
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisualization, quantification and coordination of Ag+ ions released from silver nanoparticles in hepatocytes†
G.
Veronesi‡
 *abcd,
A.
Deniaud‡
*abc,
T.
Gallon
abc,
P.-H.
Jouneau
ef,
J.
Villanova
d,
P.
Delangle
gh,
M.
Carrière
gh,
I.
Kieffer
ij,
P.
Charbonnier
abc,
E.
Mintz
abc and
I.
Michaud-Soret
*abc
*abcd,
A.
Deniaud‡
*abc,
T.
Gallon
abc,
P.-H.
Jouneau
ef,
J.
Villanova
d,
P.
Delangle
gh,
M.
Carrière
gh,
I.
Kieffer
ij,
P.
Charbonnier
abc,
E.
Mintz
abc and
I.
Michaud-Soret
*abc
aCNRS, Laboratoire de Chimie et Biologie des Métaux (LCBM) UMR 5249 CNRS-CEA-UGA, F-38054 Grenoble, France
bCEA, LCBM, F-38054 Grenoble, France
cUniversité Grenoble Alpes, LCBM, F-38054 Grenoble, France. E-mail: giulia.veronesi@esrf.fr; aurelien.deniaud@cea.fr; isabelle.michaud-soret@cea.fr
dESRF, The European Synchrotron, 71, Avenue des Martyrs, 38043 Grenoble, France
eINAC-MEM, Université Grenoble Alpes, Grenoble, France
fCEA, INAC-MEM, Grenoble, France
gUniversité Grenoble Alpes, INAC-SCIB, F-38054 Grenoble, France
hCEA, INAC-SyMMES, F-38054 Grenoble, France
iBM30B/FAME beamline, ESRF, F-38043 Grenoble cedex 9, France
jObservatoire des Sciences de l'Univers de Grenoble, UMS 832 CNRS Université Joseph Fourier, F-38041 Grenoble, France
First published on 30th August 2016
Abstract
Silver nanoparticles (AgNPs) can enter eukaryotic cells and exert toxic effects, most probably as a consequence of the release of Ag+ ions. Due to the elusive nature of Ag+ ionic species, quantitative information concerning AgNP intracellular dissolution is missing. By using a synchrotron nanoprobe, silver is visualized and quantified in hepatocytes (HepG2) exposed to AgNPs; the synergistic use of electron microscopy allows for the discrimination between nanoparticular and ionic forms of silver within a single cell. AgNPs are located in endocytosis vesicles, while the visualized Ag+ ions diffuse in the cell. The averaged NP dissolution rates, measured by X-ray absorption spectroscopy, highlight the faster dissolution of citrate-coated AgNPs with respect to the less toxic PVP-coated AgNPs; these results are confirmed at the single-cell level. The released Ag+ ions recombine with thiol-bearing biomolecules: the Ag–S distances measured in cellulo, and the quantitative evaluation of gene expression, provide independent evidence of the involvement of glutathione and metallothioneins in Ag+ binding. The combined use of cutting-edge imaging techniques, atomic spectroscopy and molecular biology brings insight into the fate of AgNPs in hepatocytes, and more generally into the physicochemical transformations of metallic nanoparticles in biological environments and the resulting disruption of metal homeostasis.
1. Introduction
Colloidal silver and more recently Ag-nanoparticles (AgNPs) are extensively used in the world as antibacterial coatings in medical implants, textiles and plastics or antibacterial agents in cosmetics or else anti-algae and anti-fungi in paints.1,2 However, this widespread use as a biocide can also turn out to be toxic for animals and plants and is therefore a matter of concern.3,4 The biocide activity of AgNPs is mainly due to Ag+ release in the vicinity of the bacteria,4,5 a phenomenon that requires oxidation.6The coordination properties of Ag+ resemble those of Cu+ but their redox properties differ, so that in addition to having no physiological role, Ag+ can enter eukaryotic cells,7 poison cuproenzymes by standing in for Cu+,8,9 and release thiol-bound Cu+ thereby inducing reactive oxygen species.10 Since AgNPs are produced in numerous and various industrial formulations, most often polyvinylpyrrolidone- (PVP) and citrate-coatings, their Ag0 core can be coated with molecules of different charges and/or by proteins forming a corona.11–13 However, whatever the AgNP coating, its influence on the uptake mechanism (endocytosis, micropinocytosis or phagocytosis) and on the dissolution rate, and therefore on the toxicity, has been pointed out many times.14 Indeed, studies on eukaryotic cell lines showed that the mechanisms involved in AgNP uptake, intracellular dissolution and toxicity are still a matter of debate (see for instance ref. 15–17). Therefore, insight must be gained into the fate of AgNPs and their dissolution into Ag+ ions in cells; the knowledge of these phenomena and of their dependence on the NP properties is paramount in order to achieve controlled ion release and produce safer-by-design AgNPs. The present study aims at understanding the transformations of AgNPs in cellulo, offering scope for new approaches in predictive toxicology.
AgNP dissolution is due to the interplay between the physicochemical properties of the dispersion medium and those of NPs themselves. In the simplest case of polymer-stabilized AgNPs dispersed in aqueous solutions at fixed pH, Ag+ release rates follow the Ostwald–Freundlich relation for size-dependent dissolution.18 However, dissolution in complex media, such as body fluids (gastric) or intracellular media (lysosomal), does not follow a simple law, and the properties of the medium like its pH and chemical composition strongly influence the Ag+ release rates. Ag+ release is fostered by molecular oxygen available in the medium, while it is hindered by the presence of chloride and sulfide that can passivate the NP surface through the formation of a surface precipitate of AgCl or Ag2S, respectively.19,20 In addition the AgCl precipitate on AgNPs can induce aggregation through van der Waals attraction.21 The interaction between AgNPs and thiol-bearing molecules is more controversial: depending on the experimental conditions, thiolate has been shown either to inhibit or to enhance Ag+ release.20,22 AgNP coating and surface charge influence as well their dissolution rates in vitro: the charge-stabilized (acidic) citrate-coated AgNPs are less stable than the sterically-stabilized (neutral) PVP-coated AgNPs in neutral solutions. Moreover, the former are more prone to aggregate in electrolyte solutions.23,24
In cellular models, and much more in vivo, AgNPs experience a wide variety of media and, consequently, undergo a rich set of transformations that are responsible for their toxicological outcome.25 Recent studies have attempted to correlate the physicochemical behavior of AgNPs in cell culture medium or in simulated biological media with their toxicity towards cultured cells: the higher the Ag+ release in vitro, the more toxic the AgNPs, supporting the hypothesis that no particle-induced toxicity exists.26,27 However, AgNP dissolution has been shown to occur into cells rather than in the extracellular medium,28–30 and dissolution in cellulo rather than in simulated biological fluids should be measured for a real appreciation of toxic effects exerted by the fraction of released Ag+ ions.
The abundance of data and the diversity of the proposed mechanisms for the toxicity exerted by AgNPs on eukaryotic cells outline a very complex scenario that fosters the need to develop methods for the detection, quantification, and assessment of Ag+ speciation in cellulo, once it is released from the AgNP surface. De Matteis et al. recently made use of a fluorescent probe that allowed one to visualize Ag+ in living cells after AgNP internalization;16 this method, although effective in ion detection, solves the problem only partially since it does not allow the simultaneous visualization of AgNPs nor the quantification of Ag species in a single cell. Therefore, we propose to make use of synchrotron-based nano-beam X-Ray Fluorescence (nano-XRF) microscopy to unravel the still open issues. XRF microscopy with sub-micrometric resolution has been previously applied to the investigation of ZnO nanoparticle uptake and dissolution in human bronchial epithelial cells and macrophages,31,32 and of the fate of AgNPs in the lungs of mice exposed through oropharyngeal aspiration.29,30
In the present work we make use of XRF imaging with the unequalled spatial resolution and sensitivity of the newly built X-ray nanoprobe ID16B-NA33 of the ESRF to visualize and quantify silver species (AgNPs as well as Ag+) in HepG2 single cells exposed to AgNPs. This hepatoma-derived cell line was chosen because the liver is the main target organ for the accumulation of silver upon exposure to AgNPs.34 The information retrieved is correlated with X-ray absorption spectroscopy (XAS) experiments under cryogenic conditions. Thanks to an analytical protocol we developed,28 these experiments allow for the determination of the fraction of Ag+ released from the NP and for the assessment of Ag+ binding properties in cellulo. The correlation between the fate of AgNPs in cellulo and their biological effects constitutes a substantial advancement with respect to previous studies correlating in vitro dissolution with cytotoxicity. To gain insights into the toxicity mechanisms, the cell responses were also followed. We show that the sensitivity of nano-XRF allows one to visualize and quantify Ag+ in a single cell and to analyse the impact of the AgNP coating on the dissolution observed after various exposure times.
2. Results
2.1 Ag nanoparticle characterization and toxicity
We compared the behavior of two commercial AgNPs, citrate- or PVP-coated. Electron microscopy (EM) showed homogeneous citrate-coated AgNPs (Fig. 1(a)) with a diameter determined by dynamic light scattering (DLS) of 26 nm (Fig. 1(c) and (d)). PVP-coated AgNPs were more heterogeneous (Fig. 1(b)) with two distinct populations with diameters of 19 and 101 nm based on DLS data (Fig. 1(c) and (d)). In all cases the polydispersity index was lower than 12% (Fig. 1(d)). Interestingly, DLS measurement performed in cell culture media showed some agglomeration for citrate-coated AgNPs that formed a relatively homogeneous population of 160 nm (polydispersity index around 15%). For the PVP-coated AgNPs a unique species at 90 nm was found with a similar polydispersity index around 15%. These species are therefore less homogeneous but one has to be aware that these are the NP states, when they are in contact with the cells. We also determined by ICP-AES the amount of Ag+ present in the initial AgNP solutions. The amount of Ag+ released from PVP-coated AgNPs after dilution at 100 μM in buffer or complete culture medium was found to be 2.8 ± 0.5% and 2.7 ± 0.1%, respectively. For citrate-coated AgNPs this amount increased from 1.1 ± 0.5% in buffer to 2.3 ± 0.5% in cell culture medium. The toxicity of these two types of AgNPs was compared with the toxicity of silver nitrate salt (AgNO3) after 24 h exposure using trypan blue exclusion assay (Fig. 2). AgNO3 is highly toxic in the micromolar range concentration with a lethal dose for 50% of the cells (LD50) at 10 μM of Ag+. | ||
| Fig. 1 Characterization of AgNPs. (a) TEM micrograph of citrate-coated AgNPs obtained on a JEOL 1200 operating at 100 kV. (b) STEM micrograph of PVP-coated AgNPs obtained on a Zeiss Merlin microscope operating at 20 kV. (c) Size distribution of citrate- and PVP-coated AgNPs was determined after dilution in buffer by dynamic light scattering, using a Wyatt Dynapro Nanostar instrument. (d) Diameter and polydispersity index (in parenthesis) of the main species observed in the dynamic light scattering distribution of the citrate- and PVP-coated AgNPs in buffer or in complete culture medium (minimum essential medium + 10% foetal bovine serum). | ||
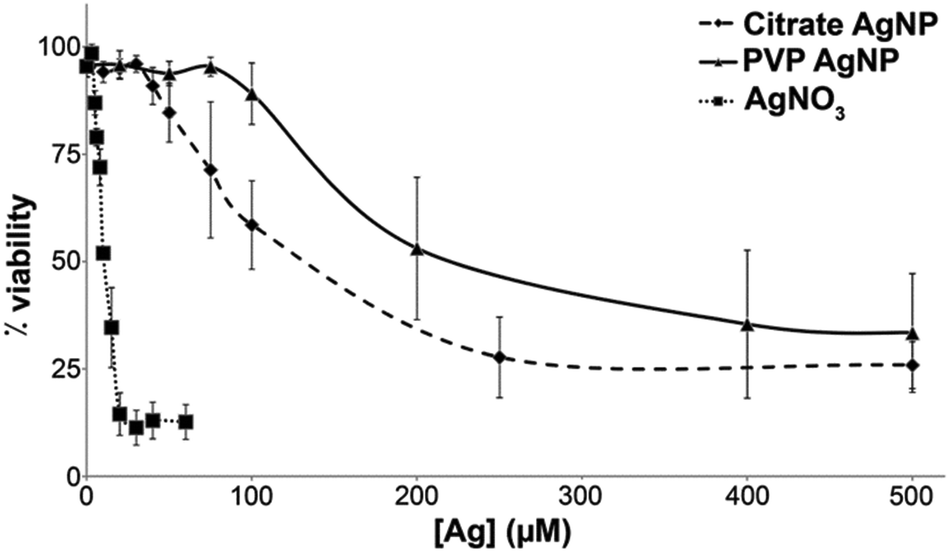 | ||
| Fig. 2 HepG2 cell viability. Hepatocyte cells were exposed to citrate-coated AgNPs (diamond), PVP-coated AgNPs (triangle) or AgNO3 (square) for 24 h before being harvested, suspended in PBS and mixed in one volume of trypan blue. Total and viable cells were counted in a TC20 Automated Cell Counter (Bio-Rad). | ||
Both AgNPs induce a much lower toxicity with LD50 about 120 and 200 μM equivalent Ag for citrate- and PVP-coated AgNPs, respectively. This is not surprising since only the NP surface could be oxidized to the Ag+ species responsible for the toxicity, and the Ag0 core is not expected to be reactive.
2.2 Nano-XRF imaging
In the first instance, entire cells were imaged with a 250 × 250 nm2 step size, in order to visualize the overall distribution of Ag. Silver distributions in one chosen cell per condition are reported in Fig. 3, both in linear (column B) and in logarithmic scales (column C); maps of all replicates are reported in Fig. S1.† In untreated cells a uniform distribution was found (Fig. 3, row 1), revealing no difference between the interior and the exterior of the cell; this clearly means that no Ag contamination was present inside control cells. When the areal density (AD) of Ag was extracted, the maximum estimated value over the three cells analysed was 0.008 ± 0.002 μg cm−2, which gives an estimation of the background noise relative to our experimental conditions. The high sensitivity of the method enabled the detection of ionic Ag+ in HepG2 cells exposed for 30 minutes to 50 μM AgNO3 (Fig. 3, row 2): Ag was detected throughout the whole cell, with the highest Ag fluorescence signal in the nuclear region. In whole cells this region is thicker than the rest of the cell because the nucleus and the cytosol pile up, accounting for the higher Ag areal density; however, this also suggests that Ag in this region was present not only in the surrounding cytosol, which would not cause the areal density to increase significantly with respect to the cell periphery, but rather throughout the whole thickness, i.e. as well as in nuclei. This rapid access to the nuclei may explain part of the deleterious effect of silver ions. The maximum areal density measured in cells exposed to AgNO3 is 0.13 ± 0.04 μg cm−2, one order of magnitude above the background level, demonstrating the strength of the method in visualizing Ag+ species distribution throughout the cell; it is worth noting that this areal density corresponds to 8 × 10−17 g in a single 250 × 250 nm2 pixel, i.e. 80 attograms. | ||
| Fig. 3 HepG2 cells analysed by nano-XRF for the chosen exposure conditions (rows 1–6). (A column) Optical micrographs; the areas of the regions scanned with X-rays are highlighted by red rectangles. (B column) Linear and (C column) logarithmic false-color representation of silver areal density, extracted from hyperspectral XRF data in the map areas. Areal densities are expressed in μg cm−2; pixel size is 250 × 250 nm2. Scale bars = 5 μm. | ||
When cells were exposed to AgNPs (Fig. 3, rows 3–6), the linear-scale visualization of Ag areal density (column B) showed mainly scattered intense spots of a few pixels (AD ≥ 1 μg cm−2), a size compatible with small Ag-dense aggregates, i.e. aggregates of AgNPs. When areal density maps were represented in a log-scale (Fig. 3, column C), a uniform weak signal due to Ag was visible throughout the whole cells for all exposure conditions with a density of about 0.1 μg cm−2 after 6 h exposure for both citrate- and PVP-coated AgNPs. This signal is comparable to the one due to ionic Ag+, both in its spatial distribution and intensity; this highlights the slow increase of Ag+ concentration inside the cell upon exposure to AgNPs, which could explain the lower toxicity of AgNPs compared to AgNO3. After 24 h exposure, the AD associated with the putative Ag+ signal ranged between 0.1 and 1 μg cm−2 (Fig. S1†), higher than the AD measured after 6 h exposure. Altogether, nano-XRF data show that dissolved Ag+ ions were not confined into vesicles but rather they diffused within the cell, and that dissolution occurred for both citrate- and PVP-coated AgNPs and progressed with the exposure time.
In order to confirm the presence of Ag, the XRF sum spectra of several pixels in the low intensity region were examined and compared with the XRF spectra of Ag hotspots and those of the extracellular background region. The results are illustrated in Fig. S2† for the citrate-6 h sample and prove that a significant Ag Kα emission peak was detected in the sum spectrum of pixels from the low intensity region. This confirms that, under all probed exposure conditions, Ag+ ions were released from AgNPs and distributed within HepG2 cells.
Secondly, in order to visualize the aggregation state of the two kinds of AgNPs in cellulo, we acquired high-resolution nano-XRF images in selected regions of exposed cells. The 70 × 70 nm2 resolution maps are reported in Fig. 4 where the same length and AD scales are used for all exposure conditions, with all AD values above 2.0 μg cm−2 represented in red. This visualization allows one to compare the size of different AgNP aggregates and gives a direct appreciation of the extent of dissolution in the different samples. Clearly, after 24 h exposure, the putative Ag+ signal was more intense in cells treated with citrate- (Fig. 4(d)) than with PVP-coated AgNPs (Fig. 4(b)). Again, it is worth noting that the lowest Ag signal detected in a single pixel was ∼0.1–0.2 μg cm−2, corresponding to a total amount of 5–10 attograms in a 70 × 70 nm2 area.
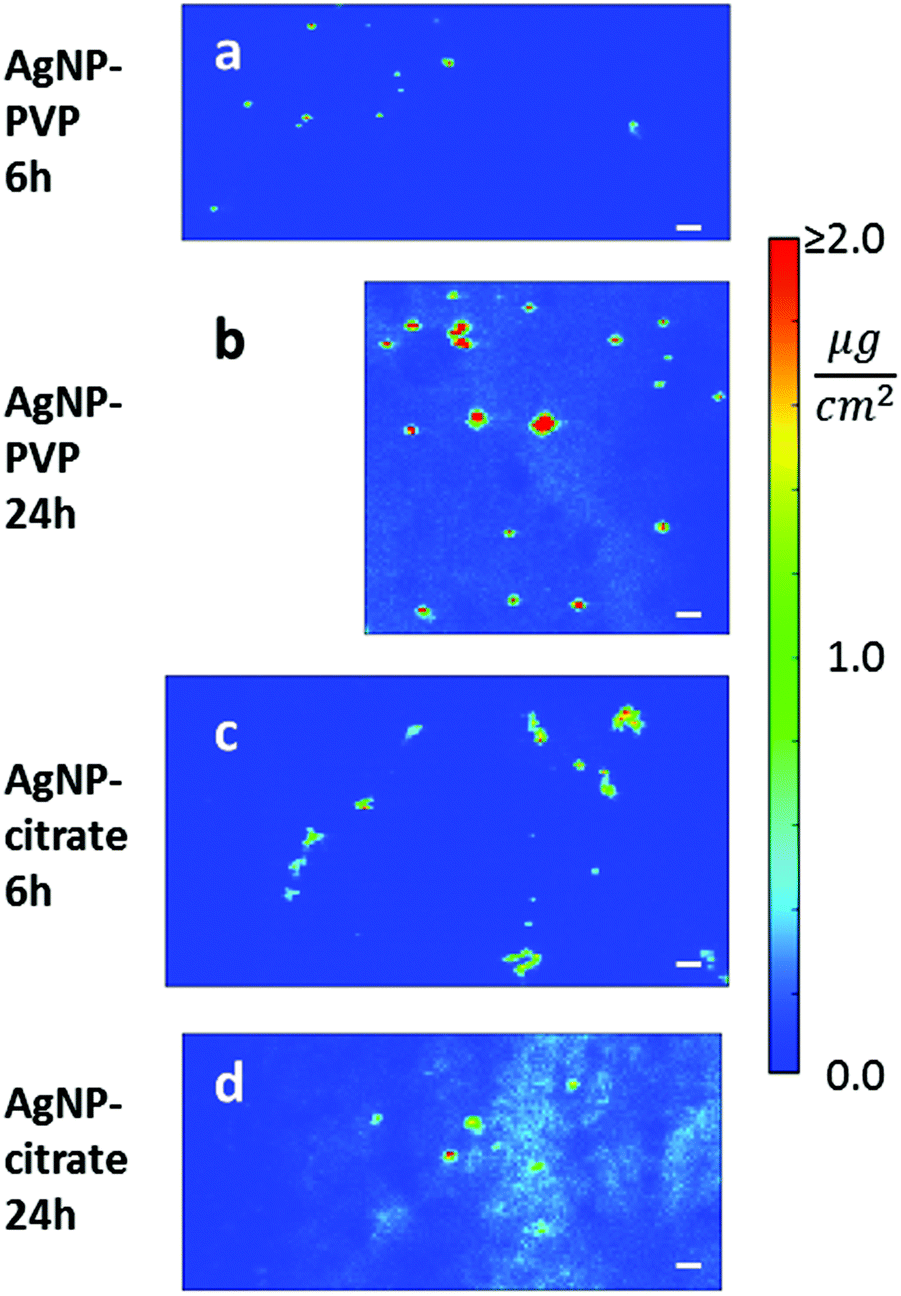 | ||
| Fig. 4 High-resolution (70 × 70 nm2) silver areal density maps in the selected regions of HepG2 cells exposed to AgNPs. (a) 6 h or (b) 24 h exposure to PVP-coated AgNPs. (c) 6 h or (d) 24 h exposure to citrate-coated AgNPs. Concentrations are represented in false color; the same color scale is used for all maps: linear between 0.0 μg cm−2 and 2.0 μg cm−2, with all values above 2.0 μg cm−2 represented in red. The same length scale is used for all maps (scale bar = 500 nm). | ||
2.3 Correlation between nano-XRF and TEM observations
The extremely high spatial resolution provided by the beamline ID16B allowed for the visualization of individual AgNPs or small aggregates in cellulo: under all conditions, localized Ag hotspots were observed (Fig. 4). The analysis of cell sections of the equivalent samples by TEM (Fig. 5) showed the presence of AgNPs in the endocytic-type of vesicles with two different behaviours for samples treated with citrate- or PVP-coated AgNPs. This is consistent with their DLS characterization performed in culture media (Fig. 1(d)). On the one hand, citrate-coated AgNPs are internalized as agglomerates (Fig. 5(a)). On the other hand, the TEM micrograph showed that PVP-coated AgNPs interacted with the cell membrane as individual particles that were internalized alone or with a few others (Fig. 5(b)). There was no observation of AgNPs free in the cytosol. Therefore, we can conclude that the Ag hotspots observed with nano-XRF imaging corresponded to intracellular vesicles holding AgNPs. | ||
| Fig. 5 TEM micrographs of 240 nm section of HepG2 cells exposed to AgNPs. (a) Cell exposed for 6 h to citrate-coated AgNPs with insets on AgNP-rich intracellular vesicles. Agglomerated NPs are observed during the entry process and inside vesicles. (b) Cell exposed for 6 h to PVP-coated AgNPs. Individual NPs interacting with the cell surface and inside vesicles are observed. In both cases, AgNPs are pinpointed with black arrows. | ||
The correlation of nano-XRF and TEM observations thus allowed insightful interpretation of the XRF data. Indeed, the larger Ag hotspots visualized for citrate-coated compared to PVP-coated AgNPs after 6 h exposure (Fig. 4(a) and (c)) were due to the larger number of particles per vesicle in the case of citrate-coated AgNPs compared to PVP-coated ones. Furthermore, Ag hotspots increased in size between 6 and 24 h exposure for both types of AgNPs (Fig. 4 and S1†). This phenomenon is due to normal cellular processes that led to the fusion of endocytic vesicles and their transformation to large lysosomal vesicles. Nano-XRF micrographs also showed the perinuclear distribution of AgNP rich vesicles after 24 h exposure to both citrate- and PVP-coated AgNPs (Fig. 3 and S1†).
2.4 X-ray absorption spectroscopy
Average information concerning coating- and time-dependent dissolution of AgNPs needs to be combined with the local single-cell information, in order to unravel the relationship between the AgNP fate and the cellular response induced in hepatocytes. We previously developed an experimental and analytical method based on X-ray Absorption Spectroscopy (XAS) that was proved to provide the fraction of Ag atoms released from AgNPs into macrophages in cellulo.28Furthermore, this method is unique because it provides the average coordination chemistry of the Ag+ species formed in cellulo upon ion release from the NP surface. Therefore we applied it to the present study on HepG2 cells. The near-edge region (XANES) of the X-ray absorption spectra was fitted as a linear combination of reference compounds in order to measure the fraction of the different Ag species encountered in HepG2 cells exposed to AgNPs. XANES spectra are shown in Fig. S3;† the spectrum of AgNPs diluted in cell culture medium does not differ from that of an Ag foil, which means that cells are initially in contact with intact AgNPs whose signal is dominated by bulk atoms in the Ag0 metallic phase. The fit results are reported in Table 1: the fraction of Ag atoms in the nanoparticles (AgNP component) decreased with the incubation time, confirming that Ag+ ions were released from the NP surface in cells. At the chosen dose of 100 μM, dissolution was faster for citrate- than for PVP-coated nanoparticles. The majority of the Ag+ ions released formed complexes with Ag–S bonds under all exposure conditions, as suggested by the estimated fraction of the AgGSH component (Table 1). The AgNO3 and AgO components were never detected and the AgCl species was far below 10%, which is not enough to conclude reliably on their presence. Despite the high intracellular chloride concentration, thiolate containing biomolecules seem to be the most important factor for Ag+ chelation following intracellular dissolution of AgNPs. The formation of Ag–thiolate complexes is consistent with the hypothesis that intracellular Ag+ binding is operated by biomolecules with thiolate-containing metal binding sites; however, this part of the analysis alone does not allow for the discrimination between different biomolecules involved in such processes. In order to corroborate our results, and to unravel the still open issues, we performed a fit of the EXAFS spectra based on ab initio models. Our analytical protocol allows measuring the Ag–S distance in the complexes formed in cellulo by Ag+ ions released from AgNPs, as previously demonstrated in primary murine macrophages.28 Taking advantage of the correlation between the Ag coordination number and the Ag–S bond length that we recently described,35 the averaged Ag–S bond length was measured in cellulo to decipher the chemical environment of the dissolved Ag+ ions and to allow for the identification of the potential binding biomolecules. A summary of the fitting model is given in the Experimental section (see ref. 28 for details). The fits are reported in Fig. 6 and the quantitative results in Table 2; the model provided an excellent agreement with the experimental data. The parameter n in Table 2 is the fraction of Ag atoms that occupy an fcc crystal site, i.e. the fraction of Ag in undissolved nanoparticles under given exposure conditions. It can be compared with the results obtained by LCF of the XANES region (column AgNP in Table 1): n values correspond within the error to the AgNP fractions estimated by LCF of XANES.
 | ||
| Fig. 6 Fourier-transformed Ag K-edge EXAFS spectra of HepG2 hepatocytes exposed to AgNPs. Experimental spectra obtained after 24 h exposure to PVP-coated (open diamonds) or citrate-coated (black circles) AgNPs, and relative best-fitting curves (red) based on an ab initio model given by a combination of Ag0 in crystalline fcc phase (i.e. Ag in nanoparticles) and ionic Ag+ recombined with thiolate groups. | ||
| NP coating | Exposure time | AgNP (%) | AgGSH (%) | AgO (%) | AgCl (%) | AgNO3 (%) | R fit (10−5) |
|---|---|---|---|---|---|---|---|
| PVP | 6 h | 84.3 (7) | 14.7 (10) | — | <2 | <2 | 0.9 |
| 24 h | 73.3 (14) | 21 (4) | 2 (4) | 3.9 (11) | — | 4.5 | |
| Citrate | 6 h | 75 (2) | 21 (3) | — | <2 | 2 (2) | 8.6 |
| 24 h | 51.0 (11) | 46.2 (17) | — | 3 (2) | — | 3.4 |
| Sample | n (%) | Ag–Ag | Ag–S | ΔE0 (eV) | R fit (%) | ||
|---|---|---|---|---|---|---|---|
| R (Å) | σ 2 (10−3 Å2) | R (Å) | σ 2 (10−3 Å2) | ||||
| PVP-coated AgNPs | 67 (5) | 2.871 (3) | 2.7 (4) | 2.44 (4) | 7 (5) | 1.6 (2) | 1.2 |
| Citrate-coated AgNPs | 51 (5) | 2.872 (4) | 2.8 (6) | 2.45 (2) | 9 (3) | 1.9 (2) | 0.9 |
The R value relative to the Ag–S contribution in Table 2 is the average bond length in the Ag–S species formed in cellulo. In primary murine macrophages this value was found in the 2.38–2.41 Å range, depending on the exposure scenario: this bond length is indicative of digonal AgS2 coordination, which led us to propose that Ag+ is mainly bound to intracellular GSH. In HepG2 cells averaged bond lengths were significantly longer (Table 2). In metallothioneins, Ag+ occupies mainly trigonal sites, resulting in a longer average Ag–S bond length (2.47–2.48 Å):35 the bond lengths of 2.44–2.45 Å found in HepG2 cells exposed to both PVP- and citrate-coated AgNPs suggest an important participation of metallothioneins in Ag+ chelation, besides the binding to GSH already encountered in other cells.
2.5 mRNA quantification
In order to corroborate the results provided by XAS, metallothionein 1 (MT1) and glutamate–cysteine ligase (GCLM) mRNA expression in HepG2 cells were determined following AgNP exposure (Fig. 7). GCLM is the rate limiting enzyme involved in GSH biosynthesis and therefore highlights the cellular need for GSH. After 6 h, the MT1 mRNA level increased 362- and 539-fold with PVP- and citrate-coated AgNPs, respectively and both AgNPs led to about 120-fold increase after 24 h exposure. The difference between citrate- and PVP-coated AgNP at 6 h was most probably due to the faster dissolution of citrate-coated AgNPs, while cells were maintaining a high but similar level after 24 h exposure for both AgNPs. Similarly, the GCLM mRNA level reached a 12 to 13-fold increase after 6 h exposure for both AgNPs and a 7-fold increase after 24 h. These overexpressions are in accordance with intracellular binding of the released Ag+ in HepG2 cells by metallothioneins and GSH. Furthermore, the two different types of AgNPs tested induced the same cellular response, the only difference being a higher MT1 overexpression in citrate-coated AgNP treated cells for the 6 h exposure time.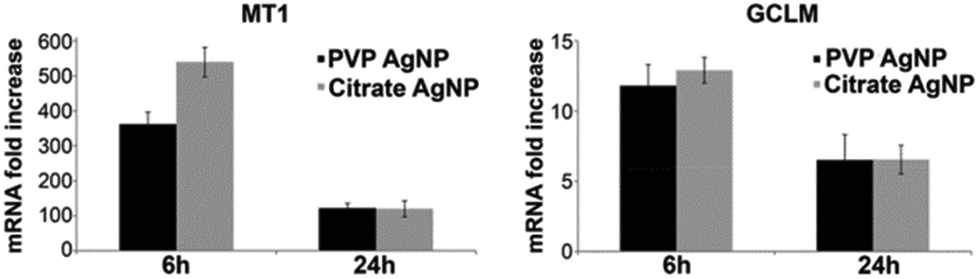 | ||
| Fig. 7 MT1 and GCLM mRNA fold increase in HepG2 cells exposed to silver nanoparticles. Cells were exposed for 6 h or 24 h to citrate- (grey bar) and PVP-coated (black bar) AgNPs. Results are expressed as the relative change in expression compared to the control. The results are expressed as means +/− standard error of the mean of at least three independent experiments. | ||
3. Discussion
In this work, we have taken advantage of the latest developments in nano-XRF in terms of resolution and sensitivity to visualize for the first time Ag+ species within hepatocytes after AgNP intracellular dissolution. Interestingly, Ag quantification highlighted the capability of the cell to handle higher silver concentrations after AgNP treatment compared with AgNO3. Indeed, Ag+ areal densities reached a maximum in the range of 0.1 μg cm−2 before dying from 30 min exposure to AgNO3 while it can increase up to 1 μg cm−2 after 24 h AgNP exposure. These results explained the lower cytotoxicity of the AgNP formulations compared to the silver salt by their slow cytosolic release of Ag+ species. Quantification by nano-XRF could even allow for Ag amount determination in a specific area in the cell or in the whole cell by integrating the areal signal. It was thus possible to show that HepG2 cells contain between 0.2 and 0.6 pg of Ag after 6 h exposure to both citrate- and PVP-coated AgNPs and between 1 and more than 2 pg after 24 h exposure (Fig. S1†). For this analysis it has to be noticed that the maps do not always contain the entire cell. However, it clearly indicates that NP uptake still continues after 6 h. Furthermore, we have substantiated nano-XRF results with other experimental methods to provide a more complete picture of the Ag fate inside hepatocytes. Using TEM, AgNPs have only been visualized in vesicles confirming their entry via an endocytosis type of mechanism. These AgNP rich vesicles correspond to the Ag hotspot observed by nano-XRF at a lower spatial resolution. We strengthen here the complementarity between TEM and nano-XRF, by exploiting the nanometric resolution of the former and the attogram elemental sensitivity of the latter. AgNP dissolution rates were determined by XAS analysis confirming nano-XRF observation; remarkably, the averaged results obtained through XAS on cellular pellets (∼107 cells per sample) were confirmed at the single-cell level. XAS also allowed for the determination of Ag speciation in cellulo, highlighting the complexation of Ag+ with thiol functions both in AgS2 and AgS3 coordination geometries. Moreover, the quantitative PCR results were consistent with GSH and metallothioneins being the main partners for Ag+ binding in AgS2 and AgS3 environments, when Ag+ is formed upon AgNP dissolution and distributed throughout the hepatocyte. Therefore, our data clearly demonstrate the role of GSH in Ag binding, while its main role is to keep the redox potential stable in the cytosol oscillating between reduced and oxidized states, acting as an antioxidant and also described as a ROS scavenger. Furthermore, XAS data showed an important difference in the behaviour observed in macrophages, where Ag–GSH species are by far the most abundant.28 It also highlights the well-known role of metallothioneins to scavenge metal ions that are in excess in the cell. Other studies also describe the transient appearance of some AgO species, a necessity for AgNP dissolution that requires a surface oxidation step.17,29 AgO species, if present, are expected to relate only to Ag atoms at the NP surface, and would represent a minor fraction of the totality of Ag atoms present in cells. This explains why XAS spectroscopy, which interrogates all Ag atoms in the specimen, does not detect AgO species.Therefore, it is not possible to conclude whether this oxidation step really occurs in vivo or if other mechanisms exist. For instance, a direct Ag+ dissolution from AgNPs assisted by thiol-containing biomolecules could arise.
4. Experimental
4.1 Nano-XRF imaging
HepG2 cells were grown on dedicated Si3N4 membranes (Silson Ltd) and exposed to 100 μM citrate- or PVP-coated AgNPs for 6 h or 24 h. After the chosen time, cells were fixed with methanol, washed and dried under an argon atmosphere. Nano-XRF hyperspectral images were acquired in air at room temperature on ID16B,33 with a pink beam fixed photon energy of 29.6 keV; the beam was focused to 60 × 75 nm2 (V × H) with Kirkpatrick–Baez mirrors; photon flux at sample was ∼2.7 × 1011 ph s−1. A single-element Vortex SDD detector and 3-elements silicon drift detector arrays from SGX Sensortech were simultaneously used to collect X-ray fluorescence emission from the sample. Elemental concentrations were extracted from fundamental parameters with the PyMCA software;36 the detector response was calibrated with an AXO reference and with bovine liver NIST SRM 1577c.4.2 XAS data acquisition
HepG2 hepatocytes were exposed to 100 μM PVP- or citrate-coated AgNPs. After 6 h or 24 h exposure, cells were washed with PBS, trypsinized, centrifuged, suspended in PBS and flash-frozen in liquid N2 in the sample holder, and then transferred into an He cryostat for measurements at 16 K. Ag K-edge XAS spectra of cellular samples and reference compounds were collected on the beamline FAME-BM30B of the ESRF;37 details of data acquisition and analysis are described in ref. 28. The AgO reference spectrum was kindly provided by Géraldine Sarret, and the details of data acquisition are reported in ref. 38.4.3 XAS data analysis
The near-edge region (XANES) of the spectra was analysed as Linear Combination Fitting (LCF) of reference compounds representative of the possible speciation of the silver atom. Reference compounds were: silver nanoparticles diluted in cell culture medium (AgNP), a Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GSH = 1:1 solution (AgGSH) representative of species containing Ag–S bonds, AgCl, AgO and AgNO3. The chosen standards were the same as in previous studies that made use of this analytical approach,17,29 except for Ag–S speciation for which we chose to use AgGSH, more representative of the species formed in cellulo than the crystalline silver sulphide (Ag2S) most often used.35 Moreover, our experiment was performed under cryogenic conditions, which ensures the preservation of silver speciation, while all previous studies were performed at room temperature on dried cellular pellets.17,29
:GSH = 1:1 solution (AgGSH) representative of species containing Ag–S bonds, AgCl, AgO and AgNO3. The chosen standards were the same as in previous studies that made use of this analytical approach,17,29 except for Ag–S speciation for which we chose to use AgGSH, more representative of the species formed in cellulo than the crystalline silver sulphide (Ag2S) most often used.35 Moreover, our experiment was performed under cryogenic conditions, which ensures the preservation of silver speciation, while all previous studies were performed at room temperature on dried cellular pellets.17,29
The extended region of the spectra (EXAFS) was interpreted as a combination of two major contributions, due to Ag0 atoms in the fcc crystal structure (Ag-fcc, i.e. atoms in AgNP) and Ag+ atoms released from the NPs and recombined with organic S (Ag-rec). The measured absorption coefficient χ(k)Ag-exp can therefore be written as:
| χ(k)Ag-exp = n·χ(k)Ag-fcc + (1 − n)·χ(k)Ag-rec | (1) |
4.4 Electron microscopy
Monolayers of HepG2 cells were fixed overnight at room temperature in a 1:1 ratio mixture of 4% paraformaldehyde, 0.4% glutaraldehyde in 0.2 M PHEM pH 7.2 and culture medium, washed in 0.1 M PHEM pH 7.2, and fixed for 30 minutes in 2% paraformaldehyde, 0.2% glutaraldehyde in 0.1 M PHEM pH 7.2, washed in 0.1 M PHEM pH 7.2 and post-fixed in 1% OsO4, 1.5% potassium ferrocyanide in 0.1 M PHEM buffer for 1 h at room temperature. After 3 washes in water, post-staining was done using 0.5% uranyl-acetate in 30% ethanol for 30 minutes at room temperature in the dark. Cells were then dehydrated in graded ethanol series, and flat-embedded using the Epoxy Embedding Medium kit (Sigma-Aldrich). Sections (240 nm) were cut on a Leica UC7 ultra-microtome using a DiATOME 35° diamond knife and collected on Formvar carbon coated 100 mesh copper grids. Sections were stained in 5% uranyl acetate (in water) for 10 minutes and in 2% lead citrate for 5 minutes. Images were obtained on a Tecnai G2 Spirit BioTwin (FEI) at 120 kV using an ORIUS SC1000 CCD camera (Gatan).
4.5 qPCR
Cells were exposed to PVP- or citrate-coated AgNPs, for 6 or 24 h. Cells were then harvested and mRNA was isolated using an Absolutely RNA miniprep kit (Agilent # 400800). The RNA concentration was determined using a NanoDrop spectrophotometer (ND-1000). One μg mRNA was reverse transcribed with the Affinity script qPCR cDNA synthesis kit (Agilent # 600559), according to the manufacturer's instructions. Gene specific primers for MET1X were as previously described.39 The other human primers were designed according to the PrimerSelect software (DNASTAR) (Table S1†). Quantitative PCR was performed with Brilliant II SYBR green qPCR master mix1 (Agilent # 600828) and a 200 nM primer. PCR reaction mixtures (10 μl) were placed in the Cfx96 instrument (Bio-Rad) where they underwent the following cycling program, optimized for a 96-well block: 95 °C for 15 min, immediately followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. At the end, PCR products were dissociated by incubating for 1 min at 95 °C and then 30 s at 55 °C, followed by a ramp up to 95 °C. qRT-PCR reactions were run in triplicate, and quantification was performed using comparative regression (Cq determination mode) using Cfx software (Bio-Rad Cfx manager) with GAPDH and hHPRT amplification signals as housekeeping genes to correct for total RNA content and labelling the untreated sample as the “calibrator”.5. Conclusions
We made use of a combination of imaging techniques, molecular and biological approaches, which enabled for the first time the simultaneous visualization of nanoparticular and ionic Ag species in the cytosol of cells exposed to silver nanoparticles, the discrimination between such species, and the quantification of Ag content in a single cell. This brought us to conclude that both citrate- and PVP-coated AgNPs were internalized in HepG2 cells in endocytic vesicles, where NPs partially dissolved into ionic species that distributed throughout the cell, mainly as Ag–GSH and Ag–metallothionein complexes. Future perspectives concern the synergistic use of these techniques to unravel the molecular processes involved in the intravesicular dissolution of AgNPs and to explore at the subcellular level the fate of Ag+, its distribution through specific compartments or its excretion from cells.Founding sources
This work was founded by the CEA-Toxicology Transversal Program through the NanoSilverSol grant and by the CEA Transversal Programs Toxicology and Nanoscience through the NanoTox-RX grant. This research is part of the LabEx SERENADE (grant ANR-11-LABX-0064) and the LabEx ARCANE (grant ANR-11-LABX-0003-01). The platforms of the Grenoble Instruct centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) were used, with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01), within the Grenoble Partnership for Structural Biology (PSB). The electron microscope facility is supported by the Rhône-Alpes Region, the Fondation Recherche Médicale (FRM), the fonds FEDER, the Centre National de la Recherche Scientifique (CNRS), the CEA, the University of Grenoble, EMBL, and the GIS-Infrastructures en Biologie Santé et Agronomie (IBISA).Acknowledgements
The authors acknowledge the ESRF for providing access to the beamlines ID16B-NA and BM30B (experiments MD-867 and LS-2331, respectively). We acknowledge Remi Tucoulou and Sylvain Bohic for help in nano-XRF data acquisition and interpretation, and Martine Cuillel, Samia Chaouni and Sophie Terrone for preliminary cytotoxicity assays. We thank Benoit Gallet, Christine Moriscot and Guy Schoehn, from the Electron Microscopy platform of the Integrated Structural Biology of Grenoble (ISBG, UMI3265). Finally, we thank Mireille Chevallet and Isabelle Worms for useful discussions.References
- M. C. Fung and D. L. Bowen, Clin. Toxicol., 1996, 34, 119 CAS.
- B. Reidy, A. Haase, A. Luch, K. A. Dawson and I. Lynch, Materials, 2013, 6, 2295 CrossRef CAS.
- A. A. Keller, S. McFerran, A. Lazareva and S. Suh, J. Nanopart. Res., 2013, 15, 1 CrossRef.
- R. Behra, L. Sigg, M. J. D. Clift, F. Herzog, M. Minghetti, B. Johnston, A. Petri-Fink and B. Rothen-Rutishauser, J. R. Soc., Interface, 2013, 10, 20130396 CrossRef PubMed.
- O. Bondarenko, A. Ivask, A. Käkinen, I. Kurvet and A. Kahru, PLoS One, 2013, 8, 1 Search PubMed.
- Z.-m. Xiu, Q.-b. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271 CrossRef CAS PubMed.
- J. Bertinato, L. Cheung, R. Hoque and L. J. Plouffe, J. Trace Elem. Med. Biol., 2010, 24, 178 CAS.
- P. S. Babich, N. V. Tsymbalenko, S. A. Klotchenko, N. A. Platonova, O. O. Masalova, E. A. Zatulovski, M. M. Shavlovskii, N. S. Sapronov and L. V. Puchkova, Bull. Exp. Biol. Med., 2010, 148, 592 CrossRef.
- N. Armstrong, M. Ramamoorthy, D. Lyon, K. Jones and A. Duttaroy, PLoS One, 2013, 8, e53186 CAS.
- A. Russell and W. Hugo, Prog. Med. Chem., 1994, 31, 351 CrossRef CAS PubMed.
- M. N. Martin, A. J. Allen, R. I. MacCuspie and V. A. Hackley, Langmuir, 2014, 30, 11442 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265 CrossRef CAS PubMed.
- R. Foldbjerg, X. Jiang, T. Miclaus, C. Chen, H. Autrup and C. Beer, Toxicol. Res., 2015, 4, 563 RSC.
- F. Herzog, M. J. Clift, F. Piccapietra, R. Behra, O. Schmid, A. Petri-Fink and B. Rothen-Rutishauser, Part. Fibre Toxicol., 2013, 10, 1 CrossRef PubMed.
- V. De Matteis, M. A. Malvindi, A. Galeone, V. Brunetti, E. D. Luca, S. Kote, P. Kshirsagar, S. Sabella, G. Bardi and P. P. Pompa, Nanomedicine, 2015, 11, 731 CAS.
- X. Jiang, T. Miclaus, L. Wang, R. Foldbjerg, D. S. Sutherland, H. Autrup, C. Chen and C. Beer, Nanotoxicology, 2015, 9, 181 CrossRef CAS PubMed.
- R. Ma, C. Levard, S. M. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900 CrossRef CAS PubMed.
- K. Loza, J. Diendorf, C. Sengstock, L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. Koller and M. Epple, J. Mater. Chem. B, 2014, 2, 1634 RSC.
- J. L. Axson, D. I. Stark, A. L. Bondy, S. S. Capracotta, A. D. Maynard, M. A. Philbert, I. L. Bergin and A. P. Ault, J. Phys. Chem. C, 2015, 119, 20632 CAS.
- A. P. Gondikas, A. Morris, B. C. Reinsch, S. M. Marinakos, G. V. Lowry and H. Hsu-Kim, Environ. Sci. Technol., 2012, 46, 7037 CrossRef CAS PubMed.
- M. Tejamaya, I. Römer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011 CrossRef CAS PubMed.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564 CrossRef CAS PubMed.
- J. Liu, Z. Wang, F. D. Liu, A. B. Kane and R. H. Hurt, ACS Nano, 2012, 6, 9887 CrossRef CAS PubMed.
- A. R. Gliga, S. Skoglund, I. Odnevall Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 1 CrossRef PubMed.
- X. Wang, Z. Ji, C. H. Chang, H. Zhang, M. Wang, Y.-P. Liao, S. Lin, H. Meng, R. Li, B. Sun, L. V. Winkle, K. E. Pinkerton, J. I. Zink, T. Xia and A. E. Nel, Small, 2014, 10, 385 CrossRef CAS PubMed.
- G. Veronesi, C. Aude-Garcia, I. Kieffer, T. Gallon, P. Delangle, N. Herlin-Boime, T. Rabilloud and M. Carrière, Nanoscale, 2015, 7, 7323 RSC.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai, B. Li, K. Zhang, Y. Zhao and C. Chen, ACS Nano, 2015, 9, 6532 CrossRef CAS PubMed.
- S. Smulders, C. Larue, G. Sarret, H. Castillo-Michel, J. Vanoirbeek and P. H. Hoet, Toxicol. Lett., 2015, 238, 1 CrossRef CAS PubMed.
- B. Gilbert, S. C. Fakra, T. Xia, S. Pokhrel, L. Mädler and A. E. Nel, ACS Nano, 2012, 6, 4921 CrossRef CAS PubMed.
- S. A. James, B. N. Feltis, M. D. de Jonge, M. Sridhar, J. A. Kimpton, M. Altissimo, S. Mayo, C. Zheng, A. Hastings, D. L. Howard, D. J. Paterson, P. F. A. Wright, G. F. Moorhead, T. W. Turney and J. Fu, ACS Nano, 2013, 7, 10621 CrossRef CAS PubMed.
- G. MartÍnez-Criado, J. Villanova, R. Tucoulou, D. Salomon, J.-P. Suuronen, S. Labouré, C. Guilloud, V. Valls, R. Barrett, E. Gagliardini, Y. Dabin, R. Baker, S. Bohic, C. Cohen and J. Morse, J. Synchrotron Radiat., 2016, 23, 344 CrossRef PubMed.
- M. van der Zande, R. J. Vandebriel, E. Van Doren, E. Kramer, Z. Herrera Rivera, C. S. Serrano-Rojero, E. R. Gremmer, J. Mast, R. J. B. Peters, P. C. H. Hollman, P. J. M. Hendriksen, H. J. P. Marvin, A. A. C. M. Peijnenburg and H. Bouwmeester, ACS Nano, 2012, 6, 7427 CrossRef CAS PubMed.
- G. Veronesi, T. Gallon, A. Deniaud, B. Boff, C. Gateau, C. Lebrun, C. Vidaud, F. Rollin-Genetet, M. Carrière, I. Kieffer, E. Mintz, P. Delangle and I. Michaud-Soret, Inorg. Chem., 2015, 54, 11688 CrossRef CAS PubMed.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63 CrossRef.
- O. Proux, X. Biquard, E. Lahera, J.-J. Menthonnex, A. Prat, O. Ulrich, Y. Soldo, P. Trévisson, G. Kapoujyan, G. Perroux, P. Taunier, D. Grand, P. Jeantet, M. Deleglise, J.-P. Roux and J.-L. Hazemann, Phys. Scr., T, 2005, 115, 970 CrossRef.
- A. E. Pradas del Real, H. Castillo-Michel, R. Kaegi, B. Sinnet, V. Magnin, N. Findling, J. Villanova, M. Carrière, C. Santaella, A. Fernández-Martínez, C. Levard and G. Sarret, Environ. Sci. Technol., 2016, 50, 1759 CrossRef CAS PubMed.
- M. Cuillel, M. Chevallet, P. Charbonnier, C. Fauquant, I. Pignot-Paintrand, J. Arnaud, D. Cassio, I. Michaud-Soret and E. Mintz, Nanoscale, 2014, 6, 1707–1715 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr04381j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |