
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterisation, degradation and regeneration of luminescent Ag29 clusters in solution†
Marte
van der Linden
 *ab,
Arjan
Barendregt
cd,
Arnoldus J.
van Bunningen
e,
Patrick T. K.
Chin
e,
Dominique
Thies-Weesie
f,
Frank M. F.
de Groot
a and
Andries
Meijerink
e
*ab,
Arjan
Barendregt
cd,
Arnoldus J.
van Bunningen
e,
Patrick T. K.
Chin
e,
Dominique
Thies-Weesie
f,
Frank M. F.
de Groot
a and
Andries
Meijerink
e
aInorganic Chemistry & Catalysis, Debye Institute for Nanomaterials Science, Universiteit Utrecht, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: M.vanderLinden1@uu.nl
bID26, European Synchrotron Radiation Facility (ESRF), 71 Avenue des Martyrs, 38000 Grenoble, France
cBiomolecular Mass Spectrometry and Proteomics, Utrecht Institute for Pharmaceutical Sciences and Bijvoet Centre for Biomolecular Research, Universiteit Utrecht, Padualaan 8, 3584 CH Utrecht, The Netherlands
dNetherlands Proteomics Center, Padualaan 8, 3584 CH, Utrecht, The Netherlands
eCondensed Matter & Interfaces, Debye Institute for Nanomaterials Science, Universiteit Utrecht, Princetonplein 5, 3584 CC Utrecht, The Netherlands
fVan 't Hoff Laboratory for Physical and Colloid Chemistry, Debye Institute for Nanomaterials Science, Universiteit Utrecht, Padualaan 8, 3584 CH, Utrecht, The Netherlands
First published on 14th November 2016
Abstract
Luminescent Ag clusters are prepared with lipoic acid (LA) as the ligand. Using a combination of mass spectrometry, optical spectroscopy and analytical ultracentrifugation, the clusters are found to be highly monodisperse with mass 5.6 kDa. We assign the chemical composition [Ag29(LA)12]3− to the clusters, where LA likely binds in a bidentate fashion. The Ag29 clusters show slow degradation, retaining their deep red emission for at least 18 months if stored in the dark. Purification or exposure to light results in faster degradation. No other cluster species are observed during the degradation process. Once degraded, the clusters could easily be regenerated using NaBH4, which is not usually observed for thiolate-capped Ag clusters.
1 Introduction
Noble metal clusters represent a class of materials with sizes between atoms and organometallic complexes on one hand and plasmonic nanoparticles (>2 nm) on the other. The small number of atoms and thus lack of long-range order leads to discrete energy levels and optoelectronic properties such as luminescence.1,2 The (near) atomic monodispersity of particular clusters can be explained by their high stability when compared to clusters of slightly different sizes.3 The most stable clusters are termed “magic size clusters” and their stability can have both electronic and geometric origins.4 The electronic origins of stability are explained by the superatom theory, where clusters are considered to have electronic shells and closed shells correspond to the most stable clusters.5,6 In addition, a compact geometric structure7 and surface passivation by ligands help to ensure stability and to avoid oxidation and aggregation, while the structure of the ligand can also enhance the stability of particular cluster sizes.8 Thiolate-protected clusters in particular have received widespread attention9–11 and include the widely-studied [Au25(SR)18]− (where SR indicates a thiolate ligand). Interestingly, for most of the thiolate-protected Au and Ag clusters with known structures, there have been no reports of luminescence, or the cluster luminescence was found to be very weak; the reported quantum yield of both [Ag44(SR)30]4− and [Au25(SR)18]− is around 0.01%.9,12 A few recent studies have focused on thiolate-protected monodisperse Au and Ag clusters with strong visible luminescence, such as Au22(SR)18 and Ag11(SR)7 (with glutathione as ligand, quantum yields 8 and 3% respectively13,14) and Au25(SR)18 treated with Au(I)thiolate (quantum yield almost 2%15). These studies suggest that the surface structure is important for cluster luminescence. By using dithiolate rather than thiolate ligands to cap clusters, clusters with different structures and thus different properties can be obtained. Indeed, several studies have shown changes in luminescence properties during ligand exchange of thiols with dithiols.16,17 Recent work has also led to the structure elucidation of a dithiolate Ag cluster.18 A promising ligand is lipoic acid (LA), a disulfide, which has been used to prepare luminescent clusters of both Au19–22 and Ag.23–28 Advantages of LA include its solubility in water, the possibility of functionalisation of its carboxylic acid terminated tail20,26 and the straightforward synthesis of LA-protected clusters. We have previously reported on the synthesis of LA-protected Ag clusters, with strong red luminescence (quantum yield ca. 5%) and on the influence of pH, solvent and presence of poly(ethylene glycol) on the luminescence properties.25 However, different sizes have been reported for this cluster, and the monodispersity has not been verified. Adhikari et al., who first prepared these clusters, report the presence of Ag4 and Ag5 in their mass spectra (estimated size <1 nm), while simultaneously showing TEM images of 2–3 nm nanoparticles with clear crystal lattices.23 We have also observed 2–3 nm nanoparticles with TEM,25 as have our co-workers.29 Particles of this size would have several hundred Ag atoms.30,31 This suggests that the synthesis does not yield monodisperse clusters. An alternative explanation was offered by Chen et al., who propose that these nanoparticles act as a scaffold for a luminescent Ag8 species.24 In a recent study, Russier-Antoine et al. report the size as Ag29(LA)12 (no core charge) and find no larger nanoparticles.28 It is clear that extensive characterisation of LA-capped Ag clusters is needed to explain these inconsistencies and correctly identify all species present. Ideally, characterisation should be done at different times after synthesis or purification, as the cluster composition and properties can change for example by removal of excess ligands and salts32 or oxidation following exposure to air and light.33In this paper, we combine sedimentation velocity analytical ultracentrifugation (SV-AUC) experiments with electrospray ionisation mass spectrometry (ESI-MS) and optical spectroscopy to investigate the monodispersity and composition of LA-capped Ag clusters. We show here that the cluster composition is most likely [Ag29(LA)12]3− (not Ag25(LA)14 as we reported earlier25). The monodispersity of the clusters is confirmed by SV-AUC experiments; thus it is expected that the 2–3 nm particles observed with TEM are formed by cluster aggregation during TEM sample preparation. Our experiments indicate bidentate bonding of LA to the Ag cluster. The clusters are found to be remarkably stable in solution, remaining luminescent for ≥18 months if stored in the dark, although purification was found to decrease the cluster stability significantly. We do not observe different cluster species as a result of purification or cluster ageing. Degradation over time of as-synthesised clusters results in aggregation and possibly oxidation. However, this degradation is reversible upon addition of NaBH4, which is not observed for other thiolate-capped Ag clusters.
2 Experimental section
2.1 Chemicals
AgNO3 (laboratory reagent grade) was obtained from Fisher Scientific. NaBH4 (99%), (±)-α-lipoic acid (≥99%) and methanol (≥99.9%) were purchased from Sigma Aldrich. Butanol (99.5%) was obtained from Acros. Water was of Milli-Q quality, purified using a Millipore Direct-Q 3 water purification system.2.2 Synthesis
The synthesis of the Ag clusters is adapted from that of Adhikari et al.23 and has been described in our previous paper.25 Full details can be found in ESI.† In the standard synthesis, the Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LA ratio is around 1:5. The synthesis was also performed with other Ag:LA ratios (from 1:0.6 to 1:28) by changing the amount of LA while the Ag concentration was kept constant. Synthesis of clusters and storage of samples took place at room temperature. Unless stated otherwise, clusters with Ag:LA ratio of ≃1:5 are used for the experiments reported in this article.
:LA ratio is around 1:5. The synthesis was also performed with other Ag:LA ratios (from 1:0.6 to 1:28) by changing the amount of LA while the Ag concentration was kept constant. Synthesis of clusters and storage of samples took place at room temperature. Unless stated otherwise, clusters with Ag:LA ratio of ≃1:5 are used for the experiments reported in this article.
2.3 Optical spectroscopy
UV-Vis spectra were recorded using a Perkin Elmer Lambda 950, a Perkin Elmer Lambda 40 or a Varian Cary 50 spectrometer. Emission spectra were recorded with a Spex 1680 (0.22 m) double beam spectrofluorometer equipped with a liquid nitrogen cooled Acton Research SpectraPro 300i CCD camera. The CCD camera was equipped with a 150 lines per mm grating blazed at 800 nm. The excitation wavelength was 425 nm. Emission spectra were corrected for the spectral response of the equipment. Luminescence lifetimes were recorded with a time-correlated single photon counting card (TimeHarp 260 PC, PicoQuant) with a laser (Opolette HE 355-II, Opotec Inc.) operating at 500 nm as excitation source. For optical spectroscopy measurements, the clusters solution was diluted with water. The dilution factor varied between 4–10×, but the dilution was the same when comparing different samples for the same experiment.2.4 Photobleaching
Photobleaching of the clusters was done with a 532 nm laser (4 mW, GDL-A004, Photop Technologies) under continuous stirring. Absorption spectra were recorded at regular intervals to follow the process.2.5 Analytical ultracentrifugation
Sedimentation velocity experiments have been performed on XL-I and XL-A analytical ultracentrifuges (Proteomelab and Optima XL-A, Beckman Coulter) using absorbance optics. Samples were centrifuged in 12 mm path length double-sector aluminium centrepieces with sapphire windows in an An60-Ti rotor; the reference sector was filled with Milli-Q water. All measurements were performed at 50000 and 60000 rpm at 20 °C. Changes in solute concentrations were detected by 300–500 absorbance scans measured at 528 nm (undiluted samples) or 423 nm (diluted samples). Analysis and fitting of the data was performed using the program Sedfit v 14.3.34 A continuous c(s) distribution model was fitted to the data. The resolution was set to 200 over a sedimentation coefficient range of 0.0–10.0 S. The meniscus and the bottom were kept at fixed values, and the frictional coefficient, the baseline and the raw data noise were floated in the fitting.
2.6 Mass spectrometry
MS measurements were performed in negative ion mode using an electrospray ionisation time-of-flight (ESI-ToF) instrument (LC-T; Micromass, Manchester, UK) equipped with a Z-spray nano-electrospray ionisation source. A nano ESI quadrupole ToF instrument (Micromass, Manchester, UK) was used for tandem mass spectrometric analysis. Needles were made from borosilicate glass capillaries (Kwik-Fil, World Precision Instruments, Sarasota, FL) on a P-97 puller (Sutter Instruments, Novato, CA), coated with a thin gold layer by using an Edwards Scancoat (Edwards Laboratories, Milpitas, CA) six Pirani 501 sputter coater. After purification, the sample was sprayed into the mass spectrometer. For tandem MS and MS analyses, the applied voltage on the needle was between 1200 and 1100 V and the sample cone voltage (SCV) was varied between −7 and 0 V for measurements of intact clusters. For in-source fragmentation of clusters, the SCV was increased (up to −50 V). Subsequently, the created fragments were used as precursor ions for tandem MS experiments. For tandem MS, the collision voltage was varied between 0 and 40 V using argon gas in the collision cell. All spectra were mass calibrated in negative ion mode, using an aqueous solution of phosphoric acid (0.1% v/v). Theoretical mass spectra were calculated using ChemCalc.35 For the intact clusters, we use FWHM = 1 for the simulation, while FWHM = 0.01 is used for theoretical spectra of fragments. In calculating theoretical mass spectra, we assume that neither of the two S-atoms in LA is present as a thiol (thus LA is C8H14O2S2).000 rpm for 25 min. The clusters remained on the filter and were washed twice with water. Both purification methods were used for MS, while tandem MS spectra were recorded of BuOH purified clusters only.
3 Results and discussion
3.1 Characterisation of Ag clusters
The as-synthesised clusters (Ag:LA ≃ 1:5) solution is orange with red luminescence, and has the same optical properties (Fig. 1) as reported earlier.25 The absorption spectrum shows three distinct peaks at 500 and 425 and 330 nm, with a shoulder at 310 nm. The emission maximum is at 680 nm with a shoulder extending into the near-infrared (NIR, ca. 800 nm). The synthesis can easily be scaled up (at least 5× without adapting the synthesis protocol), and works for different Ag:LA ratios (Fig. S1†). If low ligand concentrations (LA/Ag < 1) are used, the samples have weak (sometimes no) luminescence, although the NIR shoulder has a relatively high intensity. It is possible that the NIR emission originates from a surface state that is not fully passivated by ligands.
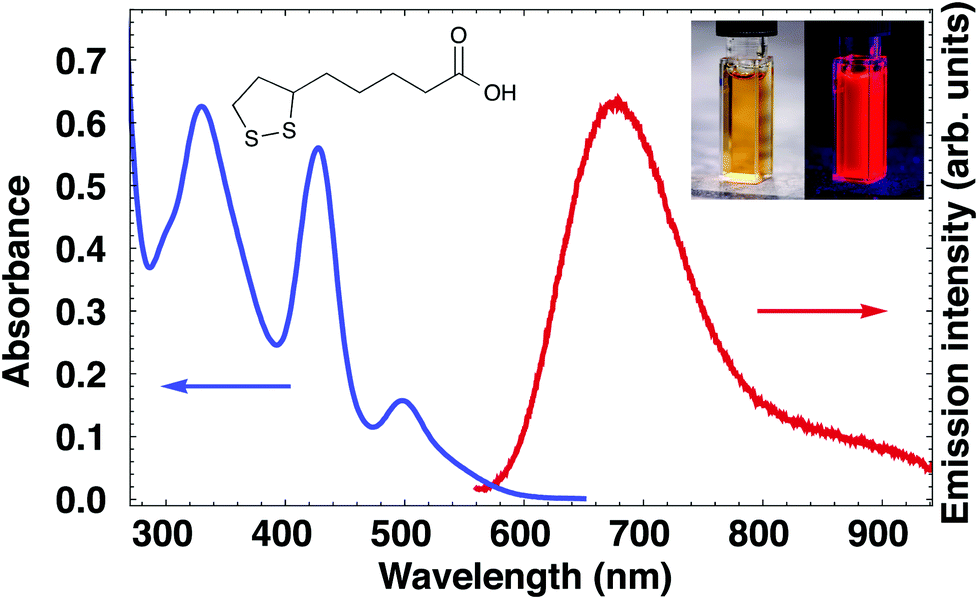 | ||
| Fig. 1 Absorption and emission spectrum of Ag cluster solution. The insets show the structure of LA and photographs of the solution under normal and UV light. | ||
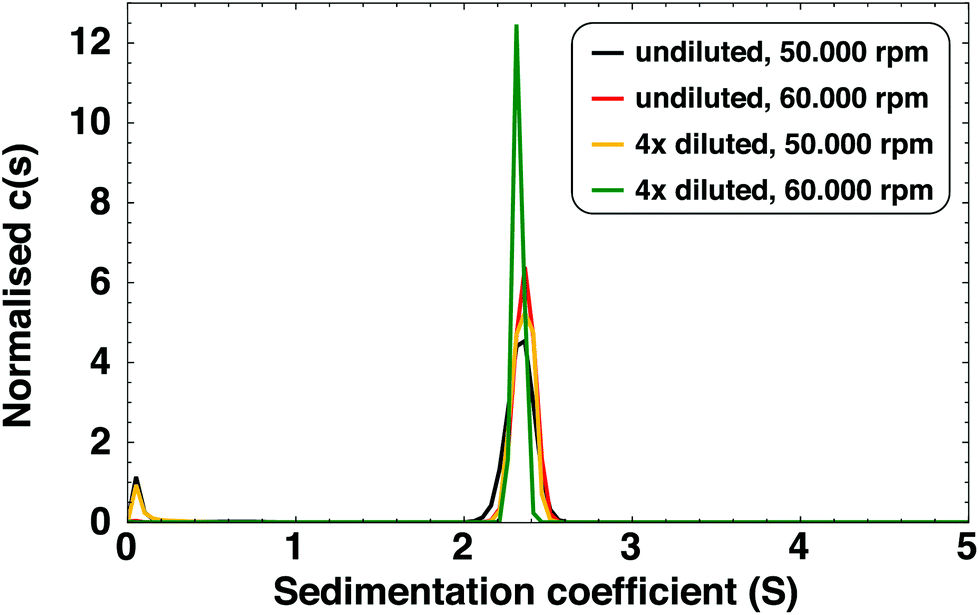 | ||
| Fig. 2 Distribution of sedimentation coefficients for Ag clusters, at various dilutions, centrifugation speeds and absorption wavelengths (black and red: 528 nm; yellow and green: 423 nm). The averaged sedimentation coefficient is 2.34 S (standard deviation 0.014 S). The small peak close to 0 S originates from free ligands. At higher S-values no significant peaks were observed. | ||
We also observed a species with an extremely low sedimentation coefficient (close to 0 S) in SV-AUC experiments. By recording an absorption spectrum of the upper part of the centrifugation cell at the end of the experiment, we identified this species as free LA (see Fig. S3†) and not small AgLA-clusters or Ag(I)–LA complexes. It can therefore be concluded that all Ag clusters are present as the 2.34 S species.
To determine the particle molar mass, we performed sedimentation equilibrium (SE) measurements (full details in ESI†). The molar mass determined in this way is, for undiluted samples, 6.6 kDa. For diluted samples, larger species were also observed (15–25 kDa). These were found to be the result of cluster aggregation due to lower stability of clusters in diluted samples (see Fig. S4† for absorption spectra of diluted clusters over time). These larger species are not present in the as-synthesised, undiluted sample. Dilution of the sample decreases the concentration of free LA which can lead to desorption of ligands from the cluster,37 resulting in lower cluster stability. Indeed, we found that purified clusters, where the excess of free LA has been removed, are less stable than as-synthesised clusters (see section 3.2.1).
Mass spectra of BuOH purified clusters (Fig. S5†) show the same features as reported earlier.25 Two groups of broad ion signals, around m/z = 1130 and 1420, originate from the same 5.6 kDa species in the z = 5− and z = 4− charge state, respectively. We previously identified this cluster as neutral Ag25(LA)14 on the basis of good agreement of theoretical mass and observed mass of the base peak in the deconvoluted spectrum. However, a subsequent detailed analysis of the spectrum before deconvolution has shown that a more likely composition is [Ag29(LA)12]3−. This cluster carries a charge of 3− on the core (which we define here as the S and Ag atoms and thus excludes any charges on the ligands themselves due to deprotonation). Deprotonation of the carboxylic acid group and subsequent association with Na+, termed H+/Na+ exchange, results in the observation of multiple ion signals for the same cluster with the same overall charge. Fig. 3 shows the theoretical mass spectrum of the cluster for all possible H+/Na+ exchanges x, in the overall charge state z = 5−. The total composition of this cluster can be written as [{Ag29(LA)12}3− − (2 + x)H+ + xNa+]5−. There is excellent agreement with the experimental spectrum, while for the previously proposed composition [Ag25(LA)14 − (5 + x)H+ + xNa+]5− (Fig. S6b†) there is no experimental ion signal for the cluster with x = 0 at m/z = 1116. Since species with low x dominate, it is unlikely that the species with x = 0 Na+ is not observed experimentally. To confirm the assignment of the cluster as [Ag29(LA)12]3−, mass spectra of clusters purified with 3 kDa centrifugal cutoff filters (Fig. S10†) were recorded. We expect that purification with the rather apolar solvent BuOH does not remove excess salts from the clusters solution, whereas purification with 3 kDa filters will, thus the two techniques should show different distributions of x. Indeed, the intensity of ion signals with low x increased when 3 kDa filters were used for purification, with the most intense ion signal at m/z = 1120 (z = 5− species), which corresponds to the Ag29 cluster with x = 0 H+/Na+ exchanges. No signal was observed at m/z = 1116 (Ag25, x = 0). Therefore we conclude that the Ag cluster is indeed Ag29.
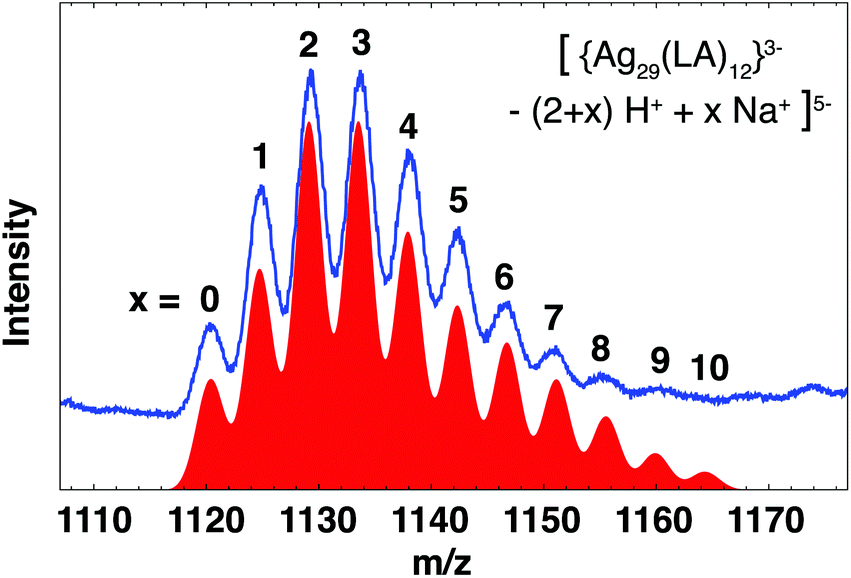 | ||
| Fig. 3 Experimental (blue) and theoretical (red) mass spectra of [{Ag29(LA)12}3− − (2 + x)H+ + xNa+]5− clusters. x refers to the number of H+/Na+ exchanges. The theoretical spectra for each possible x have been manually scaled. All features in the spectrum can be explained by the proposed composition. | ||
Our assignment broadly agrees with the recently published study of Russier-Antoine et al.28 of LA-capped Ag clusters, which were identified as neutral Ag29(LA)12. The cluster with 3− core shows better agreement with our mass spectra than a cluster with neutral core (regardless of whether we compare with the z = 5−, 4− or 3− overall charge states or the deconvoluted spectrum, see Fig. S6a and S7†). The [Ag29(LA)12]3− cluster also shows similarities with the recently discovered [Ag29(BDT)12(TPP)4]3−, where BDT is the dithiolate ligand 1,3-benzenedithiol and TPP the labile ligand triphenylphosphine.18 Besides almost identical compositions and equal core charge, this cluster and our [Ag29(LA)12]3− have comparable absorption and emission spectra. We therefore deem it very likely that [Ag29(LA)12]3− has a similar structure as [Ag29(BDT)12(TPP)4]3−, namely an icosahedral core capped with Ag-ligand units (four Ag3S6 crowns and four Ag1S3P1 units). However, in contrast to the BDT-capped cluster, [Ag29(LA)12]3− does not require additional stabilisation by phosphines, so the capping units might be slightly different or distorted to protect all Ag atoms. The increased stability of LA-capped clusters compared to BDT-capped clusters could be due to the presence of the carboxylic acid-terminated alkyl chain of LA, which provides additional steric stabilisation.
The ligand-to-metal ratio is very low for these Ag29 clusters. Recently, a scaling law (L = mN2/3) was proposed for Au clusters to relate the number of ligands (L) to the number of metal atoms (N).7 The coefficient m was found to be 2.08 for Au–thiolate clusters (1.82 for stable Au–thiolate clusters) while a higher coefficient (2.41) was found for [Ag44(SR)30]4−.38 For [Ag29(LA)12]3− the coefficient is extremely low (1.27), however this ignores the fact that LA can bind bidentate. If we assume bidentate bonding of all LA, the clusters can be considered as [Ag29(SR)24]3− with m = 2.54 (which is in good agreement with what was found for Ag44). This cluster has 8 electrons and is thus a magic number cluster with a closed electron shell.5 In [Ag29(BDT)12TPP4]3− bidentate bonding of all BDT ligands was observed. While this is not necessarily the case for our LA-capped clusters, the ligand-to-metal ratio indicates at least a significant fraction of bidentate ligands to ensure complete capping of the Ag surface. If the structure of the LA-capped cluster is indeed comparable to that of the BDT-capped Ag29 and all LA bind bindentate, this means there are 16 Ag- and 24 S-atoms in the capping layer surrounding the icosahedral core.
We occasionally observe what appears to be bidispersity (see Fig. S8†), with two species showing H+/Na+ exchange in the range m/z = 1120–1160, although all these ion signals could originate from [Ag29(LA)12]3− as they show the same m/z as this cluster. In some spectra, lighter and heavier cluster species are observed which could not be explained from the [Ag29(LA)12]3− composition (5.7–5.9 kDa). It is unlikely that clusters slightly bigger or smaller than the 5.6 kDa species ([Ag29(LA)12]3−) are present in the original sample, as size-focusing would convert them to the most stable size; Ag29. Therefore we expect that the observed polydispersity is to a large extent due to fragmentation or aggregation of the clusters, during purification or ionisation. Indeed, we found that during MS measurements (which typically consisted of a few hundred scans), the signal-to-noise ratio usually decreased and relative intensity of lighter and heavier species than Ag29 increased (Fig. S9†). The influence of heat, light or high voltage could cause degradation of the clusters in the capillary needle.
At low collision voltage (5 V), we find that the cluster loses one ligand. The fact that fragmentation already occurs at such low collision voltage suggests that even a soft ionisation method such as ESI can and will result in some cluster fragmentation, which can be mistaken for inherent polydispersity of the cluster sample. Interestingly, the ion signal for the ligand fragment is observed at m/z = 207, with z = 1− and a mass of 207 Da. LA itself has mass 206 Da, so the 207 Da species is LA with an additional hydrogen atom (termed HLA). This is possible if one of the sulphur atoms exists as a thiol group (–SH) while the other probably is a thiolate (deprotonated thiol) and thus carries the negative charge (alternatively, both sulphur atoms exist as thiols and the carboxylic acid group is deprotonated). We do not observe a fragment corresponding to LA (206 Da), and HLA is not observed in any of the fragments containing silver. We were unable to determine whether HLA is present in the cluster (which would mean that some clusters contain at least one monodentate ligand), or that it is formed from LA and a proton from a neighbouring ligand, during fragmentation.
At higher collision voltages (10–20 V), we observe HLA−, [AgLA]−, [Ag2LA − H+]−, [Ag4LA2 − H+]−, [{Ag5LA2}+ − 2H+]−, [{Ag5LA3}− − H+]2−, [Ag6LA3 − H+]− and [Ag6LA3 − 2H+]2− as well as the corresponding large fragment ions. Some of these fragments carry a core charge (on Ag and S). A number of these fragments were also observed by Russier-Antoine et al. for the neutral Ag29, although it is not clear whether those fragments carry a core charge.28
Little is known about fragmentation pathways of thiolate-protected noble metal clusters, although it appears that fragmentation occurs in the capping units while the core remains intact.39,40 There are a number of similarities between our results and reported tandem mass spectra of [Au25(SR)18]−.40 First, the electron count5 of all the small fragment ions is 0, assuming all LA binds bidentate. Second, we observe only small fragments (up to 6 Ag atoms) or large fragments (down to 23 Ag atoms) and nothing in between, suggesting the core remains intact. Note that our collision voltages are considerably lower than those used for [Ag7(DMSA)4]− and for [Au25(SR)18]−.11,40 This suggests that the Ag–thiolate capping units are relatively weakly bound to the core. However, the cluster is not stripped of its ligands (we do not observe for example [M5− − 2LA−]3−), which indicates strong bonding of the ligand to the silver atoms.
Tandem MS was also done using cluster fragments as precursor ions. These are obtained by raising the initial sample cone voltage. Of particular interest is the precursor ion [AgLA]−, which was fragmented to investigate the bonding of LA to Ag. The initial fragmentation that yielded [AgLA]− could result in rearrangement so that it is important to note that it may not accurately reflect the bonding of LA to Ag in the cluster itself. Spectra are in Fig. S16.† The mass of this precursor is 313 or 315 Da, depending on silver isotope, which means that either the carboxylic acid group or one of the sulphur atoms of LA is protonated. At low collision voltage (5 V), the main fragment ion signal appears at m/z = 269/271, which corresponds to the loss of a neutral fragment with mass 44 Da – consistent with the loss of neutral CO2 from the carboxylic acid group of LA. At higher collision voltages, intense ion signals are observed at m/z = 139–143 and 172/174. We assign these to the fragments [AgS]− + [AgHS]− + [AgH2S]− and [AgHS2]−. The presence of fragments which contain AgS2 is strong evidence that the preferential bonding of LA to Ag is bidentate.
3.2 Cluster stability, degradation and regeneration
The as-synthesised clusters are stable for many months if stored in the dark, with observable luminescence present after 18 months for some samples. The stability of the as-synthesised clusters is comparable to those prepared by Bakr et al.,9 which were later shown to be the ultrastable and atomically monodisperse [Ag44(SR)30]4−, a magic number cluster with 18 electrons.38,41 The stability of these Ag44 clusters was demonstrated by synthesising them in the presence of existing Ag44.41 We performed a similar experiment with our Ag clusters with the same result: rather than acting as seeds for nanoparticle growth, the pre-existing Ag clusters were inert under reductive (synthesis) conditions. The clusters prepared in presence of Ag clusters had the same absorption profile as a reference batch prepared simultaneously (see ESI† for details and Fig. S2† for spectra). This illustrates the high stability of these clusters and how different they are from classical nanoparticles.We further investigated the stability of the [Ag29LA12]3− clusters by studying their degradation over time. Fully degraded cluster solutions are clear, very pale yellow and non-luminescent, and are referred to as “time bleached (TB) clusters”. Full cluster degradation can take anything between 4 and more than 18 months, depending on the sample. We found that exposure to light and oxygen (opening the vial often) has a negative effect on sample stability, with these samples showing the shortest degradation times of just a few months. During degradation (time bleaching), the luminescence intensity decreases and the peaks in the absorption spectrum become less pronounced (Fig. 4 and S24†). The shape of the emission spectrum is the same regardless of sample age, and apart from a small blueshift of the 330 nm peak, the absorption features do not shift and no new features appear during degradation (Fig. S24†). We therefore conclude that no other dominant cluster species are formed as the clusters age.
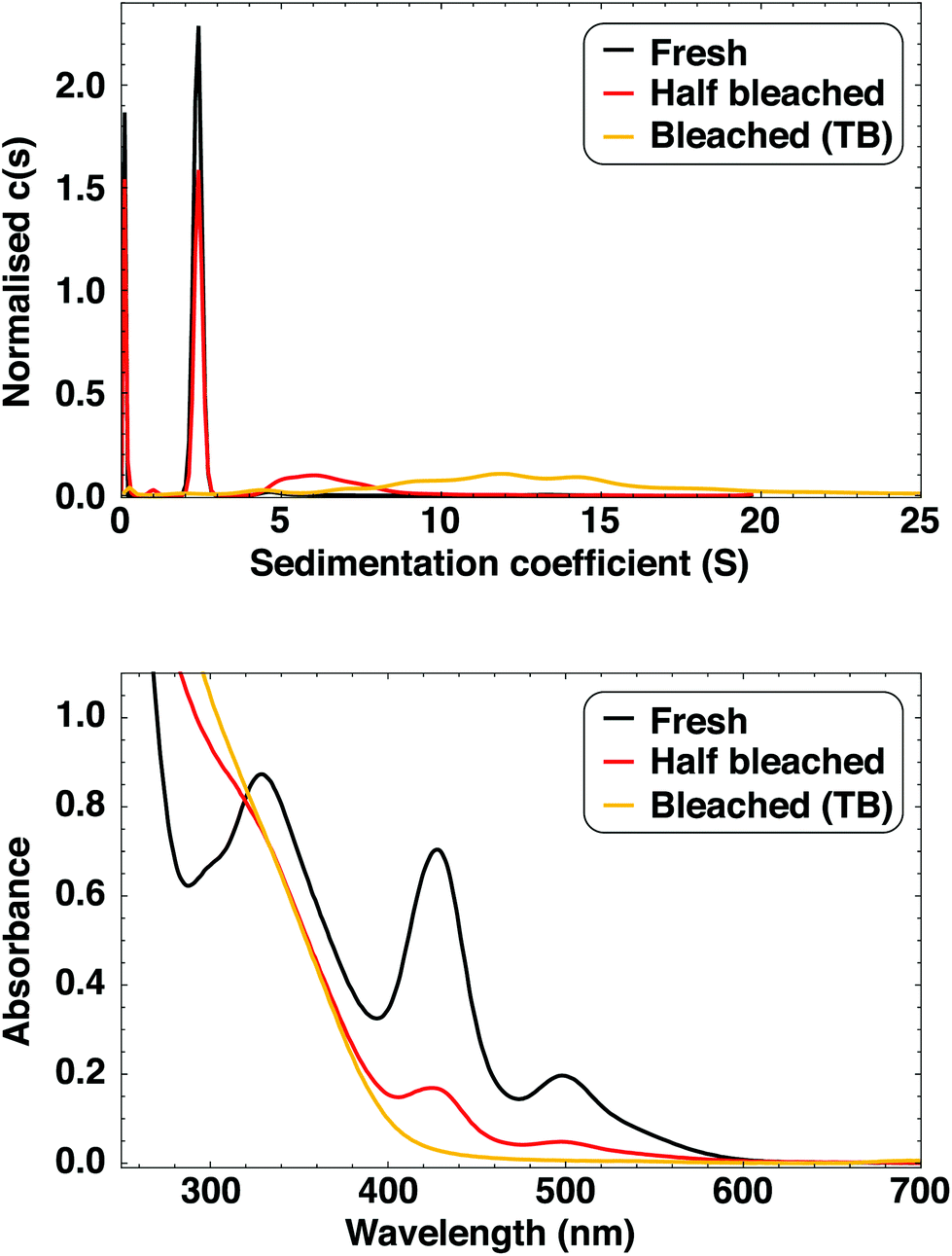 | ||
| Fig. 4 Top: sedimentation coefficient distributions (60000 rpm, 395 nm) and bottom: UV-Vis absorption spectra of fresh clusters (black), TB clusters (yellow) and clusters that are not yet fully bleached (red). The small species with sedimentation coefficient close to 0 S is free LA. | ||
The degradation coincides with a shortening in luminescence lifetime (Fig. 5). The lifetime at 680 nm is fitted to a single exponential. We use the absorbance at 500 nm as a measure for the extent of sample degradation rather than sample age, since the latter does not take into account faster degradation due to exposure to light and air. Clusters with pronounced absorption features have luminescence lifetimes of around 4.1 μs, while for almost fully degraded clusters the luminescence lifetime is 0.5 μs shorter. The dependence of lifetime on luminescence intensity shows a similar trend (Fig. S25†).
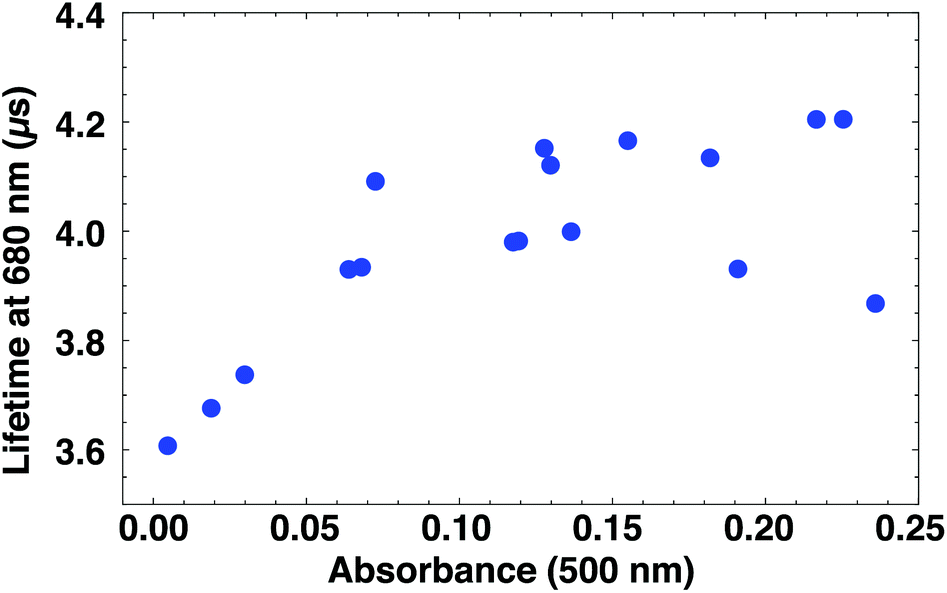 | ||
| Fig. 5 Dependence of luminescence lifetime on the sample absorbance. Fresh samples have higher absorbance and longer luminescence lifetimes than partially degraded samples. Note that quenching occurs from A = 0.15 (see Fig. S25†). | ||
As exposure to light was found to reduce Ag cluster stability, we investigated the effect of irradiation with laserlight (532 nm). Initially, the solution became brown but it lightened over time to a clear, pale yellow. 12.5 hours of laser irradiation was required to form these “laser bleached (LB) clusters”. Observable luminescence disappeared after ca. 30 min. UV-Vis spectra during photobleaching are shown in Fig. S26.† While TB and LB clusters are clearly obtained via a different process (as evidenced by changes in sample colour), the final bleached clusters are indistinguishable by eye. UV-Vis spectra (Fig. S27†) of TB and LB clusters show a decrease in absorbance with increasing wavelength which is characteristic of Mie scattering of nanoparticles.42 The spectrum shows no sharp absorption features (indicating monodisperse clusters) or a surface plasmon absorption (indicating plasmonic Ag nanoparticles).
SV-AUC experiments (Fig. 4) show that TB clusters are polydisperse and larger than as-synthesised clusters, with sedimentation coefficients in the range 5–20 S. The species with 2.34 S is not observed, i.e. the clusters are fully aggregated. We also performed SV-AUC experiments of an old cluster sample that is not fully bleached, and found a broad distribution around 7 S as well as a sharper distribution at 2.34 S, shown to originate from the Ag29 clusters. This is in excellent agreement with the UV-Vis absorption spectra (Fig. 4) where the not fully bleached clusters show the same absorption features as fresh clusters, but with lower absorbance. There is no evidence of Ag clusters that are slightly bigger or smaller than Ag29. The large size, ∼5 nm, of TB clusters is confirmed with TEM (Fig. S28†) and by purification with 3 kDa centrifugal cutoff filters, where the yellow species remained on the filter.
TB and LB clusters could be regenerated by the addition of NaBH4 (a similar amount as was used during the last step of the synthesis). Regenerated clusters have the same absorption and emission features as freshly prepared clusters (Fig. S29†). SV-AUC shows that regenerated clusters have the same sedimentation coefficient and narrow size distribution as fresh clusters (Fig. S30†). We were unable to purify regenerated clusters with BuOH for ESI-MS. However, mass spectra of regenerated clusters purified with 3 kDa filters confirm the presence of [Ag29(LA)12]3−. In addition, we observe a number of slightly lighter species (around 5.0 and 5.4 kDa). Fresh clusters appeared more polydisperse in ESI-MS when purified with 3 kDa filters than with BuOH (Fig. S10†), so it is possible that these lighter species are formed during purification. Full spectra and details of the analysis can be found in ESI (Fig. S31 and S32†).
As the TB clusters can easily be regenerated by addition of NaBH4, it is likely that they are oxidised (for example aggregates of Ag(I)–thiolates). Alternatively, bleaching could be due to ligand desorption and subsequent cluster aggregation; addition of NaBH4 reduces free LA to its dithiol form which could induce a size reduction for example via etching. The exact composition of TB clusters remains unknown; we could find no clear ion signals indicating the presence of species with Ag with ESI-MS.
Regeneration has been demonstrated earlier for DNA-capped Ag clusters43 and selenolate-capped Ag44 clusters.44 Interestingly, the authors of the latter study report that regeneration was not successful for thiolate-capped Ag44 clusters and that larger nanoparticles were formed instead. We could find no reports in the literature of any fully reversible degradation of thiolate-capped Ag clusters, suggesting that LA-capped clusters could be unique in this respect. The high LA/Ag ratio (≈ 5) could be a contributing factor to the success of regeneration. When LA/Ag < 1, the cluster stability is decreased and the samples become non-luminescent within weeks. TB clusters with such a low LA concentration were somewhat darker than the standard TB clusters, and while regeneration by NaBH4 is still possible, optical properties of regenerated clusters appear not to be fully recovered although this could be partially due to the lack of reproducibility of cluster synthesis with low LA concentration (Fig. S34†). If regeneration is performed with both NaBH4 and LA, the optical properties resemble that of a freshly prepared Ag:LA = 1:5 batch. Addition of only LA is not sufficient for cluster regeneration (see Fig. S33 and S35†).
While the initial synthesis of the Ag29 clusters is a bottom-up method, cluster regeneration appears to proceed via a top-down route. The same luminescent species are formed in both cases which is evidence for the stability of our clusters and high affinity of LA for Ag. The strong bonding of LA to Ag (and also to Au) has been demonstrated in several studies where luminescent Ag (or Au) clusters are prepared from nanoparticles (ca. 5 nm) which are etched to clusters by addition of LA – a process which could be similar to the regeneration of our TB clusters.26,45,46
3.3 Luminescence
Finally, we briefly remark on the luminescence properties of the Ag29 clusters (see Fig. 1). The quantum yield (5%25) is lower than that typically observed for clusters capped with polymers (19%47), DNA strands (64%43) or proteins (11%48). However, these ligands are large, can bind to the cluster in multiple ways and can have an inherent polydispersity, all of which makes accurate cluster size determination and structure elucidation challenging. Of Ag–thiolate clusters, those capped with glutathione show especially high quantum yields, with reports of nearly 1%39 for Ag11(SR)7 and 7%49 for mixtures of Ag11(SR)7 and Ag10(SR)6. An extraordinarily high quantum yield of 68% was also found for glutathione-capped Ag clusters, although the size and monodispersity of these clusters are unknown.50 In contrast, we present an atomically precise and highly stable cluster. The high quantum yield of the Ag29 cluster makes it a candidate for both fundamental studies into the origin of luminescence51 and for potential applications,52 for example in bioimaging.4 Conclusions
In conclusion, we have prepared stable, luminescent Ag clusters with high monodispersity. The combination of mass spectrometry, optical spectroscopy and analytical ultracentrifugation has allowed us to unequivocally identify a 5.6 kDa species (assigned to [Ag29(LA)12]3−) as the luminescent cluster. We found no evidence of other cluster sizes at different times after synthesis or purification. Tandem MS experiments indicate that LA is strongly bound to the cluster surface, likely with both sulphur atoms. Over time, the clusters degraded resulting in loss of luminescence, but the luminescent clusters could easily be regained by addition of NaBH4. The reversibility of cluster degradation has not been previously observed for thiolate-capped Ag clusters.Acknowledgements
We thank Marcel Verheijen for recording TEM images of TB clusters. This work was financially supported by the Debye Graduate Programme (The Netherlands Organisation for Scientific Research, NWO, project 022.004.016), and ESRF Graduate Programme. The mass spectrometry research was performed within the framework of The Netherlands Organisation for Scientific Research (NWO) and supported by the large scale proteomics facility Proteins@Work (project 184.032.201) embedded in The Netherlands Proteomics Centre.References
- I. Díez and R. H. A. Ras, Nanoscale, 2011, 3, 1963–1970 RSC.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- W. A. de Heer, Rev. Mod. Phys., 1993, 65, 611–675 CrossRef CAS.
- A. Dass, Nanoscale, 2012, 4, 2260–2263 RSC.
- C. Zeng, Y. Chen, G. Li and R. Jin, Chem. Mater., 2014, 26, 2635–2641 CrossRef CAS.
- O. M. Bakr, V. Amendola, C. M. Aikens, W. Wenseleers, R. Li, L. Dal Negro, G. C. Schatz and F. Stellacci, Angew. Chem., Int. Ed., 2009, 48, 5921–5926 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- Z. Wu, E. Lanni, W. Chen, M. E. Bier, D. Ly and R. Jin, J. Am. Chem. Soc., 2009, 131, 16672–16674 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- B. A. Ashenfelter, A. Desireddy, S. H. Yau, T. Goodson and T. P. Bigioni, J. Phys. Chem. C, 2015, 119, 20728–20734 CAS.
- A. Mathew, E. Varghese, S. Choudhury, S. K. Pal and T. Pradeep, Nanoscale, 2015, 7, 14305–14315 RSC.
- Z. Tang, T. Ahuja, S. Wang and G. Wang, Nanoscale, 2012, 4, 4119–4124 RSC.
- Z. Tang, B. Xu, B. Wu, D. A. Robinson, N. Bokossa and G. Wang, Langmuir, 2011, 27, 2989–2996 CrossRef CAS PubMed.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. Del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-E. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS PubMed.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- X. Le Guevel, O. Tagit, C. E. Rodríguez, V. Trouillet, M. Pernia Leal and N. Hildebrandt, Nanoscale, 2014, 6, 8091–8099 RSC.
- L. Shang, L. Yang, F. Stockmar, R. Popescu, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2012, 4, 4155–4160 RSC.
- B. Adhikari and A. Banerjee, Chem. Mater., 2010, 22, 4364–4371 CrossRef CAS.
- W.-T. Chen, Y.-J. Hsu and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 2493–2499 CrossRef CAS PubMed.
- P. T. K. Chin, M. van der Linden, E. J. van Harten, A. Barendregt, M. T. M. Rood, A. J. Koster, F. W. B. van Leeuwen, C. de Mello Donega, A. J. R. Heck and A. Meijerink, Nanotechnology, 2013, 24, 075703 CrossRef PubMed.
- M. A. H. Muhammed, F. Aldeek, G. Palui, L. Trapiella-Alfonso and H. Mattoussi, ACS Nano, 2012, 6, 8950–8961 CrossRef CAS PubMed.
- L. Shang, R. M. Dörlich, V. Trouillet, M. Bruns and G. U. Nienhaus, Nano Res., 2012, 5, 531–542 CrossRef CAS.
- I. Russier-Antoine, F. Bertorelle, R. Hamouda, D. Rayane, P. Dugourd, Z. Sanader, V. Bonačić-Koutecký, P.-F. Brevet and R. Antoine, Nanoscale, 2016, 8, 2892–2898 RSC.
- A. Bunschoten, P. T. K. Chin, T. Buckle, M. van der Linden, A. Barendregt, M. A. Verheijen and F. W. B. van Leeuwen, Eur. J. Inorg. Chem., 2016, 3030–3035 CrossRef CAS.
- Y. Levi-Kalisman, P. D. Jadzinsky, N. Kalisman, H. Tsunoyama, T. Tsukuda, D. A. Bushnell and R. D. Kornberg, J. Am. Chem. Soc., 2011, 133, 2976–2982 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- X. Yuan, Q. Yao, Y. Yu, Z. Luo, X. Dou and J. Xie, J. Phys. Chem. Lett., 2013, 4, 1811–1815 CrossRef CAS PubMed.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CAS.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS PubMed.
- L. Patiny and A. Borel, J. Chem. Inf. Model., 2013, 53, 1223–1228 CrossRef CAS PubMed.
- B. Michen, C. Geers, D. Vanhecke, C. Endes, B. Rothen-Rutishauser, S. Balog and A. Petri-Fink, Sci. Rep., 2015, 5, 9793 CrossRef CAS PubMed.
- I. Moreels, J. C. Martins and Z. Hens, ChemPhysChem, 2006, 7, 1028–1031 CrossRef CAS PubMed.
- K. M. Harkness, Y. Tang, A. Dass, J. Pan, N. Kothalawala, V. J. Reddy, D. E. Cliffel, B. Demeler, F. Stellacci, O. M. Bakr and J. A. McLean, Nanoscale, 2012, 4, 4269–4274 RSC.
- A. Baksi, M. S. Bootharaju, X. Chen, H. Häkkinen and T. Pradeep, J. Phys. Chem. C, 2014, 118, 21722–21729 CAS.
- C. A. Fields-Zinna, J. S. Sampson, M. C. Crowe, J. B. Tracy, J. F. Parker, A. M. DeNey, D. C. Muddiman and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 13844–13851 CrossRef CAS PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC.
- J. Sharma, H.-C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280–3282 RSC.
- I. Chakraborty and T. Pradeep, Nanoscale, 2014, 6, 14190–14194 RSC.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS PubMed.
- S. Huang, C. Pfeiffer, J. Hollmann, S. Friede, J. J.-C. Chen, A. Beyer, B. Haas, K. Volz, W. Heimbrodt, J. M. Montenegro Martos, W. Chang and W. J. Parak, Langmuir, 2012, 28, 8915–8919 CrossRef CAS PubMed.
- L. Shang and S. Dong, Chem. Commun., 2008, 1088–1090 RSC.
- A. Mathew, P. R. Sajanlal and T. Pradeep, J. Mater. Chem., 2011, 21, 11205–11212 RSC.
- K. Zheng, X. Yuan, K. Kuah, Z. Luo, Q. Yao, Q. Zhang and J. Xie, Chem. Commun., 2015, 51, 15165–15168 RSC.
- X. Le Guével, C. Spies, N. Daum, G. Jung and M. Schneider, Nano Res., 2012, 5, 379–387 CrossRef.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017–1053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra of samples with different Ag:LA ratios and old samples; SE-AUC data; tandem MS spectra; absorption, emission and ESI-MS spectra after cluster purification and cluster regeneration. See DOI: 10.1039/c6nr04958c |
| This journal is © The Royal Society of Chemistry 2016 |