
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of [Fe(L)(bipy)]n spin crossover nanoparticles using blockcopolymer micelles†
Ottokar
Klimm
a,
Christoph
Göbel
a,
Sabine
Rosenfeldt
b,
Florian
Puchtler
c,
Nobuyoshi
Miyajima
d,
Katharina
Marquardt
d,
Markus
Drechsler
e,
Josef
Breu
c,
Stephan
Förster
b and
Birgit
Weber
 *a
*a
aAnorganische Chemie II, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de; Web: http://www.ac2-weber.uni-bayreuth.de
bPhysikalische Chemie I, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany
cAnorganische Chemie I, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany
dBayerisches Geoinstitut, Universität Bayreuth, Universitätsstr. 30, 95440 Bayreuth, Germany
eSoft Matter Electron Microscopy, BIMF, Universität Bayreuth, Universitätsstraße 30, NW I, 95440 Bayreuth, Germany
First published on 19th October 2016
Abstract
Nowadays there is a high demand for specialized functional materials for specific applications in sensors or biomedicine (e.g. fMRI). For their implementation in devices, nanostructuring and integration in a composite matrix are indispensable. Spin crossover complexes are a highly promising family of switchable materials where the switching process can be triggered by various external stimuli. In this work, the synthesis of nanoparticles of the spin crossover iron(II) coordination polymer [Fe(L)(bipy)]n (with L = 1,2-phenylenebis(iminomethylidyne)bis(2,4-pentanedionato)(2-) and bipy = 4,4′-bipyridine) is described using polystyrene-poly-4-vinylprididine blockcopolymer micelles as the template defining the final size of the nanoparticle core. A control of the spin crossover properties can be achieved by precise tuning of the crystallinity of the coordination polymer via successive addition of the starting material Fe(L) and bipy. By this we were able to synthesize nanoparticles with a core size of 49 nm and a thermal hysteresis loop width of 8 K. This is, to the best of our knowledge, a completely new approach for the synthesis of nanoparticles of coordination polymers and should be easily transferable to other coordination polymers and networks. Furthermore, the use of blockcopolymers allows a further functionalization of the obtained nanoparticles by variation of the polymer blocks and an easy deposition of the composite material on surfaces via spin coating.
Introduction
The synthesis of nanostructured and composite materials is of growing importance for coordination polymers and (porous) coordination networks (e.g. MOFs) that are discussed for their high potential in drug delivery, sensing, catalysis, or as contrast agents.1,2 Such applications require their incorporation into composite materials or mesoscopic systems. Additionally, there is a great interest in tailoring size-dependent physical properties such as light absorption. A well-known example is the different colours of colloidal gold nanoparticles.3 Nowadays many methods for the synthesis of metal- or metalchalcogenate nanoparticles are available, e.g. decomposition of complexes, reduction of metal salts, fast precipitation, or inverse micelle techniques.4 However, for the synthesis of nanostructured coordination polymers or coordination networks (including MOFs), the number of methods is restricted and further depends strongly on the used system.1,2 The potential of blockcopolymers for the synthesis of coordination network nanoparticles is almost unexplored.5Spin crossover (SCO) coordination polymers and networks are well established model systems to develop new synthesis strategies for nanostructured coordination compounds and further to investigate size and matrix effects.6 These materials can be switched by external stimuli between a low-spin (LS) and a high-spin (HS) state.7 This switching ability is associated with changes in the chemical and physical properties, explaining the high interest for their applications in sensors,8 display devices9 or as functional contrast agents.10 For potential applications it is essential to understand the interplay between the particle size and/or matrix effects and the SCO properties. In bulk material, cooperative spin transitions with hysteresis (bistability)11 are possible due to intermolecular interactions. There are only a few systems where 1D coordination polymer nanoparticles or 3D coordination network nanoparticles were prepared, generally accompanied by the loss of the spin crossover behaviour of the bulk material.12,13–16 For most of those systems the inverse micelle technique was used13–17 and few attempts were made to entrap nanoparticles in a matrix.16,18,19 Nearly no examples preserving the hysteresis in a nanostructured system are known.18,20 Nanospheres of mononuclear spin crossover complexes can be obtained through the self-assembly of amphiphilic complexes.21 In this work we investigate the size and crystallinity of nanoparticles and show how it is possible to preserve the spin crossover properties of the bulk material down to particle sizes below 50 nm.
We use our extensive library of mononuclear and polynuclear spin crossover complexes to investigate systematically the origin of cooperative effects (e.g. thermal hysteresis loops or steps) during the spin transition.22 Recently, we reported a strong influence of a poly-4-vinylpyridine (P4VP) matrix on the spin transition properties of sub-microcrystals of the SCO coordination polymer [Fe(L)(bipy)]n.23 Inspired by the results we used P4VP based blockcopolymers (BCP) as a template for the nanoparticle synthesis. Polystyrene-poly-4-vinylprididine BCPs are known for their ability to build micellar structures via self-assembly.24 The direct synthesis of nanoparticles (NPs) in the polymer micelles is expected to bring large yields while omitting toxic surfactants and using less toxic solvents compared to the inverse micelle technique. In addition, the block morphology of the polymer offers the possibility of controlled deposition on various surfaces.24 PS-P4VP based BCPs are used for the large area deposition of inorganic nanoparticles such as gold25 or iron oxide.26 The incorporation of SCO NPs in such a polymer matrix may lead to an increased stability against degradation under aerobic conditions. Consequently we decided to use PS-P4VP based micelles as nano-reactors for the synthesis of nanoparticles of the iron(II) complex [Fe(L)(bipy)]n. A schematic representation of the general approach using a self-assembly strategy is given in Scheme 1.
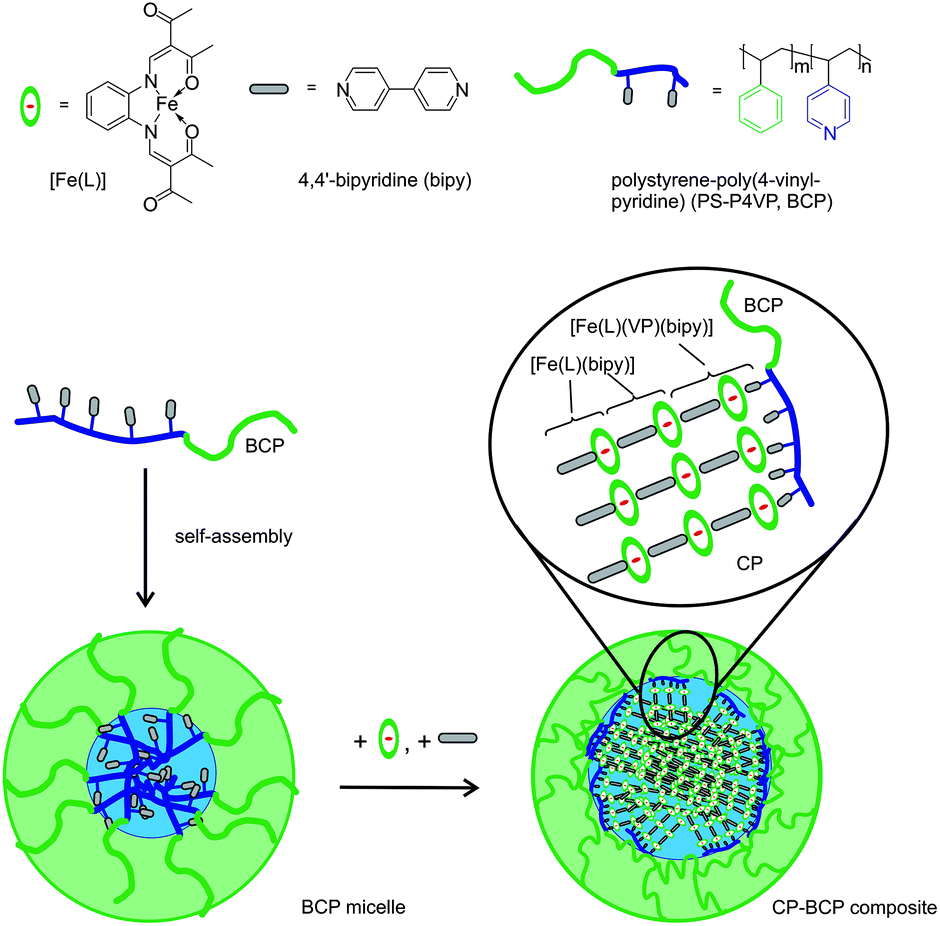 | ||
| Scheme 1 Top: Formula of the compounds with the used abbreviations. Bottom: Schematic representation of a self-assembled blockcopolymer micelle for use as a nano-reactor. Successive addition of the complex [Fe(L)] and the bridging ligand bipy to the blockcopolymer (BCP) micelle will lead to a growth of the coordination polymer (CP) in the core of the micelle. | ||
Results and discussion
Synthesis of the nanoparticles
For the synthesis of the coordination polymer (CP)–blockcopolymer (BCP) composite materials, the self-assembly approach recently reported for sub-microcrystals in a poly(4-vinylpyridine) matrix was used.23 The polystyrene-poly-4-vinylpyridine BCP (PS-P4VP, M = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 1250 PS units, 200 P4VP units, 14% P4VP) was dissolved in THF and stirred for 15 min to allow self-assembly. The resulting empty BCP micelles have a hydrodynamic radius of 63 nm (ESI Fig. S1†). For the CP–BCP composite materials, a solution of the BCP with the precursor iron(II) complex [Fe(L)] was used as the starting material. In the solid state [Fe(L)] is stabilized by two additional methanol molecules as axial ligands that are easily replaced by pyridine derivatives. Based on the size of [Fe(L)] of 12 × 9 Å, approximately one iron complex can coordinate to every third or fourth vinylpyridine (VP) unit. Thus a ratio of 1:4.5 of [Fe(L)]:VP-units is chosen to avoid an uncoordinated iron complex in the reaction mixture. It results in two iron(II) species, namely the penta-coordinated [Fe(L)(VP)] (20%, high-spin, HS) and the octahedral [Fe(L)(VP)2] (80%, low-spin, LS; see the Mössbauer spectrum of compound 1, Fig. S2†).
000 g mol−1, 1250 PS units, 200 P4VP units, 14% P4VP) was dissolved in THF and stirred for 15 min to allow self-assembly. The resulting empty BCP micelles have a hydrodynamic radius of 63 nm (ESI Fig. S1†). For the CP–BCP composite materials, a solution of the BCP with the precursor iron(II) complex [Fe(L)] was used as the starting material. In the solid state [Fe(L)] is stabilized by two additional methanol molecules as axial ligands that are easily replaced by pyridine derivatives. Based on the size of [Fe(L)] of 12 × 9 Å, approximately one iron complex can coordinate to every third or fourth vinylpyridine (VP) unit. Thus a ratio of 1:4.5 of [Fe(L)]:VP-units is chosen to avoid an uncoordinated iron complex in the reaction mixture. It results in two iron(II) species, namely the penta-coordinated [Fe(L)(VP)] (20%, high-spin, HS) and the octahedral [Fe(L)(VP)2] (80%, low-spin, LS; see the Mössbauer spectrum of compound 1, Fig. S2†).
After 2 h of reflux (66 °C) the bridging ligand 4,4′-bipyridine (bipy) was added to the solution. Due to the LS state of the majority species [Fe(L)(VP)2], the ligand exchange is expected to be slowed down. To allow crystalline growth of the CP in the BCP micelles, further successive additions of [Fe(L)] and bipy were used (= number of cycles; each with the same ratio of [Fe(L)]:bipy).
To optimize the reaction conditions for the formation of the CP, the [Fe(L)]:bipy ratio, reaction time, and reaction temperature were varied. An overview of the used reaction conditions is given in Table 1. Tables S1 and S2† summarize the results of this screening. Independent of the [Fe(L)]:bipy ratio, well defined spherical particles are obtained for the samples 7–15. For too high amounts of 4,4′-bipyridine (samples 13–15), gradual spin transitions and an increasing LS fraction were observed (see Table S2†). This hints to the formation of the mononuclear complex [Fe(L)(bipy)2] or very short detached CP chains. Consequently the [Fe(L)]:bipy ratio was fixed to 1:2.5 for the following screening to prevent the formation of [Fe(L)(bipy)2] species. For the samples stirred at room temperature (21–24), larger aggregates and less well defined spherical particles were observed in the TEM pictures (Table S2†), thus a higher reaction temperature (66 °C is the boiling point of the solvent THF) supports the formation of uniform composite nanoparticles. The increase of the reaction time from 15 min (samples 16–20) to 1 hour (samples 2–6) for each cycle improves the SCO properties of the composite materials. This can be related to the time available for crystallite growth, as will be shown in the following section.
| Sample | Cycles | [Fe(L)]:bipy [mol:mol] |
Total time [h] | Temperature [°C] |
|---|---|---|---|---|
| 1 | 0 | 1:0 |
2 | 66 |
| 2 | 1 | 1:2.5 |
3 | 66 |
| 3 | 2 | 1:2.5 |
4 | 66 |
| 4 | 3 | 1:2.5 |
5 | 66 |
| 5 | 4 | 1:2.5 |
6 | 66 |
| 6 | 5 | 1:2.5 |
7 | 66 |
| 7 | 1 | 1:1 |
3 | 66 |
| 8 | 1 | 1:3 |
3 | 66 |
| 9 | 1 | 1:4 |
3 | 66 |
| 10 | 1 | 1:5 |
3 | 66 |
| 11 | 3 | 1:6 |
5 | 66 |
| 12 | 3 | 1:7 |
5 | 66 |
| 13 | 3 | 1:8 |
5 | 66 |
| 14 | 3 | 1:9 |
5 | 66 |
| 15 | 3 | 1:10 |
5 | 66 |
| 16 | 1 | 1:2.5 |
2.25 | 66 |
| 17 | 2 | 1:2.5 |
2.50 | 66 |
| 18 | 3 | 1:2.5 |
2.75 | 66 |
| 19 | 4 | 1:2.5 |
3.00 | 66 |
| 20 | 5 | 1:2.5 |
3.25 | 66 |
| 21 | 1 | 1:2.5 |
3 | RT |
| 22 | 2 | 1:2.5 |
4 | RT |
| 23 | 3 | 1:2.5 |
5 | RT |
| 24 | 5 | 1:2.5 |
7 | RT |
Each additional cycle leads to an increasing amount of CP in the BCP micelles. This is reflected in an increase of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibrations (1640 cm−1 and 1560 cm−1) of [Fe(L)] in the composite material, followed by IR spectroscopy, see Fig. S3.† Transmission electron microscopy (TEM) was used to study the size, shape and uniformity of the obtained material and Mössbauer spectra to determine the conversion of the octahedral [Fe(L)(VP)2] LS fraction. In agreement with an increasing amount of the desired [Fe(L)(bipy)] units of the CP (see Scheme 1), the LS fraction decreases from 47% for sample 3 to 45%, 16%, and finally 0% for the samples 4, 5, and 6, respectively. The results are summarized in Table S3.† Furthermore, magnetic measurements were performed to get information about the spin crossover properties.
O) stretching vibrations (1640 cm−1 and 1560 cm−1) of [Fe(L)] in the composite material, followed by IR spectroscopy, see Fig. S3.† Transmission electron microscopy (TEM) was used to study the size, shape and uniformity of the obtained material and Mössbauer spectra to determine the conversion of the octahedral [Fe(L)(VP)2] LS fraction. In agreement with an increasing amount of the desired [Fe(L)(bipy)] units of the CP (see Scheme 1), the LS fraction decreases from 47% for sample 3 to 45%, 16%, and finally 0% for the samples 4, 5, and 6, respectively. The results are summarized in Table S3.† Furthermore, magnetic measurements were performed to get information about the spin crossover properties.
Control of the crystallite size of the coordination polymer
The investigated CP–BCP composite material consists of a BCP micelle in which the CP was incorporated. As a representative example of the morphology of the CP–BCP composite micelles, in Fig. 1 transmission electron microscopy (TEM) pictures of sample 6 (5 cycles) are given. TEM images of further samples are provided in Table S1.† In all cases a spherical core–shell morphology was obtained. Based on the differences in contrast, the iron complexes in the P4VP part form the core while the PS part is the shell. An energy dispersive X-ray spectroscopy analysis (EDX) of 6 was done to confirm the regio-selectivity of the [Fe(L)(bipy)] complex to the VP units of the BCP micelles (ESI Fig. S4†). | ||
| Fig. 1 Characterization of the CP–BCP composite micelles. (a) TEM pictures of 6 (5 cycles) at two different magnifications illustrating the core shell nature of the particles. (b) Size histogram from the TEM picture given in (a). (c) Autocorrelation function (left) of the CP–BCP particles in THF (43 wt%) and the resulting distribution of the hydrodynamic radius (right). | ||
Dynamic light scattering (DLS) was used to determine the total size of the CP–BCP composite micelles in solution. There is a slight increase in the hydrodynamic size upon loading the BCP micelle with the CP. After the first addition of the CP, the outer diameter of the CP–BCP composite micelle increases from 126 nm for the empty micelle to 147 nm for the loaded one. For the following additions of the CP, the outer diameter stays more or less constant. TEM was used to determine the size of the core and the outer diameter in the dried state. Independent of the amount of added CP, for loaded CP–BCP micelles the same core and total diameter were obtained in the range of the error. This indicates that the BCP micelle is an ideal template for the synthesis of spherical particles, in our case of the CPs. This opens up a new route to obtain CP nanoparticles with a spherical morphology instead of the usually obtained needle-like structures.
Note that the size of the CP–BCP micellar core seems to be determined by the first addition of [Fe(L)(bipy)]n. This may be explained by changes in the morphology of [Fe(L)(bipy)]n in the core of the micelle. Consequently, powder X-ray diffraction (PXRD) (see Fig. 2a and S5†) was used to estimate the crystallinity of the CP in the BCP core. The corresponding results are summarized in Table 2 together with the results for the empty BCP micelles. In Fig. 2b, the size of the core as a function of the number of cycles is compared with the size of the crystalline parts determined by PXRD. Therefore, the half width of the most prominent peak in the PXRD spectra at a 2θ value of 23°, illustrated in Fig. 2a, was analyzed using the Debye–Scherrer equation (1):27
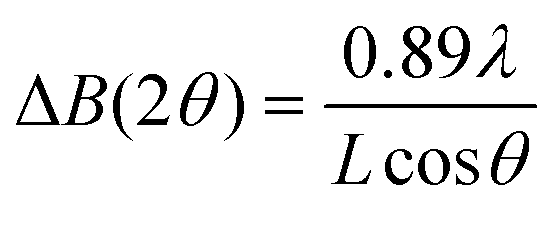 | (1) |
 | ||
| Fig. 2 Particle core size and crystallinity of the CP–BCP composite micelles. (A) PXRD spectra between 22° and 24°. The continuous red line resembles the fit used for the determination of the FWHM. (B) Particle core size determined by TEM (dots) and calculated via the Debye–Scherrer equation from the PXRD data (open circles) as a function of the number of reaction cycles. (C) Schematic representation of the CP–BCP composite micelle illustrating the growth of the crystalline CP parts in the core of the micelle. | ||
| Cycles | Sample | DLSa | TEM core | TEM shella | PXRD (23°) |
|---|---|---|---|---|---|
| a The differences in the hydrodynamic diameter (DLS) and the outer diameter determined by TEM are due to drying effects. | |||||
| 0 | BCP | 126 ± 22 | 70 ± 8 | ||
| 1 | 2 | 147 ± 22 | 52 ± 8 | 101 ± 15 | |
| 2 | 3 | 140 ± 13 | 57 ± 8 | 94 ± 15 | 33 ± 3 |
| 3 | 4 | 142 ± 12 | 62 ± 13 | 91 ± 12 | 32 ± 3 |
| 4 | 5 | 147 ± 15 | 44 ± 6 | 96 ± 12 | 47 ± 3 |
| 5 | 6 | 145 ± 13 | 49 ± 5 | 113 ± 15 | 45 ± 3 |
Magnetism
The change in the crystallinity of the CP core and therefore of n in [Fe(L)(bipy)]n in the BCP micelle should significantly influence the SCO properties of the composite material. Due to the differences in the coordination environment of the outside [Fe(L)] units of the [Fe(L)(bipy)]n coordination polymer, only the inside [Fe(L)] units are expected to undergo spin crossover (SCO). Magnetic measurements of samples 3–6 were done in the temperature range between 330 K and 50 K in the cooling and heating modes. The results are given as χMT versus T plots in Fig. 3, where χM is the molar susceptibility and T is the temperature. The ratio of high spin (HS):low spin (LS) iron centers was confirmed by Mössbauer spectroscopy at room temperature (see Fig. S6 and Table S4†).
 | ||
| Fig. 3 Plot of the χMT product versus T determined by SQUID measurements. The increase in the crystallinity is reflected by an increase of the cooperative interactions (hysteresis) during the spin transition. | ||
The room temperature χMT product of the samples with 2 and 3 cycles (3 and 4) is 1.9 cm3 K mol−1 significantly lower than the theoretical value for iron(II) in the HS state (χMT = 3.0 cm3 K mol−1) due to a significant LS fraction. Upon cooling, for both samples a drop of the χMT product to ca. 1.0 cm3 K mol−1 is observed in the temperature region between 225 K and 125 K. This can be correlated to a very gradual and incomplete spin crossover. This is in contrast to the previously described microcrystals in a P4VP matrix, where the spin transition was either quenched or a spin transition with hysteresis was observed.23 For sample 5 the room temperature χMT product has a value of 2.9 cm3 K mol−1 which is almost in the region expected for an iron(II) complex in the HS state. A gradual SCO is observed in the temperature region between 200 K and 125 K with about 40% of the iron centers involved. This indicates that the number of SCO active iron centers did increase compared to 3 and 4 (both about 30%). This trend continues for 6 with five cycles with a room temperature χMT product of 3.1 cm3 K mol−1, typical of iron(II) HS complexes. The spin transition takes place between 210 K and 125 K with 45% of the iron center involved. From 3 to 6 a significant increase in the fraction of SCO active [Fe(L)] units is observed indicating the increase of n in [Fe(L)(bipy)]n. In the case of 5 and even more pronounced in the case of 6, different transition temperatures are observed in the heating and the cooling. The T1/2 values (where 50% of the SCO active centers did undergo spin transition) of 6 are 162 K in the cooling and 170 K in the heating mode corresponding to a 8 K wide thermal hysteresis loop. With the increasing number of cycles, an increase of the hysteresis width is observed and the SCO properties converge towards those of the bulk material (20 K hysteresis for the bulk28 and 8 K for 6, approx. 1 K hysteresis for 5). This is directly linked to the crystal size of the CP core in the CP–BCP composite (see Fig. 2).
Experimental
Synthetic procedures
Polystyrene-b-poly(4-vinylpyridine) (PS-P4VP, purum, MW ≈ 150000) was synthesized as described before.24 4,4′-Bipyridine was obtained from Alfa Aesar and used as received. Tetrahydrofurane (THF) was purified as described in the literature.29 [Fe(L)(MeOH)2] was synthesized as described before.30 All syntheses were performed under inert conditions using the Schlenk technique with argon (purity ≥ 99999%, 5.0). The synthesis of all samples was repeated at least twice.
1: PS-P4VP (50 mg, 0.33 μmol) and [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) were added to a 50 ml flask. Subsequently THF (20 ml) was added and the mixture was heated to reflux for 2 h. After cooling to room temperature the solvent of the brown solution was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 63.24, H 7.78, N 1.69.
2: PS-P4VP (50 mg, 0.33 μmol) and [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) were added to a 50 ml flask. Subsequently THF (20 ml) was added and the mixture was heated to reflux for 2 h. After cooling to room temperature, 4,4′-bipyridine (5.6 mg, 36 μmol) was added to the brown solution and the reaction mixture was heated for 1 h to reflux. After cooling to room temperature the solvent was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 63.70, H 7.49, N 2.38.
3: the synthesis as described for sample 2 was repeated. Before solvent removal, [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) and 4,4′-bipyridine (5.6 mg, 36 μmol) were added in a second cycle and the mixture was heated for one further hour to reflux. After cooling to room temperature the solvent was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 65.55, H 7.81, N 1.55.
4: the synthesis described for sample 3 was repeated, with one further cycle of addition of [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) and 4,4′-bipyridine (5.6 mg, 36 μmol) followed by further heating to reflux for 1 h. After cooling to room temperature the solvent was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 68.34, H 7.05, N 4.67.
5: the synthesis described for sample 4 was repeated, with one further cycle of addition of [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) and 4,4′-bipyridine (5.6 mg, 36 μmol) followed by further heating to reflux for 1 h. After cooling to room temperature the solvent was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 60.58, H 7.06, N 3.13.
6: the synthesis described for sample 5 was repeated, with one further cycle of addition of [Fe(L)(MeOH)2] (6.4 mg, 15 μmol) and 4,4′-bipyridine (5.6 mg, 36 μmol) followed by further heating to reflux for 1 h. After cooling to room temperature the solvent was removed via cold distillation to yield a brown, polymer-like powder. Elemental anal. (%) found: C 63.91, H 6.94, N 4.85.
The colour turned increasingly darker from sample 2 to 6 with an increasing amount of iron. Elemental analyses reveal increasing nitrogen contents from sample 1 to 6 in line with the increasing amount of CP. The variations in the values are due to differences in the solvent contents and contamination of the samples with grease.
Methods
Conclusions
In this work the synthesis of spin crossover iron(II) coordination polymer nanoparticles is described using blockcopolymer micelles as microreactors. An excellent control over the size of the coordination polymer is obtained via the blockcopolymer micelle size. A strong influence of the crystallinity, tunable by the number of cycles, of the coordination polymer core on the spin crossover properties of the material is observed. This is, to the best of our knowledge, a completely new approach for the synthesis of nanoparticles of coordination polymers and should be easily transferable to other coordination polymers and networks. Furthermore, the use of blockcopolymers allows a further functionalisation of the obtained nanoparticles by variation of the polymer blocks.Acknowledgements
Financial support from the University of Bayreuth and the SFB 840 (TP A10 and B10) is gratefully acknowledged. O. K. was supported by the BayNAT program of the UBT. We thank Juliane Kary and Stella Buchmann (AC II, UBT) for their contribution to the synthesis of the CP–BCP composites.Notes and references
- E. A. Flügel, A. Ranft, F. Haase and B. V. Lotsch, J. Mater. Chem., 2012, 22, 10119–10133 RSC
.
- M. Sindoro, N. Yanai, A.-Y. Jee and S. Granick, Acc. Chem. Res., 2014, 47, 459–469 CrossRef CAS PubMed
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS
-
(a) H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC
-
(a) T.-Y. Ma, H. Li, Q.-F. Deng, L. Liu, T.-Z. Ren and Z.-Y. Yuan, Chem. Mater., 2012, 24, 2253–2255 CrossRef CAS
-
P. N. Martinho, C. Rajnak and M. Ruben, in Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013, pp. 375–404 Search PubMed
-
(a)
Spin-Crossover Materials, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2013 Search PubMed
-
(a) E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas, F. Rey and I. J. Vitórica-Yrezábal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed
-
(a)
J.-F. Létard, P. Guionneau and L. Goux-Capes, in Spin Crossover in Transition Metal Compounds I–III, ed. P. Gütlich and H. Goodwin, Springer, Berlin/Heidelberg, 2004, pp. 221–249 Search PubMed
-
(a) R. N. Muller, L. Vander Elst and S. Laurent, J. Am. Chem. Soc., 2003, 125, 8405–8407 CrossRef CAS PubMed
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC
-
(a) A. Tissot, New J. Chem., 2014, 38, 1840 RSC
- F. Volatron, L. Catala, E. Rivière, A. Gloter, O. Stéphan and T. Mallah, Inorg. Chem., 2008, 47, 6584–6586 CrossRef CAS PubMed
- T. Forestier, S. Mornet, N. Daro, T. Nishihara, S.-I. Mouri, K. Tanaka, O. Fouche, E. Freysz and J.-F. Letard, Chem. Commun., 2008, 4327–4329 RSC
- T. Forestier, A. Kaiba, S. Pechev, D. Denux, P. Guionneau, C. Etrillard, N. Daro, E. Freysz and J.-F. Letard, Chem. – Eur. J., 2009, 15, 6122–6130 CrossRef CAS PubMed
- V. Martínez, I. Boldog, A. B. Gaspar, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Chem. Mater., 2010, 22, 4271–4281 CrossRef
- I. Boldog, A. B. Gaspar, V. Martínez, P. Pardo-Ibañez, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Angew. Chem., Int. Ed., 2008, 47, 6433–6437 CrossRef CAS PubMed
- J. Larionova, L. Salmon, Y. Guari, A. Tokarev, K. Molvinger, G. Molnár and A. Bousseksou, Angew. Chem., Int. Ed., 2008, 47, 8236–8240 CrossRef CAS PubMed
-
(a) A. Tissot, J.-F. Bardeau, E. Rivière, F. Brisset and M.-L. Boillot, Dalton Trans., 2010, 39, 7806 RSC
-
(a) S. Cobo, G. Molnár, J. A. Real and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 5786–5789 CrossRef CAS PubMed
- Y.-H. Luo, Q.-L. Liu, L.-J. Yang, Y. Sun, J.-W. Wang, C.-Q. You and B.-W. Sun, J. Mater. Chem. C, 2016, 4, 8061–8069 RSC
-
(a) B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed
- C. Göbel, T. Palamarciuc, C. Lochenie and B. Weber, Chem. – Asian J., 2014, 9, 2232–2238 CrossRef PubMed
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef
-
(a) S.-H. Yun, S. I. Yoo, J. C. Jung, W.-C. Zin and B.-H. Sohn, Chem. Mater., 2006, 18, 5646–5648 CrossRef CAS
- R. D. Bennett, G. Y. Xiong, Z. F. Ren and R. E. Cohen, Chem. Mater., 2004, 16, 5589–5595 CrossRef CAS
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS
- B. Weber, R. Tandon and D. Himsl, Z. Anorg. Allg. Chem., 2007, 633, 1159–1162 CrossRef CAS
-
H. G. O. Becker, Organikum. Organisch-chemisches Grundpraktikum, Johann Ambrosius Barth, Berlin, 19th edn, 1993 Search PubMed
- B. Weber and E.-G. Jäger, Z. Anorg. Allg. Chem., 2009, 635, 130–133 CrossRef CAS
-
K. Lagarec and D. G. Rancourt, Recoil, Mössbauer spectral analysis software for windows 1.0, Department of Physics, University of Ottawa, Canada, 1998 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06330f |
| This journal is © The Royal Society of Chemistry 2016 |