the 2-oxo products, thereby increasing the regioselectivity for the desired 2-oxo isomers, in addition to the aforementioned expansion in the substrate scope to other aminoazine substrates.
Synthesis of pyrido[1,2-a]pyrimidin-2-ones and related nitrogen-rich bicyclic heterocycles
Using 2-aminopyridine 23 as a model substrate, it was found that deprotonation of the amine group with an excess of n-butyllithium (nBuLi), lithium diisopropylamide (LDA), or lithium bis(trimethylsilyl)amide (LiHMDS) at low temperature followed by addition of alkynoate 24 delivered the desired amide intermediate 25 (Table 1). In all cases, there was no evidence for conjugate addition to the alkynoate or bis-acylation of the amino group (as determined by LC-MS analysis of the of the crude reaction mixtures). Compound 26 was detected as a by-product in all cases, which presumably resulted from the conjugate addition of ethoxide to 24.31 The highest level of conversion was observed with 1.2 equivalents of 24 (Table 1, entry 3) and 2.1 equivalents of nBuLi; further increases in the equivalents of 24 (Table 1, entries 4–6) used had little impact upon reaction conversion, but led to larger amounts of the by-product 26. Thus, the conditions described in Table 1 entry 3 were chosen for further application.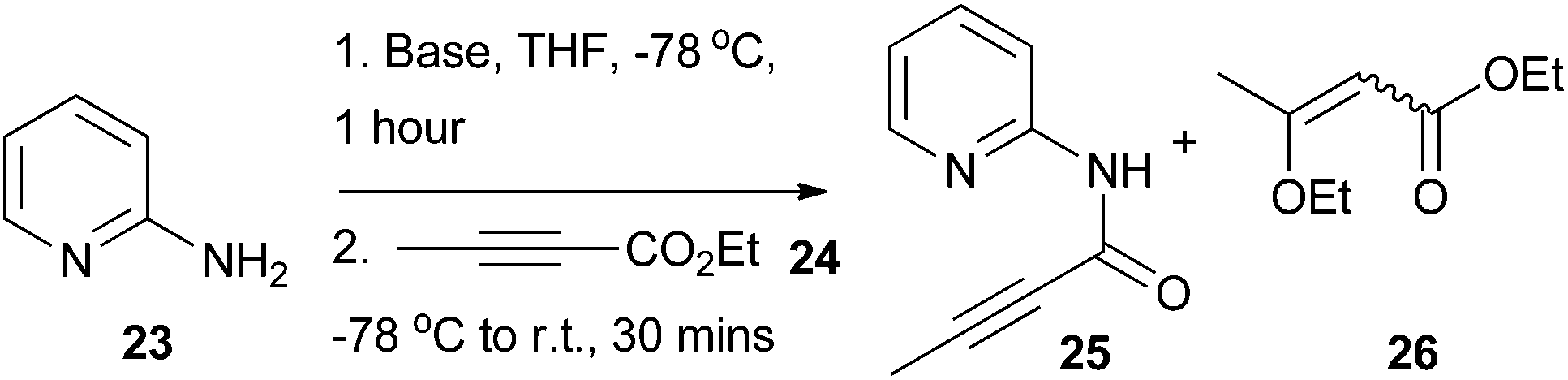
A selection of readily-available 2-aminopyridines and aminoazine substrates was then subjected to the optimised conditions for acylation with ethyl 2-butynoate 24 (Scheme 3). Pleasingly, amidation was successful with a range of heteroaromatic systems 23, 27–42, leading to products 25, 43–58. For some substrates, treatment with nBuLi led to side reactions (which appeared to involve displacement or reduction of the halide functionality), which impacted upon reaction yields; in these cases, the use of LDA as the base led to cleaner reaction profiles. For compound 23 and cases where the heteroaromatic unit bore an electron-releasing substituent (aminoazines 28 and 32), some apparent spontaneous cyclisation of the presumed amide intermediates was observed under the reaction conditions (which could be a consequence of increased relative nucleophilciity of each of the corresponding ring nitrogen atoms). It was found that the yields for the acylation step were highest and most reproducible when reaction and purification were carried out within a six hour time period and solvents were removed under reduced pressure without heating. For example, under these circumstances the average yield for the synthesis of compound 48 over three repeats was 35 ± 4%. A lower yield (25%) for compound 48 was obtained when the acylation reaction and purification was done over a longer time period. It was also found that trituration with heptane was a useful way of obtaining analytically pure material if column chromatography proved insufficient.
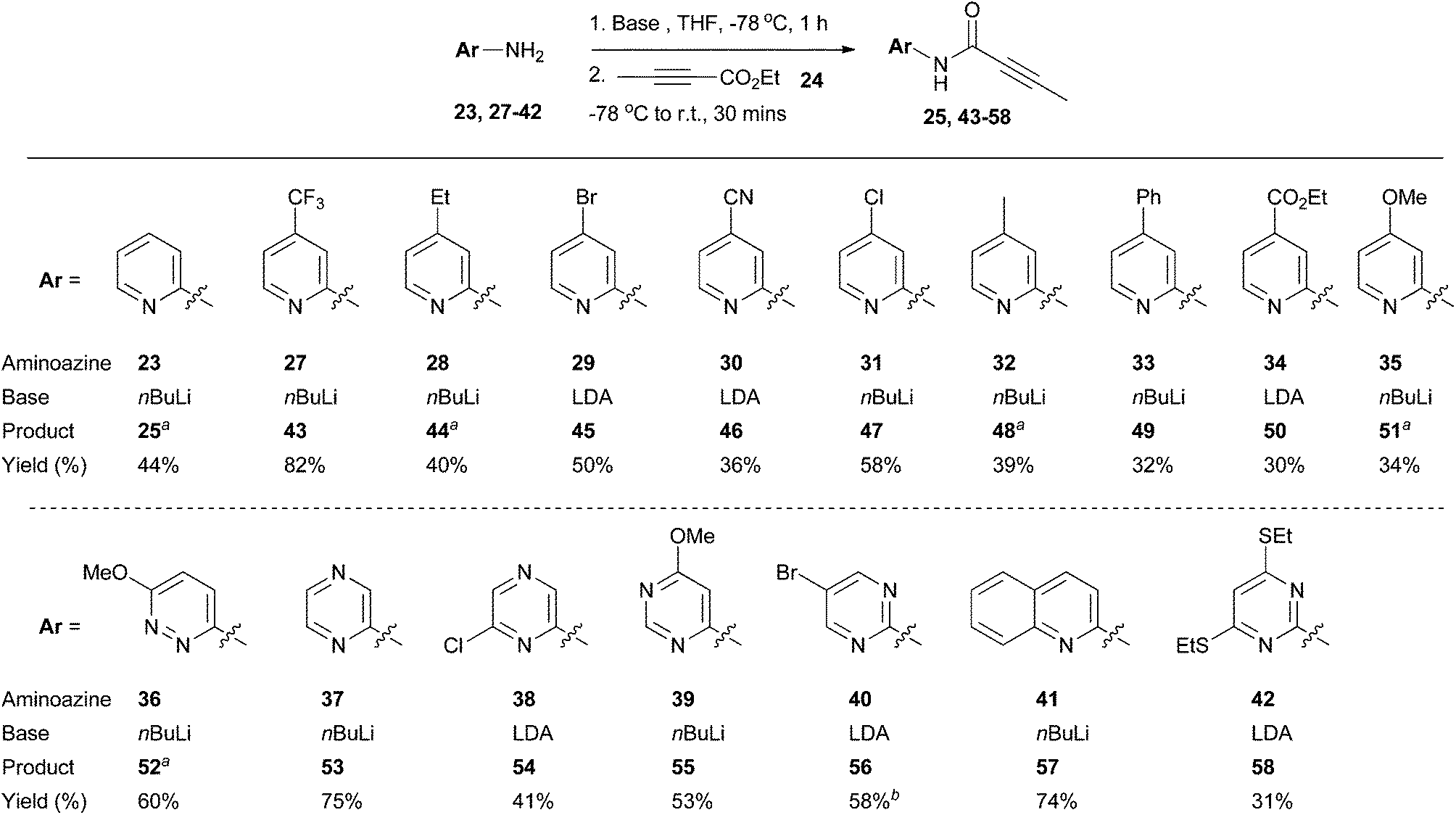 | ||
| Scheme 3 Synthesis of amides 25 and 43–58. aSome cyclised material was observed in the crude material obtained after reaction work-up but this could not be isolated. bImpure product mixture isolated. Further purification by HPLC possible. See ESI† for more details. | ||
For aminoazine substrates 59–61 the corresponding final target azabicyclic derivatives 62–64 were isolated exclusively under the amidation reaction conditions (that is, the presumed amide intermediates were not detected, Scheme 4). The yields of these derivatives were low, which could be mainly attributed to their high aqueous solubilities (likely a result of a degree of zwitterionic character by analogy with the quinolizin-2-one scaffold, see Fig. 1) and thus their poor recoveries after aqueous work-ups. The facile in situ cyclisation of the presumed amide intermediate generated from substrate 59 (to form 62) might be due to the steric influence of the benzyloxy group, which could potentially force the acetylenic moiety into close proximity to the pyridine nitrogen and thus encourage cyclisation. In the case of the amide intermediate generated from 60 the high propensity to cyclise (to form 63) might be a kinetic effect, as both nitrogen atoms in the ring could presumably induce cyclisation (and thus increase the likelihood of cyclisation). The reaction of 2-aminopyridine 23 with the other readily available alkynoates ethyl 3-phenylbutynoate, diethyl acetylenedicarboxylate and ethyl 4,4,4-trifluorobut-2-ynoate (65) was briefly explored. Disappointingly, the acylation reaction failed with ethyl 3-phenylbutynoate and diethylacetylenedicarboxylate, leading to complex intractable mixtures of unidentifiable products.32 In contrast, the reaction of 23 with 65 led to the formation of the final target pyrido[1,2-a]pyrimidin-2-one 66 (Scheme 4). This was not altogether unexpected, as Harriman et al.9 have previously reported that the thermal cyclisative condensation of 23 and 65 occurs readily at room temperature, presumably as a consequence of the electrophilicity of the CF3-bearing carbon.
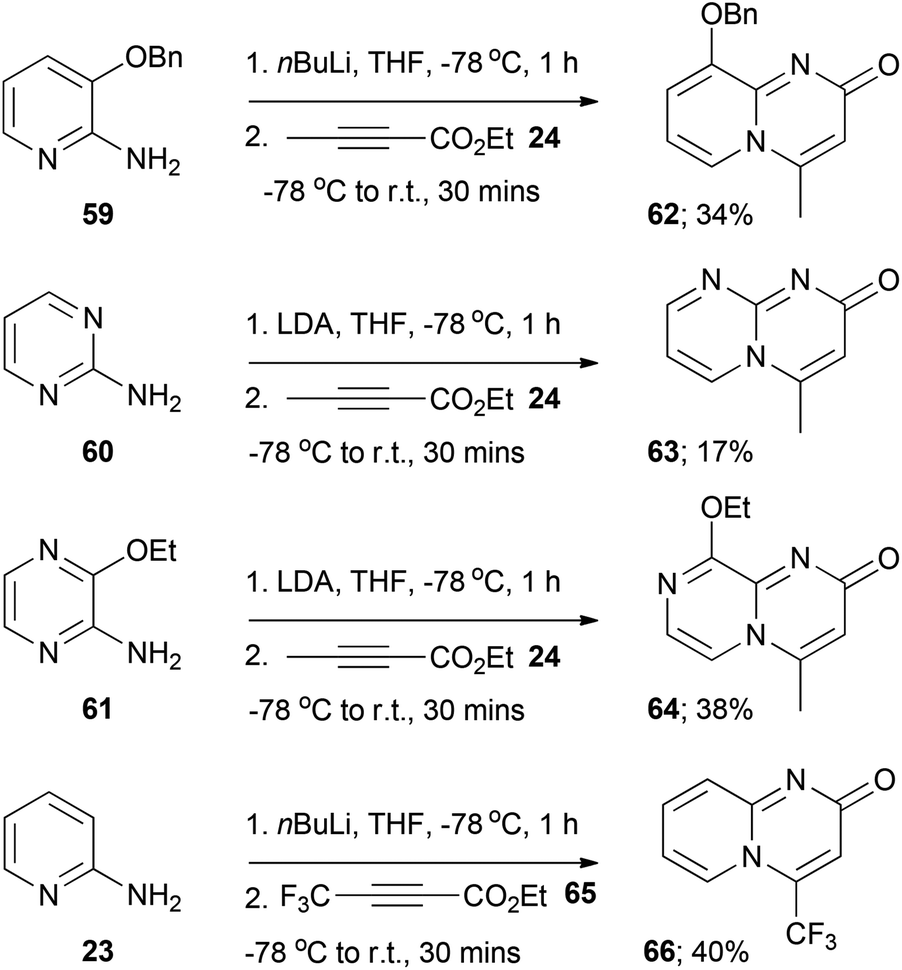 | ||
| Scheme 4 Formation of azabicyclic derivatives 62–64 and 66 under amidation reaction conditions. | ||
With amide intermediates 25 and 43–58 in hand, we were ready to examine the 6-endo-dig cyclisation step in an attempt to access the corresponding target azabicyclic compounds (Scheme 5). It was anticipated that cyclisation could be triggered by thermal heating. This was indeed found to be the case for the majority of the amide substrates, with the rate of reaction dependant upon both the solvent and temperature used (reactions were generally found to proceed more quickly as both the temperature and the dielectric constant of the solvent was increased). Thermal heating in DMSO at 85 °C was found to be optimal for the 2-aminopyridine derivatives 25 and 43–51. In the majority of cases reaction times were under five hours and the yields of the target pyrido[1,2-a]pyrimidin-2-ones 67–76 were typically good-to-excellent. In all cases there was no evidence for the formation of the undesired 4-oxo-isomeric derivatives. The cyclisation of amide intermediates with higher nitrogen contents was comparatively sluggish, which was taken to be indicative of higher activation energy barriers for these reactions. Pleasingly however, the use of elevated reaction temperatures and times did enable access to quinoline derivative 80 and compounds 77–79 which are based around extremely rare nitrogen-rich bicyclic scaffolds, again typically in good-to-excellent yields. In all these reactions there was no evidence for the formation of the undesired 4-oxo-isomeric derivatives. Unfortunately, the cyclisation of aminoazine amides 54 and 56, which each contain an electron withdrawing substituent, could not be achieved (which presumably can be explained by the decreased nucleophilicty of the respective endocyclic nitrogen atoms). Amide 58 also failed to cyclise.33
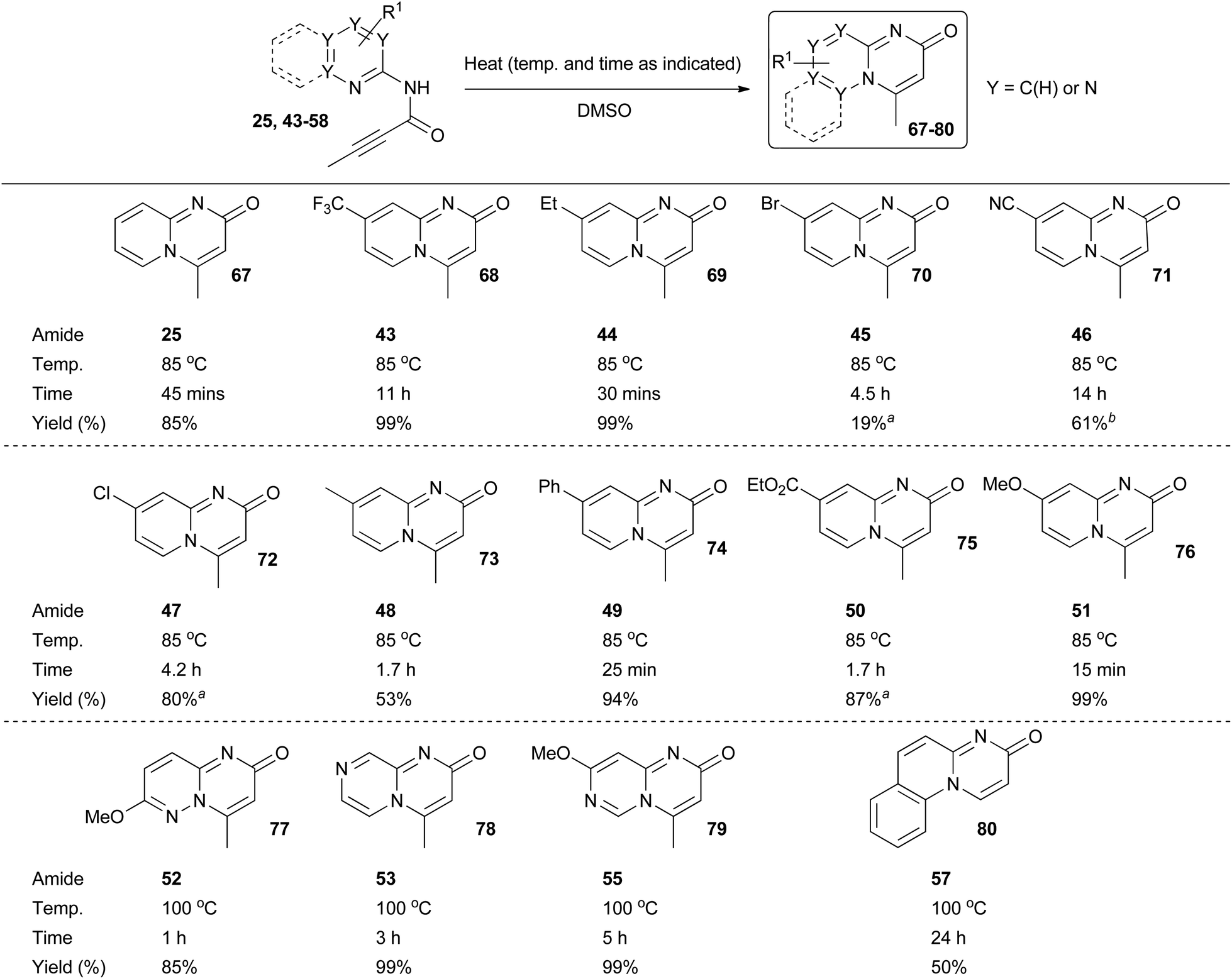 | ||
| Scheme 5 Formation of azabicyclic derivatives 67–80. aImpure product mixture isolated. Further purification by HPLC possible. See ESI† for more details. bSpectroscopically pure material isolated but some degradation observed over time. | ||
Conclusions
In conclusion, we have developed a new and modular strategy for the concise synthesis of pyrido[1,2-a]pyrimidin-2-ones and related compounds based around bicyclic heterocyclic scaffolds with a ring-junction nitrogen. These rare heterocylic scaffolds have elicited interest from the pharmaceutical industry owing to reports of biological activities and favourable anticipated physico–chemical properties, yet they remain largely unexplored, primarily due to problems with their synthetic tractability.Our strategy is based around the combination of aminoazines and alkynoate esters, a known and general approach towards compounds of this sort. The novelty of this new strategy centres on the adoption of a two-step procedure that involves the use of the lithium amide anions of the aminoazines. This ensures excellent regioselectivity for the desired 2-oxo-isomers of the bicyclic scaffolds over the undersired 4-oxo-isomers, a notable advantage over some existing methodologies. Furthermore, the new protocol allows for an expansion of the substrate scope of this general synthetic approach; in addition to 2-aminoazines, we have demonstrated, for the first time, the utilisation of a range of aminoazines with higher nitrogen contents (aminopyrimidines, aminopyrazines and aminopyridazines). Consequently, the new protocol allows for access not only to pyrido[1,2-a]pyrimidin-2-ones, but also a number of very unusual aza-bicyclic systems with higher nitrogen contents.
Overall, this new methodology was found to be widely applicable to a range of aminoazines and we believe that it represents an important contribution towards the synthesis of pyrido[1,2-a]pyrimidin-2-ones and other rare azabicycles with a ring-junction nitrogen. It is envisaged that these scaffolds could serve as attractive structural templates in drug discovery endeavours (for example, as starting fragments in fragment-based approaches). Crucially in this regard, our methodology was shown to be tolerant of some functionality in the aminoazine component. This allows for the installation of some synthetic handles in the final aza-bicycles for further potential elaboration. Studies examining the biological and physico–chemical properties of the compounds generated in this report are underway and results will be reported in due course.
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement n° [279337/DOS]. The authors also thank AstraZeneca, the European Union (EU), the Engineering and Physical Sciences Research Council (EPSRC), the Biotechnology and Biological Sciences Research Council (BBSRC), the Medical Research Council (MRC), and the Wellcome Trust for funding. Data accessibility: all data supporting this study are provided as ESI† accompanying this paper.Notes and references
- C. W. Muir, A. R. Kennedy, J. M. Redmond and A. J. Watson, Org. Biomol. Chem., 2013, 11, 3337–3340 CAS.
- M. S. Frei, M. K. Bilyard, T. A. Alanine, W. R. J. D. Galloway, J. E. Stokes and D. R. Spring, Bioorg. Med. Chem., 2015, 23, 2666–2679 CrossRef CAS PubMed.
- J. J. Lie, Chapter 1: Introduction, in Heterocyclic Chemistry in Drug Design, ed. J. J. Lie, John Wiley & Sons, Inc, Hoboken, New Jersey, 2013, pp. 1–16 Search PubMed.
- M. Dow, M. Fisher, T. James, F. Marchetti and A. Nelson, Org. Biomol. Chem., 2012, 10, 17–28 CAS.
- S. R. Langdon, N. Brown and J. Blagg, J. Chem. Inf. Model., 2011, 51, 2174–2185 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
- J. J. Lai, D. B. Salunke and C. M. Sun, Org. Lett., 2010, 12, 2174–2177 CrossRef CAS PubMed.
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- G. C. B. Harriman, S. Chi, M. Zhang, A. Crowe, R. A. Bennett and I. Parsons, Tetrahedron Lett., 2003, 44, 3659–3662 CrossRef CAS.
- S. R. Natarajan, M. H. Chen, S. T. Heller, R. M. Tynebor, E. M. Crawford, M. X. Cui, K. Z. Han, J. C. Dong, B. Hu, W. Hao and S. H. Chen, Tetrahedron Lett., 2006, 47, 5063–5067 CrossRef CAS.
- T. A. Alanine, W. R. J. D. Galloway, T. M. McGuire and D. R. Spring, Eur. J. Org. Chem., 2014, 5767–5776 CrossRef CAS.
- A. R. Katritzky, J. W. Rogers, R. M. Witek and S. K. Nair, ARKIVOC, 2004, 52–60 CAS.
- S. K. Guchhait and G. Priyadarshani, J. Org. Chem., 2015, 80, 6342–6349 CrossRef CAS PubMed.
- Number ‘substances’ retrieved from a ‘Chemical Structure substructure’ search with ‘Lock ring fusion or formation’ selected (August 2015): pyrido[1,2-a]pyrimidin-4-one 62032 (143220, 20, 1); pyrido[1,2-a]pyrimidin-2-one 338 (162728, 17, 0). Data reported in the form: conventional substructure (closely associated tautomers and zwitterions, loosely associated tautomers and zwitterions, other).
- S. Matsutani and Y. Mizushima, Eur. Pat. Appl, EP89-10263519890216, 1989 Search PubMed.
- H. Suga and J. Igarashi, PCT Pat, WO2009063901A1, 2009 Search PubMed.
- V. A. Dorokhov, S. V. Baranin, A. Dib, V. S. Bogdanov, I. P. Yakovlev, G. A. Stashina and V. M. Zhulin, Bull. Acad. Sci. USSR Div. Chem. Sci. (Engl. Transl.), 1990, 39, 1918–1923 CrossRef.
- G. Roma, M. Dibraccio, A. Balbi, M. Mazzei and A. Ermili, J. Heterocycl. Chem., 1987, 24, 329–335 CrossRef CAS.
- O. P. Suri, K. A. Suri, B. D. Gupta and N. K. Satti, Synth. Commun., 2002, 32, 741–746 CrossRef CAS.
- S. Rapolu, M. Alla, R. J. Ganji, V. Saddanapu, C. Kishor, V. R. Bommena and A. Addlagatta, MedChemComm, 2013, 4, 817–821 RSC.
- H. N. Aljallo and I. A. Albiaty, J. Heterocycl. Chem., 1978, 15, 801–805 CrossRef CAS.
- G. R. Lappin, J. Org. Chem., 1961, 26, 2350–2353 CrossRef CAS.
- K. A. Wheeler and B. M. Foxman, Chem. Mater., 1994, 6, 1330–1336 CrossRef CAS.
- J. G. Wilsonn and W. Bottoley, J. Heterocycl. Chem., 1967, 4, 360–364 CrossRef.
- The majority of reports on pyrido[1,2-a]pyrimidin-2-one synthesis by the cyclisative condensation method do not mention the 4-oxo derivative as a possible side-product or comment upon selectivity between the isomeric 2-oxo and 4-oxo derivatives. Thus, it is difficult to ascertain the significance of this issue. Some reports on related systems are worthy of note. Agarwal et al. (see: (a) S. K. Agarwal, A. K. Saxena and P. C. Jain, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1981, 20, 67–68 Search PubMed); have reported that the condensation of aminopyridoindole with alkynoates to form pyrimidopyridoindoles. When ethyl propiolate was used, the 4-oxo-isomer of the product was obtained in 20% yield. Yan et al. (see: (b) H. Yan, Y. Ma, Y. Sun, C. Ma, Y. Wang, X. Ren and G. Huang, Tetrahedron, 2014, 70, 2761–2765 CrossRef CAS ) have reported that the reaction of 2-aminopyridines with symmetrical dialkyl ethlenedicarboxylates leads to the formation of alkyl 4-oxo-4H-pyrido[1,2-a]pyrimidines rather than the 2-oxo isomers.
- Lappin (see ref. 22) studied the addition of 2-aminopyridines to methyl propiolate under neutral conditions and reported that both nitrogen atoms can participate in conjugate addition.
- R. de la Rosa, V. Martinez-Barrasa, C. Burgos and J. Alvarez-Builla, Tetrahedron Lett., 2000, 41, 5837–5840 CrossRef CAS.
- V. B. Kurteva, L. A. Lubenov, D. V. Nedeltcheva, R. P. Nikolova and B. L. Shivachev, ARKIVOC, 2012, 282–294 CAS.
- A. J. Elliott, H. Guzik and J. R. Soler, J. Heterocycl. Chem., 1982, 19, 1437–1440 CrossRef CAS.
- A. R. Katritzky and A. F. Pozharski, Handbook of heterocyclic chemistry, Elsevier Science Ltd, Oxford, UK, 2nd edn, 2000 Search PubMed.
- Katritzky et al. (see ref. 12) have reported that the thermal heating of 1-benzotriazol-1-yl-3-phenylpropynone with 2-aminopyridne leads to the formation of an analogous by-product (formed by the counter attack of benzotriazole(in addition to the desired pyrido[1,2-a]pyrimidin-2-one derivative).
- Harriman et al. (see ref. 9) have also reported a similar lack of success for thermal cyclizative condensation reactions involving these alkynoates and 2-aminopyridine derivatives.
- For compound 58 only starting material was recovered. Raising the temperature led to decomposition. For materials 54 and 56 heating led to decomposition (tarring).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data. See DOI: 10.1039/c5ob01784j |
| This journal is © The Royal Society of Chemistry 2016 |