
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoengineering a library of metallic nanostructures using a single microfluidic reactor†
Víctor
Sebastián
ab and
Klavs F.
Jensen
 *a
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: kfjensen@mit.edu
bDepartment of Chemical Engineering, Aragon Institute of Nanoscience (INA), University of Zaragoza, Campus Rio Ebro, 50018 Zaragoza, Spain and Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
First published on 8th August 2016
Abstract
Microfluidic synthesis in a microfabricated reactor enables fast and facile synthesis of a wide library of metallic nanostructures: monometallic, bimetallic, anisotropic growth and heterostructures. Specific nanostructures are realized by selection of flow pattern and synthesis parameters. The technique is shown to have advantages over conventional batch technologies. Not only does it allow faster scalable synthesis, but also realization of nanostructures hitherto not reported such as Pt–Ru, Pt–Ni and Pt–Co nanodendrites, Pt–Pd heterostructures, Ag–Pd core–shell NPs, Au–Pd nanodumbbells and Au–Pd nanosheets.
Introduction
The synthesis of nanomaterials for electronic, optical and medical applications requires tight control of nucleation and growth processes to realize materials with desired features and properties.1,2 Significant efforts have been devoted to improving size distributions and quality beyond those obtained with conventional batch reactors.3,4 Highly monodisperse nanoparticles (NPs) are commonly produced by two stage synthesis: polydisperse nanoparticles are first synthesized using standard synthetic routes and afterwards, NPs of a given particle size are isolated by size classification methods and repeated recrystallizations.5 These approaches may be appropriate for small-scale production of nanostructures, but they are typically too laborious and have too low yields for large-scale production.3Continuous flow technologies offer potential advantages over batch synthesis, including feedback control of temperature and feed streams, reproducibility, potential for sensor integration for in situ synthesis monitoring, rapid screening of parameters and easier scale-up.6,7 The larger surface to volume ratio of microfluidic reactors (MR) enhance mass and heat transfer,8 but present challenges in terms of wall deposition and particle agglomeration and blockage of the flow path.9 The ability to work at elevated temperatures and pressures present opportunities for using environmentally benign solvent and accelerating nanoparticle synthesis.6
Herein we report a versatile synthesis platform based in a single microfluidic reactor to obtain a broad library of metal nanostructures in a continuous fashion. Although micro and meso scale flow reactors have been used to improve particle size distribution of nanomaterials,10,11 to promote the formation of nanocoatings12,13 and to effect shape control,14–21 this report is the first time, to our knowledge, that same microfluidic system is applied to control shape, size and chemical composition of nanomaterials. The library of nanostructures obtained comprises monodisperse monometallic and bimetallic NPs, Pt-based nanodendrites, nano core–shell structures, nanocubes, Janus nanostructures, dumbbells, nanorods and nanosheets. The microfluidic continuous synthesis at elevated temperature and pressures produces nanostructures on the time scales of seconds to minutes in contrast to the hours reported for traditional batch. These results are realized by modification of precursor chemistry and synthesis conditions (temperature, pressure, reactants, residence time and flow conditions). Moreover, new nanostructures are synthesized under conditions difficult to achieve in batch.
Results and discussion
Microfabrication techniques produced the silicon-Pyrex MR (Fig. 1) used in the continuous synthesis of the library of NPs.22 The unit could withstand high pressures (6 MPa) and high temperature (350 °C), and allowed easy optical access from Pyrex side. In order to control the nucleation and growth steps, the device had two zones, a cold inlet/outlet mixing area and a hot reaction area.23,24 Mixing of precursors in the cold area before entering to the reaction zone enabled us to achieve monodisperse nanostructures. The outlet stream also passed through the cold area to quench further nanomaterials growth. The reaction zone (100 μL) was a long spiral channel (400 μm width) designed to prevent solid accumulation and clogging. Segmented flow was generated by the separation of small liquid droplets at the nanoliter scale with gas slugs to enhance the proper reagents mixing (Fig. S1 in ESI†).23 The flow pattern in the reactor could be switched from laminar to segmented flow through the inlet labelled as carrier, in order to overcome dispersion problems associated with laminar flow at short residence times.25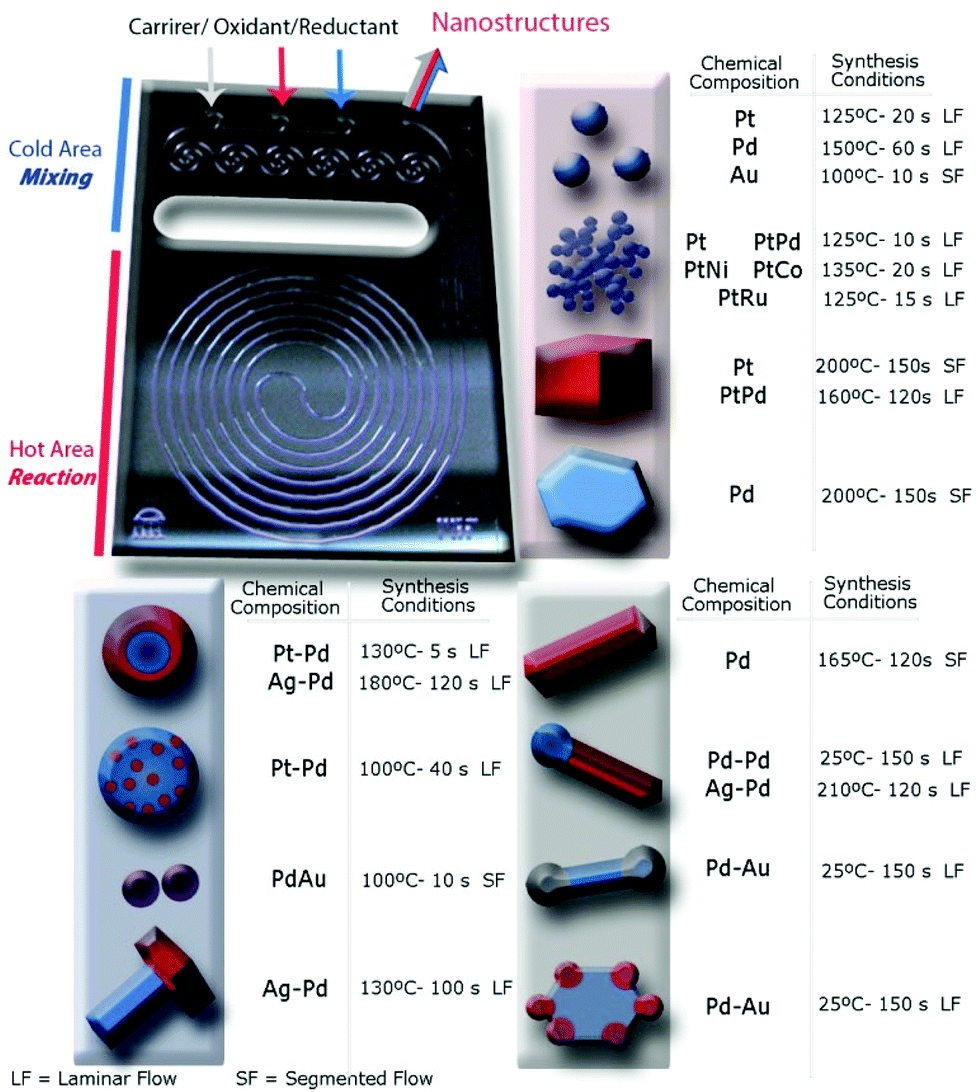 | ||
| Fig. 1 Silicon-Pyrex microfluidic reactor device used to synthesize the library of nanostructures. Schematic of the library of metallic nanostructures obtained and their synthesis conditions: monometallic and bimetallic spherical nanoparticles, nanodendrites, nanocubes, core–shell nanoparticles, Janus nanostructures, dumbbells, nanorods and nanosheets. | ||
Synthesis of metallic nanoparticles
Access to active sites is a key issue in production of NP for catalysis. Strong capping agents promote monodispersed nanoparticle synthesis, but also can compete with reactants and products for adsorption sites, altering the catalytic action of the particle.26 In that case, the use of a weak capping agent is preferred, but synthesis of small monodisperse NPs becomes challenging because uncontrolled mixing promotes particle agglomeration and heterogeneous size distribution. We were able to synthesize different monodisperse noble metal NPs (<5 nm) by using the MR and a weak capping agent tetradecyltrimethylammonium bromide – TTAB26 (see ESI† for experimental details).Reduction of Pt2+ ions with a strong agent such as sodium borohydride promoted a fast nucleation process that produced individual small NPs at reaction temperatures over 100 °C and reaction times shorter than 60 s (Fig. S2 and S3†). Monodisperse Pt NPs, 3.3 nm ± 0.2, were obtained at 125 °C and 20 s residence time, tR, (Fig. S2a and b†). The nucleation and growth stages were sensitive to synthesis temperature and residence time, with the size distribution of Pt NPs broadening as the temperature and residence time increased in the range of 125 °C–200 °C and 5 s–60 s, respectively (Fig. S3a and b†). By the controlled addition of ethanol in the reaction mixture, the size of the Pt NPs could be tuned even to small dimensions (2.0 nm ± 0.2) (Fig. 2a, S2b and S3c†). Ethanol is a mild reducing agent that promote monodisperse distribution of small nanoparticles by both modifying the saturation concentration during the nucleation process and increasing the reduction rate of sodium borohydride.27
 | ||
| Fig. 2 TEM images of metallic spherical NPs produced under different reaction conditions. (a) Pt NPs collected in laminar flow at 125 °C and tR = 20 s. (b) Pd NPs collected in laminar flow at 150 °C and tR = 60 s. (c) Au NPs. (d) AuPd alloy NPs, both collected in air segmented flow at 100 °C and tR = 10 s. | ||
Using the polyol method with ethylene glycol as reducing agent at 150 °C and 60 s residence time produced monodisperse Pd NPs under 3 nm (Fig. 2b and S2d†). Sodium borohydride as reducing agent instead of ethylene glycol, yielded palladium agglomerates over the entire range of reaction conditions tested (Fig. S2c†). These findings demonstrate the importance of using the proper solvent and reducing agent to realize continuous MR synthesis of monodisperse NPs without agglomeration and clogging.
Growth of Au NPs is normally achieved by an initial and rapid stage of gold precursor reduction into gold nuclei within less than 200 ms, after which the nuclei grow due to a coalescence process.28 Thus, the mixing time must be shorter than 200 ms to obtain monodisperse Au NPs. However, representative TEM images show broad size distributions of Au NPs produced across the entire range of synthesis conditions (Fig. S2e†). Thus, the fast reactions in a MR are not sufficient to realize narrow size distributions. The relative values of the kinetic rate and the rate of mixing, together with axial dispersion in single phase flow, decide the extent of polydispersity in the nanocrystals formation.23 Flow segmentation increases the cross stream mixing, reducing dispersion, to generate uniform sized Au NPs.29,30Fig. 2c shows a monodisperse distribution of gold nanoparticles, 2.8 ± 0.2 nm, obtained at 100 °C with an air segmented flow and a residence time of 10 s.23 The particle size of collected Au NPs at a residence time larger than 10 s were not controlled because the low slip velocity between slugs lead to poor mixing inside the slugs, which as a consequence leads to a wider particle size distribution (Fig. S3d†).23
Gold–palladium bimetallic NPs have received significant attention for their special catalytic properties, but it is challenging not end up with segregated populations of monometallic NPs when synthesizing small monodisperse NPs (<5 nm).31 By using the previous mentioned gas segmented flow, monodisperse AuPd bimetallic NPs (3.7 ± 0.3 nm) resulted at the same conditions for the pure Au counterpart. The UV-visible spectra of the bimetallic NPs did not show the surface plasmon signature of pure Au nanoparticles in the 500–520 nm range, suggesting the synthesis of bimetallic nanoparticles (Fig. S2f†). This phenomenon is consistent with previous observations19,32,33 of a group 10 metal (d8s2) in the bimetallic alloyed NPs suppressing the surface plasmon resonance energies of group 11 metals (d10s1) and implies the formation of Au/Pt alloyed NPs.
Synthesis of platinum and palladium heterostructures
Platinum and palladium are important catalysts for many industrial processes.34 When Pd is added to Pt, a change in the electronic structure can lead to activity enhancement for particular reactions.35 Both metals adopt the highly symmetrical face-centered-cubic (fcc) crystal structure, and the two metals have a very close lattice match promoting alloying (Pd and Pt have a lattice mismatch of only 0.77%). Thus, it is challenging to obtain nanostructures with Pt and Pd as segregated phases in a single step. In this work, we realized for the first time different Pt–Pd nanostructures in a continuous and single step process and using the advantages of microfluidics and some shape control strategies36 (Fig. 3). Growth at 130 °C with residence time as short as 5 s produced Pt–Pd core–shell nanostructures, where Pt is located in the core (Fig. 3a). Modifying the Pt/Pd precursor flow ratios tuned the shell thickness and particles size distribution, obtaining a thick shell at a large Pt/Pd ratio (Fig. S4†). At these conditions, Pt with a reduction potential higher that Pd nucleated faster and Pd grew heterogeneously on the Pt cores. The high-speed flows in the microchannels yielded fast multi-fluid lamination, which appeared to be a requirement for obtaining the core–shell structure. Pt and Pd agglomerates were obtained at residence times larger than 20 s (Fig. S4a†). The addition of ethanol to the crystallization mixture slowed down the platinum nucleation, switching the crystallization pattern between Pt and Pd. Under those conditions, Pd crystallized rapidly and Pd spherical particles decorated with small 2.1 nm Pt NPs resulted (Fig. 4b). The molar composition of both metallic precursors controlled the density of the Pt NPs on the Pd cores (Fig. S5a and b†). | ||
| Fig. 3 TEM images of metallic platinum–palladium nanostructures produced under different reaction conditions. (a), PtPd core–shell nanostructure, platinum located at the core, collected at 130 °C and tR = 5 s. (b), Pd NPs decorated with Pt NPs collected at 100 °C and tR = 40 s. (c) PtPd alloy nanocubes collected at 160 °C and tR = 120 s. (d) PtPd nanodendrites, collected at 125 °C and tR = 20 s. | ||
 | ||
| Fig. 4 TEM images of metallic silver–palladium nanostructures produced under different reaction conditions. (a) AgPd semi-Janus nanostructure, collected at 130 °C and tR = 100 s. (b) and (c) AgPd core–shell nanostructures collected at tR = 120 s, and 160 °C and 180 °C, respectively. (d) Ag nanowire with a Pd head, nanostructure collected at 210 °C and tR = 120 s. | ||
Pt–Pd alloyed nanocubes were obtained using the before mentioned polyol method under an air segmented flow to promote the oxidative etching (Fig. 3c and S5c†).19 The oxidative etching enables the selective etching of twinned particles, promoting the formation of cubic shape NPs.37 Further details about oxygen slug flow and oxidative etching appear below in the anisotropic growth section.
Bimetallic single crystal PtPd nanodendrites (17 ± 2 nm) were obtained by regulating the reduction kinetics with a mild reducing agent, ascorbic acid, which promotes dendritic growth (Fig. 3d and S5d†). This synthesis approach will be discussed in the section on synthesis of 3D dendritic nanostructures.
Synthesis of palladium and silver heterostructures
Palladium and silver nanostructures were also nanoengineered with good morphology control by using the same MR. Ag–Pd Janus-like NPs were obtained in continuous flow at 130 °C and 100 s residence time, using the polyol synthesis method (Fig. 4a). Segregation of individual palladium and silver particles resulted when the synthesis temperature was lower than 130 °C or with use of another reducing agent, such as sodium borohydride or ascorbic acid (Fig. S6a and b†). The HRTEM micrograph shows that Pd and Ag Janus-like nanostructure form via epitaxial growth and with high crystallinity (Fig. S5c and d†).Core–shell morphology resulted by increasing the Ag to Pd molar ratio at high temperature, 160 °C (Fig. 4b). Under those conditions, Pd nucleated faster and Ag nucleated on it to form a shell ultimately. At 180 °C, we observed both a hollow halo between the core and the shell and porosity in the Ag shell, suggesting a galvanic replacement reaction (Fig. 4c).
“Tadpole” like Ag–Pd NPs were observed to form at a temperature higher than 210 °C, where silver grew as a nanowire and palladium was the seed activating the growth (Fig. 4d). These results are in agreement with the work of Xia et al.38 who have recently reported the production of Ag nanorods using Pd decahedral seeds. Silver atoms only deposited along one side of the Pd decahedral seed to generate Ag nanorods with the Pd seed being positioned at one of its two ends.38 For reaction temperatures exceeding 220 °C, the microfluidic channels completely clogged, presumably because of the tadpole dimensions (Fig. 4d and S6e and f†).
Synthesis of 3D dendritic nanostructures
The synthesis of metal 3D dendritic nanostructures has received attention for catalytic applications because of these nanoarchitectures combine a high surface area with active edges.39–43 However, availability of scalable procedures to realize large-scale synthesis has limited their applications. Here we report, for the first time, the continuous flow synthesis of Pt and Pt–M bimetallic dendritic nanostructures of Pd, Ni, Co and Ru (Fig. 3d and 5). The metallic nanodendrites (NDs) were obtained via a one-step aqueous phase reaction with a mild reducing agent (ascorbic acid) and by controlling the nucleation and growth through three critical variables: reaction temperature, residence time and pH. The excellent synthesis control afforded by microfluidics produced NDs with an acceptable size distribution in residence times as small as 20 s. The slow nucleation rate of Pt in the presence of ascorbic acid could be an explanation for the differences from monodisperse Pt NPs obtained with NaBH4 as reducing agent (Fig. 2a). Lim et al.44 stated that the growth mechanism of Pt NDs is based on oriented attachment of the initially Pt formed particles, suggesting that discrete Pt nanoparticles were fused to form branches. | ||
| Fig. 5 TEM images of bimetallic platinum nanodendrites (NDs). (a) Pt NDs, produced at 125 °C and tR = 20 s. (b) PtNi NDs, collected at 135 °C and tR = 20 s., (c) PtCo NDs obtained at 135 °C and tR = 20 s. (d) PtRu NDs collected at 125 °C and tR = 15 s. | ||
The continuous flow synthesis occurred in seconds, much faster than the hour long time scales reported for batch synthesis.40,41 The NDs were characterized by HRTEM to gain insight into the crystallinity and nanobranches arrangement. HRTEM micrographs reveal that the ND morphologies depend on the metals incorporated. The branching is less favoured in Pt–Pd NDs(Fig. S5d†). In contrast, Pt–Ru and Pt–Ni NDs are highly branched (Fig. S7e and f†). Pt–Ni and Pt–Co NDs were magnetic so they can in principle be collected and recycled by applying a magnetic field.24 The bimetallic composition was confirmed by energy-dispersive X-ray spectroscopy analysis (Fig. S7g†). The lattice fringes reveal the high crystallinity of the NDs and show a continuous lattice in Pt, Pt–Ni, Pt–Co and Pt–Ru (Fig. S7†). Domain boundaries were clearly observed in Pt–Ni NDs, supporting the idea that two different mechanisms could occur: epitaxial growth by oriented attachment44 in Pt, Pt–Co, Pt–Ru and Pt–Pd and random aggregation of Pt and Ni nuclei in Pt–Ni NDs.
Synthesis of anisotropic nanostructures
The microfluidic reactor was also applied in kinetically controlled synthesis of nanostructures under short synthesis times.19 Anisotropic growth of palladium can be obtained in two different environments, air and carbon monoxide, obtaining nanostructures with a different morphology.45 The combination of oxygen from air and Cl− ions are a powerful etchant to selectively control the growth of single crystals seeds rather than twinned seeds.46 The oxidative etching preferentially affect twin seeds because of the presence of defect zones on the surface, being the zero-valent metal atoms oxidized back to ions. Nevertheless, not only the oxidative etching is required to anisotropically grow Pd nanostructures, but also a proper reduction rate and a selective capping agent to promote both of them the atomic addition on a specific facet.47The gas–liquid segmented approach was proposed as a tool to control the oxidative etching19 because air slugs promoted the oxygen transfer from the gas to the liquid phase and it enables an easy phase-product separation after synthesis. Furthermore the reactant injection in separated streams allowed obtaining a fine control on the oxidative etching and reducing rate. The palladium atomic addition was also controlled by the synthesis temperature, achieving an atomic addition sufficiently fast to produce elongated nanostructures and preventing the formation of low aspect nanostructures or cubooctahedrons.19Fig. 6a shows the Pd nanorods obtained at 165 °C and 2 minutes residence time, using Br– ions as capping agents to promote the formation of {100} and {110} facets. The nanorod aspect ratio could be varied to obtain approximately a value of 4.5–6.19
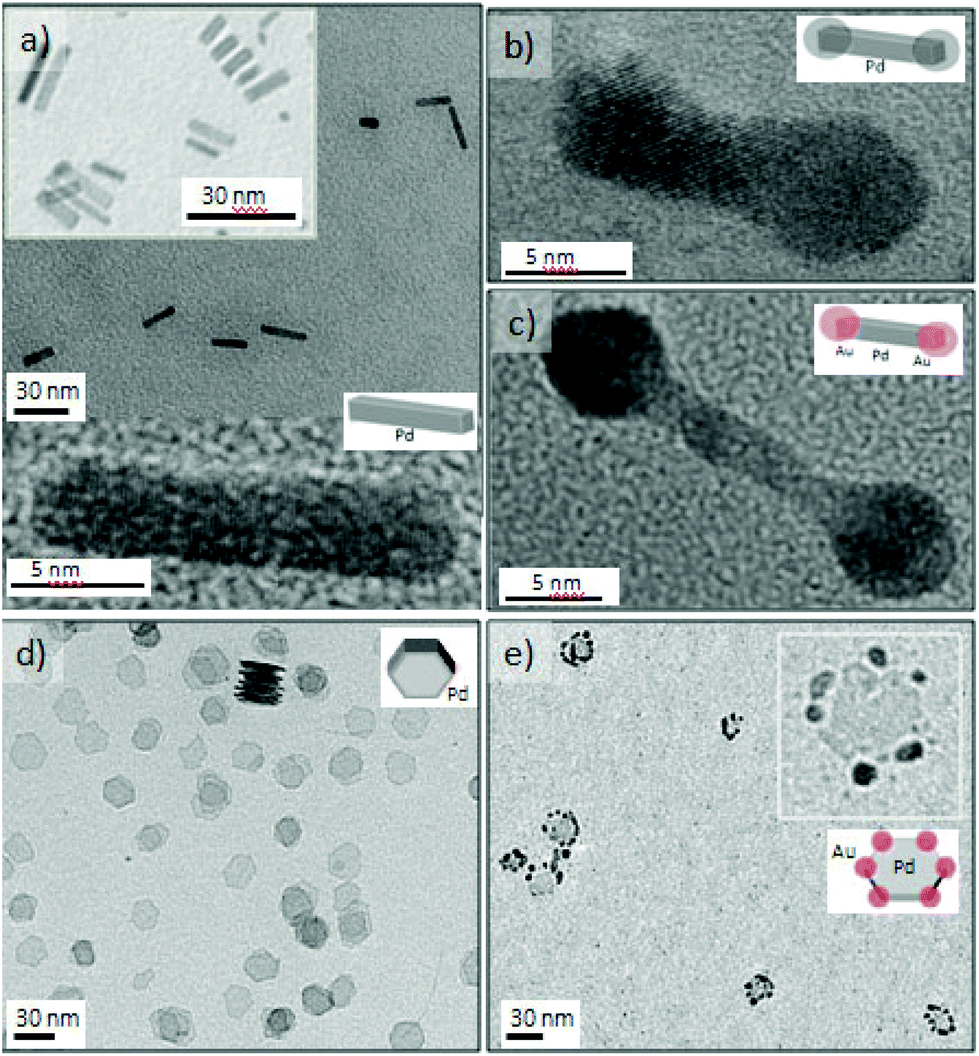 | ||
| Fig. 6 TEM images of metallic Pd nanostructures produced under different reaction conditions in a segmented flow. (a) Pd nanorods collected in air segmented flow at 165 °C and tR = 120 s (b) Pd nanodumbbells collected under ultrasound radiation at 25 °C and tR = 150 s. (c) PdAu nanodumbbells, Au located at the sides , collected under ultrasound radiation at 25 °C and tR = 150 s. (d) Pd nanosheets collected at 100 °C and tR = 150 s. (e) PdAu nanostars obtained at 25 °C and tR = 150 s. | ||
Carbon monoxide–liquid segmented flow was also applied to modify the anisotropic growth of palladium into a sheet-like shape.45 Carbon monoxide has the double role of being a reducing agent and a selective capping agent.15 The chemisorption of CO monoxide on {111} palladium facets can provide a means for controlling the atomic addition, hindering the growth along the [111] direction.48 Such hindered growth promotes the formation of a sheet-like structure.48,49 Carbon monoxide reduce Pd ions forming a Pd carbonyl complex, which collapses into clusters that are added onto given facets.49,50 Bromide ions were selected to define the sheet-like structure in a hexagonal shape. Fig. 6d shows a TEM micrograph of the palladium nanosheets obtained at 100 °C and 150 s residence time. The nanosheets presented sharp edges with a homogenous size distribution, edge lengths of 14 ± 1.4 nm. Fig. 6d shows a stack of nanosheets with ultrathin sheets of 1.2 nm. The high aspect ratio between the edge length and thickness of the palladium nanosheets determine the optical properties of this nanostructure,48 producing a surface plasmon resonance (SPR) absorption at 740 nm, (Fig. S8d†). The SPR absorption of this nanostructure arises from the ultrathin nature because palladium nanorods obtained in air–liquid segmented flow did not exhibit any optical properties, contrarily to other noble metals4 (Fig. S8d†). Consistent with effect of reactive gases, simple spherical Pd NPs resulted when N2 was used instead of O2 or CO in the slug flow experiments (Fig. S8a and b†).
Interfacial interactions that originate from electron transfer across the nanometer contact at the interface of two different materials can induce new properties that are not present in the individual components. In addition to the hetero nanostructures reported in this work by a single step synthesis, new nanostructures were obtained by a secondary growth method merging the same microfluidic reactor technology and ultrasound.51 Applying ultrasound, palladium nanorods were modified to obtain a dumbbell-like structure under a time scale process of 150 s and room temperature. The ultrasound induced cavitation promotes the synthesis of new nanostructured materials.52 Extreme physical and chemical conditions are created at localized spots while maintaining the bulk temperature. This property was crucial to conserve the metastable structure of nanorods and nanosheets, considering that these morphologies will be structurally modified at temperatures exceeding 80 °C.
The MR was immersed in an ultrasonic bath operating at 42 kHz and two streams of Pd nanorods and Au or Pd precursors, respectively, were injected. Au and Pd grew on the tips of the palladium nanorods by a heterogeneous nucleation-epitaxial growth process (Fig. 6b and c). The selective growth at the ends of the Pd nanorods was accomplished by the growth of Pd and Au at the areas not covered by the capping agent anchored during the rod growth, facilitating the dumbbell like morphology. Au grew faster that Pd because the difference of reduction potentials between Pd and Au additionally promotes a galvanic replacement reaction.8,53 Regarding the etching effect of the galvanic replacement reaction, the palladium rod structure was still preserved and gold was simultaneously deposited at the ends, (Fig. 6c). This type of complex nanostructure could not be obtained using the galvanic replacement method at high temperature and in batch type reactor because the Pd rod would be split in several units and Oswald ripening would promote a semi-dumbbell structure.53
The combination of microfluidic and ultrasound resulted in growth of gold nanoparticles at the edges of the palladium nanosheets, producing a star-like nanostructure with potentially interesting optical and catalytic properties (Fig. 6e).
Carbon monoxide–liquid segmented flow also controlled the growth kinetics of platinum to produce cube-shaped nanostructures The critical variables for the formation of faceted platinum particles in short synthesis time were metal precursor and temperature.45 Mono-disperse Pt nanocubes with an average edge length of 5.6 nm and sharp corners were obtained using platinum(II) acetylacetonate at 200 °C and a residence time as small as 150 seconds (Fig. 7a). While faceted nanocrystals were obtained with CO segmented flow, only shapeless nanoparticles were obtained if N2 gas replaced CO (Fig. 7b). The binding strength and adsorption of CO on Pt facets were found to be crucial variables in formation of Pt nanocrystals with (100) facets. It worth noting that the synthesis of Pt cube shaped nanocrystals batch requires synthesis times ranging from 60 minutes54 to 20 hours,55 demonstrating that microfluidic technology enable fast production of faceted nanocrystals.
 | ||
| Fig. 7 TEM images of metallic platinum nanostructures obtained with platinum(II) acetylacetonate at 200 °C, Pco = 120 psi and residence time = 150 s. under: (a) CO segmented flow. (b) N2 segmented flow. | ||
Experimental
Reagents
Potassium bromide (KBr, Aldrich); dipotassium tetrachloroplatinate(II) (K2PdCl4, Aldrich); platinum(II) acetylacetonate (Pt(acac)2, Aldrich); sodium tetracholoropalladate (Na2PdCl4, Aldrich); silver nitrate (AgNO3, Aldrich); nickel chloride(II) (NiCl2·6H2O, Aldrich); cobalt chloride(II) (CoCl2·6H2O, Aldrich); ruthenium chloride(III) (RuCl3·3H2O, Aldrich), ascorbic acid (AA, Aldrich); sodium borohydride (NaBH4, Aldrich); polyvinylpyrrolidone (PVP, MW = 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aldrich); tetradecyltrimethylammonium bromide (TTAB, Aldrich); ethylene glycol (EG, Aldrich); N,N-dimethylformamide (DMF, Omnisolv EMD); oleic acid (OLA, Sigma Aldrich), oleylamine (OAm, Across Organics). All chemical were of analytical grade and were used without further purification.
000, Aldrich); tetradecyltrimethylammonium bromide (TTAB, Aldrich); ethylene glycol (EG, Aldrich); N,N-dimethylformamide (DMF, Omnisolv EMD); oleic acid (OLA, Sigma Aldrich), oleylamine (OAm, Across Organics). All chemical were of analytical grade and were used without further purification.
Synthesis setup
The reactants were introduced in the microfluidic reactor by three separated inlets, oxidant, reductant and a spare inlet that was blocked for case not requiring additional reagents. The reagents were introduced in the mixing zone by means of two high pressure syringe pumps (ISCO 100 DM), insuring good control of the flow rates and residence time. Before a reaction between the reagents, intimate contact between the component molecules must be realized through mixing either in laminar or segmented flow approach. The mixing zone was maintained at room temperature by circulation of cold water. The product stream exited the microreactor through the cold zone, quenching the nanoparticles growth. A back-pressure regulator (Jasco, Model BP-1580) maintained a set pressure inside the reactor while enabling samples to be collected in a continuous mode without depressurizing the system.The microfluidic channels and ports were etched into silicon wafers by deep reactive ion etching (DRIE). The silicon surface was then oxidized to silica (glass) and anodically bonded to a Pyrex wafer to cap the channels, providing a glass surface that is compatible with reagents used. The reactor was designed to get a reaction volume of 100 microL. The nanostructures were analyzed by transmission electron microscopy (TEM, JEOL Model 200CX, HRTEM JEOL 2010 and STEM JEOl 2011F). The UV-Vis spectra was measured using an Agilent 8453 UV-Visible spectrophotometer.
Further details about the synthesis procedure of each type of nanostructure are given in the ESI.†
Conclusions
In conclusion, we have demonstrated facile, fast and shape-selective synthesis of an extensive library of nanostructures in continuous flow process by using a microfluidic reactor platform. The microreactor separated the mixing and reaction in order to control the nucleation and growth kinetics, and it produced monodisperse NPs by controlling the residence time distribution. We avoided wall-induced nucleation and clogging by selection of flow pattern, reagents concentration, solvent, ligand, reducing agent and reaction conditions (temperature and residence time). Similar nanostructures to those reported for conventional batch-type reactor were realized at considerably reduced synthesis time along with new complex nanostructures: Pt–Ru, Pt–Ni and Pt–Co nanodendrites, Pt–Pd heterostructures, Ag–Pd core–shell NPs, Au–Pd nanodumbbells and Au–Pd nanosheets. The fast synthesis with a fine control of size, shape and metal distributions was made possible by proper selection of metal precursors, reducing agents, stabilizing-capping agents, flow patterns, synthesis temperature and reactants residence time.Acknowledgements
Authors acknowledge US NSF grant (CHE-0714189) for funding this research, and the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807 V. S. acknowledges the support of the Fulbright Commission and the Ministry of Education in Spain (Programa Nacional de Movilidad de Recursos Humanos. del Plan Nacional de I + D + I 2008–2011), as well as People Program (CIG-Marie Curie Actions, REA grant agreement no. 321642) to develop this research.Notes and references
- S. Kumar and T. Nann, Small, 2006, 2, 316–329 CrossRef CAS PubMed.
- G. J. Zhou, M. K. Lu, Z. L. Xiu, S. F. Wang, H. P. Zhang, Y. Y. Zhou and S. M. Wang, J. Phys. Chem. B, 2006, 110, 6543–6548 CrossRef CAS PubMed.
- V. Sebastian, M. Arruebo and J. Santamaria, Small, 2014, 10, 835–853 CrossRef CAS PubMed.
- Y. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS PubMed.
- L. Gomez, V. Sebastian, S. Irusta, A. Ibarra, M. Arruebo and J. Santamaria, Lab Chip, 2014, 14, 325–332 RSC.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347–1357 CrossRef CAS.
- J. Baek, P. M. Allen, M. G. Bawendi and K. F. Jensen, Angew Chem., Int. Ed., 2011, 50, 627–630 CrossRef CAS PubMed.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef CAS.
- A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones and S. Nair, Science, 2014, 345, 72–75 CrossRef CAS PubMed.
- F. Cacho-Bailo, G. Caro, M. Etxeberria-Benavides, O. Karvan, C. Tellez and J. Coronas, Chem. Commun., 2015, 51, 11283–11285 RSC.
- Y. H. Kim, L. Zhang, T. Yu, M. S. Jin, D. Qin and Y. N. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- A. Larrea, V. Sebastian, A. Ibarra, M. Arruebo and J. Santamaria, Chem. Mater., 2015, 27, 4254–4260 CrossRef CAS PubMed.
- S. E. Lohse, J. R. Eller, S. T. Sivapalan, M. R. Plews and C. J. Murphy, ACS Nano, 2013, 7, 4135–4150 CrossRef CAS PubMed.
- L. Zhang and Y. N. Xia, Adv. Mater., 2014, 26, 2600–2606 CrossRef CAS PubMed.
- L. Zhang, G. D. Niu, N. Lu, J. G. Wang, L. M. Tong, L. D. Wang, M. J. Kim and Y. N. Xia, Nano Lett., 2014, 14, 6626–6631 CrossRef CAS PubMed.
- V. Sebastian, S. Basak and K. F. Jensen, AIChE J., 2016, 62, 373–380 CrossRef CAS.
- M. Thiele, J. Z. E. Soh, A. Knauer, D. Malsch, O. Stranik, R. Muller, A. Csaki, T. Henkel, J. M. Kohler and W. Fritzsche, Chem. Eng. J., 2016, 288, 432–440 CrossRef CAS.
- A. Knauer, A. Csaki, F. Moller, C. Huhn, W. Fritzsche and J. M. Kohler, J. Phys. Chem. C, 2012, 116, 9251–9258 CAS.
- S. Marre, A. Adamo, S. Basak, C. Aymonier and K. F. Jensen, Ind. Eng. Chem. Res., 2010, 49, 11310–11320 CrossRef CAS.
- V. S. Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef PubMed.
- S. K. Lee, X. Y. Liu, V. S. Cabeza and K. F. Jensen, Lab Chip, 2012, 12, 4080–4084 RSC.
- S. A. Khan, A. Gunther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS PubMed.
- P. D. Yang, H. Lee, S. E. Habas, S. Kweskin, D. Butcher and G. A. Somorjai, Angew. Chem., Int. Ed., 2006, 45, 7824–7828 CrossRef PubMed.
- H. I. Schlesinger, H. C. Brown, D. L. Mayfield and J. R. Gilbreath, J. Am. Chem. Soc., 1953, 75, 213–215 CrossRef CAS.
- J. Polte, R. Erler, A. F. Thunemann, S. Sokolov, T. T. Ahner, K. Rademann, F. Emmerling and R. Kraehnert, ACS Nano, 2010, 4, 1076–1082 CrossRef CAS PubMed.
- A. Gunther, M. Jhunjhunwala, M. Thalmann, M. A. Schmidt and K. F. Jensen, Langmuir, 2005, 21, 1547–1555 CrossRef PubMed.
- J. M. Kohler, S. N. Li and A. Knauer, Chem. Eng. Technol., 2013, 36, 887–899 CrossRef.
- L. Uson, V. Sebastian, A. Mayoral, J. L. Hueso, A. Eguizabal, M. Arruebo and J. Santamaria, Nanoscale, 2015, 7, 10152–10161 RSC.
- M. L. Wu, D. H. Chen and T. C. Huang, Langmuir, 2001, 17, 3877–3883 CrossRef CAS.
- W. B. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
- C. R. Henry, Surf. Sci. Rep., 1998, 31, 235–325 CrossRef.
- R. Adzic, M. H. Shao, P. Liu and J. L. Zhang, J. Phys. Chem. B, 2007, 111, 6772–6775 Search PubMed.
- R. D. Tilley, S. S. Cheong and J. D. Watt, Nanoscale, 2010, 2, 2045–2053 RSC.
- Y. J. Xiong, J. Y. Chen, B. Wiley, Y. N. Xia, Y. D. Yin and Z. Y. Li, Nano Lett., 2005, 5, 1237–1242 CrossRef CAS PubMed.
- M. Luo, H. W. Huang, S. I. Choi, C. Zhang, R. R. da Silva, H. C. Peng, Z. Y. Li, J. Y. Liu, Z. K. He and Y. N. Xia, ACS Nano, 2015, 9, 10523–10532 CrossRef CAS PubMed.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2009, 131, 9152–9153 CrossRef CAS PubMed.
- L. Wang and Y. Yamauchi, Chem. Mater., 2009, 21, 3562–3569 CrossRef CAS.
- H. Lee, S. E. Habas, S. Kweskin, D. Butcher, G. A. Somorjai and P. D. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824–7828 CrossRef CAS PubMed.
- A. Mohanty, N. Garg and R. C. Jin, Angew. Chem., Int. Ed., 2010, 49, 4962–4966 CrossRef CAS PubMed.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. A. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- B. Lim, M. J. Jiang, T. Yu, P. H. C. Camargo and Y. N. Xia, Nano Res., 2010, 3, 69–80 CrossRef CAS.
- V. Sebastian, C. D. Smith and K. F. Jensen, Nanoscale, 2016, 8, 7534–7543 RSC.
- B. Lim, M. J. Jiang, J. Tao, P. H. C. Camargo, Y. M. Zhu and Y. N. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS.
- Y. J. Xiong, H. G. Cai, B. J. Wiley, J. G. Wang, M. J. Kim and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 3665–3675 CrossRef CAS PubMed.
- X. Q. Huang, S. H. Tang, X. L. Mu, Y. Dai, G. X. Chen, Z. Y. Zhou, F. X. Ruan, Z. L. Yang and N. F. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed.
- P. F. Siril, L. Ramos, P. Beaunier, P. Archirel, A. Etcheberry and H. Remita, Chem. Mater., 2009, 21, 5170–5175 CrossRef CAS.
- J. B. Wu, A. Gross and H. Yang, Nano Lett., 2011, 11, 798–802 CrossRef CAS PubMed.
- D. G. Shchukin, D. Radziuk and H. Mohwald, Annu. Rev. Mater. Res., 2010, 40, 345–362 CrossRef CAS.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS PubMed.
- P. H. C. Camargo, Y. Xiong, L. Ji, J. M. Zuo and Y. Xia, J. Am. Chem. Soc., 2007, 129, 15452–15453 CrossRef CAS PubMed.
- B. H. Wu, N. F. Zheng and G. Fu, Chem. Commun., 2011, 47, 1039–1041 RSC.
- J. T. Ren and R. D. Tilley, J. Am. Chem. Soc., 2007, 129, 3287–3291 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and experimental information. See DOI: 10.1039/c6nr04104c |
| This journal is © The Royal Society of Chemistry 2016 |