
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the AB ring system of clifednamide utilizing Claisen rearrangement and Diels–Alder reaction as key steps†
Inga
Loke
a,
Guillaume
Bentzinger
a,
Julia
Holz
a,
Aruna
Raja
b,
Aman
Bhasin
b,
Florenz
Sasse
b,
Andreas
Köhn
*c,
Rainer
Schobert
d and
Sabine
Laschat
*a
aInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: sabine.laschat@oc.uni-stuttgart.de
bAbteilung Chemische Biologie, Helmholtz-Zentrum für Infektionsforschung, Inhoffenstr. 7, D-38124 Braunschweig, Germany
cInstitut für Theoretische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
dLehrstuhl für Organische Chemie, Universität Bayreuth, Universitätsstr. 30, D-95447 Bayreuth, Germany
First published on 24th November 2015
Abstract
In order to construct the functionalized AB ring system of clifednamide, member of the class of macrocyclic tetramic acid lactams, a synthesis was developed which utilized an Ireland–Claisen rearrangement and an intramolecular Diels–Alder reaction. Starting from di-O-isopropylidene-D-mannitol the allyl carboxylate precursor for the sigmatropic rearrangement was prepared. This rearrangement proceeded diastereoselectively only in the presence of an allyl silyl ether instead of the parent enone in the side chain, as suggested by deuteration experiments. A subsequent Diels–Alder reaction yielded the target ethyl hexahydro-1H-indene-carboxylate with high diastereoselectivity. Quantum-chemical investigations of this intramolecular Diels–Alder reaction support the proposed configuration of the final product.
Introduction
Macrocyclic tetramic acid lactams are a subfamily among the 2,4-pyrrolidinedione (tetramic acid) natural products1 with a wide range of biological activities. This class of structurally complex molecules is characterized by a macrocyclic lactam ring with incorporated tetramic acid moiety, fused to carbocycles based on either bicyclo[3.3.0]octane or a dodecahydro-as-indacene or a related system. Representative examples are ikarugamycin (1), maltophilin (2), clifednamide A (3a) and B (3b) as well as cylindramide (4) (Fig. 1).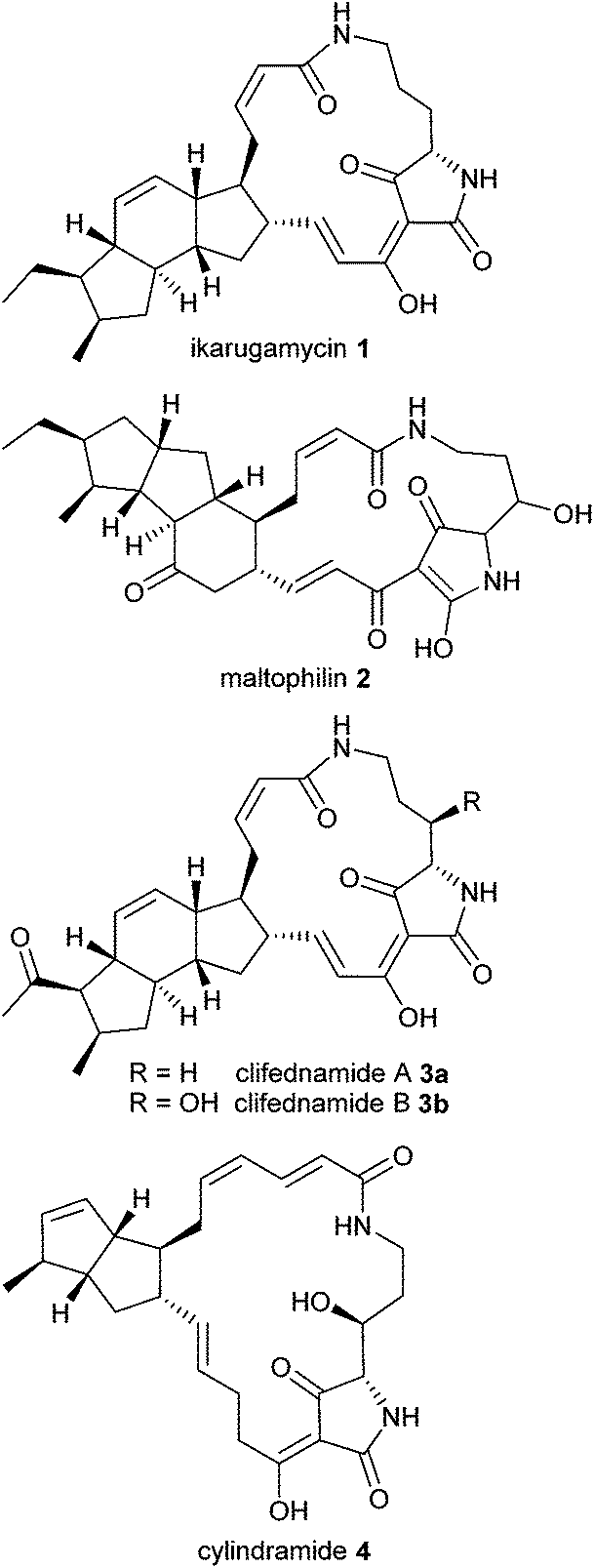 | ||
| Fig. 1 Representative polycyclic tetramate macrolactams 1–4. | ||
Ikarugamycin (1) which was isolated in 1972 from a culture broth of Streptomyces phaeochromogenes2 shows strong specific antiprotozoal activity, antiamoebic activity and activity against some Gram-positive bacteria.2 The structure and absolute stereochemistry of 1 was assigned by chemical degradation and spectroscopic methods,3 and recently, its biosynthesis has been reconstituted in Escherichia coli.4 Maltophilin (2), bearing a cyclopenta[a]indene core, was obtained from strains of Stenotrophomonas maltophilia R3089.5 It is an antifungal compound which is active against various human-pathogenic and phytopathogenic fungi.5
In 2010 Clardy and coworkers isolated clifednamide A (3a) and B (3b) from a strain Streptomyces sp. JV178.6 They postulated a biosynthetic pathway and common biosynthetic origins for polycyclic tetramate macrolactams.7 Cylindramide (4) originally isolated in 1993 from the marine sponge Halichondria cylindrata exhibits pronounced cytotoxicity against B16 melanoma cells8 and other mammalian cell lines, as was published in 2007.9 The cytotoxicity of 4 was found to be correlated with its complexation of Ca2+.9
Both the challenging molecular architecture and the broad variety of biological activities of these natural products have kindled an interest in their total synthesis. Two total syntheses of ikarugamycin (1) have been independently developed by Boeckman10 and by Paquette11 employing an intramolecular Diels–Alder reaction and an anionic oxy-Cope rearrangement as key steps, respectively. Convergent, highly stereoselective total syntheses of cylindramide (4) were developed by the group of Phillips12 and Laschat,13 and furthermore, numerous synthetic routes to the carbocyclic dodecahydro-as-indacene fragment of 1 have been established.14–17 However, surprisingly no synthetic strategies for the related clifednamides 3 have been reported, so far. Motivated by Boeckman's initial success on ikarugamycin (1) we anticipated that a related approach might be suitable for 3 as well. Herein we report on our route towards the bicyclic AB system of clifednamide 3.
Results and discussion
The construction of the AB system 5 in ikarugamycin (1) according to Boeckman's synthesis was based on an intramolecular Diels–Alder reaction of triene 6 prepared from the two subunits 7 and 8a (Scheme 1).10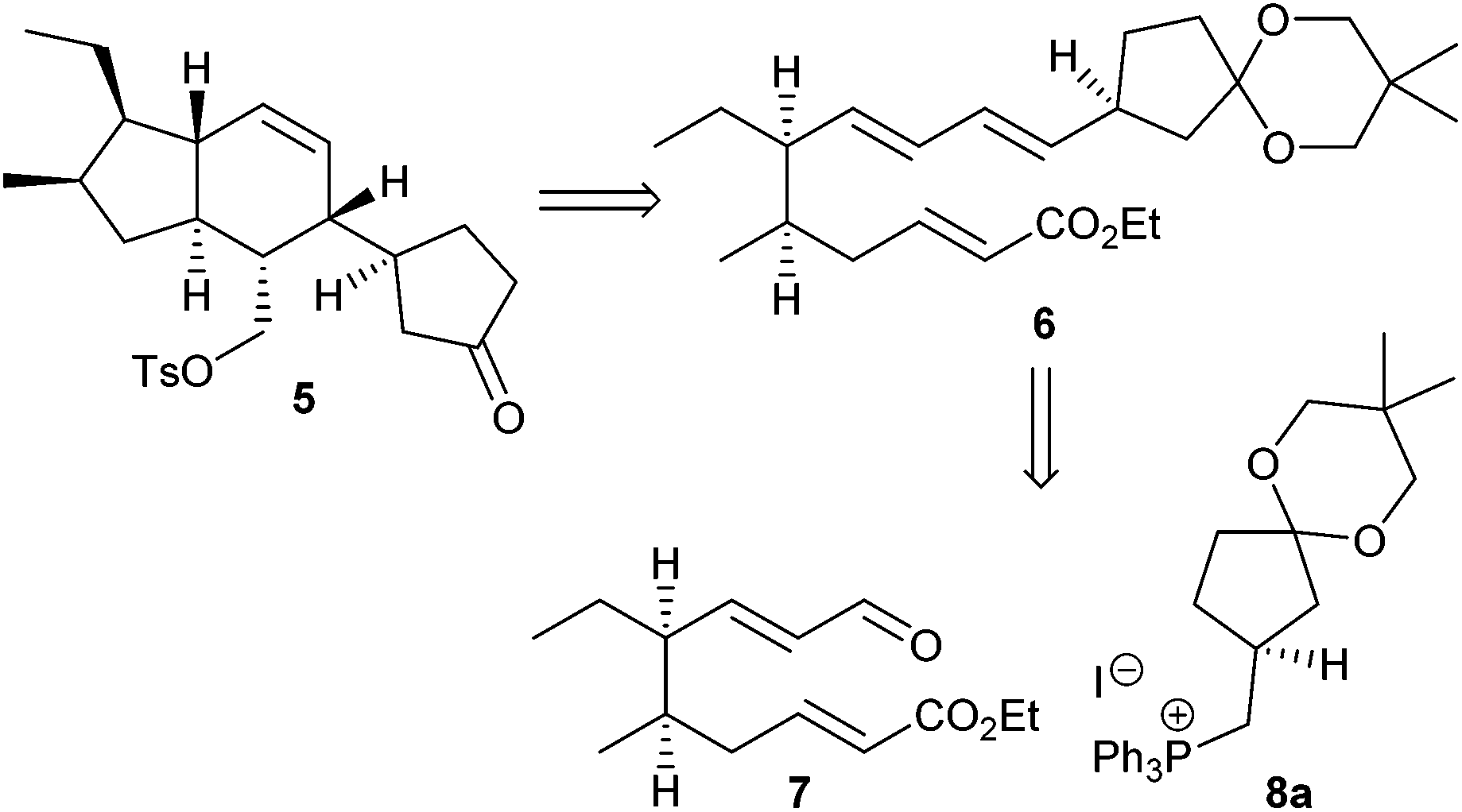 | ||
| Scheme 1 Subunits for Boeckman's key intramolecular Diels–Alder reaction to the AB system of ikarugamycin (1).10 | ||
We initially intended to adopt the ikarugamycin route for clifednamide A (3a). However, all attempts failed to obtain phosphonium salt 8a from the precursor iodide (see ESI† for details). Thus, we envisaged a modified Boeckman route to 3 using 8b instead of 8a and encompassing a functionalization of the C-ring of tricyclic compound 9 at a later stage as well as an intramolecular enolate alkylation of 10 (Scheme 2).
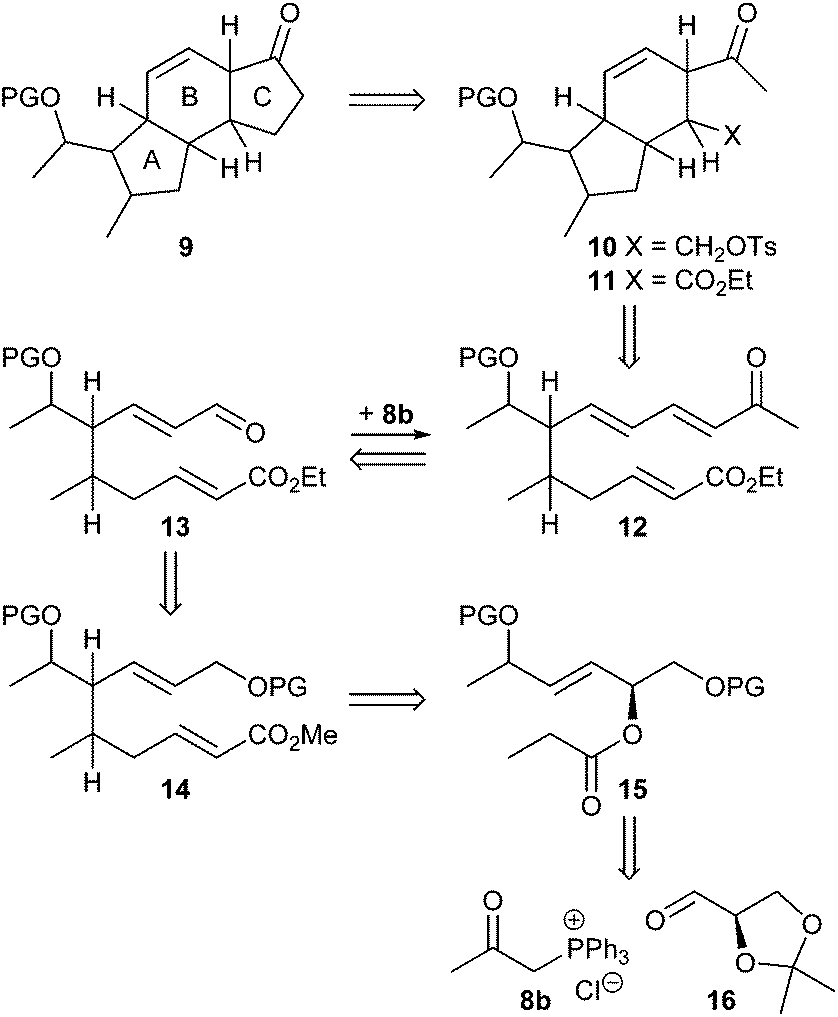 | ||
| Scheme 2 Retrosynthetic pathway to the AB system of clifednamide 3. | ||
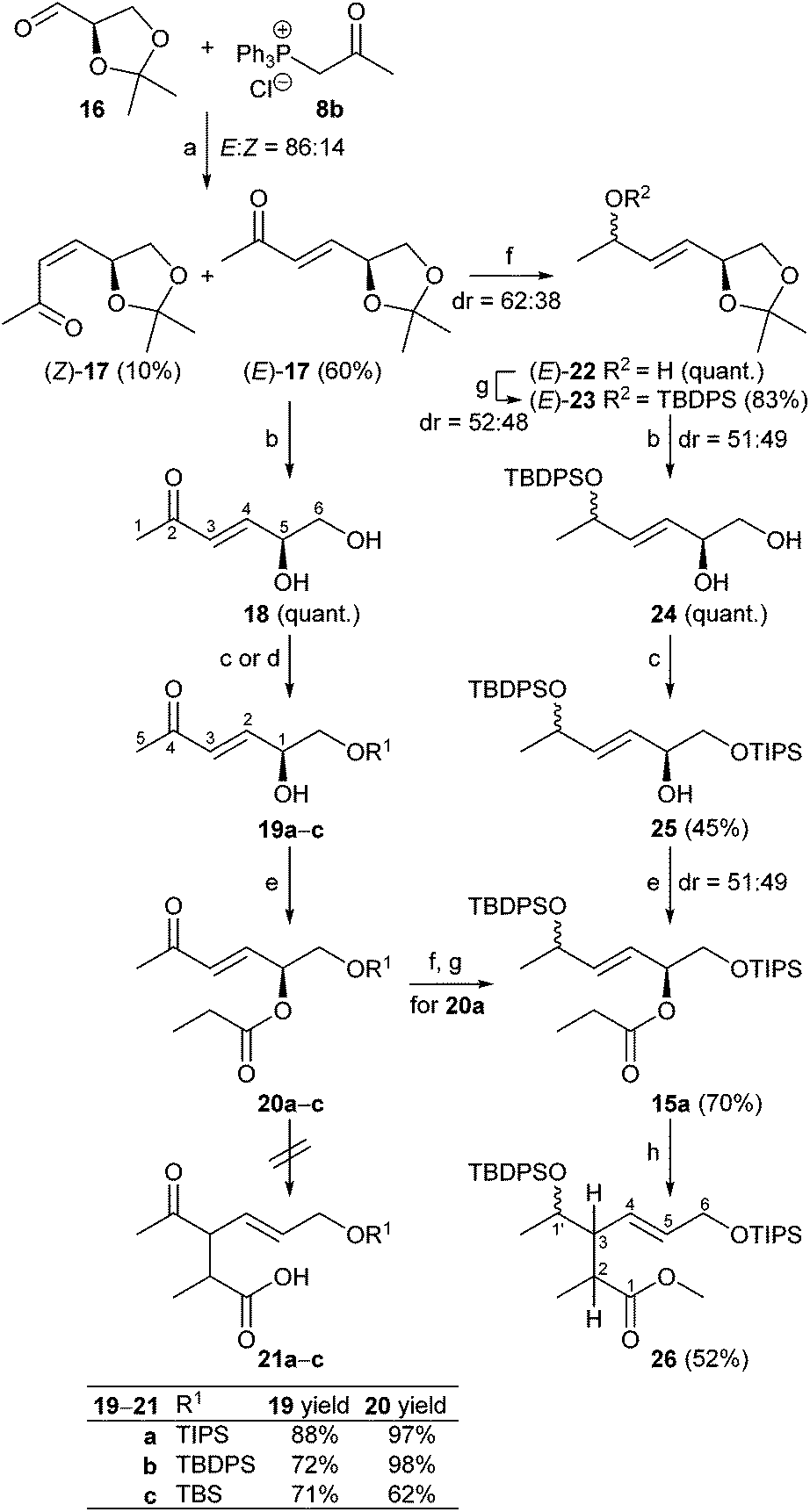
As shown in Scheme 3, (R)-glyceraldehyde acetonide 16 prepared from di-O-isopropylidene-D-mannitol as described in the literature18 was submitted to a Wittig reaction with 8b giving a (86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14) mixture of E/Z-isomers 17.19 Chromatographic separation yielded (E)-17 and (Z)-17 in 60% and 10%, respectively. Acetal cleavage of (E)-17 with a mixture of acetic acid/H2O/THF (4:2:1) according to Horiguchi20 gave the diol 18 quantitatively. Silylation to 19a with only a small excess of TIPSCl and base to avoid double silylation and subsequent esterification of the secondary hydroxy group with propionyl chloride afforded compound 20a in 97% yield. Silyl ethers 20b and c were prepared analogously in 98% and 62% yield, respectively.
:14) mixture of E/Z-isomers 17.19 Chromatographic separation yielded (E)-17 and (Z)-17 in 60% and 10%, respectively. Acetal cleavage of (E)-17 with a mixture of acetic acid/H2O/THF (4:2:1) according to Horiguchi20 gave the diol 18 quantitatively. Silylation to 19a with only a small excess of TIPSCl and base to avoid double silylation and subsequent esterification of the secondary hydroxy group with propionyl chloride afforded compound 20a in 97% yield. Silyl ethers 20b and c were prepared analogously in 98% and 62% yield, respectively.
 | ||
| Scheme 3 Reagents and conditions: (a) NEt3, CH2Cl2, reflux, 4 h; (b) AcOH/H2O/THF (4:2:1), reflux, 2 h; (c) TIPSCl (1.1 equiv.), imidazole (1.1 equiv.), DMF, 0 °C → r.t., 20 h; (d) TBDPSCl or TBSCl (1.1 equiv.), NEt3, DMF, r.t., 20 h; (e) propionyl chloride (1.2 equiv.), pyridine (1.2 equiv.), CH2Cl2, 0 °C → r.t., 4 h; (f) NaBH4 (0.6 equiv.), EtOH, r.t., 3 h; (g) TBDPSCl (1.2 equiv.), imidazole (2 equiv.), DMF, r.t., 6 h; (h) (1) LDA (1.8 equiv.), TMSCl (1.1 equiv.), THF, −100 °C, 1 h, r.t., 20 h; (2) CH2N2 (2 equiv.), Et2O, 0 °C, 1 h, for details concerning dr of 26 see Scheme 5. Numbering for NMR assignment. | ||
The Ireland–Claisen rearrangement was investigated in detail with silyl ethers 20b,c (Table 1). For example, deprotonation of 20c with LDA in THF at −78 °C and addition of TMSCl for 4 days following the Ireland protocol21 did not provide any trace of the rearranged product 21c according to the crude NMR spectra (entry 1). Using LiHMDS with NEt3 in toluene according to the method by Collum22 also failed to give 21b,c (entries 2, 4, and 11). It should be mentioned that a substrate closely related to 20c but without the ketone moiety was converted to the rearranged product using Collum's conditions.23 Next a method by Kishi24 was tested, employing LiHMDS and TIPSCl in THF. Alternatively LDA was used, but neither method worked (entries 3, 5, 7, and 10).
| Entry | Compd | Base 1 | Base 2 | Additive | Solvent | T (°C) | t (h) |
|---|---|---|---|---|---|---|---|
| 1 | 20c | LDA | — | TMSCl | THF | −78 → 60 | 96 |
| 2 | 20c | LiHMDS | Et3N | — | Toluene | −78 → r.t. | 20 |
| 3 | 20c | LiHMDS | — | TIPSCl | THF | −78 → 0 | 24 |
| 4 | 20c | LiHMDS | Et3N | TIPSCl | Toluene | −78 → r.t. | 24 |
| 5 | 20b | LDA | — | TIPSCl | THF | −78 → 0 → r.t. → 40 | 24 |
| 6 | 20b | LDA | — | TESCl | THF | −78 → 0 → r.t. → 40 | 24 |
| 7 | 20b | LiHMDS | — | TIPSCl | THF | −78 → 0 | 24 |
| 8 | 20b | LDA | Et3N | TMSCl | THF | −100 → r.t. → 50 | 24 |
| 9 | 20b | LDA | Et3N | TIPSCl | THF | −100 → r.t. | 24 |
| 10 | 20b | LiHMDS | — | TIPSCl | THF | −100 → r.t. | 4 |
| 11 | 20b | LiHMDS | Et3N | — | Toluene | −100 → r.t. | 4 |
Finally, a protocol by Burke was employed,25 which he had been successfully applied to a Claisen substrate carrying an enone moiety in a similar fashion as compared to 20c (entries 8 and 9). Even this method failed. Presumably, the enolizable enone (or ketone) might interfere with the Claisen rearrangement under basic conditions. However, it should be noted that Paterson realized the Claisen rearrangement with a complex polyketide substrate in high yield despite the presence of an additional enolizable ketone.26
In order to prove the hypothesis of competing enolate formation, a deuteration experiment was carried out. tert-Butyldiphenylsilyl ether 20b was deprotonated with LDA and the reaction was quenched with D2O. Chromatographic purification gave a pure fraction suitable for 1H NMR investigation, where the signals of the olefinic protons H-2 and H-3 which remain unaffected by the deuteration were set to 1 as reference (see ESI† for details).
Integration of the H-5 protons resulted in a value of 2.01 instead of the expected 3. That means, the derivative with fully deuterated CD3 (no signal in the 1H NMR spectrum) was obtained with 33% percentage (Scheme 4). In contrast, double deuteration at C-6 proceeded with 9%, as shown by the integration value of 1.82 instead of 2 for the H-6 proton. Furthermore, the spectra were analyzed regarding a H/D isotope effect.27 Indeed, the H/D exchange caused an upfield shift of the H-5 proton signals, allowing independent integration of the triplet for CH2D and the quintet for CHD2 formed by coupling between H and D. Thus, integration of signals for the deuteration at position 5 gave a percentage of 13% (20b), of 25% monodeuterated and of 28% double deuterated derivative. The degree of deuteration is 87%.
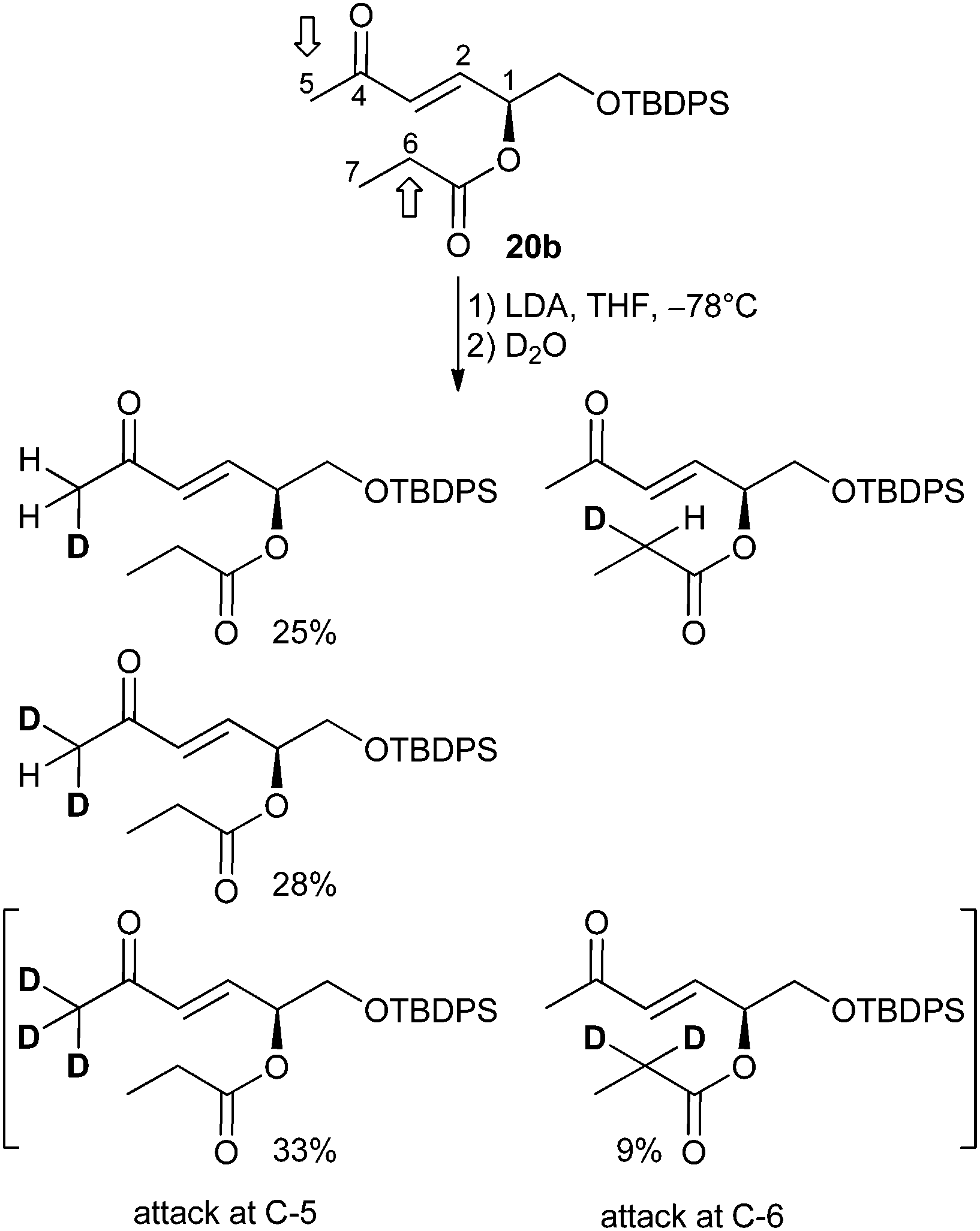 | ||
| Scheme 4 Deuteration experiment of 20b and percentage obtained for differently deuterated derivatives by integration of 1H NMR signals. The percentage of the C-6 monodeuterated derivative could not be determined due to signal overlap. | ||
A comparison of the fully deuterated CD3 group (C-5) with the double deuterated CD2 group at C-6 indicates a more facile H/D exchange of H-5 protons than of H-6 ones. Thus the attack of the base at the CH3 group is preferred.
The results revealed that indeed under kinetic deprotonation conditions competing enolate formation of both ester and methylvinylketone had taken place. In order to circumvent this problem, the enone moiety in 20a was first reduced with NaBH4 to a (52:48) mixture of diastereomeric allylic alcohols (88%) that was subsequently protected with TBDPSCl, imidazole to afford ester 15a with a primary OTIPS and secondary OTBDPS group in 70% yield with dr 51:49 (Scheme 3). Alternatively, a (51:49) mixture of ester 15a was accessible starting from (E)-17 as depicted in Scheme 3 (see ESI† for details). Then ester 15a was treated at −100 °C with LDA in THF in the presence of TMSCl as trapping agent, followed by esterification of the rearranged product with diazomethane in Et2O at 0 °C. After workup, the ester 26 was obtained in 52% yield (Scheme 3). As outlined in Scheme 5, the Claisen rearrangement resulted in four diastereomeric esters 26a–d (dr = 50:47:2:1). This ratio, however, varied throughout the follow-up reaction sequence (Scheme 6) presumably due to partial enrichment of diastereomers during chromatographic purification steps.
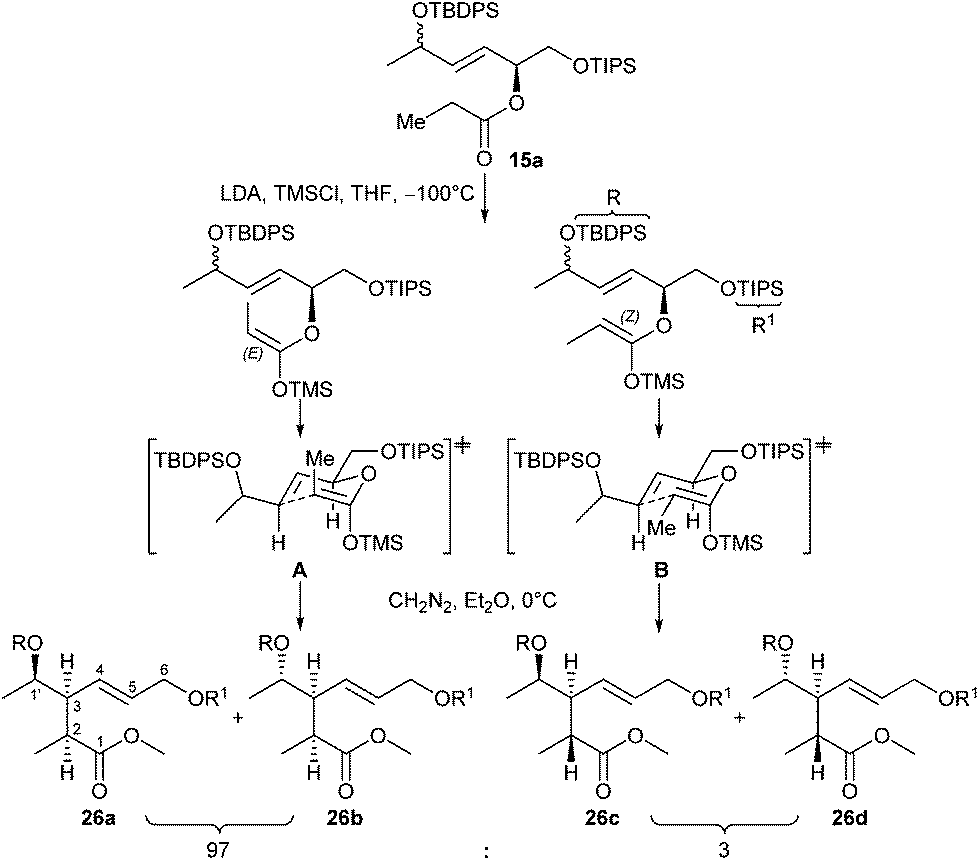 | ||
| Scheme 5 Ireland–Claisen rearrangement of ester 15a with LDA in the presence of TMSCl via assumed preferred transition states for the (E)-enolate (A) and (Z)-enolate (B), resulting in the diastereomeric products 26a–d in a ratio of 50:47:2:1 in 52% total yield. | ||
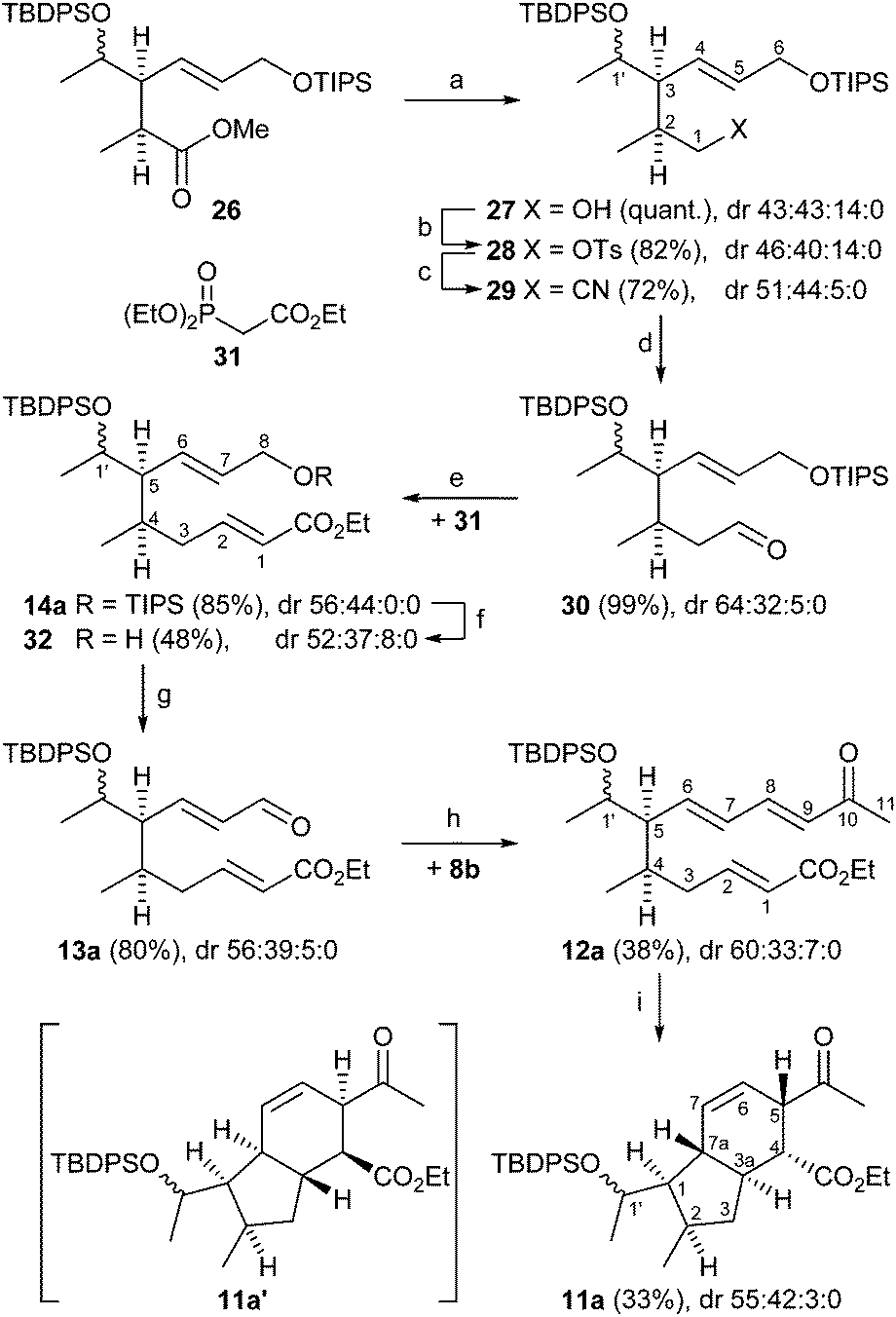 | ||
| Scheme 6 Reagents and conditions: (a) DIBAL (3.0 equiv.), THF, 0 °C, 3 h; (b) pTsCl (1.1 equiv.), NEt3, DMAP, CH2Cl2, 0 °C → r.t., 20 h; (c) KCN (5.5 equiv.), DMF, 80 °C, 3 h; (d) DIBAL (1.06 equiv.), hexane, −78 °C, 1 h; (e) 31 (1.4 equiv.), DBU, LiCl, MeCN/CH2Cl2 (1:1), 0 °C → r.t., 15 h; (f) TBAF (3 equiv.), THF, 0 °C, 1 h; (g) DMP (1.3 equiv.) CH2Cl2, 0 °C, 3 h; (h) 8b (1.1 equiv.), NEt3 (2 equiv.), CH2Cl2, reflux, 23 h; (i) BHT (0.1 equiv.), toluene, reflux, 4 d. Numbering for NMR assignment. | ||
The observed diastereoselectivity concerning the newly generated stereogenic centres C-2, C-3 (26a,b:26c,d = 97:3) is due to the known preference of LDA to generate the (E)-enolate, which resulted in the formation of 26a,b as major diastereomers via transition state A, whereas 26c,d were generated from small amounts of (Z)-enolate via transition state B. In contrast, nearly equimolar mixtures were obtained regarding stereocentre C-1′.
Reduction of diastereomeric esters 26 with DIBAL afforded alcohols 27 quantitatively, which were tosylated to 28 (82%). Nucleophilic substitution with KCN in DMF at 80 °C provided nitriles 29 in 72% yield (Scheme 6). Reduction of the nitriles 29 with DIBAL in hexane gave aldehydes 30 (99%), which were submitted to a Horner–Wadsworth–Emmons olefination with phosphonate 31 in the presence of LiCl and DBU to yield (E)-crotonates 14a in 85%. Conversion to enals 13a (dr 56:39:5:0) was effected in 38% yield over two steps by treatment of 14a with TBAF in THF at 0 °C followed by Dess–Martin oxidation of the resulting allylic alcohol 32. By running the desilylation on a small scale product 32 could be obtained quantitatively. However, performing the reaction on a preparative scale led to a decreased yield presumably due to the large excess of basic TBAF resulting in partial deprotection of the TBDPS group or attack at the enoate.
Assembly of the key E,E-ketotriene ester 12a (38%) was accomplished by Wittig olefination of 13a with the ylide generated in situ from phosphonium salt 8b and NEt3. Triene 12a underwent an intramolecular Diels–Alder cycloaddition by heating in the presence of 3,5-di-tert-butyl-4-hydroxytoluene (BHT) in toluene28 under reflux for 4 days providing, after aqueous workup, the desired hexahydroindene 11a in 33% yield. It should be noted that triene 12a is rather sensitive towards polymerization during workup and purification. This problem was also encountered in the Diels–Alder reaction of 12a to 11a. Even in the presence of radical inhibitor BHT polymerization could not be completely suppressed, resulting in a moderate yield of 33%. The NMR spectra of 11a showed only two sets of signals in a 1:1 ratio due to the undefined stereogenic centre at C-1′ in the side chain. Since no further sets of NMR signals were observed, we anticipated that the cycloaddition proceeded in stereospecific fashion as reported by Boeckman10b yielding only one relative configuration.
In order to understand the stereoselectivity of this Diels–Alder reaction, we carried out a set of quantum-chemical calculations using density functional theory (DFT) (see ESI† chapter 7 for details of the computations). The transition state structures leading to the two diastereomers 11a and 11a′ shown in Scheme 6 have been determined (Fig. 2). According to the computations, the reaction barriers of the two pathways differ by more than 30 kJ mol−1, furthermore the final product 11a is energetically more favourable than 11a′ by nearly the same amount. While the computations are not fully exhaustive in terms of exploring the entire conformational space (in particular concerning the OTBDPS residue) and full thermal averaging, the energy differences appear sufficiently significant to understand the selectivity of the reaction and to claim that indeed 11a is the preferred product.
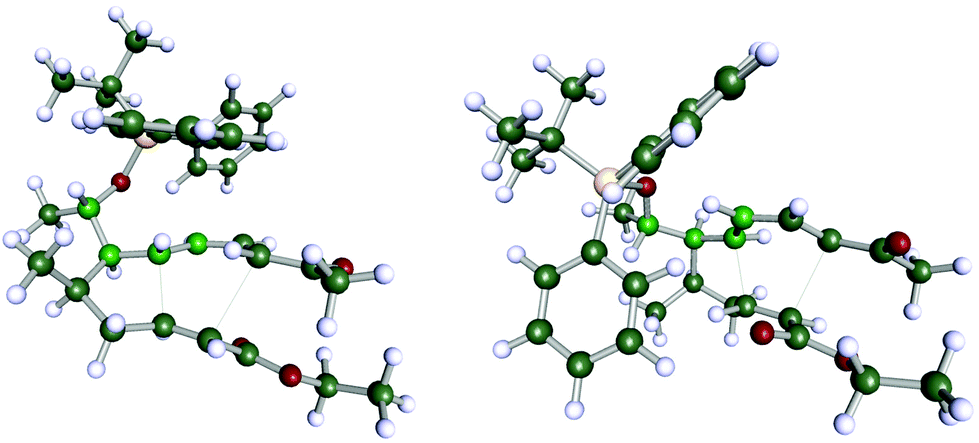 | ||
| Fig. 2 Computed transition structures leading to 11a (left) and 11a′ (right), respectively (see ESI† for details of computations). The centres C-1′, C-5, C-6, C-7 (cf. Scheme 6) are highlighted to emphasize the main conformational difference of the two structures. | ||
Investigation of the structural differences of the two transition states confirms the proposal by Boeckman.10 In the less favourable pathway, the diene group is forced into a nearly eclipsed conformation with respect to one of the residues at centre C-5 (Fig. 2). The selectivity of the reaction is maintained for less bulky substituents, as well. In a set of computations in which the OTBDPS residue was replaced by OMe we found the same energetic preference for the reaction path leading to the OMe equivalent of 11a. However, the difference between the activation energies was somewhat smaller (around 10 kJ mol−1), suggesting that large residues at the C-1′ position increase the selectivity of the reaction.
So far no informations about the biological properties of clifednamide 3 are available. As there are several examples in the literature that truncated natural products and fragments often still retain biological activity,29 we preliminary investigated some synthetic precursors and bicyclic ester 11a with respect to antiproliferative activities (see ESI†).
Conclusions
In summary, a stereoselective route to the AB-ring system 11a of clifednamide 3 was developed with Ireland–Claisen rearrangement and intramolecular thermal Diels–Alder reaction as the key steps in the sequence. In comparison with Boeckman's ikarugamycin synthesis our strategy circumvents the use of optically pure cyclopentane-derived ylide 8a and utilizes the simpler ylide 8b instead. The study clearly showed that the Ireland–Claisen rearrangement required a silyl ether in the side chain of the allyl carboxylate 15a rather than a ketone (20a). Compound 11a was obtained in 17 steps and 1.3% overall yield from D-mannitol. Quantum-chemical calculations explain the selectivity of the final Diels–Alder reaction and confirm the proposed conformation of 11a. Further work is already underway to convert compound 11a to the tricyclic ABC system of clifednamide.Experimentals
General information
NMR spectra were recorded on a Bruker Avance 300 or a Bruker Avance 500 spectrometer in CDCl3 with TMS as an internal standard. IR spectra were recorded on a Bruker FT-IR-spectrometer Vektor 22 with MKII golden gate single reflection Diamant ATR-system. Mass spectra were recorded on a Varian MAT 711 (EI, 70 eV) and a Bruker Daltonics micrOTOF_Q (ESI) with nitrogen as carrier gas. Optical rotations were measured with a Perkin-Elmer 241 polarimeter at 20 °C. Flash chromatography was performed on silica gel, grain size 40–63 μm (Fluka). Moisture-sensitive reactions were performed under nitrogen atmosphere in oven-dried glassware. All reagents were used as purchased unless otherwise noted. Solvents used for chromatography were distilled. THF was distilled from potassium/benzophenone, CH2Cl2 and toluene from CaH2. The reactions were monitored by TLC (Merck 60 F254 plates).:48 by 13C NMR), which was used without further purification. FT-IR (ATR) (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) cm−1) 3433 (w), 2943 (s), 2866 (vs), 1740 (s), 1463 (m), 1367 (w), 1186 (vs), 1129 (vs), 1068 (s), 1014 (w), 970 (w), 920 (w), 882 (vs), 788 (m), 682 (s). 1H NMR (300 MHz, CDCl3) δ 0.99–1.10 [m, 21H, CH(CH3)2], 1.15 (t, J = 7.5 Hz, 3H, CH2CH3), 1.27 (d, J = 6.5 Hz, 3H, H-5), 1.87 (br, 1H, OH), 2.35 (q, J = 7.5 Hz, 2H, CH2CH3), 3.66–3.85 (m, 2H, CH2), 4.26–4.38 (m, 1H, H-4), 5.32–5.42 (m, 1H, H-1), 5.68 (dddd, J = 15.6, 6.2, 2.2, 1.1 Hz, 1H, H-2), 5.83 (dddd, J = 15.6, 5.7, 3.2, 0.9 Hz, 1H, H-3). 13C NMR (75 MHz, CDCl3) δ 9.1 (CH2CH3), 11.9 [CH(CH3)2], 17.9 [CH(CH3)2], 23.12, 23.15 (C-5), 27.8 (CH2CH3), 65.1 (CH2), 68.1, 68.2 (C-4), 74.19, 74.23 (C-1), 125.2, 125.3 (C-2), 137.52, 137.54 (C-3), 173.7 (COO). MS (ESI) m/z 367.2 [M + Na]+, 271.2 [M + H − C3H6O2]+, 253.2 [M + H − C3H6O2 − H2O]+, 213.2, 191.1, 175.2, 163.1, 137.1, 119.1, 97.1. HRMS (ESI) obsd 367.2259, calc. for C18H36O4SiNa+: 367.2275.
cm−1) 3433 (w), 2943 (s), 2866 (vs), 1740 (s), 1463 (m), 1367 (w), 1186 (vs), 1129 (vs), 1068 (s), 1014 (w), 970 (w), 920 (w), 882 (vs), 788 (m), 682 (s). 1H NMR (300 MHz, CDCl3) δ 0.99–1.10 [m, 21H, CH(CH3)2], 1.15 (t, J = 7.5 Hz, 3H, CH2CH3), 1.27 (d, J = 6.5 Hz, 3H, H-5), 1.87 (br, 1H, OH), 2.35 (q, J = 7.5 Hz, 2H, CH2CH3), 3.66–3.85 (m, 2H, CH2), 4.26–4.38 (m, 1H, H-4), 5.32–5.42 (m, 1H, H-1), 5.68 (dddd, J = 15.6, 6.2, 2.2, 1.1 Hz, 1H, H-2), 5.83 (dddd, J = 15.6, 5.7, 3.2, 0.9 Hz, 1H, H-3). 13C NMR (75 MHz, CDCl3) δ 9.1 (CH2CH3), 11.9 [CH(CH3)2], 17.9 [CH(CH3)2], 23.12, 23.15 (C-5), 27.8 (CH2CH3), 65.1 (CH2), 68.1, 68.2 (C-4), 74.19, 74.23 (C-1), 125.2, 125.3 (C-2), 137.52, 137.54 (C-3), 173.7 (COO). MS (ESI) m/z 367.2 [M + Na]+, 271.2 [M + H − C3H6O2]+, 253.2 [M + H − C3H6O2 − H2O]+, 213.2, 191.1, 175.2, 163.1, 137.1, 119.1, 97.1. HRMS (ESI) obsd 367.2259, calc. for C18H36O4SiNa+: 367.2275.
(b) To a solution of 4-hydroxy-1-{[(triisopropylsilyl)oxy]methyl}pent-2-enyl propionate (4.00 g, 11.6 mmol) in DMF (60 mL) imidazole (1.58 g, 23.2 mmol) was added followed by dropwise addition of TBDPSCl (3.62 mL, 3.83 g, 13.9 mmol) and the reaction mixture stirred at r.t. for 6 h. The solvent was removed under vacuum and the residue purified by chromatography on SiO2 with hexanes/EtOAc (100:1) to give 15a (4.76 g, 8.17 mmol, 70%) as a colorless oil (dr 51:49 by 13C NMR). Rf = 0.36 (hexanes/EtOAc, 50:1). FT-IR (ATR) ( cm−1) 2942 (m), 2864 (m), 1740 (s), 1463 (m), 1428 (m), 1367 (w), 1184 (m), 1111 (vs), 1081 (s), 997 (m), 967 (m), 882 (m), 822 (w), 789 (w), 738 (m), 701 (vs), 688 (s), 612 (m). 1H NMR (500 MHz, CDCl3) δ 1.02–1.06 [m, 60H, CH(CH3)2, C(CH3)3], 1.11–1.16 (m, 12H, H-5, CH2CH3), 2.31 (q, J = 7.3 Hz, 2H, CH2CH3), 2.32 (q, J = 7.7 Hz, 2H, CH2CH3), 3.60–3.68 (m, 4H, CH2), 4.26–4.33 (m, 2H, H-4), 5.29–5.35 (m, 2H, H-1), 5.49 (ddd, J = 15.4, 1.6, 1.6 Hz, 1H, H-2), 5.50 (ddd, J = 15.6, 1.4, 1.4 Hz, 1H, H-2), 5.75 (ddd, J = 15.4, 1.5, 1.5 Hz, 1H, H-3), 5.76 (ddd, J = 15.6, 1.3, 1.3 Hz, 1H, H-3), 7.31–7.44 (m, 12H, o-H, p-H), 7.60–7.69 (m, 8H, m-H). 13C NMR (125 MHz, CDCl3) δ 9.1 (CH2CH3), 11.9 [CH(CH3)2], 17.9 [CH(CH3)2], 19.2 [C(CH3)3], 24.0 (C-5), 27.0 [C(CH3)3], 27.8 (CH2CH3), 65.17, 65.20 (CH2), 69.4, 69.5 (C-4), 74.2, 74.4 (C-1), 124.11, 124.13 (C-2), 127.4, 127.5 (o-C), 129.50, 129.51 (p-C), 134.1, 134.4 (i-C), 135.8, 135.9 (m-C), 137.7, 137.9 (C-3), 173.55, 173.58 (COO). MS (ESI) m/z 605.4 [M + Na]+, 509.3 [M − C3H5O2]+, 431.3, 391.3, 327.2, 293.2, 253.2, 223.1, 193.1, 163.1, 145.1, 127.1, 97.1. HRMS (ESI) obsd 605.3446, calc. for C34H54O4Si2Na+: 605.3453.
:1 → 100:1) to give 26 (2.11 g, 3.45 mmol, 52%) as a colorless oil (dr = 50:47:2:1 by 1H NMR). Rf = 0.33 (hexanes/EtOAc, 50:1). FT-IR (ATR) ( cm−1) 2930 (m), 2863 (m), 1736 (s), 1698 (w), 1461 (m), 1428 (m), 1376 (w), 1257 (m), 1130 (s), 1105 (vs), 1053 (s), 975 (m), 882 (m), 821 (m), 766 (w), 739 (m), 701 (vs), 686 (s), 610 (m). 1H NMR (500 MHz, CDCl3) δ 0.93 (d, J = 6.3 Hz, 3H, 1′-CH3*), 0.976 (d, J = 5.8 Hz, 3H, 1′-CH3), 0.981 (d, J = 7.3 Hz, 3H, 2-CH3), 0.96–1.03 [m, 60H, CH(CH3)2, C(CH3)3], 1.00 (d, J = 7.1 Hz, 3H, 2-CH3*), 2.17–2.24 (m, 1H, H*-3), 2.60–2.66 (m, 1H, H-3), 2.84 (qd, J = 7.3, 7.1 Hz, 1H, H-2), 2.86 (qd, J = 10.4, 7.1 Hz, 1H, H*-2), 3.51 (s, 3H, OCH3), 3.54 (s, 3H, OCH3*), 3.73 (qd, J = 6.9, 5.8 Hz, 1H, H-1′), 3.79 (qd, J = 6.3, 2.1 Hz, 1H, H*-1′), 4.19 (ddd, J = 4.2, 3.0, 1.4 Hz, 2H, H-6), 4.30 (ddd, J = 4.2, 3.0, 1.5 Hz, 2H, H*-6), 5.41 (ddt, J = 15.3, 9.8, 1.4 Hz, 1H, H-4), 5.59 (ddt, J = 15.4, 4.2, 0.8 Hz, 1H, H*-5), 5.60 (ddt, J = 15.3, 4.2, 0.9 Hz, 1H, H-5), 5.73 (ddt, J = 15.4, 10.0, 1.5 Hz, 1H, H*-4), 7.30–7.46 (m, 12H, o-H, p-H), 7.60–7.73 (m-H). 13C NMR (125 MHz, CDCl3) δ 10.99, 11.04 [CH(CH3)2], 11.8 (2-CH3), 15.8 (2-CH3*), 16.98, 17.04 [CH(CH3)2], 18.31, 18.35 [C(CH3)3], 19.6 (1′-CH3), 21.3 (1′-CH3*), 26.0 [C(CH3)3], 39.2, (C-2), 39.4 (C-2*), 50.3 (OCH3), 51.8 (C-3), 52.3 (C-3*), 62.5 (C-6), 62.7 (C-6*), 68.9 (C-1′), 69.7 (C-1′*), 124.9 (C-4), 125.2 (C-4*), 126.1, 126.3, 216.5, 126.6 (o-C), 128.2, 128.4, 128.55, 128.59 (p-C), 132.0, 132.4, 133.8, 134.1 (i-C), 132.7 (C-5), 133.1 (C-5*), 134.88, 134.91, 134.96, 134.99 (m-C), 175.3 (C-2), 176.3 (C-2*). MS (EI) m/z 581.4 [1%, (M − CH3)+], 565.4 [4, (M − CH3O)+], 553.3 [56, (M − C3H7)+], 539.3 [100, (M − C4H9)+], 495.3 (3), 466.4 (5), 448.5 (4), 399.2 (7), 355.2 (8), 335.3 (2), 297.2 (3), 283.1 (39), 253.2 (4), 248.2 (8), 213.0 (18), 182.4 (13), 139.1 (5), 135.0 (27), 111.1 (12), 97.1 (14), 71.0 (18), 57.0 [26, (C4H9)+], 55.0 (14). HRMS (ESI) obsd 619.3616, calc. for C35H56O4Si2Na+: 619.3609.
:43:14:0 by 1H NMR). FT-IR (ATR) ( cm−1) 3396 (m), 2931 (w), 2862 (w), 1464 (w), 1428 (w), 1111 (m), 1050 (s), 1024 (vs), 881 (m), 821 (m), 704 (s), 607 (m). 1H NMR (300 MHz, CDCl3) δ 0.72 (d, J = 7.1 Hz, 3H, 2-CH3*), 0.81 (d, J = 6.8 Hz, 3H, 2-CH3), 0.99 (d, J = 6.1 Hz, 3H, 1′-CH3*), 1.01–1.14 [m, 63H, CH(CH3)2, C(CH3)3, 1′-CH3], 1.73 (br, 2H, OH), 1.95–2.03 (m, 1H, H-3), 2.30–2.11 (m, 2H, H*-2, H*-3), 3.39–3.45 (m, 2H, H-1), 3.47–3.50 (m, 2H, H-1), 3.82–3.88 (m, 1H, H*-1′), 3.94 (qd, J = 6.3, 3.6 Hz, 1H, H-1′), 4.14–4.30 (m, 4H, H-6), 5.36 (dt, J = 15.4, 4.6 Hz, 1H, H-5), 5.47 (ddt, J = 17.8, 9.3, 2.0 Hz, 1H, H*-4), 5.53–5.73 (m, 2H, H*-5, H-4), 7.30–7.52 (m, 12H, o-H, p-H), 7.59–7.80 (m, 8H, m-H). 13C NMR (75 MHz, CDCl3) δ 12.1 (2-CH3), 12.2 [CH(CH3)2], 15.9 (2-CH3*), 18.2 [CH(CH3)2], 19.3, 19.5 [C(CH3)3], 20.4 (1′-CH3*), 22.2 (1′-CH3), 27.17, 27.20 [C(CH3)3], 35.3 (C-2), 35.6 (C-2*), 52.2 (C-3), 52.9 (C-3*), 63.77 (C-6*), 63.84 (C-6), 67.3 (C-1*), 67.4 (C-1), 70.2 (C-1′), 72.4 (C-1′*), 127.5 (m-C), 127.52 (C-4), 127.6, 127.7, 127.9 (m-C), 129.5 (C-4*), 129.73, 129.74, 129.9 (p-C), 132.3 (C-5*), 133.0 (C-5), 133.8, 134.4 (i-C), 136.1, 136.2 (o-C). MS (ESI) m/z 591.4 [M + Na]+, 317.2, 273.2, 239.1, 199.1, 139.1, 121.1, 95.1. HRMS (ESI) obsd 591.3671, calc. for C34H56O3Si2Na+: 591.3660.
:1) to give 28 (83.0 mg, 0.11 mmol, 82%) as a colorless oil (dr 46:40:14:0 by 1H NMR). Rf = 0.55 (hexanes/EtOAc, 10:1). FT-IR (ATR) ( cm−1) 2941 (m), 2864 (m), 1462 (m), 1428 (w), 1363 (s), 1259 (w), 1188 (m), 1177 (s), 1103 (vs), 1059 (m), 966 (vs), 882 (m), 814 (s), 738 (m), 702 (vs), 686 (s), 666 (vs), 611 (m), 554 (s). 1H NMR (300 MHz, CDCl3) δ 0.63 (d, J = 7.1 Hz, 3H, 2-CH3), 0.77–0.95 (m, 9H, 1′-CH3, 1′-CH3*, 2-CH3*), 1.10–0.97 [m, 60H, C(CH3)3, CH(CH3)2], 1.87 (ddd, J = 10.6, 7.5, 3.3 Hz, 1H, H*-3), 2.01–2.12 (m, 1H, H*-2), 2.19 (ddd, J = 9.6, 8.5, 3.3 Hz, 1H, H-3), 2.43 (s, 3H, CH3Tos), 2.44 (s, 3H, CH3Tos*), 2.45–2.52 (m, 1H, H-2), 3.66–3.77 (m, 4H, H-1′, H-1, Ha*-1), 3.77–3.82 (m, 1H, H*-1′), 3.95–4.14 (m, 3H, Hb*-1, H-6), 4.10–4.20 (m, 2H, H*-6), 5.24 (ddt, J = 15.4, 9.8, 1.4 Hz, 1H, H-4), 5.31–5.42 (m, 2H, H-5, H*-5), 5.50–5.63 (m, 1H, H*-4), 7.23–7.48 (m, 16H, o-H, p-H, o-HTos*), 7.57–7.82 (m, 12H, m-H, m-HTos). 13C NMR (75 MHz, CDCl3) δ 11.3 (2-CH3), 12.1, 12.2 [CH(CH3)2], 15.0 (2-CH3*), 18.1, 18.2 [CH(CH3)2], 19.4, 19.5 [C(CH3)3], 21.4 (1′-CH3*), 21.8 (CH3Tos), 22.4 (1′-CH3), 27.1, 27.2 [C(CH3)3], 32.1 (C-2), 33.2 (C-2*), 51.4 (C-3, C-3*), 63.4 (C-6*), 63.5 (C-6), 69.8 (C-1′), 70.7 (C-1′*), 74.0 (C-1*), 74.0 (C-1), 125.3 (C-4), 127.0 (C-4*), 127.5, 127.8 (m-C), 127.8, 128.05, 128.08 (o-CTos), 129.56, 129.60, 129.8, 129.9 (p-C), 129.87, 129.89 (m-CTos), 133.41, 133.43 (i-CTos), 133.8 (C-5*), 134.0 (C-5), 134.7, 134.9 (i-C), 136.0, 136.10, 136.13 (o-C), 144.6 (p-CTos).
:1) to give 29 (57.8 mg, 0.10 mmol, 72%) as a colorless oil (dr 51:44:5:0 by 1H NMR). Rf: 0.71 (hexanes/EtOAc, 10:1). FT-IR (ATR) ( cm−1) 2940 (m), 2863 (m), 1462 (m), 1427 (m), 1382 (m), 1104 (s), 1058 (m), 1012 (w), 978 (m), 918 (w), 882 (m), 821 (m), 739 (s), 701 (vs), 686 (s), 660 (m), 611 (m). 1H NMR (300 MHz, CDCl3) δ 0.77 (d, J = 6.9 Hz, 3H, 2-CH3), 0.96–1.18 [m, 69H, C(CH3)3, CH(CH3)2, 1′-CH3, 2-CH3*, 1′-CH3*], 1.77–1.88 (m, 1H, H*-3), 1.96–2.29 (m, 6H, H-1, H*-1, H*-2, H-3), 2.48–2.61 (m, 1H, H-2), 3.69–3.81 (m, 1H, H-1′), 3.82–3.93 (m, 1H, H*-1′), 4.19 (dd, J = 4.4, 1.5 Hz, 2H, H-6), 4.23 (dd, J = 4.0, 1.1 Hz, 2H, H*-6), 5.30 (dd, J = 15.3, 10.3 Hz, 1H, H-4), 5.52 (dt, J = 15.3, 4.2 Hz, 1H, H-5), 5.58–5.73 (m, 2H, H*-4, H*-5), 7.30–7.51 (m, 12H, o-H, p-H), 7.59–7.78 (m, 8H, m-H). 13C NMR (75 MHz, CDCl3) δ 12.1, 12.2 [CH(CH3)2], 14.4 (2-CH3), 18.1, 18.2 [CH(CH3)2], 19.4, 19.5 [C(CH3)3], 21.9 (1′-CH3*), 22.6 (1′-CH3), 22.9 (C-1*), 23.5 (C-1), 27.0, 27.2 [C(CH3)3], 30.2 (C-2*), 30.5 (C-2), 54.6 (C-3*), 54.8 (C-3), 63.51 (C-6*), 63.53 (C-6), 69.95 (C-1′*), 70.0 (C-1′), 119.1 (CN*), 119.5 (CN), 125.0 (C-4), 126.8 (C-4*), 127.5, 127.8, 128.0 (o-C), 129.6, 129.7, 129.9, 130.0 (p-C), 133.6, 133.7 (i-C), 133.9 (C-5*), 134.6 (i-C), 134.7 (C-5), 134.8 (i-C), 136.02, 136.06, 136.13, 136.15 (m-C). MS (ESI) m/z 578.4 [M + H]+, 404.2 [M − C9H22OSi]+, 360.2, 283.2 [C18H23OSi]+, 265.1, 241.1, 163.1, 137.1, 105.1. HRMS (ESI) obsd 600.3661, calc. for C35H55NO2Si2Na+: 600.3664.
:32:5:0 by 1H-NMR, CHO). FT-IR (ATR) ( cm−1) 2941 (m), 2864 (m), 1707 (m), 1462 (w), 1427 (w), 1382 (w), 1105 (s), 1058 (m), 977 (m), 882 (m), 832 (m), 738 (s), 701 (vs), 686 (s), 610 (m). 1H NMR (300 MHz, CDCl3) δ 0.72 (d, J = 6.9 Hz, 3H, 2-CH3), 0.83 (d, J = 6.8 Hz, 3H, 2-CH3*), 0.95–1.19 [m, 66H, C(CH3)3, CH(CH3)2, 1′-CH3, 1′-CH3*], 1.70–1.79 (m, 1H, H*-3), 1.93–2.46 (m, 6H, H-1, H*-1, H*-2, H-3), 2.66–2.80 (m, 1H, H-2), 3.72–3.85 (m, 1H, H-1′), 3.85–3.97 (m, 1H, H*-1′), 4.19 (dd, J = 4.7, 1.7 Hz, 2H, H-6), 4.25 (dd, J = 4.7, 1.8 Hz, 2H, H*-6), 5.34 (ddt, J = 15.4, 10.0, 1.7 Hz, 1H, H-4), 5.42–5.59 (m, 2H, H-5, H*-5), 5.60–5.73 (m, 1H, H*-4), 7.29–7.50 (m, 12H, o-H, p-H), 7.59–7.79 (m, 8H, m-H), 9.59 (dd, J = 2.9, 1.0 Hz, 1H, CHO*), 9.69 (t, J = 2.4 Hz, 1H, CHO). 13C NMR (75 MHz, CDCl3) δ 12.15, 12.20 [CH(CH3)2], 15.0 (1′-CH3*), 18.1 (1′-CH3), 18.2 [CH(CH3)2], 19.5, 19.6 [C(CH3)3], 22.2, 22.4 (2-CH3, 2-CH3*), 27.2 [C(CH3)3], 27.5 (C-2*), 28.3 (C-2), 49.4 (C-1), 50.1 (C-1*), 55.3 (C-3), 55.6 (C-3*), 63.7, 63.8 (C-6), 70.0 (C-1′*), 70.2 (C-1′), 126.2 (C-4*), 127.4, 127.5, 127.75, 127.80 (o-C), 127.9 (C-4), 129.6, 129.8, 129.9 (p-C), 133.3 (C-5), 133.8 (C-5*), 133.9, 134.0, 134.9, 135.0 (i-C), 136.11, 136.15, 136.18 (m-C), 203.20, 203.22 (CHO, CHO*). MS (ESI) m/z 581.4 [MH]+, 407.3 [M − OTIPS]+, 393.2, 360.2, 325.3 [M − OTBDPS]+, 301.1, 283.2, 227.1, 211.2, 183.1, 151.1 [M − OTIPS − OTBDPS]+, 133.1, 105.1. HRMS (ESI) obsd 603.3660, calc. for C35H56O3Si2Na+: 603.3660.
:1) to give 14a (77.8 mg, 0.12 mmol, 85%) as a colorless oil (dr 56:44 by 13C NMR). Rf = 0.56 (hexanes/EtOAc, 10:1). FT-IR (ATR) ( cm−1) 2931 (w), 2864 (w), 2341 (w), 1714 (m), 1428 (w), 1261 (w), 1105 (m), 978 (w), 907 (s), 882 (w), 804 (w), 730 (vs), 702 (s), 686 (m), 648 (w). 1H NMR (300 MHz, CDCl3) δ 0.66 (d, J = 6.8 Hz, 3H, 4-CH3), 0.79 (d, J = 6.7 Hz, 3H, 4-CH3*), 0.95–1.00 (m, 6H, 1′-CH3, 1′-CH3*), 1.01–1.13 [m, 60H, C(CH3)3, CH(CH3)2], 1.29 (t, J = 7.0 Hz, 3H, CH2CH3*), 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 1.68–1.77 (m, 1H, H*-3), 1.94–2.28 (m, 6H, H-1, H*-1, H*-2, H-2, H-3), 3.72–3.84 (m, 1H, H-1′), 3.89–3.98 (m, 1H, H*-1′), 4.20 (q, J = 7.0 Hz, 4H, CH2CH3, CH2CH3*), 5.32 (ddt, J = 15.3, 10.1, 1.7 Hz, 1H, H-6), 5.40–5.59 (m, 2H, H-7, H*-7), 5.65 (dd, J = 15.4, 1.4 Hz, 1H, H*-6), 5.68 (dt, J = 15.4, 2.0 Hz, 1H, H*-2), 5.77 (dt, J = 15.6, 1.6 Hz, 1H, H-2), 6.77–6.98 (m, 2H, H-1, H*-1), 7.30–7.48 (m, 12H, o-H, p-H), 7.61–7.76 (m, 8H, m-H). 13C NMR (75 MHz, CDCl3) δ 12.17, 12.22 [CH(CH3)2], 14.4 [CH2CH3], 15.0 (1′-CH3), 17.6 (1′-CH3*), 18.15, 18.21 [C(CH3)3], 19.49, 19.54 [CH(CH3)2], 22.29–22.38 (4-CH3, 4-CH3*), 27.1 [C(CH3)3], 31.8 (C-4), 32.5 (C-4*), 37.9 (C-3*), 38.6 (C-3), 55.1 (C-5), 55.2 (C-5*), 60.3 (CH2CH3), 63.9 (C-8), 70.1 (C-1′), 70.2 (C-1′*), 122.4 (C-1), 122.6 (C-1*), 126.8 (C-6), 127.4, 127.6, 127.71, 127.74 (o-C), 128.3 (C-6*), 129.5, 129.7, 129.8 (m-C), 132.9 (C-7*), 133.4 (C-7), 134.0, 135.06, 135.10 (i-C), 136.1, 136.2 (m-C), 148.3 (C-2*), 148.8 (C-2), 166.8 (COO*), 166.9 (COO). MS (ESI) m/z 673.4 [M + Na]+, 477.3 [M − OTIPS]+, 433.3 [M − OTIPS − OC3H5]+, 399.2. HRMS (ESI) obsd 673.4057, calc. for C39H62O4Si2Na+: 673.4079.
:1 → 1:1) to give 32 (558 mg, 1.13 mmol, 48%) as a colorless oil (dr 52:37:8:0 by 1H NMR). FT-IR (ATR) ( cm−1) 2929 (m), 2856 (m), 1720 (s), 1651 (w), 1461 (w), 1427 (m), 1367 (w), 1313 (w), 1261 (m), 1176 (m), 1109 (s), 976 (s), 937 (w), 821 (m), 739 (s), 702 (vs), 686 (s), 610 (m). MS (ESI) m/z 517.3 [M + Na]+, 400.2, 284.2, 221.2, 149.1, 105.1. HRMS (ESI) obsd 517.2726, calc. for C30H42O4SiNa+: 517.2745. Diastereomer 1: Rf = 0.46 (hexanes/EtOAc, 3:1). 1H NMR (500 MHz, CDCl3) δ 0.76 (d, J = 6.5 Hz, 3H, 4-CH3), 1.02 (d, J = 6.3 Hz, 3H, 1′-CH3), 1.04 [s, 9H, C(CH3)3], 1.29 (t, J = 7.1 Hz, 3H, CH2CH3), 1.50 (br, 1H, OH), 1.76–1.81 (m, 1H, H-5), 1.84–1.94 (m, 2H, Ha-3, H-4), 2.16–2.24 (m, 1H, Hb-3), 3.96 (qd, J = 6.3, 4.4 Hz, 1H, H-1′), 4.09–4.11 (m, 2H, H-8), 4.18 (q, J = 7.1 Hz, 2H, CH2CH3), 5.50–5.58 (m, 2H, H-6, H-7), 5.68 (dt, J = 15.6, 1.6 Hz, 1H, H-1), 6.83 (dt, J = 15.6, 7.6 Hz, 1H, H-2), 7.33–7.46 (m, 6H, o-H, p-H), 7.63–7.71 (m, 4H, m-H). 1H NMR (500 MHz, C6D6) δ 0.68 (d, J = 6.6 Hz, 3H, 4-CH3), 0.99 (t, J = 7.1 Hz, 3H, CH2CH3), 1.00 (d, J = 6.3 Hz, 3H, 1′-CH3), 1.15 [s, 9H, C(CH3)3], 1.32 (br, 1H, OH), 1.64–1.74 (m, 2H, H-4, H-5), 1.79–1.87 (m, 1H, Ha-3), 2.05–2.12 (m, 1H, Hb-3), 3.87 (dt, J = 5.5, 1.6 Hz, 2H, H-8), 3.98 (qd, J = 6.3, 4.3 Hz, 1H, H-1′), 4.06 (q, J = 7.1 Hz, 2H, CH2CH3), 5.36 (dt, J = 15.4, 5.5 Hz, 1H, H-7), 5.55 (ddt, J = 15.4, 9.9, 1.6 Hz, 1H, H-6), 5.88 (dt, J = 15.5, 1.5 Hz, 1H, H-1), 7.09 (ddd, J = 15.5, 8.2, 6.8 Hz, 1H, H-2), 7.12–7.27 (m, 6H, o-H, p-H), 7.73–7.80 (m, 4H, m-H). 13C NMR (125 MHz, CDCl3) δ 14.3 (CH2CH3), 16.9 (4-CH3), 19.4 [C(CH3)3], 22.1 (1′-CH3), 27.0 [C(CH3)3], 32.2 (C-4), 37.9 (C-3), 55.0 (C-5), 60.2 (CH2CH3), 63.8 (C-8), 70.0 (C-1′), 122.6 (C-1), 127.4, 127.6 (o-C), 129.5, 129.7 (p-C), 131.1 (C-7), 132.4 (C-8), 133.9, 134.7 (i-C), 135.9, 136.0 (m-C), 147.9 (C-3), 166.6 (COEt). Diastereomer 2: Rf = 0.45 (hexanes/EtOAc, 3:1). 1H NMR (500 MHz, CDCl3) δ 0.68 (d, J = 6.9 Hz, 3H, 4-CH3), 0.98 (d, J = 6.1 Hz, 3H, 1′-CH3), 1.04 [s, 9H, C(CH3)3], 1.30 (t, J = 7.2 Hz, 3H, CH2CH3), 1.58 (br, 1H, OH), 1.93 (dddd, J = 14.5, 7.4, 7.4, 1.5 Hz, 1H, Ha-3), 2.03–2.12 (m, 2H, Hb-3, H-5), 2.16–2.26 (m, 1H, H-4), 3.79 (dq, J = 7.6, 6.1 Hz, 1H, H-1′), 4.06–4.10 (m, 2H, H-8), 4.20 (q, J = 7.2 Hz, 2H, CH2CH3), 5.35 (ddt, J = 15.4, 10.2, 1.4 Hz, 1H, H-6), 5.61 (dt, J = 15.4, 5.7 Hz, 1H, H-7), 5.77 (dt, J = 15.5, 1.5 Hz, 1H, H-1), 6.91 (dt, J = 15.5, 7.4 Hz, 1H, H-2), 7.34–7.46 (m, 6H, o-H, p-H), 7.65–7.72 (m, 4H, m-H). 13C NMR (125 MHz, CDCl3) δ 14.3 (CH2CH3), 15.1 (4-CH3), 19.4 [C(CH3)3], 22.1 (1′-CH3), 27.1 [C(CH3)3], 31.6 (C-4), 38.4 (C-3), 54.9 (C-5), 60.2 (CH2CH3), 63.6 (C-8), 69.8 (C-1′), 122.4 (C-1), 127.4, 127.6 (o-C), 129.4 (p-C), 129.7 (C-6), 132.8 (C-7), 133.8, 134.7 (i-C), 135.95, 136.00 (m-C), 148.3 (C-2), 166.7 (COEt).
:1) and a small quantity of CH2Cl2 to dissolve the remaining DMP to give 13a (356 mg, 0.72 mmol, 80%) as colorless oil (dr 56:39:5:0 by 1H NMR, CHO). Rf = 0.33 (hexanes/EtOAc, 10:1). [α]20D −190 (c 1.0 in CHCl3). FT-IR (ATR) ( cm−1) 2962 (w), 2931 (w), 2857 (w), 2253 (w), 1716 (m), 1689 (m), 1653 (w), 1473 (w), 1427 (w), 1390 (w), 1367 (w), 1314 (w), 1265 (w), 1225 (w), 1178 (w), 1110 (m), 1043 (w), 979 (m), 906 (s), 821 (w), 729 (vs), 702 (vs), 648 (m), 610 (m). 1H NMR (500 MHz, CDCl3) δ 0.74 (d, J = 6.9 Hz, 3H, 4-CH3), 0.79 (d, J = 6.3 Hz, 3H, 4-CH3*), 1.00–1.06 [m, 24H, C(CH3)3, C(CH3)3*, 1′-CH3, 1′-CH3*], 1.30 (t, J = 7.1 Hz, 6H, CH2CH3, CH2CH3*), 1.81–1.88 (m, 1H, Ha*-3), 1.88–1.95 (m, 1H, Ha-3), 1.99–2.05 (m, 2H, H*-4, H*-5), 2.05–2.10 (m, 1H, Hb-3), 2.13–2.19 (m, 1H, Hb*-3), 2.19–2.27 (m, 1H, H-4), 2.31–2.38 (m, 1H, H-5), 3.90–3.97 (m, 1H, H*-1′), 4.03–4.09 (m, 1H, H-1′), 4.15–4.23 (m, 4H, CH2CH3, CH2CH3*), 5.67 (d, J = 15.5 Hz, 1H, H*-1), 5.77 (d, J = 15.7 Hz, 1H, H-1), 6.00–6.10 (m, 2H, H-7, H*-7), 6.57 (dd, J = 15.7, 10.4 Hz, 1H, H-6), 6.72–6.80 (m, 1H, H*-6), 6.80–6.88 (m, 2H, H-2, H*-2), 7.33–7.49 (m, 12H, o-H, p-H), 7.59–7.72 (m, 8H, m-H), 9.47 (d, J = 7.8 Hz, 1H, CHO), 9.54 (d, J = 8.1 Hz, 1H, CHO). 13C NMR (125 MHz, CDCl3) δ 14.4 (CH2CH3), 15.8 (4-CH3), 17.3 (4-CH3*), 19.45, 19.48 [C(CH3)3], 21.7 (1′-CH3), 22.7 (1′-CH3*), 27.15, 27.17 [C(CH3)3], 32.2 (C-4), 32.4 (C-4*), 37.4 (C-3*), 38.1 (C-3), 55.4 (C-5), 55.9 (C-5*), 60.4 (CH2CH3), 69.26, 69.30 (C-1′, C-1′*), 123.2 (C-1*), 123.4 (C-1), 127.59, 127.63, 127.90, 127.96 (o-C), 129.81, 129.84, 130.04 (m-C), 133.3, 133.5, 134.3, 134.4 (p-C), 136.06, 136.08 (C-7, C-7*), 146.6 (C-6*), 145.0 (C-2, C-2*), 156.1 (C-6), 157.8 (COO*), 166.5 (COO), 193.5, 193.8 (CHO, CHO*). MS (ESI) m/z 515.26 [M + Na]+, 284.15 [M − C18H23OSi + H]+, 259.13, 233.11. HRMS (ESI) obsd 515.2582, calc. for C30H40O4SiNa+: 515.2588.
:1) to give 12a (20 mg, 0.04 mmol, 38%) as a colorless liquid (dr 60:33:7:0 by 1H NMR). Rf = 0.24. FT-IR (ATR) ( cm−1) 3071 (w), 2960 (m), 2930 (m), 2893 (w), 2857 (w), 1717 (s), 1870 (w), 1668 (s), 1630 (w), 1593 (m), 1473 (w), 1427 (m), 1364 (m), 1312 (w), 1251 (s), 1222 (w), 1176 (w), 1107 (vs), 1043 (w), 999 (s), 980 (w), 938 (w), 909 (w), 822 (m), 770 (w), 735 (s), 702 (vs), 686 (w), 646 (w), 608 (m). 1H NMR (500 MHz, CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H, 4-CH3), 0.77 (d, J = 6.9 Hz, 3H, 4-CH3*), 0.99 (d, J = 6.1 Hz, 3H, 1′-CH3), 1.02 (d, J = 6.5 Hz, 3H, 1′-CH3*), 1.03–1.06 (m, 18H, C(CH3)3, C(CH3)3*), 1.30 (t, J = 7.1 Hz, 3H, CH2CH3), 1.31 (t, J = 7.1 Hz, 3H, CH2CH3*), 1.80–2.00 (m, 4H, Ha-3, Ha*-3, H*-4, H-5), 2.00–2.10 (m, 1H, Hb-3), 2.11–2.24 (m, 3H, Hb*-3, H-4, H*-5), 2.26 (s, 3H, H-11), 2.28–2.32 (m, 3H, H*-11), 3.83–3.90 (m, 1H, H-1′), 3.96–4.02 (m, 1H, H*-1′), 4.19 (q, J = 7.1 Hz, CH2CH3), 4.20 (q, J = 7.1 Hz, CH2CH3*), 5.66 (dt, J = 15.8, 1.5 Hz, H*-1), 5.76 (dt, J = 15.6, 1.4 Hz, 1H, H-1), 5.89 (dd, J = 15.5, 9.9 Hz, 1H, H-6), 6.05–6.12 (m, 4H, H-6, H*-7, H-9, H*-9), 6.15 (dd, J = 15.3, 10.8 Hz, 1H, H-7), 6.79 (ddd, J = 15.1, 8.4, 6.6 Hz, 1H, H*-2), 6.87 (dt, J = 15.6, 7.4 Hz, 1H, H-2), 7.05 (dd, J = 15.6, 10.8 Hz, 1H, H-8), 7.08–7.21 (m, 1H, H*-8), 7.33–7.47 (m, 12H, o-H, p-H), 7.60–7.73 (m, 8H, m-H). 13C NMR (125 MHz, CDCl3) δ 14.4 (CH2CH3, CH2CH3*), 15.6 (4-CH3), 17.5 (4-CH3*), 19.4, 19.5 [C(CH3)3], 22.0 (1′-CH3), 22.6 (1′-CH3*), 27.2, 27.5, 27.49, 27.52 [C(CH3)3], 32.2 (C-4), 32.6 (C-4*), 37.7 (C-3), 38.4 (C-3*), 56.0 (C-5), 56.2 (C-5*), 60.4 (CH2CH3, CH2CH3*), 69.7 (C-11, C-11*), 122.9 (C-1*), 123.0 (C-1), 127.5, 127.6, 127.80, 127.83 (o-C), 129.3 (C-9, C-9*), 129.5, 129.7, 129.9, 130.0 (m-C), 131.9, 132.2 (C-7, C-7*), 133.7, 133.8, 134.6, 134.7, 136.06, 136.1, 136.13 (p-C), 142.6 (C-6, C-6*), 143.2 (C-8), 143.6 (C-8*), 147.4 (C-2*), 147.8 (C-2), 166.6, 166.7 (CO2Et), 198.8, 199.0 (C-10). MS (ESI) m/z 283.15 [M − C18H23OSi]+, 265.14, 231.14, 105.08. HRMS (ESI) obsd 555.2890, calc. for C33H44O4SiNa+: 555.2901.
:1) to give 11a (25 mg, 0.05 mmol, 33%) as a colorless oil (dr 55:42:3:0 by 1H NMR, H-1′). Rf = 0.39. FT-IR (ATR) ( cm−1) 3072 (w), 2959 (m), 2930 (m), 2856 (m), 2359 (w), 2255 (w), 1729 (s), 1713 (s), 1589 (w), 1473 (m), 1461 (m), 1427 (m), 1392 (w), 1375 (m), 1354 (m), 1308 (w), 1277 (br), 1206 (m), 1178 (m), 1155 (m), 1109 (s), 1054 (w), 1030 (w), 1007 (w), 975 (w), 939 (w), 908 (s), 861 (w), 821 (m), 731 (vs), 702 (vs), 648 (w), 609 (m). 1H NMR (500 MHz, CDCl3) δ 0.94 (d, J = 7.2 Hz, 3H, 2-CH3*), 0.98 (d, J = 7.3 Hz, 3H, 2-CH3), 1.02 (d, J = 6.4 Hz, 3H, 1′-CH3), 1.03–1.05 (m, 20H, C(CH3)3, Ha-3, Ha*-3), 1.12 (d, J = 6.3 Hz, 3H, 1′-CH3), 1.24–1.30 (m, 6H, CH2CH3, CH2CH3*), 1.54–1.71 (m, 4H, H-3a, H*-3a, H-1, H*-1), 1.89–2.00 (m, 2H, Hb-3, Hb*-3), 2.07–2.22 (m, 4H, H-7a, H*-7a, H-2, H*-2), 2.23 (s, 3H, COCH3), 2.24 (s, 3H, COCH3*), 2.82–2.94 (m, 2H, H-4, H*-4), 3.64–3.72 (m, 2H, H-5, H*-5), 4.02–4.08 (m, 1H, H-1′), 4.08–4.12 (m, 1H, H*-1′), 4.12–4.21 (m, 4H, CH2CH3, CH2CH3*), 5.62–5.70 (m, 2H, H-7, H*-7), 6.03–6.08 (m, 1H, H-6), 6.24–6.30 (m, 1H, H-6), 7.34–7.45 (m, 12H, o-H, p-H), 7.64–7.71 (m, 8H, m-H). 13C NMR (125 MHz, CDCl3) δ 14.4 (CH2CH3, CH2CH3*), 18.0 (2-CH3*), 19.28 (2-CH3), 19.31, 19.4 [C(CH3)3], 22.2, 24.0 (1′-CH3, 1′-CH3*), 27.2, 27.3 [C(CH3)3], 28.54, 28.56 (COCH3, COCH3*), 32.7, 34.1 (C-2, C-2*), 38.3, 38.5 (C-3, C-3*), 43.2 (C-7a), 44.1, 44.6 (C-3a, C-3a*), 44.7 (C-7a*), 46.76, 46.83 (C-4, C-4*), 51.0, 51.6 (C-1, C-1*), 55.4, 55.5 (C-5, C-5*), 60.7 (CH2CH3, CH2CH3*), 69.3, 70.7 (C-2, C-2*), 122.5, 123.0 (C-7, C-7*), 127.4, 127.5, 127.6, 127.7 (o-C), 129.5, 129.7, 129.75 (m-C), 132.5 (C-6, C-6*), 133.5, 133.9, 134.2 (i-C), 134.9 (C-6, C-6*), 135.1 (i-C), 136.0, 136.07, 136.10 (p-C), 174.78, 174.84 (CO2Et), 207.32, 207.39 (COCH3). MS (ESI) m/z 321.13, 283.15 [M − C18H23OSi]+, 265.14, 231.14, 203.15 [M − C16H19OSi − C14H20O2 − H]+, 187.15, 145.10, 105.08. HRMS (ESI) obsd 555.2895, calc. for C33H44O4SiNa+: 555.2901.
Acknowledgements
The authors would like to thank the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, Hoffmann-La Roche Basel (industrial internship for I. L.), the European School of Chemistry, Polymers and Materials Science (ECPM) in Strasbourg (French-German Double Master program for G. B.) and the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg for generous financial support.References
- Reviews: (a) R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed; (b) B. J. L. Royles, Chem. Rev., 1995, 95, 1981–2001 CrossRef CAS; (c) H.-G. Henning and A. Gelbin, Adv. Heterocycl. Chem., 1993, 57, 139–185 CrossRef CAS; (d) T. Rosen, Drugs Future, 1989, 14, 153–163 Search PubMed.
- K. Jomon, Y. Kuroda, M. Ajisaka and H. Sakai, J. Antibiot., 1972, 25, 271–280 CrossRef CAS PubMed.
- S. Ito and Y. Hirata, Bull. Chem. Soc. Jpn., 1977, 50, 1813–1820 CrossRef CAS.
- J. Antosch, F. Schaefers and T. A. M. Gulder, Angew. Chem., 2014, 126, 3055–3058 ( Angew. Chem., Int. Ed. , 2014 , 53 , 3011–3014 ) CrossRef.
- (a) M. Jakobi, G. Winkelmann, D. Kaiser, C. Kempter, G. Jung, G. Berg and H. Bahl, J. Antibiot., 1996, 49, 1101–1104 CrossRef CAS PubMed; (b) P. R. Graupner, S. Thornburgh, J. T. Mathieson, E. L. Chapin, G. M. Kemmitt, J. M. Brown and C. E. Snipes, J. Antibiot., 1997, 50, 1014–1019 CrossRef.
- S. Cao, J. A. V. Blodgett and J. Clardy, Org. Lett., 2010, 12, 4652–4654 CrossRef CAS PubMed.
- J. A. V. Blodgett, D.-C. Oh, S. Cao, C. R. Currie, R. Kolter and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11692–11697 CrossRef CAS PubMed.
- S. Kanazawa, N. Fusetani and S. Matsunaga, Tetrahedron Lett., 1993, 34, 1065–1068 CrossRef CAS.
- N. Cramer, S. Helbig, A. Baro, S. Laschat, R. Diestel, F. Sasse, D. Mathieu, C. Richter, G. Kummerlöwe, B. Luy and H. Schwalbe, ChemBioChem, 2008, 9, 2474–2486 CrossRef CAS PubMed.
- (a) R. K. Boeckman, C. H. Weidner, R. B. Perni and J. J. Napier, J. Am. Chem. Soc., 1989, 111, 8036–8037 CrossRef CAS; (b) R. K. Boeckman, J. J. Napier, E. W. Thomas and R. I. Sato, J. Org. Chem., 1983, 48, 4152–4154 CrossRef CAS.
- (a) L. A. Paquette, D. Macdonald and L. G. Anderson, J. Am. Chem. Soc., 1990, 112, 9292–9299 CrossRef CAS; (b) L. A. Paquette, D. Macdonald, L. G. Anderson and J. Wright, J. Am. Chem. Soc., 1989, 111, 8037–8039 CrossRef CAS; (c) L. A. Paquette, J. L. Romine and H.-S. Lin, Tetrahedron Lett., 1987, 28, 31–34 CrossRef CAS.
- A. C. Hart and A. J. Phillips, J. Am. Chem. Soc., 2006, 128, 1094–1095 CrossRef CAS PubMed.
- (a) N. Cramer, M. Buchweitz, S. Laschat, W. Frey, A. Baro, D. Mathieu, C. Richter and H. Schwalbe, Chem. – Eur. J., 2006, 12, 2488–2503 CrossRef CAS PubMed; (b) N. Cramer, S. Laschat, A. Baro, H. Schwalbe and C. Richter, Angew. Chem., 2005, 117, 831–833 ( Angew. Chem., Int. Ed. , 2005 , 44 , 820–822 ) CrossRef.
- W. R. Roush and C. K. Wada, J. Am. Chem. Soc., 1994, 116, 2151–2152 CrossRef CAS.
- M. J. Kurth, D. H. Burns and M. J. O'Brien, J. Org. Chem., 1984, 49, 731–733 CrossRef CAS.
- J. K. Whitesell and M. A. Minton, J. Am. Chem. Soc., 1987, 109, 6403–6408 CrossRef CAS.
- G. Mehta, A. N. Murthy and D. S. K. Reddy, Tetrahedron Lett., 1987, 28, 1467–1468 CrossRef CAS.
- (a) K. A. Ahrendt and R. M. Williams, Org. Lett., 2004, 6, 4539–4541 CrossRef CAS; (b) C. Niu, T. Pettersson and M. J. Miller, J. Org. Chem., 1996, 61, 1014–1022 CrossRef CAS; (c) C. R. Schmid and J. D. Bryant, Org. Synth., 1998, 9, 450 Search PubMed; C. R. Schmid and J. D. Bryant, Org. Synth., 1995, 72, 6 Search PubMed; (d) J. Jurczak, S. Pikul and T. Bauer, Tetrahedron, 1986, 42, 447–488 CrossRef CAS.
- Y.-J. Hu, X.-D. Huang, Z.-J. Yao and Y.-L. Wu, J. Org. Chem., 1998, 63, 2456–2461 CrossRef CAS PubMed.
- T. Horiguchi, T. Oritani and H. Kiyota, Tetrahedron, 2003, 59, 1529–1538 CrossRef CAS.
- R. E. Ireland, R. H. Mueller and A. K. Willard, J. Am. Chem. Soc., 1976, 98, 2868–2877 CrossRef CAS.
- P. F. Godenschwager and D. B. Collum, J. Am. Chem. Soc., 2008, 130, 8726–8732 CrossRef CAS PubMed.
- T. N. Snaddon, P. Buchgraber, S. Schulthoff, C. Wirtz, R. Mynott and A. Fürstner, Chem. – Eur. J., 2010, 16, 12133–12140 CrossRef CAS PubMed.
- C.-L. Chen, K. Namba and Y. Kishi, Org. Lett., 2009, 11, 409–412 CrossRef CAS PubMed.
- S. D. Burke, W. F. Fobare and G. J. Pacofsky, J. Org. Chem., 1983, 48, 5221–5228 CrossRef CAS.
- I. Paterson and A. N. Hulme, J. Org. Chem., 1995, 60, 3288–3300 CrossRef CAS.
- S. N. Smirnov, N. S. Golubev, G. S. Denisov, H. Bendict, P. Schah-Mohammedi and H.-H. Limbach, J. Am. Chem. Soc., 1996, 118, 4094–4101 CrossRef CAS.
- J.-C. Han, L.-Z. Liu, Y.-Y. Chang, G.-Z. Yue, J. Guo, L.-Y. Zhou, C.-C. Li and Z. Yang, J. Org. Chem., 2013, 78, 5492–5504 CrossRef CAS PubMed.
- (a) M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 RSC; (b) J.-Y. Wach and K. Gademann, Synlett, 2012, 163–170 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Attempted preparation of phosphonium salt 8a, preparation of derivatives 16, 17, 19 and 20, preparation of 15b and its rearrangement, alternative route to 15a, quantum-chemical investigation of the intramolecular Diels–Alder cycloaddition to 11a, biological activity studies as well as 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c5ob01491c |
| This journal is © The Royal Society of Chemistry 2016 |