
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain-induced effects in the electronic and spin properties of a monolayer of ferromagnetic GdAg2†
A.
Correa
abc,
B.
Xu
def,
M. J.
Verstraete
 de and
L.
Vitali
*acg
de and
L.
Vitali
*acg
aDepartamento de fisica de materiales, Universidad del Pais Vasco, 20018 San Sebastián, Spain. E-mail: lucia.vitali@ehu.es
bDonostia International Physics Center, 20018 San Sebastian, Spain
cCentro de Fisica de Materiales (CSIC-UPV/EHU) y Material Physics Center, 200.18 San Sebastián, Spain
dCESAM and Département de Physique, Université de Liège, B-4000 Sart Tilman, Belgium
eEuropean Theoretical Spectroscopy Facility, Belgium Web: http://www.etsf.eu
fDepartment of Physics and Institute for Nanoscience and Engineering, University of Arkansas, Fayetteville, Arkansas 72701, USA
gIkerbasque Foundation for Science, 48013 Bilbao, Spain
First published on 2nd November 2016
Abstract
We report on the structural, electronic and magnetic properties of a monolayer of GdAg2, forming a moiré pattern on Ag(111). Combining scanning tunneling microscopy and ab initio spin-polarized calculations, we show that the electronic band structure can be shifted linearly via thermal controlled strain of the intra-layer atomic distance in the range of 1–7%, leading to lateral hetero-structuring. Furthermore, the coupling of the incommensurable GdAg2 alloy layer to the Ag(111) substrate leads to spatially varying atomic relaxation causing subsurface layer buckling, texturing of the electronic and spin properties, and inhomogeneity of the magnetic anisotropy energy across the layer. These results provide perspectives for control of electronic properties and magnetic ordering in atomically-thin layers.
Geometrical structure, lattice periodicity and atomic arrangement are subtly intertwined with the electronic properties of materials. Sub-angstrom changes in the atomic distance are sufficient to modify the physical and chemical properties, such as the band-structure, carrier mobility and the chemical reactivity.1–15 The deposition of two-dimensional layered crystals on mechanically stretchable or bendable substrates can produce one-dimensional strained structures with intriguing properties.1,2 Similarly, biaxial-strained two-dimensional layers have been obtained exploiting the interface energy between lattice mismatched epitaxial layers.3–9,15 The efficiency of these strategies has generated considerable progress in tailoring the electronic and optical properties.3,8–12,15 Few sparse works have demonstrated instead the role of surface-strain in bulk or thick layers of ferromagnetic materials.4–7,12–14 On the other hand, in mesoscopic systems formed by few-layers thick ferromagnetic materials, structure relaxation processes lead to interesting effects in the electronic and magnetic properties.15 This suggests strain as a good method to manipulate the electronic properties, and consequently the magnetic order, also of thin layers of strong magnetic materials. Nonetheless, a general picture on the influence of atomistic structure on the local magnetic order is still missing.
Here, we will show that the local atomic arrangement critically affects the electronic properties of two-dimensional layer and we predict its effect on the magnetic properties of the system. Surface strain inherent in epitaxially-grown two-dimensional layers is sufficient to texture the electronic and the magnetic properties of a ferromagnetic structure stable up to 85 K.16 Specifically, we will characterize a monolayer structure based on rare-earth surface stoichiometric alloy GdAg2 forming a weakly interacting moiré superstructure on Ag(111). This monolayer is particularly suitable to investigate the relation between the structural, electronic and magnetic properties. Indeed, in rare-earth based materials, the exchange interaction process and, consequently, the magnetic order are critically sensible to structural variations and orbital hybridizations5–7,12–14 which affects the delicate interplay between the 5d, 6s nearly-free conduction and the highly-localized 4f electrons.
Furthermore, we will demonstrate that in-plane lattice strain conveys to the formation of hetero-structures with variable alloy unit-cell (in the range of 5.15–5.6 Å) and distinct density of states. Moreover, the incommensurate alloy layer induces an out-of-plane atomic buckling of the supporting substrate with the periodicity of the moiré structure. This prompts a position dependent coupling interaction to the Ag(111) surface, steers a modulation in the density of states and leads to a local weakening of the ferromagnetic order resulting in a spin-texture across the layer. The direct comparison of scanning tunneling microscopy (STM) and spectroscopy (STS), with state-of-the-art spin-polarized numerical simulations based on density functional theory (DFT), facilitate the understanding of the role of strain, structural relaxation and coupling interaction on the physical properties of the system.
A monolayer of GdAg2 forms an incommensurate structure on Ag(111), similar to the previously reported GdAu2 on the Au(111) surface16,17 (see ESI1†). This results in topographic STM images (Fig. 1a) as a moiré superstructure, whose apparent minima and maxima reflect the variation of the geometrical registry of the atoms of the alloy layer with respect to the Ag(111) surface. Three of the canonical geometrical configurations, namely hcp, fcc and top, through which the atoms of the continuous alloy layer must pass periodically are shown in Fig. 1b. Considering each of these stacking configurations separately, we provide a picture of the structural and electronic properties across the alloy layer and at its interface with Ag(111).
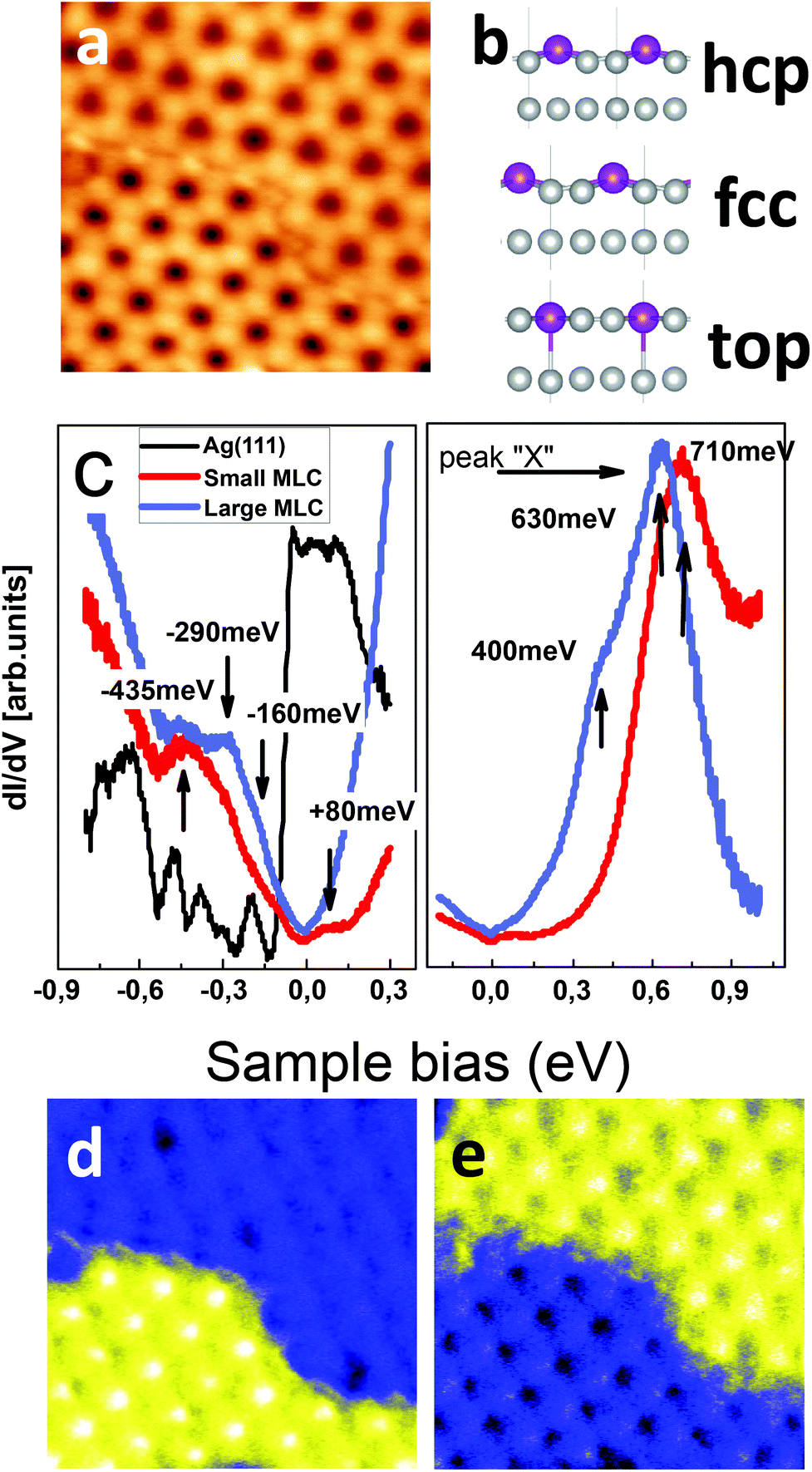 | ||
| Fig. 1 Structure and electronic properties of a monolayer thick GdAg2 on Ag(111). (a) Two GdAg2 moiré superstructures differing in lattice constants are seen in topographic images. Upper: large moiré lattice constant (LMLC); lower: small moiré lattice constant (SMLC). Image size 20 nm × 20 nm (b) Calculated relaxed geometry for the alloy layer in hcp, fcc and top configuration of one monolayer of GdAg2 on Ag(111). (c) dI/dV spectra acquired on the two moiré superstructures. (d–e) Conductance maps at 380 meV and 700 meV, showing the localization of the electronic peaks in LMCL and SMLC, respectively. All experimental data presented here have been acquired using a low temperature scanning tunneling microscope operated at 77 K.20 | ||
Using first-principles calculations18,19 (details in ESI2†), we show that the formation of the superstructure induces different relaxations of the supporting substrate according to the stacking registry of the alloy layer. The Gd atoms are found 2.872 Å (2.870 Å) above the substrate in the fcc (hcp) stacking configuration, while for the top configuration the preferred position is slightly lower, i.e. 2.794 Å. Correspondingly, the Ag atoms of the substrate below the Gd atoms are pushed into the bulk of the crystal (Fig. 1b). The modeling of these configurations allows us to assign the top stacking to the valleys of the topographic image and the fcc and hcp to the hills (i.e., to the “dark” and “bright” sites), respectively. The clearly distinct relaxation of the system in top stacking has a critical role in the electronic properties and in the local spin order of the alloy, as will be shown here.
A closer analysis of Fig. 1a shows the presence of two neighboring moiré superstructures differing in their relative orientation and periodicity where the upper-right one has a large moiré lattice constant (hereafter, LMLC) and the other a smaller one (SMLC). The local density of states, measured using standard lock-in techniques on the hills of the two super-structures, is shown as red and blue lines in Fig. 1c. The corresponding dI/dV spectra achieved on the valley positions and space-resolved energy maps are shown in ESI3–ESI5.† The local spectroscopic data shown in Fig. 1 demonstrate clearly that the two superstructures differ both in their occupied as well as the empty states. A characteristic feature at 400 meV, visible as a shoulder of a higher energy peak occurring at about 630 meV, characterizes the LMLC pattern. This peak (hereafter, labeled “X”) shifts to higher energy (710 meV) as the lattice constant of the moiré pattern is reduced. An unequivocal localization of these states in either of the two moiré areas can be evinced by the conductance maps (Fig. 1c and d), corroborating the formation of lateral electronic hetero-structures. Further differences between the two superstructures can be observed at lower-energy, where an additional small peak can be observed at 80 meV and at higher-energy (see ESI3 and ESI4†). Also the occupied density of state differs in the two superstructures. Their reduced intensity and many shoulders impede, however, a clear quantification of their energy positions. The overall emerging picture is that a reorganization of the electronic properties takes place as a function of the superstructure lattice size. Notably, the surface state of Ag(111) cannot be further observed through the moiré pattern. This suggests that a modification of the electronic properties of the noble metal surface has occurred despite the formation of moiré patterns is indicative of a weak coupling to the supporting substrate.
The spectra shown in Fig. 1b are representative of a series of dI/dV measurements achieved on different moiré superstructures. A careful data analysis of these shows that the energy position of peak X varies as a function of the lattice constant of the superstructure according to the statistical distribution shown in Fig. 2. The observed energy values cluster around two mean values of the superstructure periodicity, 32 and 34 Å. The close relation between the electronic structure and lattice constant is further corroborated by a fit of the data (blue line in Fig. 2), which suggests a linear dependence. This is confirmed by first principle calculations, as it will be shown in the following.
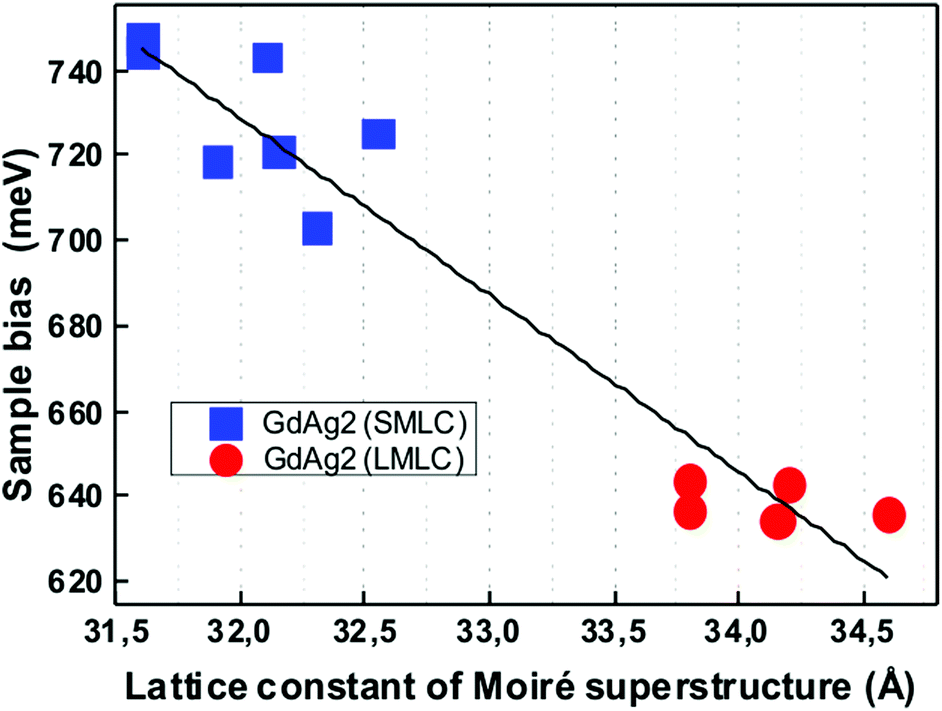 | ||
| Fig. 2 Correlation between the lattice constant of the moiré pattern and the energy of electronic structure. Energy shifts around equilibrium positions are observed monitoring peak X both in the SMLC (i.e. large alloy unit cell) and LMLC (i.e. small alloy unit cell) patterns (blue and red symbols). The line is a linear fit of the experimental data points. | ||
To understand the observed correlation of the lattice constant of the superstructure and the electronic properties, we have applied to the system the coincidence model proposed by Hermann to describe moiré patterns.21 This model envisions the moiré super-structure as a coincidence network formed by the atoms of the overlayer that periodically match those of the substrate. Outcomes of the model are the size of the unit cell of the alloy and its angle of rotation α with respect to the Ag(111) high symmetry directions (see ESI6† for further details). The structural parameters of two moiré patterns among all predictable for this system, (Table 1) allow a direct correspondence to the experimentally observed high-resolution images on the two moiré superstructures (see ESI3†). Experimental results and modeling consistently show that the unit cell size of the alloy monolayer and its orientation differ in the two moiré patterns seen in Fig. 1. The two periodicities of 32 and 34 Å respectively, correspond to alloy unit cells differing in size by a relative strain of 1%, while the angle α is almost constant. The superstructure with larger periodicity, i.e. with the larger coincidence distance, is formed by the alloy layer with the smaller lattice constant, in good agreement with the experimental observations21–23 and intuitive expectations.
| Experimentally observed | Values expected [Hermann] | |||
|---|---|---|---|---|
| Moiré periodicity | 32 Å | 34 Å | 32.12 ± 0.1 Å | 34.2 ± 0.1 Å |
| Rotation angle γ | 28 ± 1° | 20 ± 1° | 28.7 ± 0.1° | 20.9 ± 0.1° |
| Atomic distance between Gd atoms | 5.23 ± 0.2 Å | 5.13 ± 0.2 Å | 5.247 ± 0.001 Å | 5.156 ± 0.001 Å |
| Nearest neighbor distance | 3.033 Å | 2.981 Å | ||
| β | 3.1 ± 0.2° | 13.7 ± 0.2° | ||
| α | 34.62 ± 0.02° | 34.67 ± 0.02° | ||
Guided by this understanding of the structural origin of the two moiré patterns, we simulated their electronic properties with density functional theory (see ESI2†). The band structure of the GdAg2 alloy is first calculated with the supporting layer Ag(111) and then compared with the free standing case. In this way, we separate the effect of strain on the alloy from the interaction with the substrate, achieve a base line for the band character, estimate the importance of surface interactions, and of the magnetic anisotropy energy. The electronic structure of the GdAg2/Ag(111) was calculated using a commensurate √3 × √3 supercell of the noble metal (111) surface in the plane in fcc stacking, which is the configuration with lowest-energy (Fig. 3a). The hcp and fcc stackings (ESI2–ESI7† share most properties, but differ from the top case in the band energy positions and dispersion.
 | ||
| Fig. 3 Density functional theory calculations of the band structure of GdAg2: (a) Calculated band structure of the GdAg2 alloy layer with (√3 × √3)R30° periodicity, in fcc configuration on Ag(111). Color lines highlight the band structure responsible for the main observed density of states in Fig. 1. (b) Calculated band structure for the free-standing GdAg2 alloy layer with the same lattice constant as the Ag (111) substrate (c) Calculated band structures of the free standing GdAg2 alloy layer for different lattice constants. The main bands that are observed in STS spectroscopy are highlighted. | ||
The free-standing GdAg2 alloy layer, calculated using the same parameters as in the supported case, is shown in Fig. 3b. Upon the removal of the supporting substrate few bands shift towards lower energies considerably more than the other bands of the system (C and C′). This is an artefact of the free-standing approximation, which highlights their coupling potential with the Ag(111) surface. The effective coupling potential acting on the surface-supported alloy monolayer is expected to be intermediate between the free standing and the theoretical commensurate case shown in Fig. 3a and b, respectively.
The free standing alloy is then progressively strained to survey the impact on the band structure. Relaxed and strained structures are compared in Fig. 3b and c. The color code of the lines highlights in both cases the correspondence of the two bands around the Γ point, namely X and D, whose contribution dominates the dI/dV spectra. These have a mixed Gd-d, Ag p and Ag d character and play a critical role in the magnetic character of the system.16 Outcome of these calculations is the relative position of the D and X bands as a function of strain, while their absolute energy position is shifted towards negative energies by the approximation used as discussed above. By increasing the size of the alloy unit cell, i.e., decreasing the periodicity of the moiré supercell, the empty states band X shifts towards higher energies (Fig. 3c) explaining quite straightforwardly the shift of peak X shown in Fig. 1 and 2. A similar effect was observed also in graphene layers and in MoS2 where a linearly increasing gap of about 100 meV per strain percentage was reported.24,25 Energy shifts are also predicted for the occupied states (red line). The clearly diminishing intensity at low energy and the appearance of a peak at 80 meV with the increased lattice size confirm the predicted trend of an upward shift of the D band. Bearing in mind the shortcomings of the used approximation and the onset of Ag(111) bulk state,26,27 not visible in the present calculations, the agreement between experimental and theoretical results can explain quite straightforwardly the main feature of the density of states as a function of lattice structure.
Remarkably, on Au(111) the GdAu2 alloy do not form moiré hetero-structures. This can be explained considering the calculated surface binding energies using Esurf = Ehetero − EML − Eslab, where Ehetero, EML, and Eslab are the total energies of the hetero-structure, the free standing monolayer, and of the 7-layer noble metal substrate, respectively. Whereas, the binding energy of GdAg2 on Ag(111) is in the range 3.10–3.97 eV per unit cell in the three stacking configurations, a considerably higher binding energy (between 9.96–10.48 eV) is found for GdAu2 on Au(111). Such energy limits the spontaneous arrangements and orientation of the GdAu2 alloy layer on the Au surface compared with the case of Ag.
The GdAg2 alloy layer can be strained further by depositing Gd at a slightly lower temperature (240 °C). As shown in Fig. 4, at this temperature large portions of the surface are tessellated by hexagonal cells of uniform size, formed by a GdAg2 layer with a lattice constant of 5.5 Å. Each hexagonal tile is 5–6 GdAg2 unit cells wide (Fig. 4b). At this critical size strain cannot be sustained further and discommensuration lines appear as sketched in Fig. 4c. Despite the clear change in the STS density of states measured at the discommensuration lines (Fig. 4d), the spectrum measured on the tile closely resembles the one of the moiré superstructures reported in Fig. 1. The main difference is a further decreased of the low-energy density of occupied states, where the peak at −440 meV remain almost unaffected and the lowest unoccupied electronic feature shift further to +270 meV. This agrees with the predicted trend of the onset of the D band as a function of lattice periodicity already discussed for the moiré superstructures.
 | ||
| Fig. 4 Hexagonal network of GdAg2 under large strain. (a–b) Topographic images of the alloy layer showing hexagonal patterns tessellating the Ag(111) surface. (a. 25 × 25 nm, 1 eV; b. 13 × 13 nm, −31 meV). The alloy unit cell (black) and hexagons (white) are drawn on the figure as guide to the eye. (c) Graphical representation of the hexagonal network. (d) dI/dV spectra taken at different positions moving from the center towards the disconmensutration line. | ||
The magnetic character of the GdAg2 alloys can be probed by considering the energies of the in-plane and out-of-plane magnetic orientations for the three stacking shown in Table 2. The in-plane magnetization is energetically preferred with respect to the out-of-plane direction by 168 meV for hcp and fcc stacking. On the contrary, the top stacking configuration has no anisotropy energy and, is energetically much less stable (0.7–0.8 eV). An in-plane ferromagnetic character was experimentally observed in magnetic measurements averaging over the moiré superstructure of GdAg2.16 As the continuity of the layer imposes that GdAg2 alloy will be found also in top position, our calculations suggest the formation of a magnetic texture along the layer, where top positions behave as paramagnetic dots embedded in an otherwise in-plane ferromagnetic layer. Even if the magnetic hardness here described were overestimated by the imposition of commensurate boundary conditions, which increases the atomic orbitals overlap of the alloy and of the Ag(111) substrate, we expect that the trend in the magnetic anisotropy will be preserved.
| Calculated magnetization anisotropy energy | In-plane | Out-of-plane | ||||
|---|---|---|---|---|---|---|
| Surface alloy position | hcp | fcc | top | hcp | fcc | top |
| Energy relative to in-plane fcc (meV) | 24 | 0 | 852 | 192 | 168 | 852 |
The calculated magnetic anisotropy energies between in-plane and out-of-plane spin orientation are quite large in fcc and hcp, and would correspond to a Curie temperature of 1950 K according to mean field theory. The observed critical temperature of 85 K (ref. 16) can be understood in terms of in-plane magnetic disorder, where the spin orientation is parallel to the surface but fluctuates between different in-plane orientations. The very low out-of-plane anisotropy in the top configuration should further weaken the total magnetic state and contribute to reduce the observed Tc. Free standing alloy layers subjected to strain present similar anisotropy energies (see Table S1 of ESI†).
Conclusions
In conclusion, the intimate relation between atomic structure, electronic and magnetic properties allows for controlling the physical properties of materials and for modulating in space the electronic properties and the magnetic order. We compared the structural and local spectroscopic measurements with the theoretical prediction to survey two major electronic contributions in the occupied and empty band structure of the incommensurate GdAg2 monolayer grown on Ag(111) as a function of in-plane lattice strain. This leads to surface electronic hetero-structuring. Furthermore, out-of-plane structural relaxation and buckling of the atomic structure result in a variation of the interlayer distance, texturing the electronic and magnetic properties of the GdAg2 monolayer with the periodicity of the moiré superstructure. We predict that the magnetic hardness changes across the moiré superstructure leading to a ferromagnetic layer with embedded paramagnetic dots corresponding to the top stacking configuration of the GdAg2 alloy. We believe that the intimate relation found here between structural, electronic and magnetic properties have a general validity in weakly interacting layered systems, most of which form moiré superstructures.Acknowledgements
AC and LV acknowledge the financial support of the Spanish ministry of economy (MAT2013-46593-C6-4-P).MJV and BX acknowledge two ARC grants (TheMoTherm #10/15-03 and AIMED # 15/19-09) from the Communauté Française de Belgique, a PDR project (GA T.1077.15) from the Fonds National pour la Recherche Scientifique (Belgium). Computer time was provided by CECI, SEGI, Zenobe/CENAERO (Walloon region GA 1117545), and PRACE-2IP and 3IP (EU FP7 GA RI-283493 and RI-312763) on EPCC Archer.
References
- H. J. Conley, B. Wang, J. I. Ziegler, R. F. Haglund Jr., S. T. Pantelides and K. I. Bolotin, Nano Lett., 2013, 13, 3626 CrossRef CAS PubMed.
- A. Castellanos-Gomez, R. Roldan, E. Cappelluti, M. Buscema, F. Guinea, H. S. J. van der Zant and G. A. Steele, Nano Lett., 2013, 13, 5361 CrossRef CAS PubMed.
- S. Cheng, J. Yu, T. Ma and N. M. R. Peres, Phys. Rev. B: Condens. Matter, 2015, 91, 075410 CrossRef.
- G. Neuhold and K. Horn, Phys. Rev. Lett., 1997, 78, 1327 CrossRef CAS.
- I. N. Yakovkin, T. Komesu and P. A. Dowben, Phys. Rev. B: Condens. Matter, 2002, 66, 035406 CrossRef.
- C. Waldfried, T. McAvoy, D. Welipitiya, T. Komesu, P. A. Dowben and E. Vescovo, Phys. Rev. B: Condens. Matter, 1998, 58, 7434 CrossRef CAS.
- A. VI Andrianov, O. A. Savel'eva, E. Bauer and J. B. Staunton, Phys. Rev. B: Condens. Matter, 2011, 84, 132401 CrossRef.
- M. Bokdam, T. Amlaki, G. Brocks and P. J. Kelly, Phys. Rev. B: Condens. Matter, 2014, 89, 201404R CrossRef.
- B. Amorim, A. Cortijo, F. de Juan, A. G. Grushin, F. Guinea, A. Gutiérrez-Rubio, H. Ochoa, V. Parente, R. Roldán, P. San-Jose, J. Schiefele, M. Sturla and M. A. H. Vozmediano, Phys. Rep., 2016, 617, 1 CrossRef CAS.
- M. M. Fogler, F. Guinea and M. I. Katsnelson, Phys. Rev. Lett., 2008, 101, 226804 CrossRef CAS PubMed.
- N. N. Klimov, S. Jung, S. Zhu, T. Li, C. A. Wright, S. D. Solares, D. B. Newell, N. B. Zhitenev and J. A. Stroscio, Science, 2010, 336, 1557 CrossRef PubMed.
- E. Vescovo, C. Carbone and O. Rader, Phys. Rev. B: Condens. Matter, 1993, 48, 7731 CrossRef CAS.
- M. Getzlaff, Surface magnetism, Springler Verlag, Berlin, 2010 Search PubMed.
- C. Waldfried, D. N. McIlroyy and P. A. Dowben, J. Phys.: Condens. Matter, 1997, 9, 10615 CrossRef CAS.
- O. O. Brovko, D. Bazhanov, H. L. Meyerheim, D. Sander, V. S. Stepanyuk and J. Kirschner, Surf. Sci. Rep., 2014, 69, 159 CrossRef CAS.
- M. Ormaza, L. Fernandez, M. Ilyn, A. Magaña, B. Xu, M. J. Verstraete, M. Gastaldo, M. A. Valbuena, P. Gargiani, A. Mugarza, A. Ayuela, L. Vitali, M. Blanco-Rey, F. Schiller and J. E. Ortega, Nano Lett., 2016, 16, 4230 CrossRef CAS PubMed.
- L. Fernandez, M. Blanco-Rey, M. Ilyn, L. Vitali, A. Magaña, A. Correa, P. Ohresser, E. Ortega, A. Ayuela and F. Schiller, Nano Lett., 2014, 14, 2977 CrossRef CAS PubMed.
- X. Gonze, Z. Kristallogr., 2005, 220, 558 CAS.
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval, D. Caliste, R. Caracas, M. Côté, T. Deutsch, L. Genovese, Ph. Ghosez, M. Giantomassi, S. Goedecker, D. R. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet, M. J. T. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf, M. Torrent, M. J. Verstraete, G. Zerah and J. W. Zwanziger, Comput. Phys. Commun., 2009, 180, 2582 CrossRef CAS.
- The non-working performances of the TYTO-STM (SPECS) impeded to reach lower temperatures.
- K. Hermann, J. Phys., 2012, 24, 314210 Search PubMed.
- L. Vitali, M. G. Ramsey and F. P. Netzer, Surf. Sci., 2000, 452, L281 CrossRef CAS.
- P. Sutter, J. T. Sadowski and E. Sutter, Phys. Rev. B: Condens. Matter, 2009, 80, 245411 CrossRef.
- H. J. Conley, B. Wang, J. I. Ziegler, R. F. Haglund, S. J. Pantelides and K. I. Bolotin, Nano Lett., 2013, 13, 3626 CrossRef CAS PubMed.
- H. Peelaers and C. G. Van de Walle, Phys. Rev B: Condens. Matter, 2012, 86, 241401(R) CrossRef.
- L. Vitali, P. Wahl, M. A. Schneider, K. Kern, V. M. Silkin, E. V. Chulkov and P. M. Echenique, Surf. Sci., 2003, 523, L47 CrossRef CAS.
- R. Paniago, R. Matzdorf, G. Meister and A. Goldmann, Surf. Sci., 1995, 336, 113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06398e |
| This journal is © The Royal Society of Chemistry 2016 |