
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ordered gyroidal tantalum oxide photocatalysts: eliminating diffusion limitations and tuning surface barriers†
Alexey S.
Cherevan
a,
Spencer
Robbins
b,
Dennis
Dieterle
a,
Paul
Gebhardt
a,
Ulrich
Wiesner
b and
Dominik
Eder
*a
aInstitut für Materialchemie, Technische Universität Wien, Getreidemarkt 9/BC/2, A-1060, Vienna, Austria. E-mail: dominik.eder@tuwien.ac.at
bDepartment of Materials Science and Engineering, 330 Bard Hall, Cornell University, Ithaca, NY 14853, USA
First published on 30th August 2016
Abstract
In this work we synthesized well-ordered, Ta2O5 films with a 3D-interconnected gyroid mesopore architecture with large pore sizes beyond 30 nm and extended crystalline domains through self-assembly of tailor-made triblock-terpolymers. This has effectively eliminated diffusion limitations inherent to previously reported mesoporous photocatalysts and resulted in superior hydrogen evolution with apparent quantum yields of up to 4.6% in the absence of any cocatalyst. We further show that the injection barrier at the solid–liquid interface constitutes a key criterion for photocatalytic performance and can be modified by the choice of the carbon template. This work highlights pore and surface engineering as a promising tool towards high-performance mesoporous catalysts and electrodes for various energy-related applications.
Introduction
Photocatalysis/photoelectrochemistry is central to a range of cutting-edge applications that address important socioeconomic areas such as energy (e.g. solar fuels, photovoltaics), environment (e.g. water and air purification, CO2 fixation) and health (e.g. disinfection, antibacterial coatings). Among the currently most sought-after applications is the photocatalytic oxidation and reduction of water (“artificial water splitting”), which is a process that converts energy of light photons into energy of chemical bonds1 and constitutes an important step for a potential hydrogen-based economy of the future.2–5 This requires a semiconductor with a suitable bandgap capable of absorbing light photons with sufficient energy to initiate the desired redox reactions. Another criterion is the lifetime of photoexcited charge carriers, which need to transfer to the catalyst's surface before they recombine. This can be addressed by improving crystallinity (i.e. diminishing grain boundaries and other structural defects, which can act as recombination centres)6 and through nanostructuring (i.e. shortening their diffusion paths to the solid–liquid interface).7 Most importantly, the photocatalyst needs to provide direct access to a large surface area without diffusion limitations.Mesoporous materials engineering has emerged as a promising strategy for designing high-performance (photo)catalysts and (photo)electrodes in various energy related applications.8 Among the benefits of a mesoporous matrix are an increased active surface area and shorter diffusion paths for charge carriers.9 In addition, in contrast to random agglomerates of nanoparticles, materials with continuous porous networks are stable towards agglomeration in aqueous solutions.10
Various mesostructures have been produced by block copolymer self-assembly (e.g. with pluronics),11 ligand- or surfactant-assisted templating12 or by using anodized alumina as hard templates.13 Such structures have shown great promise in applications, such as dye-sensitized and perovskite solar cells,15 in which efficient pore penetration and charge transport are required. So far, films have typically been reported with 0D cubic micellar and 1D hexagonally-ordered cylindrical architectures and were limited to comparatively small pore sizes (e.g. 5 nm). The 1D access and small pore size generally induce kinetic limitations for reactant diffusion in liquid environments14 and as a consequence restrict the accessibility of the entire surface area for photocatalytic conversion.
In this work we aim to overcome these challenges by synthesizing well-ordered photocatalyst films with large, 3D-interconnected mesopores through self-assembly of tailor-made triblock-terpolymers. We chose tantalum oxide (Ta2O5) as a model compound, which is a highly active photocatalyst in the UV16 and – after conversion into (oxy)nitride – in the visible light range.17 So far, very few studies have been dedicated to mesoporous Ta2O5 albeit with limited success.18–22 In particular, we aim to design 3D-gyroidal Ta2O5 structures with (a) pore sizes beyond 20 nm to facilitate reactant diffusion and enable direct access to a large inner surface without kinetic limitations, and (b) thin, highly-crystalline walls with large crystal domains to enhance charge transport to the solid–liquid interface and reduce charge recombination. We further envision that these structures will benefit other energy-related applications including (photo)electrochemical cells and batteries.
Experimental section
Materials
Hydrochloric acid (Aristar BDH, 37 wt%, ACS/NF/FCC), tantalum(V) ethoxide (Sigma-Aldrich, 99.98%, trace metals basis), tetrahydrofuran (Sigma-Aldrich, anhydrous, ≥99.9%, inhibitor-free), chloroplatinic acid (Sigma-Aldrich, 8 wt% in water solution), toluene (Sigma-Aldrich, 99.8%, anhydrous), ferrocene (Sigma-Aldrich, 98%) and benzyl alcohol (Sigma-Aldrich, 99.8%, anhydrous) were all used as received.Methods
The block copolymer, poly(isoprene-b-styrene-b-ethylene oxide) (PI-b-PS-b-PEO; ISO), was synthesized by sequential anionic polymerization and characterized by a combination of gel permeation chromatography and nuclear magnetic resonance.23 In particular, the ISO used in this work had an overall molar mass of 69 kg mol−1 and consisted of 29.6 wt% polyisoprene (I), 64.8 wt% polystyrene (S), and 5.6 wt% poly(ethylene oxide) (O) with a polydispersity index of 1.04.Gyroid Ta2O5 samples were prepared via the sol–gel process using hydrochloric acid as the catalyst. ISO was dissolved in tetrahydrofuran (THF) to yield a 2.7 wt% solution. The inorganic sol was prepared in a separate vial; 1 mL of THF (24.66 mmol) was added to 0.343 mL HCl and 1 mL (3.86 mmol) of Ta(OEt)5 was quickly injected while stirring vigorously. After 5 min, 1.28 mL of THF (15.78 mmol) were quickly added to the solution, which was stirred for another 2 minutes. An aliquot of this sol was then added to the ISO solution to obtain a ratio of 0.34 mL sol per 100 mg ISO. The solution was stirred again for 90 minutes and then poured into a PTFE dish to cast an ISO/Ta2O5 film. The PTFE dish was placed on a shallow glass crystallization dish, placed on a hotplate set to 50 °C and covered by a glass hemisphere to allow slow solvent evaporation and to initiate self-assembly. After complete solvent evaporation, the ISO/Ta2O5 hybrid film was aged in a vacuum oven at 130 °C for several hours and subsequently calcined in ambient air at 700 °C for 2 h (ramp rate of 1 K min−1) to completely remove the polymer and induce crystallization. The reference sol–gel Ta2O5 (SG Ta2O5) was synthesized following a similar procedure as described for the gyroid Ta2O5, however, without the addition of the polymer. The procedure typically yielded around 30 mg of the gyroid Ta2O5 (per batch), which was optimized to ensure uniform pore characteristics throughout the entire film (∼1 cm2, confirmed by statistics with SEM). In order to obtain comparatively large amounts of samples required for material characterization (e.g. 150 mg for BET) and for photocatalytic tests (50 mg), we prepared several batches and combined each of them after careful characterization with TEM/SEM, XRD and SAXS.
Ta2O5 nanotubes were prepared using CNTs as sacrificial templates. At first, MWCNTs were hybridized with Ta2O5 by means of a hydrothermally assisted sol–gel process to yield a CNT–Ta2O5 hybrid with a thin layer of the oxide (see Fig. S2†). Importantly, benzyl alcohol was used to link the precursor molecules to the hydrophobic CNT surface.24 To obtain Ta2O5 NTs the nanocarbon was subsequently removed via heat treatment in air at 700 °C for 2 h (1 K min−1) upon which the sample color turned white indicating oxidation of the CNTs.
Photocatalytic experiments
Water splitting experiments were performed using an inner irradiation gas flow slurry type home-build reactor consisting of two cylindrical parts, each double walled, the inner cylinder completely made of quartz (HSQ300 type from Heraeus) to be transparent to the UV portion of light. A medium pressure immersion type 700 W Hg TQ 718 lamp (output power was adjusted to 500 W for all experiments) purchased from UV Consulting Company with an output range of 200–600 nm was inserted into the inner cylinder. In a single experiment 50 mg of a powdered photocatalyst was dispersed in 10 vol% MeOH–water solution by ultrasonication. The reaction medium was then transferred to the reactor. During the experiment, the reactor was continuously purged with argon 5.8 (flow rate of 100 ml min−1, controlled with a mass flow controller Q-Flow 140 Series from MCC-Instruments). It was also used as a carrier gas for the online gas analyzer (X-Stream®, Emerson Process Management). H2 was detected and quantified using the in-built thermal conductivity detector (TCD) that was calibrated with H2/Ar mixtures. The temperature of the reactor was kept at 10 °C through a water cooling system from Lauda (Variocool 1200 W). Calculation of the apparent quantum yields (AQY) of the photocatalytic reaction is described in the ESI.†Results and discussion
Our strategy involved the preparation of ordered mesoporous Ta2O5 films with a 3D co-continuous gyroidal pore architecture (gyroid Ta2O5) using evaporation-induced self-assembly (EISA) of block copolymer directed sol–gel structures as schematically shown in Fig. 1a. Upon addition of an inorganic sol precursor, the hydrophilic block of the polymer swells, consequently altering the resulting polymer assembly relative to the parent polymer. The final pore structure can be controlled by synthesis parameters such as the water concentration, precursor/copolymer ratio and evaporation kinetics.25 This process has been demonstrated for a wide range of metal oxides using commercial pluronic copolymers, e.g. P123 or F127. The synthesis of large, >20 nm, pores with 3D pore architectures, however, requires specifically designed terpolymers. In this work, we prepared a poly(isoprene)-b-poly(styrene)-b-poly(ethylene oxide) (PI-b-PS-b-PEO, ISO) terpolymer via anionic polymerization following Stefik et al.23 Volume fractions of the three blocks were optimized to yield a gyroid architecture upon addition of an inorganic sol according to the phase diagram of ISO.26,27 | ||
| Fig. 1 (a) Illustration of the synthesis procedure of mesoporous Ta2O5 using ISO for self-assembly including casting the ISO/Ta2O5 film from a precursor solution, slow solvent evaporation enabling ordered self-assembly, and calcination to remove the terpolymer and yield mesoporous Ta2O5. (b) SEM, (c) TEM, and (d), (e) HRTEM images of Ta2O5 samples with an ordered gyroid like structure with large pores and extended crystalline domains with monolithic character. The inset in (c) shows the ED pattern of the corresponding area. Dashed circles in (d) highlight the pore-to-pore distance (i.e. strut length) with uniform lengths of about 25 nm. These areas share the same crystallographic orientation and are thus part of a larger single-crystalline domain. | ||
The ISO terpolymer was used to structure-direct the synthesis of mesoporous Ta2O5 films. As summarized in Fig. 1a, the terpolymer was first dissolved in THF and specific amounts of tantalum(V) ethoxide were added as a precursor for Ta2O5. After low-temperature solvent evaporation, the resulting films were calcined at 700 °C to remove the polymer scaffold (its oxidation occurs at about 400 °C) and, consequently, convert the network into crystalline Ta2O5. We varied the composition of the sol as well as the polymer-to-inorganic ratio in order to achieve ordered mesopore structures.
Fig. 1b shows a typical top-view SEM image of the Ta2O5 film, which reveals the presence of large, ordered pores. Both the pore diameter and pore arrangements are surprisingly uniform and representative of the macroscopic film (see Fig. S1†). TEM images in Fig. 1c and d suggest a 3D-interconnected network structure consistent with the alternating gyroid (GA) and pore sizes around 30 nm. The walls are uniform in thickness of about 10 nm, a preferred length scale for efficient light absorption and charge transport. Most importantly, the HRTEM image in Fig. 1e shows that the lattice fringes extend beyond the length of individual struts (indicated by the dashed circles in Fig. 1d), suggesting that the walls are composed of extended single-crystalline domains.
Small-angle X-ray scattering (SAXS) provides additional details on the mesostructure (Fig. 2a). The pattern of the ISO/Ta2O5 hybrid exhibits reflections at (d/d100)2 = 2, 6, 12, 14, and 22, consistent with the alternating gyroid (GA) structure, and a d100 spacing of 53.1 nm.28 The SAXS data of the 700 °C treated gyroid Ta2O5 sample show significant changes in the ISO/Ta2O5 hybrid. The primary reflection is shifted to a higher scattering vector, q, indicating a smaller d100 of 29.9 nm. This is associated with network shrinkage of about 40% as expected from comparable systems.14 Diminishing of higher order reflections and peak broadening suggest the loss of a long-range mesoscale order.
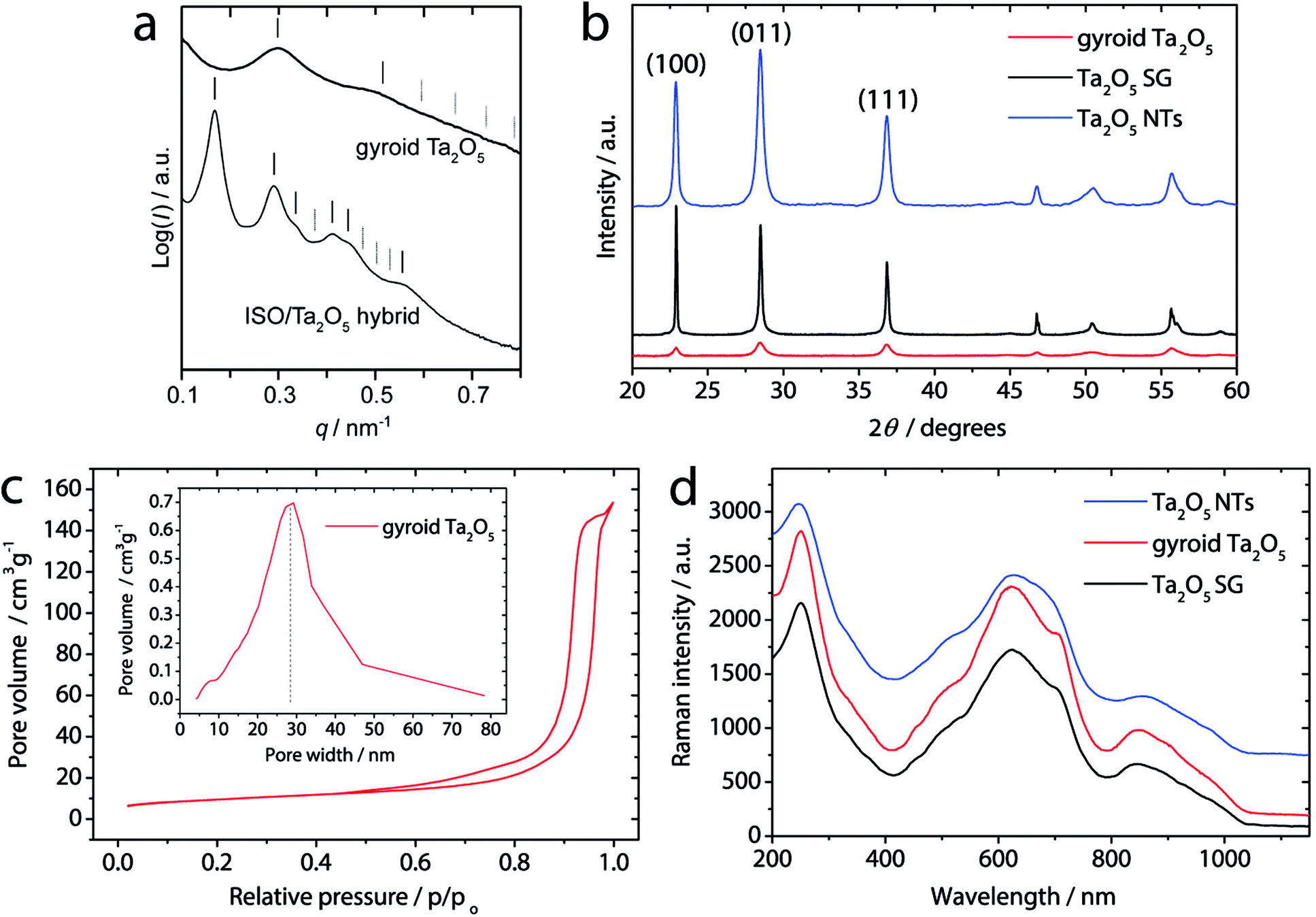 | ||
| Fig. 2 (a) Azimuthally integrated SAXS patterns of the ISO/Ta2O5 hybrid (bottom) and mesoporous gyroid Ta2O5 (top) samples. Solid vertical lines indicate the expected peak positions for the alternating gyroid (GA) morphology. (b) Comparison of the XRD patterns, and (d) Raman spectra (stacked) for the mesoporous gyroid Ta2O5, Ta2O5 NTs, and sol–gel Ta2O5 samples. (c) N2 sorption isotherms and the sorption isotherm derived pore size distribution of the mesoporous gyroid Ta2O5 sample. | ||
X-ray diffraction (XRD, Fig. 2b) confirms that the network is crystalline without any noticeable amorphous content and consists of the β-Ta2O5 phase.29 The average crystal size, calculated by Scherrer's equation using the (100) diffraction peak, is about 26 nm, which corresponds well to the average length of struts connecting network junctions of the Ta2O5 gyroid as observed by HRTEM (Fig. 1d). The dashed circles therefore indicate those sections within the extended crystalline domains that share the same crystallographic orientation and whose dimensions are guided by the wall thickness as well as the pore-to-pore distance. N2 sorption studies reveal a type IV isotherm with H2 hysteresis (Fig. 2c), confirming the presence of mesopores. The sample exhibits a narrow BJH pore size distribution centred at 28 nm. The specific surface area of 32 m2 g−1, according to BET, is considerably larger than that of a sol–gel derived reference sample (Ta2O5 SG) produced without the terpolymer (2.2 m2 g−1).
The Raman spectra in Fig. 2d and S4† show the typical peaks of β-Ta2O5 without any secondary or impurity phases (e.g. tantalum carbides, carbon layer) as well as the absence of any peak shifts and/or broadening that are typically associated with the presence of defects.32
These results confirm the successful synthesis of crystalline Ta2O5 networks with alternating gyroidal pore structures, uniformly large pore sizes, and single-crystalline domains that extend beyond the strut limitations of the gyroidal network. Moreover, the combined results reveal no carbon residues within the detection limits.
The photocatalytic performance of the samples was evaluated for hydrogen evolution via sacrificial water splitting conducted in an inner irradiation type flow reactor, each using 50 mg of the photocatalyst (see Experimental section for details). For comparison, a conventional non-porous Ta2O5 SG powder was used as a reference (see Fig. S1† and Experimental section). Fig. 3a reveals the remarkable photocatalytic activity of gyroid Ta2O5 samples towards water reduction without the use of any co-catalyst (e.g. peak and stabilized hydrogen evolution rates of 1188 and 955 μmol h−1, respectively). The calculated apparent quantum yields (AQY) for the gyroid Ta2O5 are 3.7 and 4.6% for the stabilized and peak rates, respectively – among the highest values reported for binary metal oxides optimized only via morphology and structure engineering (see Table S1 and Fig. S7† for calculations).30 Most importantly, the peak hydrogen production rate of the mesoporous gyroid Ta2O5 is about 15 times higher than that of Ta2O5 SG. This relative increase complies remarkably well with the increase in the BET surface area (∼15 times, see Table 1). This linear correlation suggests that activity is not limited by pore diffusion as observed in mesoporous Ta2O5 photocatalysts with smaller pore sizes.21,22 An additional contribution stems from the presence of the gyroid mesoporous network that provides 3D access to the inner surface (Fig. 1a). The gyroid Ta2O5 also exhibits excellent chemical and mechanical stability towards harsh UV and ultrasound-assisted treatments (Fig. S5†) and retains remarkably high activity even at long experimental times, albeit with a gradual slight decrease in the rate, which is not yet fully understood (Fig. S6†).
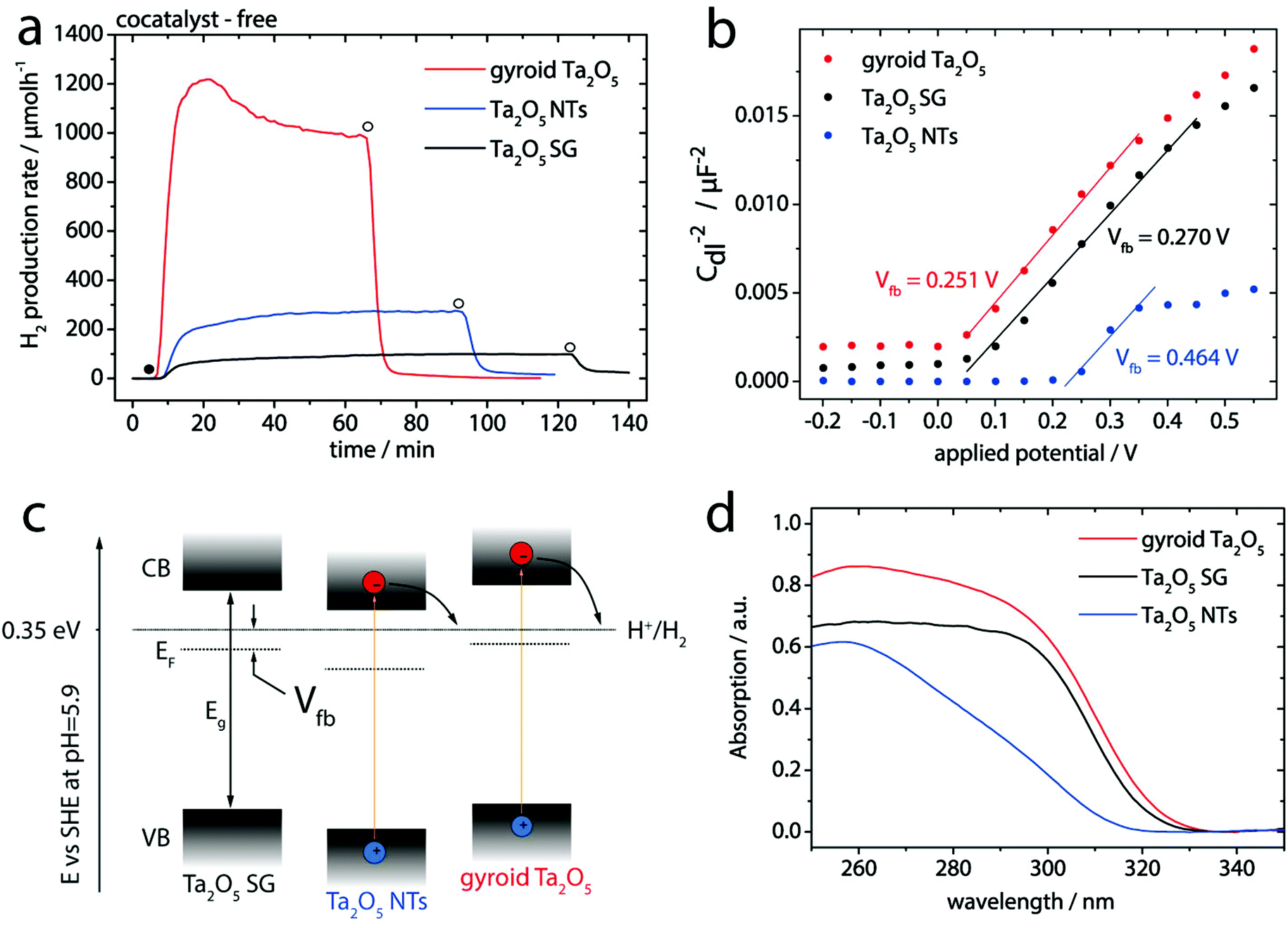 | ||
| Fig. 3 (a) Hydrogen evolution rates in photocatalytic water splitting of the mesoporous gyroid-Ta2O5, Ta2O5 SG, and Ta2O5 NT samples without any cocatalyst. The filled circle indicates the start of light irradiation, open circles – the end. (b) Mott–Schottky plots with linear extrapolation of the mesoporous gyroid Ta2O5, Ta2O5 SG, and Ta2O5 NT samples obtained from EIS measurements performed at different potentials (for more information see the ESI†). (c) Energy diagram based on the EIS data and (d) diffuse reflectance spectra of Ta2O5 SG, Ta2O5 NTs and mesoporous gyroid Ta2O5. | ||
For comparison we synthesized mesoporous Ta2O5 nanotubes (Ta2O5 NTs) with a similar surface area and large accessible inner channels, yet different surface termination by using thick multi-walled carbon nanotubes (CNTs) as a sacrificial hard template as schematically shown in Fig. 4a. In short, CNTs were first coated with Ta2O5 by means of a hydrothermally-assisted sol–gel process using tantalum(V) ethoxide as a precursor (Fig. S2†).31 The hybrid was then subjected to heat treatment in ambient air at 700 °C in order to simultaneously remove CNT templates (their oxidation occurs above 600 °C) and crystallize the metal oxide, yielding Ta2O5 NTs. It is important to note that, although the same calcination procedure was chosen for all samples in this study, crystallization of the Ta2O5 NTs was strongly affected by the presence of the CNT templates compared to the gyroid Ta2O5 sample where ISO oxidation occurred at a lower temperature range.
 | ||
| Fig. 4 (a) Schematic of Ta2O5 NT synthesis using CNTs as sacrificial templates involving sol–gel deposition of oxide onto the CNT surface and subsequent calcination in air at 700 °C oxidizing away the nanocarbon template and converting the oxide into its crystalline phase. SEM (b) and TEM (c) images of Ta2O5 NTs. The insets in (c) show the ED pattern of the area in (c) and the magnified region of the sample illustrating fringes of the crystalline structure. | ||
Fig. 4b reveals a uniform tubular morphology of Ta2O5 NTs, well-preserved after the nanocarbon oxidation. The tubes exhibit an inner channel width of about 80 nm and a wall thickness of about 10 nm – similar to gyroid Ta2O5. TEM and electron diffraction (Fig. 4c) confirm complete removal of the nanocarbon (Fig. S3†). From HRTEM it is observed that the tubes consist of large crystalline domains, as indicated by the clear lattice fringes and the single-crystalline nature of the ED (inset). The XRD results in Fig. 2b confirm that the nanotubes consist of the crystalline β-Ta2O5 phase,29 as for gyroidal Ta2O5. The Raman spectra in Fig. 2d show the typical peaks of β-Ta2O5 without any secondary or impurity phases. The absence of any peak shifts and/or broadening, typically associated with the presence of defects,32 corroborates comparable crystal structures in Ta2O5 NTs and gyroid Ta2O5 samples. Finally, the specific surface area of Ta2O5 NTs is 35 m2 g−1 according to BET, i.e. also very similar to that of gyroid Ta2O5. Importantly, the inner surface of the nanotubes is almost entirely accessible for the reactants due to the presence of sufficiently large inner channels.
From these studies we can confirm that Ta2O5 NTs are similar to gyroid Ta2O5 in terms of crystal structure, wall thickness/charge transport distance and accessible BET area, yet they differ in terms of their overall morphology and pore structure.
In contrast to the gyroid, the photocatalytic activity for H2 production of Ta2O5 NTs was only 3–4 times higher (262 μmol h−1) than that of Ta2O5 SG and thus did not correlate with the increase in the surface area. Furthermore, the Ta2O5 NTs exhibited 4 times lower activity than the gyroid Ta2O5 (Fig. 3 and Table 1), despite exhibiting a comparable surface area. Consequently, the performance of the NTs with respect to the gyroid is limited by contributions other than pore diffusion.
It is well known that different dominating facets exhibit different surface chemistry/potentials and thus energy barriers (i.e. the flat band potential, Vfb) at the solid–liquid interface.33 We therefore evaluated the flat band potentials of our materials from the Mott–Schottky plots (Fig. 3b) derived from electrochemical impedance spectroscopy (EIS).
The respective values of the flat band potentials, Vfb, are summarized in Table 1. From this analysis the various Ta2O5 samples exhibit significantly different energy barriers, suggesting a considerable shift in the respective positions of the conduction (CB) and valence (VB) bands.34 This is shown schematically in Fig. 3c. For gyroid Ta2O5 exhibiting the lowest energy barrier, both VB and CB are shifted upwards by ca. 0.2 eV compared to Ta2O5 NTs, resulting in a stronger driving force for H+ reduction and, concomitantly, a higher photocatalytic rate.
The difference in the flat band potential, i.e. surface chemistry, can be explained by a closer look at XRD (Fig. 2b). The data reveal that the relative intensities of the (100), (011) and (111) peaks are different between the two samples. This suggests the different surface termination as a result of different predominant growth directions within the crystalline walls, with a higher proportion towards (111) in gyroid Ta2O5 than in the NTs. HRTEM further confirms a different surface termination, i.e. a rather smooth surface for gyroid Ta2O5 compared with many sharp edges for Ta2O5 NTs (Fig. 1e and 4c, dashed lines). We believe that this difference originates from the strong influence of the CNT template on the crystallization during the heat treatment of Ta2O5 NTs by e.g. affecting crystal nucleation and growth.31
We speculate that the observed differences in surface termination are directly linked to the Vfb values. In this picture, the superior activity for the mesoporous gyroid Ta2O5 would, at least in part, be due to a higher fraction of more active surface facets at the solid–liquid interface.
Another effect that may influence the performance could stem from the ordered mesoporous gyroidal network. Diffuse reflectance spectroscopy (DRS; Fig. 3d) revealed that all three Ta2O5 samples share a similar absorption edge at about 320 nm, which corresponds to a previously reported band gap value of about 3.9 eV.35 However, the gyroid Ta2O5 exhibits a considerably stronger absorption around the UVB range (250–310 nm), which accounts for high energy photons of 4 to 5 eV. Superior light trapping in the UVB region and thus photoexcitation to higher-energy levels can additionally enhance catalyst's activity towards H+ reduction.36 Although the origin of this surprising optical effect is not yet well understood and clearly warrants more detailed investigations, it is likely connected to the presence of well-ordered gyroidal mesopores.
Conclusion
We report on the synthesis of well-ordered mesoporous tantalum oxides with an alternating gyroidal pore structure exhibiting large pore diameters (>20 nm) with a 3D accessible pore matrix and extended single-crystalline domains as well as of unique Ta2O5 nanotubes using tailor-made block-copolymers and carbon nanotubes as templates, respectively. In particular, we have developed a synthetic protocol using a tailor-made triblock-terpolymer, i.e. poly(isoprene)-b-poly(styrene)-b-poly(ethylene oxide), as a structure-directing agent that allows for tuning of the pore size and architecture.The resulting Ta2O5 gyroids have yielded high photocatalytic activities of up to 20 mmol h−1 g−1 towards hydrogen evolution with the apparent quantum yield of 4.6% without the use of a cocatalyst. In particular, we observed that the peak rate of the gyroid yielded about the same value as the sol–gel sample when corrected to the specific surface area, while the stabilized rate still retained about 80% of its peak value after several hours. This confirms that the pore diffusion has been successfully eliminated.
We further demonstrate that the nature of the template affects the crystallization and surface chemistry of Ta2O5 (e.g. injection barriers) and consequently the photocatalytic performance of the resulting mesostructures.
This work utilizes a fundamental materials design strategy to evaluate and eliminate diffusion limitations that are inherent to mesoporous materials and play a crucial role in liquid phase photocatalysis and photoelectrochemistry with both UV- and visible-light active materials. It further provides invaluable mechanistic insight into key criteria for photocatalyst design that is applicable also to visible-light active photocatalysis. We believe that this work will help in establishing pore and surface engineering as a powerful tool for designing high-performance catalysts and electrodes for various applications related to energy, environment and health.
Acknowledgements
This work was supported by the National Science Foundation (NSF) Single Investigator Award (DMR-1104773). This work made use of the Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR-1120296). This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under the NSF award DMR-0936384. The research leading to these results has received funding from the European Union Seventh Framework Programme under the grant agreement No. 310184, CARINHYPH project. AC is grateful for the funding provided by the graduate school of chemistry at the University of Münster (GSC-MS). We further thank Huating Hu for Raman measurements.References
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- L. Barreto, A. Makihira and K. Riahi, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS.
- S. S. Penner, Energy, 2006, 31, 33–43 CrossRef CAS.
- W. McDowall and M. Eames, Energy Policy, 2006, 34, 1236–1250 CrossRef.
- M. Ball and M. Wietschel, The Hydrogen Economy: Opportunities and Challenges, Cambridge University Press, 2009 Search PubMed.
- M. Nedelcu, S. Guldin, M. C. Orilall, J. Lee, S. Hüttner, E. J. W. Crossland, S. C. Warren, C. Ducati, P. R. Laity, D. Eder, U. Wiesner, U. Steiner and H. J. Snaith, J. Mater. Chem., 2010, 20, 1261–1268 RSC.
- J. Zhu and M. Zäch, Curr. Opin. Colloid Interface Sci., 2009, 14, 260–269 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- A. A. Ismail and D. W. Bahnemann, J. Mater. Chem., 2011, 21, 11686–11707 RSC.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- D. M. Antonelli and J. Y. Ying, Chem. Mater., 1996, 8, 874–881 CrossRef CAS.
- W. Lee, R. Scholz, K. Nielsch and U. Gösele, Angew. Chem., Int. Ed., 2005, 44, 6050–6054 CrossRef CAS PubMed.
- L. Li, M. Krissanasaeranee, S. W. Pattinson, M. Stefik, U. Wiesner, U. Steiner and D. Eder, Chem. Commun., 2010, 46, 7620–7622 RSC.
- P. Docampo, M. Stefik, S. Guldin, R. Gunning, N. A. Yufa, N. Cai, P. Wang, U. Steiner, U. Wiesner and H. J. Snaith, Adv. Energy Mater., 2012, 2, 676–682 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- M. Hara, G. Hitoki, T. Takata, J. N. Kondo, H. Kobayashi and K. Domen, Catal. Today, 2003, 78, 555–560 CrossRef CAS.
- Y. Takahara, J. N. Kondo, T. Takata, D. Lu and K. Domen, Chem. Mater., 2001, 13, 1194–1199 CrossRef CAS.
- J. N. Kondo, Y. Takahara, B. Lee, D. Lu and K. Domen, Top. Catal., 2002, 19, 171–177 CrossRef CAS.
- G. Guo and J. Huang, Mater. Lett., 2011, 65, 64–66 CrossRef CAS.
- Y. Noda, B. Lee, K. Domen and J. N. Kondo, Chem. Mater., 2008, 20, 5361–5367 CrossRef CAS.
- L. Guo, H. Hagiwara, S. Ida, T. Daio and T. Ishihara, ACS Appl. Mater. Interfaces, 2013, 5, 11080–11086 CAS.
- M. Stefik, S. Wang, R. Hovden, H. Sai, M. W. Tate, D. A. Muller, U. Steiner, S. M. Gruner and U. Wiesner, J. Mater. Chem., 2012, 22, 1078–1087 RSC.
- D. Eder and A. H. Windle, Adv. Mater., 2008, 20, 1787–1793 CrossRef CAS.
- M. Stefik, J. Song, H. Sai, S. Guldin, P. Boldrighini, M. C. Orilall, U. Steiner, S. M. Gruner and U. Wiesner, J. Mater. Chem. A, 2015, 3, 11478–11492 CAS.
- J. Chatterjee, S. Jain and F. S. Bates, Macromolecules, 2007, 40, 2882–2896 CrossRef CAS.
- C. A. Tyler, J. Qin, F. S. Bates and D. C. Morse, Macromolecules, 2007, 40, 4654–4668 CrossRef CAS.
- S. W. Robbins, H. Sai, F. J. DiSalvo, S. M. Gruner and U. Wiesner, ACS Nano, 2014, 8, 8217–8223 CrossRef CAS PubMed.
- L. A. Aleshina and S. V. Loginova, Crystallogr. Rep., 2002, 47, 415–419 CrossRef CAS.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 CAS.
- A. S. Cherevan, P. Gebhardt, C. J. Shearer, M. Matsukawa, K. Domen and D. Eder, Energy Environ. Sci., 2014, 7, 791–796 CAS.
- D. Eder, M. S. Motta and A. H. Windle, Acta Mater., 2010, 58, 4406–4413 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- W.-J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures (additional SEM and TEM images, long photocatalytic run), and the details of characterization methods and quantum yield calculations. See DOI: 10.1039/c6nr04430a |
| This journal is © The Royal Society of Chemistry 2016 |