
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInflammation-sensitive in situ smart scaffolding for regenerative medicine†
Hirak K.
Patra‡
ab,
Yashpal
Sharma‡
c,
Mohammad Mirazul
Islam
d,
Mohammad Javad
Jafari
e,
N. Arul
Murugan
f,
Hisatoshi
Kobayashi
c,
Anthony P. F.
Turner
a and
Ashutosh
Tiwari
*acgh
aBiosensors and Bioelectronics Centre, Department of Physics, Chemistry and Biology (IFM), Linköping University, S-58183, Linköping, Sweden. E-mail: ashutosh.tiwari@liu.se; Fax: (+46) 13-13-7568; Tel: (+46) 13-28-2395
bDepartment of Cell Biology, Experimental and Clinical Medicine (IKE), Linköping University, S-58185, Linköping, Sweden
cInternational Center for Materials Nanoarchitectonics, National Institute for Materials Science (NIMS), 1-2-1, Sengen, Tsukuba, Ibaraki 305-0047, Japan
dSwedish Nanoscience Center, Karolinska Institute, S-171 77 Stockholm, Sweden
eDivision of Molecular Physics, Department of Physics, Chemistry and Biology (IFM), Linköping University, Sweden
fVirtual Laboratory for Molecular Probes, Division of Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology (KTH), S-106 91 Stockholm, Sweden
gTekidag AB, UCS, Teknikringen 4A, Mjärdevi Science Park, Linköping 58330, Sweden
hVinoba Bhave Research Institute, Sirsa Road, Saidabad, Allahabad 221508, India
First published on 22nd August 2016
Abstract
To cope with the rapid evolution of the tissue engineering field, it is now essential to incorporate the use of on-site responsive scaffolds. Therefore, it is of utmost importance to find new ‘Intelligent’ biomaterials that can respond to the physicochemical changes in the microenvironment. In this present report, we have developed biocompatible stimuli responsive polyaniline–multiwalled carbon nanotube/poly(N-isopropylacrylamide), (PANI–MWCNT/PNIPAm) composite nanofiber networks and demonstrated the physiological temperature coordinated cell grafting phenomenon on its surface. The composite nanofibers were prepared by a two-step process initiated with an assisted in situ polymerization followed by electrospinning. To obtain a smooth surface in individual nanofibers with the thinnest diameter, the component ratios and electrospinning conditions were optimized. The temperature-gated rearrangements of the molecular structure are characterized by FTIR spectroscopy with simultaneous macromolecular architecture changes reflected on the surface morphology, average diameter and pore size as determined by scanning electron microscopy. The stimuli responsiveness of the nanofibers has first been optimized with computational modeling of temperature sensitive components (coil-like and globular conformations) to tune the mechanism for temperature dependent interaction during in situ scaffolding with the cell membrane. The nanofiber networks show excellent biocompatibility, tested with fibroblasts and also show excellent sensitivity to inflammation to combat loco-regional acidosis that delay the wound healing process by an in vitro model that has been developed for testing the proposed responsiveness of the composite nanofiber networks. Cellular adhesion and detachment are regulated through physiological temperature and show normal proliferation of the grafted cells on the composite nanofibers. Thus, we report for the first time, the development of physiological temperature gated inflammation-sensitive smart biomaterials for advanced tissue regeneration and regenerative medicine.
Introduction
Non-spherical anisometric nanostructures such as nanopillars, nanotubes, nanofibers and nanorods offer out of the box opportunities for tissue engineering (TE), regenerative medicine (RM), advanced diagnosis and wound healing.1–3 The foremost advantage of choosing such an anisometric nanostructure is due to its significantly reduced health risk compared to that of the spherical one.4–6 New synthetic strategies can decorate the surface with biocompatible functionalization of such fibres, rods or tubes at the nano-scale dimension with very high precision.7,8 The functional features of the anisometric nanoobjects can be adjusted for a wide range of biomedical applications via designing the different nanoarchitectures.9–13 Carbon nanotubes (CNTs) have widely been under investigation in TE scaffolds not only due to their excellent mechanical chattels but also their exceptionally large surfaces that allow plenty of biocompatible functionalization and tunable electronic and ionic mobility that could facilitate TE.14,15 Carboxylated or hydroxylated CNTs are more water-soluble and could be cleared from systemic circulation through renal excretion. Multiwalled CNTs (MWCNTs) are reported with higher mechanical strengths, and are relatively more biodegradable, and evidence shows that they can foster intra and extracellular responses on their surfaces.16 Furthermore, the multifold anisometry of MWCNTs allow the transportation and mobilization of ions or molecules including drugs.17Recent approaches offer to develop biocompatible nanocomposites which either assemble with human body parts or are degradable and by-products can harmlessly be expelled out from the body.18 Semantic therapies always directly or indirectly affect all body parts irrespective of the disease site and thus alternative smarter strategies are now much desired. In the present study, we are attempting to build a responsive nano-architecture based in situ strategy for loco-regional therapy (treatment on site; such as wound healing). TE being an interdisciplinary field, we applied the principles of materials engineering using stimuli-responsive polymers as the interfacial component of the composite to address, restore and improve the tissue functions. The logic behind developing such smart materials is to respond to slight variations in temperature, pH, light, and ionic strength. Such a response will help regenerate or replace biologically damaged tissue or generate replacement cells for a range of medical conditions where precise in situ grafting is required such as burns, diabetes, cirrhosis wound, osteoarthritis, spinal cord injury etc.19
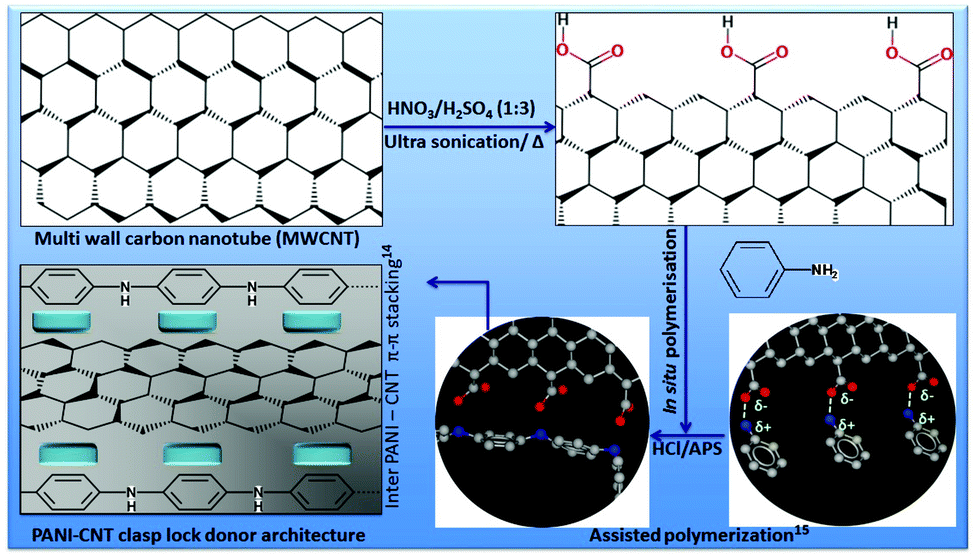
It is only quite recently that polyaniline (PANI) and its conjugates have been explored in TE for tunable electronic/ionic commotions for diverse biological applications.20 Furthermore, Mattioli-Belmonte et al. first reported PANI as a biocompatible material investigated through in vitro as well as in vivo long term animal studies21 followed by several other groups in different systems.22,23 PANI allows for in situ polymerization in the presence of CNTs and followed by electro-spinning processes to obtain composite nanofibers. Wang et al. reported that such CNT/PANI nanofibers showed that CNTs were embedded and aligned along the fiber axis with excellent thermoelectric properties.24 We have adapted and combined related logic with a bottom up approach of assisted growth25 for oriented PANI around MWCNTs for developing our primary clasp lock donor architecture. PANI can form a stretched tubular structure with such template-assisted growth and due to the robust chemical interfaces between PANI and MWCNTs.
Poly(N-isopropylacrylamide) (PNIPAm) is one such well-studied smart biocompatible and temperature responsive polymer, which exhibits a lower critical solution temperature (LCST) at 32 °C. At temperature above the LCST, the intermolecular hydrogen bonding between the PNIPAm chains and water molecules is replaced by intramolecular hydrogen bonding between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups along the PNIPAm chains resulting in compact and collapsed chains that interact minimally with water and thus exhibit hydrophobicity. On the other hand, below the LCST the ionic moieties interact strongly with the media and result in hydrophilicity.
O and N–H groups along the PNIPAm chains resulting in compact and collapsed chains that interact minimally with water and thus exhibit hydrophobicity. On the other hand, below the LCST the ionic moieties interact strongly with the media and result in hydrophilicity.
The advanced tissue engineering approach proposed here not only offers appropriate scaffolding within which cells will be seeded and consequently regenerate the tissue and eventually will mature but also their appropriate 3D structure will provide the hydrophobicity/hydrophilicity balance that sufficiently supplies the mechanical strength with nutrients and growth factors to the grafted cells. Furthermore, we have attempted the most challenging aspect to develop microenvironment controlled 3D scaffolds for easy attachment, proliferation, and differentiation of cells.26,27 Taking into account the stimuli-responsive behaviour of PNIPAm,28 the great ionic doping properties of polyaniline29 to combat loco-regional acidosis and the good mechanical properties of MWCNTs,30 we have fabricated three-dimensional smart nanofiber networks namely PANI–MWCNT/PNIPAm for the in situ regeneration of tissue and wound healing.
Experimental details
Materials
MWCNTs (99%) were procured from Nanostructured and Amorphous materials Inc. (NanoAmor, USA) with exterior diameters of 20–40 nm and length of 1–2 μm. PNIPAm, Mn = 5500 from Aldrich. 1,1,1,3,3,3-Hexafluoro-2-propanol and N,N-dimethyl formamide (DMF) were procured from Aldrich. Fetal calf serum, penicillin–streptomycin (Pen–Strep), and L-glutamine (complete medium) was purchased from Sigma. Dojin supplied propidium iodide and calcein-AM. Twenty four-well plates were purchased from B.D. Falkon. Aniline (99.5%) was purchased from Aldrich and vacuum distillation was performed before using it. All other chemicals such as HCl, APS, ethanol and sulphuric acid used were all analytical grade. PBS (pH 7.4) was supplied by Sigma.Synthesis of PANI–MWCNT nanocomposites: template-assisted polymerization
A balanced amount of 200 mg MWCNTs was ultra-sonicated under highly clean conditions with the combination of nitric and sulphuric acid for 6 h at 50 °C to functionalize the surface with an acidic group on it for better further processability of the MWCNTs. The mixture was then cooled, diluted, filtered and washed several times with water to remove excess acidic content. The obtained MWCNTs were dried under vacuum at 60 °C for at least 12 h and used for the preparation of PANI–MWCNT composites. A weighed amount of 100 mg functionalized MWCNTs was soaked in an acidic solution of 1 M HCl. Then 0.05 M aniline monomer was added to it and ultra-sonicated for 2 h. Approximately, 30 mL freshly prepared ammonium peroxydisulfate (1 M) in 1 M HCl was added gradually into the solution for about half an hour. The mixture was then left for polymerization for 6 h at 0–5 °C under constant stirring. Finally, filtering and rinsing the reaction mixture with deionized water and methanol resulted in the greenish black PANI–MWCNT composite. Methanol washing is desirable to remove the oligomer impurities if any present within the solution. The powder was dried under vacuum at 60 °C for 24 h at least. In this process, the PANI exists in its doped emeraldine salt form.Fabrication of three-dimensional nanofibrous smart scaffolds
Nanofibrous scaffolds were fabricated using different compositions of PANI–MWCNT composites and PNIPAm through electrospinning to obtain hybrid nanocomposites along with several controls such as PANI–MWCNT, PNIPAm only etc. The process has been standardized with different concentrations (18, 20, 22 and 24 wt%) of each polymer solution in a mixture of 1,1,1,3,3,3-hexafluoro-2-propanol and N,N-DMF (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).31 In the PANI–MWCNT/PNIPAm hybrid nanocomposite, the ratio of PANI–MWCNT to PNIPAm that can produce the best nanofibrous 3D networks was 1:1. Assorted homogeneous polymer solution having different concentrations was placed in the respective syringe fitted with 22 gauze metallic needles. The syringe was fixed horizontally on the syringe pump, and an electrode of high voltage power supply was clamped to the metal needle tip. The electrospinning parameters like flowing rate, voltage supply and distance from the tip of the needle to the collector surface (aluminum) for different polymeric materials are described in Fig. 1 and 2. Finally the nanofibers were collected from the aluminium surface and dried in a vacuum desiccator for 24 h at room temperature.
:2).31 In the PANI–MWCNT/PNIPAm hybrid nanocomposite, the ratio of PANI–MWCNT to PNIPAm that can produce the best nanofibrous 3D networks was 1:1. Assorted homogeneous polymer solution having different concentrations was placed in the respective syringe fitted with 22 gauze metallic needles. The syringe was fixed horizontally on the syringe pump, and an electrode of high voltage power supply was clamped to the metal needle tip. The electrospinning parameters like flowing rate, voltage supply and distance from the tip of the needle to the collector surface (aluminum) for different polymeric materials are described in Fig. 1 and 2. Finally the nanofibers were collected from the aluminium surface and dried in a vacuum desiccator for 24 h at room temperature.
 | ||
| Fig. 1 PANI–MWCNT clasp lock donor architecture: schematic representation of the synthesis of PANI–MWCNT composite. | ||
 | ||
| Fig. 2 Fabrication, synthesis and structural description of the hybrid composite nanofiber networks. (a–d) Synthesis and fabrication strategy with the help of (b) NANON electrospinning setup equipped with (c) a devised drum type collector in NIMS, Japan and with (e) optimized electrospinning parameters. (f) Three dimensional composite fiber networks with an extremely porous interface (g) before the final steps of the formation of the stimuli-responsive PANI–MWCNT/PNIPAm (h) with a complete control on the surface morphology. | ||
Functional validation of the smart scaffold
Fourier-transform infrared measurements were carried out by means of a Bruker Vertex 70 spectrometer using the KBr pellet method; the system was continuously purged with nitrogen before and during the measurements and temperature controlled by a heater mounted on a sample holder. All spectra were acquired at 2 cm−1 resolutions with a total of 200 scans, and wavenumbers ranging between 4000 and 800 cm−1. The surface morphology of the composites was examined by high resolution scanning electron microscopy (SEM, JEOL EDSEM) at an accelerating voltage of 20 kV. SEM also examined the average diameter of the pores present within the nanofibrous scaffolds. The aluminium collector with the nanofibers was coated with platinum (ELIONEX platinum coater) prior to SEM measurement.Biocompatibility
The fibroblast cells (L929) were supplied by Riken cell bank, Tsukuba, Japan, cultured in medium supplemented with 10% fetal calf serum, penicillin–streptomycin and L-glutamine (complete medium) at 37 °C with 5% CO2 environment. Cells were seeded on to the nanofiber network surfaces in a 24-well tissue culture plate. 10000 cells were seeded on each nanofiber-based surface. Cell morphology and growth were assessed at 24, 48, 72, 96, 120, 144 and 168 h after cell seeding. Cell viability was evaluated at regular intervals after cell seeding. For cell viability assays cell-loaded fibers were washed with PBS (pH 7.4) at the indicated time points, then stained with standard calcein-AM and propidium iodide (PI) concentration for 30 min at 37 °C. All living cells were labelled with calcein-AM, while dead cells were labeled with PI. The cells were imaged using a fluorescence and phase contrast light microscope (Olympus). To analyse cell growth, the number of cells present within a 0.55 mm2 area was determined in three representative images from each sample. Due to the three-dimensional nature of the fibers, it was necessary to set a brightness threshold to exclude cells outside the focal plane (threshold set to omit diffuse light). All analysis was complete under identical conditions. If cell clumps were encountered they were counted as one, unless separate cells were resolved.
In vitro inflammation sensitivity study
Before starting the culture of human umbilical vascular endothelium cells (HUVEC) empty tissue culture flasks (Corning or equivalent) were coated with gelatin (0.1%), drained and the excess was removed, then the flasks were allowed to dry (at room temperature in a laminar flow hood). The cells are then cultured with nutrient medium (M199), consisting of 20% fetal calf serum buffered with 25 mM HEPES, and supplemented with fresh 2 mM L-glutamine (final concentration), and with standard penicillin–streptomycin solution. The cells require 50 mcg ml−1 Endothelial Cell Growth Supplement (ECGS), and 100 mcg ml−1 heparin for optimal growth.We have developed the assay system using the CytoSelect™ leukocyte transmigration kit (Cell Biolabs Inc., US). Approximately 100000 HUVEC in 300 μL medium were placed in each insert in a 24-well plate containing 500 μL of previously mentioned culture medium. HUVEC were cultured for 72 h until the monolayer is formed. We then treated the monolayer endothelial cells with TNFα as per the kit instructions.
In a separate set up, leukocytes were harvested from a healthy donor and a cell suspension of 106 cells per ml in serum free RPMI 1640 with 0.5% BSA, 2 mM CaCl2 and 2 mM MgCl2 was prepared. LeukoTracker™ solution has been added to a final concentration of 1× (2 μL of 500× LeukoTracker™ to 1 mL of leukocyte suspension). The leukocytes were incubated for 60 min at 37 °C under tissue culture conditions. The stained cells were then spun down at 1000 rpm for 2 minutes, and then the leukocytes were washed twice with serum free RPMI.
The medium from the migration insert was carefully removed without disturbing the HUVEC monolayer and then the inserts were transferred to new wells containing 500 μL of complete medium containing composites as described below. The composite nanofibers were suspended and sonicated in PBS for 30 min at 37 °C and then the experimental assay medium was prepared with the complete M199 medium as described above with 5 mg mL−1 nanofiber composites for the leukocyte transmigration assay. Around 300 μL of leukocyte suspension was placed inside each insert and incubated for 24 h in a complete tissue culture environment. After carefully aspirating the media inside the insert (kept separately), cotton-tipped swabs were used to gently remove non-migratory cells from the interior of the inserts without puncturing the polycarbonate membrane. The bottom well where the leukocytes migrated, 400 μL of the 500 μL medium solution containing migratory cells was placed in a clean well that contains 150 μL of 4× lysis buffer (supplied with the kit) and the swabbed insert was placed in the same well and incubated for 15 minutes at room temperature with shaking. This present step combined leukocytes that migrated through the membrane into the medium and migratory cells still attached to the bottom surface of the membrane. Exactly 150 μL of the mixture has been transferred to a 96-well plate for fluorescence measurement at 480/520 nm (as per the kit instructions).
Results and discussion
Design, synthesis and characterization of smart precursor
Fig. 1 demonstrates the strategy to obtain the PANI–MWCNT composite clasp lock donor architecture. In brief, we have combined the bottom up approach of template-assisted growth25 for the as synthesized PANI around MWCNTs with the help of a favorably ordered arrangement of the PANI backbone. This will not only reduce the p–p conjugated defects in the polymer backbone, but will also increase the inter PANI–MWCNT π–π stacking for efficient degree of electron delocalization and therefore enriches the electronic carrier mobility with a similar logic as described by Wang et al.24Individual component polymer concentrations were optimized to obtain fibres with a fine surface morphology and the thinnest diameter. The strategy and surface morphology of the composite hybrid was characterized by scanning electron microscopy (SEM) and is shown in Fig. 2. We have first studied the surface morphology with PNIPAm having different concentrations of 18, 20, 22 and 24 wt% and found that at lower concentration (18 wt%) thick fibres were obtained having an average diameter of ∼1.4–1.5 μm. With the increase in concentration from 18 wt% to 20 and 22 wt%, the fiber diameter decreases but above this concentration the fiber diameter again starts increasing with the appearance of irregular beads on the fibers.
Finally, we found that the optimum concentration for PNIPAm to obtain fibers with a smooth surface morphology and the lowest diameter (500–400 nm) is 22 wt% (Fig. S1†). In a similar way, the electrospun PANI–MWCNT composites of different concentrations of PANI with 18, 20, 22 and 24 wt% are optimized. The electrospun PANI–MWCNT composite fibres showed similar surface morphology as shown in Fig. 2 with PNIPAm. The average diameter of the fibers electrospun from a solution having different concentrations of 18, 20, 22 and 24 wt% is 1.5–1.7 μm, 1.1–1.3 μm, 0.5–0.4 μm and 1–1.1 μm respectively.
Here we found that the optimum concentration of the PANI–MWCNT composite to obtain fibers with the thinnest diameter (500–400 nm) and smoothest surface morphology is 22 wt% (Fig. S2†). The surface morphology and average diameters of the fibers electrospun from a solution of the PANI–MWCNT/PNIPAm hybrid composite having different concentrations of 18, 20, 22 and 24 wt% are given in Fig. S3 and S4† respectively. Fibers having the thinnest diameter were obtained using 20 wt% of the composite solution and hence it was found to be the optimal concentration. Beyond that with 22 wt% polymer solution of PANI–MWCNT/PNIPAm hybrid composite also produces fibers in the nano range but the fibers appear with irregular beads and wrinkles. The average diameters of the fibers electrospun from different polymer solutions with varying concentrations is given in Fig. S4.† The fibers are in 3D form and are of several meters in length. Due to the 3D nature they provide a respectable surface for cell support. For further study on cell culture we have used the fibers obtained from the optimum concentration of the polymer solutions. The SEM also examined the average pore size of the nanofibers electrospun from the optimum concentration of PNIPAm, the PANI–MWCNT composite and the PANI–MWCNT/PNIPAm hybrid composite. The average pore size of the PNIPAm and the PANI–MWCNT composite nanofibers ranged from 5 μm to 15 μm. The PANI–MWCNT/PNIPAm hybrid composite nanofibers however had a pore size of 2 to 7 μm.
Stimuli responsive molecular rearrangement
The transmission FT-IR spectra of the materials at 21 °C in the 3800–800 cm−1 region are shown in Fig. 3(a). For PANI several bands in the spectrum can be identified: N–H stretch (3236 cm−1), aromatic (3063 cm−1) and methylene (2938 cm−1) C–H stretch, quinonoid (Q) (1569 cm−1) and benzenoid (B) (1502 cm−1) ring vibrations, CC stretch of aromatic rings (1445 cm−1), C–N vibrations of secondary (1290 cm−1) and primary (1248 cm−1) aromatic amines, B–NH–B (1145 cm−1) and B–NH–Q (shoulder at 1114 cm−1) and C–H out-of plane vibrations of the 1,4-disubstituted ring (824 cm−1).
 | ||
| Fig. 3 Chemical bonding of the hybrid composite and their stimuli-responsive molecular rearrangements. (a) Transmission FTIR spectra of PNIPAm, PANI–MWCNT and PANI–MWCNT/PNIPAm during step-by-step formation of the hybrid composite acquired. (b) One step hybrid composite (red 80% PNIPAm + 20% PANI–MWCNT) experimental (blue) and calculated spectra at <LCST of PNIPAm (at 21 °C). FTIR window for stimuli-responsive covalent structural rearrangement spectra of the hybrid composite at application (<LCST) and in situ wound recovery (>LCST) temperatures at (c) 3800–2700 cm−1 and (d) 1800–1400 cm−1. | ||
The spectrum of the PANI–MWCNT composite is totally the same as PANI's spectrum, but an extra band appears at 1043 cm−1, which can be attributed to the C–O vibration of MWCNTs. Furthermore, in the spectrum of PNIPAm, the amide group – secondary amide N–H stretch (3295 cm−1), amide II bond overtone (3076 cm−1), amide I (1648 cm−1), amide II (1548 cm−1) and amide III (1277 cm−1) – can be clearly seen; other bands of PNIPAm are related to the C–H stretch of saturated carbons (2973, 2934, 2876 and 2850 cm−1), asymmetric (1466 cm−1) and symmetric (1387 cm−1) deformation of the isopropyl (CH3) group, asymmetric (1459 cm−1) and symmetric (1367 cm−1) deformation of the CH2 group and C–H out-of-plane bending and rocking (1172, 1157 and 1131 cm−1).32 The spectrum of the PANI–MWCNT/PNIPAm hybrid composite shows the contribution of all components (Fig. 3b) and can confirm the formation of a composite.
After that, the spectra of all materials were obtained at 37 °C. For PANI and PANI–MWCNT, the spectra did not show any temperature dependent change, but band shifts related to the amide group appeared for PNIPAm and hybrid composites. As shown in Fig. 3c, the bands about 3300 and 3070 cm−1 (hydrogen bonded N–H stretch in the amide group) respond to change of temperature, and the frequency of the band around 3300 cm−1 increases with the increase of temperature (bands shift 8 and 13 cm−1 for PNIPAm and composite, respectively) and the frequency of the band around 3070 cm−1 decreases with the increase of temperature (bands shift 5 and 6 cm−1 for PNIPAm and composite, respectively). Fig. 3d shows the other bands related to the amide group; the carbonyl group (amide I around 1650 cm−1) does not show any change with temperature but the frequency of the band attributed to the coupling of a C–N stretch and N–H bending vibration (amide II around 1545 cm−1) decreases with the increase of temperature (bands shift 5 and 8 cm−1 for PNIPAm and composite, respectively). The frequencies of amide I (∼3300 cm−1) and amide II (∼1545 cm−1) bonds reflected the strength of the hydrogen bond,33 the shift of frequencies for these bonds is caused by the loss of some hydrogen bonds in the material structure. This phenomenon can confirm the loss of hydrogen bonds with the increase in temperature.
Computational modeling and optimization of molecular architecture of the thermo responsive component
The computational modeling study has been carried out to get insight into the temperature responsive behavior of PNIPAm. Modeling has been carried out only for the PNIPAm polymer with 60 units. For this, 60 repeating units of isopropyl acramide (IPA) have been arranged in a linear fashion (refer to Fig. 4). | ||
| Fig. 4 Lipid bilayer cell membrane–hybrid composite molecular modeling. The dynamic behavior of the composite and lipid bilayer cell membrane is essential for stimuli-responsive in situ scaffold formation for the proliferation of the tissue. The image representation (a–c) of the calculations show remarkable temperature dependence of secondary structures (a to b coil–globular interchange) of the PNIPAM unit that modulates the interaction between the lipid bilayer cell membrane and nanofiber in a temperature responsive way (c). | ||
The dangling groups of the terminal IPA units were capped with methyl groups. The charges for the monomer units other than at the terminal ends are based on the charges calculated for the single unit of IPA which has been obtained using density functional theory calculation by employing B3LYP exchange correlation functional34,35 and 6-31G* basis set as implemented in Gaussian09 software.36 In particular, the partial atomic charges for the terminal IPA groups were obtained through fitting to electrostatic potential (and the procedure is referred to as RESP37) with the constraints that the methyl groups are neutral. The non-bonded interaction parameters for the PNIPAm are based on the general amber force-field.38 For water as a solvent, the TIP3P force-field has been employed. Initially, the molecular dynamics (MD) calculations for PNIPAm has been carried out in an implicit solvent, the final structure of which has been used to carry out explicit solvent MD. The PNIPAm in explicit solvent simulations were carried out in an isothermal–isobaric ensemble using Amber 12 software.39 The time step used for the integration of equation of motion was 1 fs and the total time scale for the production run was around 50 ns. In modeling, the transition temperature for coil–globular structural transition depends on the system size (i.e. polymer length) and the rate of heating40 that are very different compared to experiments. For this reason, the transition temperature from simulations is overestimated than the experimental values.41 Moreover, the transition temperature also strongly depends on the force-field employed.41 Since our aim is not to study this aspect in detail, we have studied the system in two different well-separated temperatures. The structures for the input geometry and the representative structures for low and high temperature PNIPAm polymer structures are shown in Fig. 3. As can be seen, indeed the PNIPAm exhibits a coil–globular structural transition with increasing temperature (4a and 4b) and is illustrated in Fig. 4c suggesting that the temperatures chosen to study these phases are fairly good enough. The solvent molecules that are involved in hydrogen bonding with PNIPAm are also shown in Fig. 4a and b. The red spheres show the oxygen atoms of water solvent that are involved in hydrogen bonding with the NH group of the IPA unit while the white spheres refer to the hydrogen atoms of water solvent that are involved in the hydrogen bonding with the CO group of the IPA unit. As can be clearly seen, with the change the from coil to globular structure, the number of water molecules involved in hydrogen bonding with PNIPAm is drastically reduced which is also an indication that its surface behavior changed from hydrophilic-like to hydrophobic. At elevated temperature, this should be the reason for the increased interaction of PNIPAm with the membrane bilayer that is known to have stabilizing interactions with hydrophobic molecular and bio-molecular units. When the temperature is reduced, the PNIPAm is supposed to regain its hydrophilic surface (through the globular to coil-like conformation) and this should help in the regeneration of fiber nanocomposites from the cell surface. It is also worth recalling that recently we have reported that the hydrophobic interaction between the probe–membrane subsystems is the driving-force for the probe–membrane association process.42 We have not explicitly studied the interaction between the lipid bilayer and PNIPAm units in its coil-like and globular conformations due to computational demands associated with modeling. However, the temperature dependent study on PNIPAm alone gives insight into the possible mechanism for the temperature dependent behavior of ANI/CNT–PNIPAm hybrid composite fiber–membrane system.
Cellular proliferation and regeneration
To evaluate the feasibility of employing PANI–MWCNT/PNIPAm hybrid composite nanofibers as in situ cell matrices for cell adhesion using the mice L929 fibroblast cell line, the cell growth and viability on the surface of these polymer fibers was assessed. Fibroblasts are widely distributed in many types of tissues, such as tendons, ligaments and the skin. Fibroblast cells generally produce collagens and regulate the extracellular matrix (ECM) of the cells. They play an important role in wound healing. The hybrid composite electrospun nanofibers are suitable for such applications because they have a uniform 3D structure that allows the cell attachment and proliferation. Ten thousand cells were seeded on the surface of nanofibers directly in a 24 well tissue culture plate and then incubated at 37 °C with 5% CO2 supply. The cell growth on the surface of the nanofibers was checked by counting the number of cells in 0.55 mm2 area. The growth of the cells on the nanofibers surface was compared with the respective control. Fig. 5a is a histogram indicating the cell growth on the control, PNIPAm, the PANI–MWCNT composite and the PANI–MWCNT/PNIPAm hybrid composite nanofiber surface after each day of cell seeding for 7 days. The histogram indicates that the number of cells increases each day and the fiber surface shows healthy cell growth. It can be seen that the cell number increases continuously, including a higher proliferation rate after 72 h.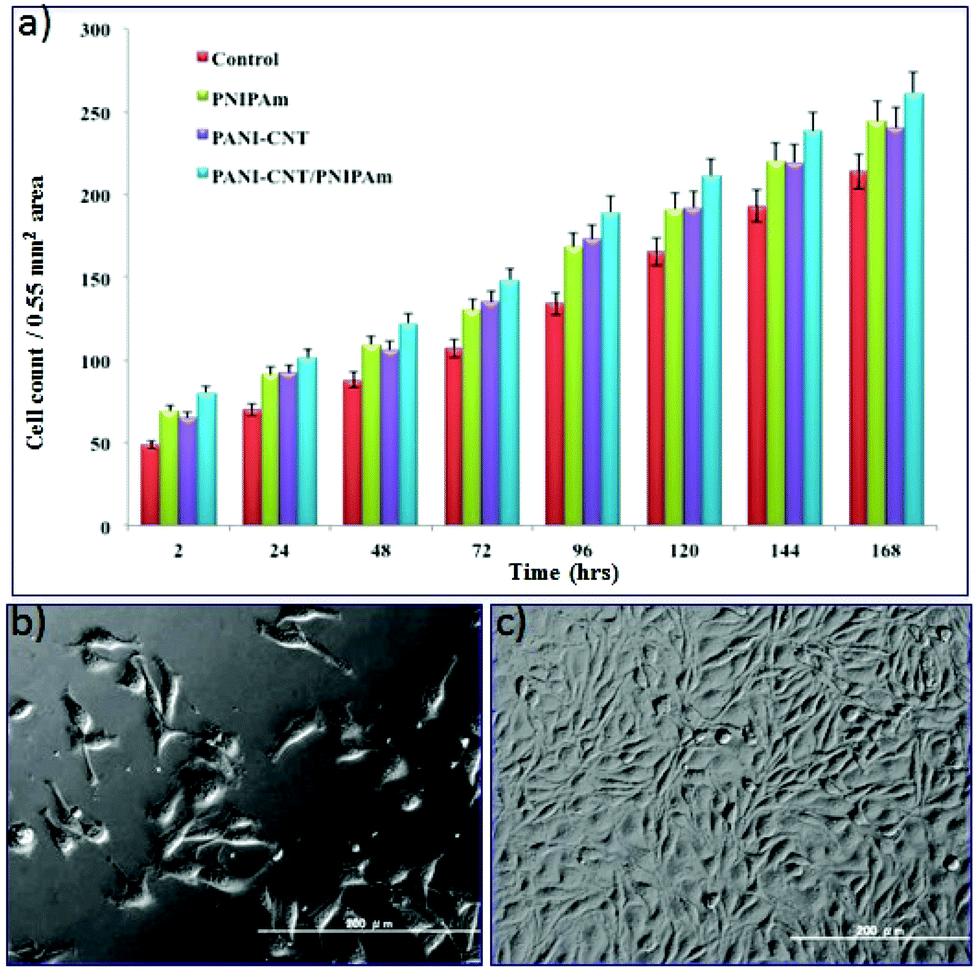 | ||
| Fig. 5 Fibroblast growth and proliferation on hybrid composite. Rate of proliferation of L929 fibroblast on PANI–MWCNT/PNIPAm hybrid and components. The respective count differences were significant; *p < 0.05, unless otherwise indicated (a). Cellular morphology of L 929 fibroblast cells after successful grafting on the scaffold (b) and after 168 h of proliferation (c). | ||
The result indicates that the nanofibers provide a biocompatible surface for cell growth. Normal confluence was also observed on the 7th day. Comparing the cell growth on the nanofiber surface of different polymeric materials, it can be said that the PANI–MWCNT/PNIPAm hybrid composite nanofiber surface provides a 3D substrate for great cell growth and proliferation that in turn can be attributed to the mechanical strength provided by the CNT to the composite nanofibers and the electrical stimuli provided by the conducting polymer.43 The highly porous morphology of these nanofibers also results in good cell growth and attachment. These results are in good agreement with the cell morphology on the surface of the PANI–MWCNT/PNIPAm hybrid composite nanofibers as shown in Fig. 4b. In our study we investigated that the hybrid composite had higher cell growth and attachment compared with the control, PANI–MWCNT composite and PNIPAm nanofibers although the PANI–MWCNTs and PNIPAm nanofibers were thinner in diameter than the PANI–MWCNT/PNIPAm hybrid composite nanofibers. This may be due to the combined effect of the individual materials in the composite.
In continuation with this, the cell viability on the surface of the PANI–MWCNT/PNIPAm hybrid composite nanofibers was checked on the 7th day of cell culture by staining the cells with calcein-AM and propidium iodide (PI) dye. The cell viability on the PANI–MWCNT/PNIPAm hybrid composite nanofibers shows a percentage viability of 98% that is as high as that obtained in the natural system. Higher cell growth and cell viability shown by the PANI–MWCNT/PNIPAm hybrid composite can be attributed to the fact that the composite exerted a synergistic effect that combines the overall efficiency of the individual materials, and thus produced a composite with great biocompatibility that supports the cell growth and attachment. It was supposed that in the PANI–MWCNT/PNIPAm hybrid composite nanofibers, the conducting polyaniline provides a charge transport phenomenon between the cells and the substrate that results in higher cell growth with great attachment on the 3D surface. PNIPAm provides temperature sensitivity to the growing cells. The mechanical strength provided by the CNT to the composite nanofibers also plays a critical role in the three-dimensional cell cultures on their surface. Combining the temperature sensitivity and biocompatibility of the PNIPAm with the electrical stimulus of PANI and mechanical strength of CNTs, the composite exerts a synergistic effect and excellent cell growth can be seen on their surface. Thus in total the composite provided a 3D surface for great cell growth and attachment. Fig. 6 shows the image of cells after staining with calcein and PI dye. The living cells are indicated by green colour and dead cells by red colour. The results indicate that the PANI–MWCNT/PNIPAm hybrid composite is biocompatible and the highly porous nature of the nanofibers, and the nanoscale dimension of the scaffolds provide a 3D surface for cell growth and cell culture. The polyaniline provides great electrical stimulus to the cells for their growth.
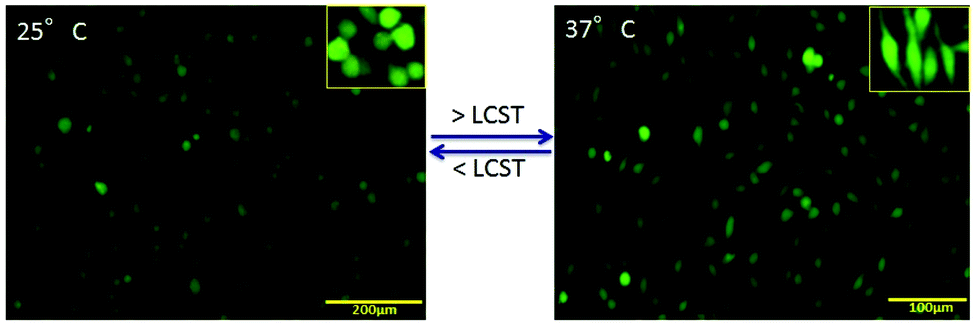 | ||
| Fig. 6 In situ regeneration of fibroblast on hybrid composite: the functional activity and respective morphology of the cells showing with living (green) and dead (red) indicator at 168 h of cell seeding after stained with calcein AM and PI dye. The comparative study shows the grafted cells on PANI–MWCNT/PNIPAm hybrid composite nanofibers scaffolds at 25 °C and 37 °C. | ||
Cellular attachment
PNIPAm is a well-known temperature sensitive polymer and exhibits a lower critical solution temperature (LCST) at around 32 °C. At temperature above the LCST, the hydrogen bonding in the PNIPAm chains become weakened or destroyed and hence the hydrophobic interaction increases due to which the water molecules come out resulting in a collapsed structure as described in Fig. 3d. This collapsed structure will help in the formation of the gel-based scaffold for attachment of the cells and help in cell growth through inter-cellular communication. Due to this temperature sensitivity, the cells grown on the surface can also be detached from the substrate and easily transplanted inside the body without the scaffolds. This can be achieved easily by lowering the temperature of the carrier.The phenomenon of in situ scaffold generation and cellular attachment can be seen in Fig. 6 which shows the cellular adhesion and proliferation on the surface at different temperatures. We have investigated the cellular regeneration behavior on the surface of the PANI–MWCNT/PNIPAm hybrid composite nanofibers by lowering and increasing the temperature of the culture dish at 25 °C and 37 °C respectively. The images of cells are shown in Fig. 5 after staining with live (green)–dead (red) dye as described above. At temperature below the LCST (Fig. 5a), the hydrophilic moieties of the individual materials in the composite i.e. N–H, COOH, CONH, become active and induce hydrophilicity in the composite. Due to increased hydrophilicity, cells interact minimally with media and their cytoskeleton structure becomes reorganized due to which they are less proliferative on the surface. When the temperature reaches or is above the LCST (here at physiological temperature), the hydrophobic moieties (isopropyl group) become dominant which favours the generation of the desired scaffold to support the cells to grow and proliferate.44
Inflammation-sensitivity
The wound healing process is divided into several complex interlinked phases including haemostasis, inflammation, proliferation and remodelling of tissue called maturation. Inflammation associated loco-regional hypoxia and low pH play major roles during wound healing and tissue regeneration. During the systemic recovery process, leukocyte extravasation into the perivascular compartment plays a major role in inflammatory cases. This multistep process is organized by sequential activation-dependent cross talk between endothelial cells and leukocytes. In Fig. 7, we have evaluated the inflammation sensitivity of the synthesized nanofiber composite network by the modified leukocyte transmigration assay. The detailed specification of the multi-component inflammation system used is described in Fig. 7, panel a, where the schematic and real description of the insert has been given with the respective HUVEC monolayer with live/dead stains (calcein-M stains green to live cells and live cell-impermeant ethidium homodimer stains red). The porous membrane around 3 μ inside the insert is sufficient to hold the monolayer of HUVEC with >95% cell viability but is porous enough to allow the cells to pass through the membrane during leukocyte extravasation. Fig. 7b describes the relative leukocyte extravasation in the presence and absence of TNFα with respect to control. The inset of Fig. 7b describes the relative sensitivity of the nanofiber composite networks during inflammation with respect to control.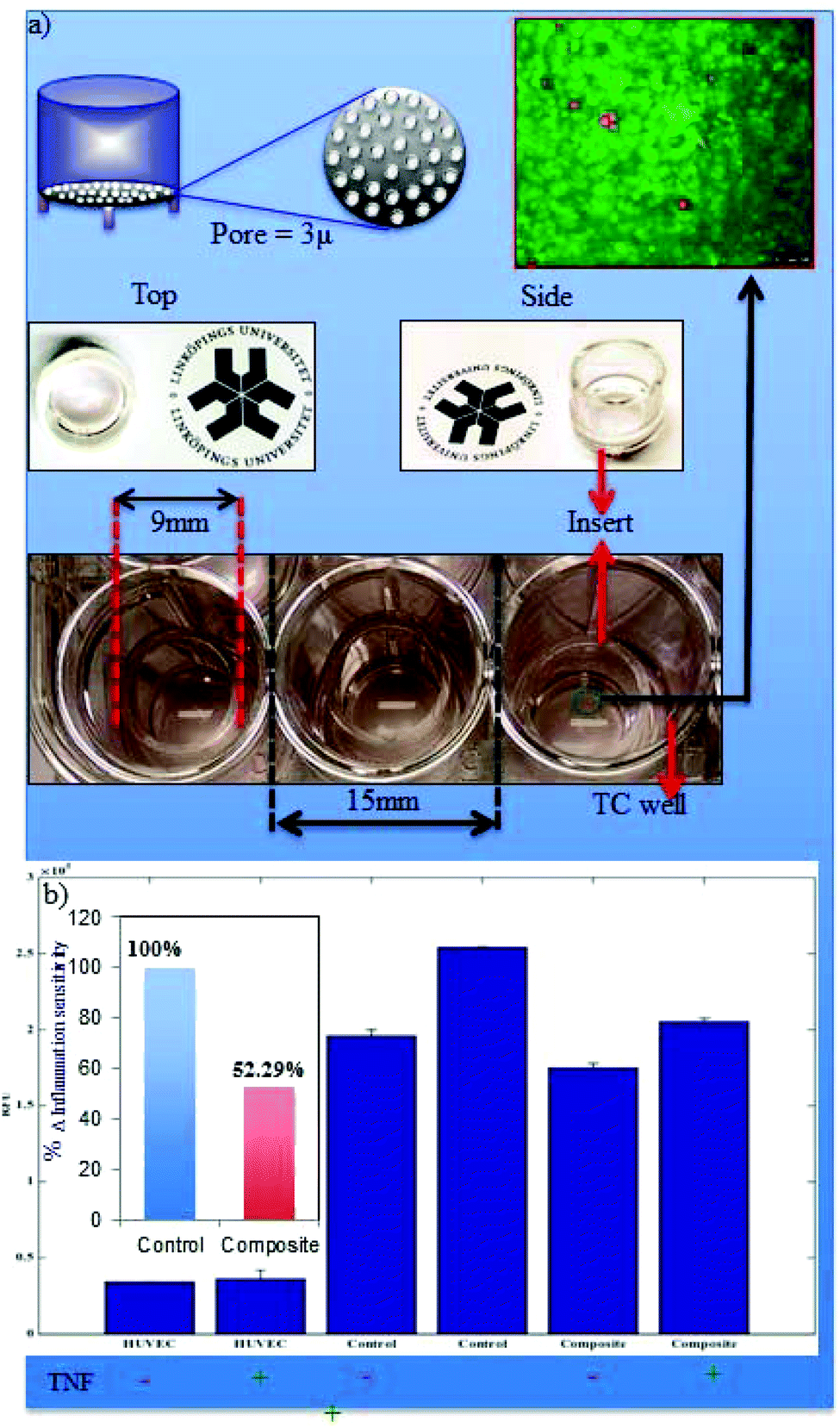 | ||
| Fig. 7 Inflammation-sensitivity. The in vitro inflammation system is designed with the help of an insert with a polycarbonate porous membrane with a precise pore size (enough for the leukocyte to pass but impermeable for HUVEC cells). The two-compartment set up has been shown in the lower panel of (a), and the HUVEC monolayer has been shown with live/dead analysis having >90% cell viability (red circles indicate the dead cells). (b) The LeukoTracker™ stained leukocytes are allowed for extravasation through the HUVEC in the presence (+) and absence (−) of tumor necrosis factor-alpha (TNFα) with respect to the control (without leukocyte) and the transmigrated leukocytes were measured through relative fluorescence (RFU). The relative inflammation-sensitivity has been estimated by taking the percentile RFU differences (Δ, of −TNFα and +TNFα) of control (absence of nanofiber composite networks) and composite (in the presence of nanofiber composite networks) where the normal extravasation of leukocytes through the endothelium has been considered as 100% (inset of b). | ||
The inflammation enrolment requires leukocyte interaction with vascular endothelium and consists of several successive steps including circulating leukocyte capture, rolling, arrest, firm adhesion and subsequent transmigration as shown in Fig. 8 (upper panel). The real time leukocyte extravasation in the absence (left panel) and presence (right panel) is shown by fluorescence microscopy (leukocytes stained with stained with LeukoTracker™ green) in Fig. 8.
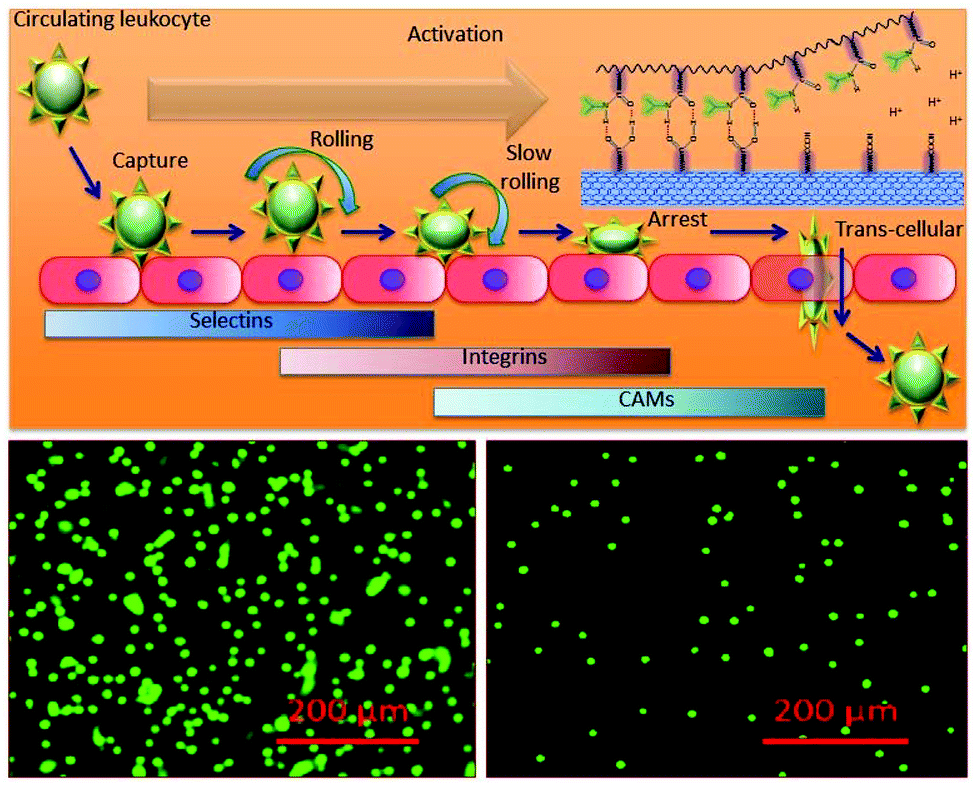 | ||
| Fig. 8 Inflammation and response to the composite. (a) The keys and regulators in real inflammation and the dynamics of leukocyte transmigration. (b) The real time transmigrated leukocytes in the absence (left panel) and presence of the composite (right panel). | ||
Recently in our lab on/off switchable nano-architectures that can sense the precise microenvironment in a well-controlled fashion have been established.45–47 In this present report, we have effectively built a stimuli-responsive nano-architecture based in situ methodology for loco-regional therapeutic management. The present strategy will not only develop the current state-of-the-art of emerging bio friendly scaffold materials but also their fortitude and computational optimization of the applicable 3D structure that will provide appropriate hydrophobicity/hydrophobicity balance to adequately supply mechanical strength with nutrients and growth factors to the newly grafted cells. The current concept and logic of developing such anisotropic nifty materials that can respond to very slight changes in the physicochemical environment such as pH, temperature and ionic strength to regenerate and replace biologically non-functional tissue or generate replacement tissues for a wide spectrum of medical circumstances where defined in situ grafting is required such as cirrhosis wound, burnt tissue, wound of diabetic patients, spinal cord injury, osteo-arthritis etc. has been demonstrated. We have an advanced plan to explore the present concept with the use of peptides that are sensitive to matrix metallo proteases (MMP), which are known to be over-expressed during infection and inflammation. This is the first report where the nanotechnology based approach has been used to address loco-regional inflammation.
Conclusion
We have proposed and designed stimuli receptive bio-friendly composite nanofibrous scaffolds using template-assisted growth and electrospinning. The molecular architectural connections and their shifts have been established through FTIR spectroscopy. We found the stimuli-responsive ability of PANI–MWCNT/PNIPAm hybrid composite nanofiber networks, and achieved a high rate of cell growth and attachment due to the nano-porous 3D architecture with a high effective surface area. The present method involves the encapsulation of cells in 3D structures in the area of interest. The present in situ formation of the cell/scaffold regenerative construct compared to the in vitro formation of the construct will allow delivery of regenerative cells, nutrients and growth factors to the wound site of any undefined shape using minimally invasive techniques. The stimuli responsive nano architecture was mixed with the regenerative cells at room temperature and then incubated at 37 °C. Due to the increase in temperature to 37 °C (above LCST), the nano architecture helps to form a physical gel. The encapsulated cells within the 3D structure of the gel show a high rate of cellular attachments, followed by proliferation. Thus, the present in situ stimulus can be used to regulate the regenerative cells for tissue engineering at the wound surface. Further computational modeling studies on the coil-like and globular PNIPAm conformations and the analysis of the network of solvents involved in intermolecular hydrogen bonding with PNIPAm suggest that hydrophobic interaction stabilizes the PNIPAm-lipid bilayer complex system above LCST. The regeneration of PNIPAm nano-fibers can be achieved by lowering the temperature since the PNIPAm subsystem regains its hydrophilic surface through reverse globular to coil-like conformation transition. Finally, a biocompatible, inflammation-sensitive, stimuli-responsive nanofiber network has been synthesized that can respond to the loco-regional inflammation to form regenerative scaffolds for cell grafting and tissue regeneration.Funding sources
The authors would like to acknowledge the IGEN (Post-Doctoral Fellowship), Swedish Research Council (VR-2011-6058357) and LIST, for generous financial support to carry out this research.Acknowledgements
We are thankful to Prof. May Griffith for her generous support to use the laboratory and cell biology facility at LiU.References
- P. Zhang, L. Chen, T. Xu, H. Liu, X. Liu, J. Meng, G. Yang, L. Jiang and S. Wang, Adv. Mater., 2013, 25, 3566–3570 CrossRef CAS PubMed.
- X. Liu, L. Chen, H. Liu, G. Yang, P. Zhang, D. Han and L. Jiang, NPG Asia Mater., 2013, 5, e63 CrossRef CAS.
- X. Liu and S. Wang, Chem. Soc. Rev., 2014, 43, 2385–2401 RSC.
- R. A. Van Santen, Acc. Chem. Res., 2008, 42, 57–66 CrossRef PubMed.
- H. K. Patra, S. Banerjee, U. Chaudhuri, P. Lahiri and A. K. Dasgupta, Nanomedicine, 2007, 3, 111–119 CAS.
- H. K. Patra and A. K. Dasgupta, Nanomedicine, 2012, 8, 842–852 CAS.
- M. Celebrano, C. Rosman, C. Sönnichsen and M. Krishnan, Nano Lett., 2012, 12, 5791–5796 CrossRef CAS PubMed.
- H. Liu, X. Liu, J. Meng, P. Zhang, G. Yang, B. Su and L. Jiang, Adv. Mater., 2013, 25, 922–927 CrossRef CAS PubMed.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- Y. Zhou, X. Zhou, D. J. Park, K. Torabi, K. A. Brown, M. R. Jones and C. A. Mirkin, Nano Lett., 2014, 14, 2157–2161 CrossRef CAS PubMed.
- G. Li, G. Yang, P. Zhang, Y. Li, J. Meng, H. Liu and S. Wang, Small, 2015, 11, 5642–5646 CrossRef CAS PubMed.
- H. K. Patra and A. K. Dasgupta, Adv. Sci. Lett., 2011, 4, 3770–3775 CrossRef CAS.
- F. Zhang, Y. Jiang, X. Liu, J. Meng, P. Zhang, H. Liu and J. S. Hu, Nano Lett., 2015, 16, 766–772 CrossRef PubMed.
- B. S. Harrison and A. Atala, Biomaterials, 2007, 28, 344–353 CrossRef CAS PubMed.
- J. Venkatesam, Z. Qian, B. Ryu, N. Ashok and S. Kim, Int. J. Biol. Macromol., 2012, 50, 393–402 CrossRef PubMed.
- L. P. Pan, X. Pei, R. He, Q. Wan and J. Wang, Colloids Surf., B, 2012, 93, 226–234 CrossRef CAS PubMed.
- N. Wu, W. Quan and A. Behrouz, Carbon, 2012, 50, 4945–4952 CrossRef CAS.
- I. Armentano, M. Dottori, E. Fortunati, S. Mattioli and J. M. Kenny, Polym. Degrad. Stab., 2010, 95, 2126–2146 CrossRef CAS.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Q. H. Taimoor, R. Rai and A. R. Boccaccini, Biomaterials, 2014, 35, 9068–9086 CrossRef PubMed.
- M. Mattioli-Belmonte, G. Giavaresi, G. Biagini, L. Virgili, M. Giacomini and M. Fini, Int. J. Artif. Organs, 2003, 26, 1077–1085 CAS.
- S. Battistoni, et al., International Conference on Memristive Systems (MEMRISYS) 2015, IEEE, 2015 Search PubMed.
- P. Faeze, M. Farahmand and A. Ardeshirylajimi, J. Paramed. Sci., 2016, 7(1), 1–6 Search PubMed.
- Q. Wang, Y. Qin, C. Jiang and C. Lidong, J. Mater. Chem., 2012, 22, 17612–17618 RSC.
- H. Xia, C. Daming, L. Peisuan and S. C. Hardy, Nanotechnology, 2006, 17, 3957 CrossRef CAS.
- B. Hu, W. Shi, Y. L. Wu, W. R. Leow, P. Cai, S. Li and X. Chen, Adv. Mater., 2014, 26, 5786–5793 CrossRef CAS PubMed.
- P. Cai, M. Layani, W. R. Leow, S. Amini, Z. Liu, D. Qi and X. Chen, Adv. Mater., 2016, 28, 3102–3110 CrossRef CAS PubMed.
- A. Galperin, T. J. Long, S. Garty and B. D. Ratner, J. Biomed. Mater. Res., Part A, 2013, 101, 775–786 CrossRef PubMed.
- T. H. Qazi, R. Rai and A. R. Boccaccini, Biomaterials, 2014, 35, 9068–9086 CrossRef CAS PubMed.
- B. S. Harrison and A. Atala, Biomaterials, 2007, 28, 344–353 CrossRef CAS PubMed.
- A. Tiwari, D. Terada, C. Yoshikawa and H. Kobayashi, Talanta, 2010, 82, 1725–1732 CrossRef CAS PubMed.
- Y. Katsumoto, T. Tanaka, H. Sato and Y. Ozaki, J. Phys. Chem. A, 2002, 106, 3429–3435 CrossRef CAS.
- T. Hasegawa, S. Tatsuta and Y. Katsumoto, Anal. Bioanal. Chem., 2010, 398, 2203–2209 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch et al. , Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. A. Case, et al., Amber 12, University of California, San Francisco, 2010 Search PubMed.
- Z. H. Jin, P. Gumbsch, K. Lu and E. Ma, Phys. Rev. Lett., 2001, 87, 055703 CrossRef CAS PubMed.
- N. A. Murugan, J. Phys. Chem. B, 2005, 109, 23955–23962 CrossRef CAS PubMed.
- N. A. Murugan, R. Apostolov, Z. Rinkevicius, J. Kongsted, E. Lindahl and H. Ågren, J. Am. Chem. Soc., 2013, 135, 13590–13597 CrossRef CAS PubMed.
- Y. Sharma, A. Tiwari, S. Hattori, D. Terada, A. K. Sharma, M. Ramalingam and H. Kobayashi, Int. J. Biol. Macromol., 2012, 51, 627–631 CrossRef CAS PubMed.
- E. Farahani, H. K. Patra, J. R. Jangamreddy, I. Rashedi, M. Kawalec, R. K. Rao Pariti, P. Batakis and E. Wiechec, Carcinogenesis, 2014, 35, 747–759 CrossRef CAS PubMed.
- H. K. Patra, R. Imani, J. R. Jangamreddy, M. Pazoki, A. Iglič, A. P. Turner and A. Tiwari, Sci. Rep., 2015 Search PubMed , 14571.
- H. K. Patra and A. P. Turner, Trends Biotechnol., 2014, 32, 21–31 CrossRef CAS PubMed.
- H. K. Patra, N. Ul Khaliq, T. Romu, E. Wiechec, M. Borga, A. P. Turner and A. Tiwari, Adv. Healthcare Mater., 2014, 3, 526–535 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06157e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |