
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2D molybdenum disulphide (2D-MoS2) modified electrodes explored towards the oxygen reduction reaction†
Samuel J.
Rowley-Neale
a,
Jamie M.
Fearn
a,
Dale A. C.
Brownson
a,
Graham C.
Smith
b,
Xiaobo
Ji
c and
Craig E.
Banks
*a
aFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
bFaculty of Science and Engineering, Department of Natural Sciences, University of Chester, Thornton Science Park, Pool Lane, Ince, Chester CH2 4NU, UK
cCollege of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, PR China
First published on 11th July 2016
Abstract
Two-dimensional molybdenum disulphide nanosheets (2D-MoS2) have proven to be an effective electrocatalyst, with particular attention being focused on their use towards increasing the efficiency of the reactions associated with hydrogen fuel cells. Whilst the majority of research has focused on the Hydrogen Evolution Reaction (HER), herein we explore the use of 2D-MoS2 as a potential electrocatalyst for the much less researched Oxygen Reduction Reaction (ORR). We stray from literature conventions and perform experiments in 0.1 M H2SO4 acidic electrolyte for the first time, evaluating the electrochemical performance of the ORR with 2D-MoS2 electrically wired/immobilised upon several carbon based electrodes (namely; Boron Doped Diamond (BDD), Edge Plane Pyrolytic Graphite (EPPG), Glassy Carbon (GC) and Screen-Printed Electrodes (SPE)) whilst exploring a range of 2D-MoS2 coverages/masses. Consequently, the findings of this study are highly applicable to real world fuel cell applications. We show that significant improvements in ORR activity can be achieved through the careful selection of the underlying/supporting carbon materials that electrically wire the 2D-MoS2 and utilisation of an optimal mass of 2D-MoS2. The ORR onset is observed to be reduced to ca. +0.10 V for EPPG, GC and SPEs at 2D-MoS2 (1524 ng cm−2 modification), which is far closer to Pt at +0.46 V compared to bare/unmodified EPPG, GC and SPE counterparts. This report is the first to demonstrate such beneficial electrochemical responses in acidic conditions using a 2D-MoS2 based electrocatalyst material on a carbon-based substrate (SPEs in this case). Investigation of the beneficial reaction mechanism reveals the ORR to occur via a 4 electron process in specific conditions; elsewhere a 2 electron process is observed. This work offers valuable insights for those wishing to design, fabricate and/or electrochemically test 2D-nanosheet materials towards the ORR.
1. Introduction
The effects of anthropogenic induced climate change are beginning to be realised on both a local and global scale, which has created a demand for the development and implementation of new “clean” methods of energy generation.1–7 Replacing the typical combustion of fossil fuels (FFs) with the utilisation of hydrogen fuel cells in the world's energy economy could dramatically decrease the production of anthropogenic greenhouse emissions.8,9 The most widely used fuel cell is the proton exchange membrane (PEM) fuel cell (also known as a polymer electrolyte membrane fuel cell) which is potentially viable in a vast number of applications, from vehicles to combined heat and power units.10,11 Their implementation is advantageous over typical FF engines due to their zero carbon emissions and ability to undergo long periods of inactivity without detrimental implications on energy output.10 The reason why they are not currently a viable alternative to FF engines in the majority of applications is a greater cost per unit energy.10 Resultantly, there is a need to lower the cost of energy production associated with fuel cells. This can be done via lowering the cost of a PEM fuel cell's fuel, typically H2, as well as increasing the energy output per unit of fuel utilised. It is therefore essential that research producing alternative/cheaper electrocatalysts (in order to increase the efficiency of PEM fuel cell energy generation) is performed.The essential reactions which allow a fuel cell to produce a current are the hydrogen oxidation reaction (HOR) and the oxygen reduction reaction (ORR).12–14 The HOR occurs on the anode and typically has a negligible overpotential, whilst the ORR occurs at the cathode and has a large kinetic inhibition given the strong (di)oxygen double bond resulting in a large energy input to initiate the reaction.12,15 This results in the ORR being the rate determining step in the production of output energy from the initial H2 fuel source. Taking this into account, by reducing the overpotential at which ORR occurs at the cathode, the process will be “more energetically favourable” and it is possible to make a significant increase within fuel cell efficiency.16,17 Ideally this reaction combines O2 (typically atmospheric, in the case of PEM fuel cells) with hydrogen (H2) in order to produce H2O; however, the reaction mechanism is dependent upon the pH of the electrode material and/or electrolyte used.18 The ORR has proven to be problematic in fuel cells due to membrane degradation and electrode fouling which occurs when the electrode utilised reduces O2via a 2 electron pathway (see below) resulting in the unfavourable production of H2O2.16–18,21 PEM fuel cell degradation via H2O2 induced electrode fouling is the predominate factor limiting the lifespan this PEM fuel cell, potentially limiting the voltage output by up to 50% as a result of cathode corrosion (causing slow ORR kinetics).19 The exact mechanism for H2O2 poisoning of the cathode is unclear with direct20 and indirect21 attack mechanisms proposed in the literature. The ORR processes in alkaline and acidic media are as follows:22,23
1.1 Acidic media
| O2 + 4H+ + 4e− → 2H2O direct (4 electron pathway) |
| O2 + 2H+ + 2e− → H2O2 indirect (2 electron pathway) |
| H2O2 + 2H+ + 2e− → 2H2O |
1.2 Alkaline media
| O2 + 2H2O + 4e− → 4OH− direct (4 electron pathway) |
| O2 + H2O + 2e− → HO2− + OH− indirect (2 electron pathway) |
| HO2− + H2O + 2e− → 3OH− |
In an attempt to achieve this goal, researchers have investigated the electrocatalytic activity of various 2D materials towards the ORR.23,26,27 Recent interest has been directed towards 2D-MoS2; Table 1 presents a thorough literature overview of 2D-MoS2 based electrocatalysts explored towards the ORR. 2D-MoS2 comprises a single layer of two monoatomic planes of hexagonally arranged sulphur atoms linked to molybdenum atoms.1,28 The electrochemical properties of 2D-MoS2 are anisotropic in nature, with the basal plane of the 2D-MoS2 being relatively inert and the exposed edges being reported as the active sites of electron transfer.1,29,30 Resultantly, highly defected sheets of 2D-MoS2 have a greater catalytic activity due to the larger number of exposed edges.31 Interestingly, in its bulk form MoS2 exhibits poor electrochemical activity due to a low ratio of exposed edge to basal planes.3,32,33 2D-MoS2 has been shown to be an effective electrocatalyst towards the ORR, for example Huang et al.34 utilised MoS2 ultra-thin nanosheets drop cast onto a rotating disk glassy carbon electrode and observed a 7.8 fold increase in current density and a ca. 170 mV positive shift in ORR onset, exhibiting a strong 4 electron mechanism selectivity for the ORR in alkaline media. Note that the terminated edges of the 2D-MoS2 will comprise of both Mo and S atoms, each having distinct electrocatalytic properties in certain scenarios. In this case, it is the electropositive charge on the Mo atoms (induced by a polarization effect of the electronegative S atoms present) found at the edge planes that are the binding sites for the electronegative O atoms within the electrolyte, thus making them the sites responsible for 2D-electrocatalytic reactions towards the ORR.34
| Catalyst | Electrode/supporting material | Comparison electrodes/supporting material | Loading (μg cm−2) | Electrolyte | ORR onset (V) | Ref. |
|---|---|---|---|---|---|---|
| Key: —: value unknown, rGO: reduced graphene oxide. NP: nanoparticle, O–MoS2-87: O–MoS2 which had 87 μl of aqueous hydrogen peroxide used in its synthesis, BDD: boron doped diamond, EPPG: edge plane pyrolytic graphite, GC: glassy carbon, SPE: screen printed electrode, RHE: reversible hydrogen electrode, SCE: saturated calomel electrode.a Homogeneous solution containing 0.3 mg of catalyst per 10 μl.b Optimal mass of 2D-MoS2 (range tested: 252 to 2533 ng cm−2). | ||||||
| Flower like MoS2 | GC | 20% Pt/C | — | 0.1 M KOH | −0.14 (vs. Ag/AgCl) | 35 |
| CO(OH)2–MoS2/rGO | GC | 40 wt% Pt/C | 510 | 0.1 M KOH | +0.86 (vs. RHE) | 65 |
| MoS2–rGO | GC | — | ca. 1529a | 0.1 M KOH | +0.80 (vs. RHE) | 66 |
| O–MoS2-87 | GC | 20% Pt/C | 283 | 0.1 M KOH | +0.94 (vs. RHE) | 34 |
| AuNP/MoS2 films | GC | 20% Pt/C | 50 | 0.1 M KOH | −0.10 (vs. SCE) | 67 |
| 2D-MoS2 | BDD | EPPG, GC, SPE and Pt | 1524b | 0.1 M H2SO4 | +0.10 (vs. SCE) | This work |
| 2D-MoS2 | EPPG | EPPG, GC, SPE and Pt | 1009b | 0.1 M H2SO4 | +0.10 (vs. SCE) | This work |
| 2D-MoS2 | GC | EPPG, GC, SPE and Pt | 1009b | 0.1 M H2SO4 | +0.10 (vs. SCE) | This work |
| 2D-MoS2 | SPE | EPPG, GC, SPE and Pt | 1009b | 0.1 M H2SO4 | +0.10 (vs. SCE) | This work |
Current literature reports are thoroughly overviewed in Table 1 and are sophisticated in their approaches towards the ORR; however, they are limited since they follow typical conventions found within the literature when MoS2 materials are explored as electrocatalysts towards the ORR, those being: (1) the use of glassy carbon (GC) almost exclusively as a supporting electrode material, with few or no attempts made to use/explore alternative carbon based supports. Note that the performance of MoS2 can only be truly understood via immobilisation using a range of supporting materials with varied electrode kinetics (electrochemical activities); (2) within the literature, electrodes are modified with only one mass (coverage) of a given MoS2 based material, which again makes it difficult to extrapolate a true understanding of the electrochemical behaviour of 2D-MoS2; (3) the use of only KOH as an electrolyte, which makes the results relevant for alkaline fuel cells, however not applicable to PEM fuel cells (with the latter using an acidic electrolyte).35 These three conventions commonly practised within the literature, neglect the ability to de-convolute the true electrochemical performance of 2D-MoS2 materials whilst also making their findings non-applicable to real world applications in PEM fuel cells.
This work breaks from academic convention (see points 1–3 above) performing diligent control experiments which have been overlooked within the current academic literature, namely: exploring different supporting electrode substrates used to electrically wire 2D-MoS2 and different immobilised masses towards the ORR, all of which are reported for what we believe to be the first time, performed in an acidic electrolyte. The use of acidic conditions mimic those found within a typical PEM fuel cell, providing a greater validity to real world PEM fuel cell applications.36
2. Experimental section
All chemicals used were of analytical grade and used as received from Sigma-Aldrich without any further purification. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm. The sulfuric acid solutions utilised are of the highest possible grade available from Sigma-Aldrich (99.999%, double distilled for trace metal analysis). The sulfuric acid (0.1 M) solution used to explore the HER was vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen. All ORR measurements were performed in 0.1 M sulfuric acid that was oxygenated and subject to rigorous bubbling of 100% medical grade oxygen for one hour, resulting in a 0.9 mM concentration of oxygen, assuming this to be a completely saturated solution at room temperature which is common practice in the literature.22,23 Where ORR onset potentials are denoted within the manuscript, note that this is defined as the potential at which the current initially deviates from the background current by a value of 25 μA cm−2, thus signifying the commencement of the faradaic current associated with the ORR redox reaction.Electrochemical measurements were performed using an Ivium Compactstat™ (Netherlands) potentiostat. Measurements were carried out using a typical three electrode system with a Pt wire counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The working electrodes used were as follows: an edge plane pyrolytic graphite (EPPG) (Le Carbone, Ltd Sussex, UK) electrode, which was machined into a 4.9 mm diameter, with the disc face parallel with the edge plane as required from a slab of highly ordered pyrolytic graphite (HOPG); a glassy carbon (GC) electrode (3 mm diameter, BAS, USA); a boron-doped diamond (BDD) electrode (3 mm diameter, BAS, USA); a Pt electrode (3 mm diameter, BAS, USA); and screen-printed graphite electrodes (SPEs), which have a 3 mm diameter working electrode. The SPEs were fabricated in-house with an appropriate stencil using a DEK 248 screen-printing machine (DEK, Weymouth, UK).37 These electrodes have been used extensively in previous studies.1,38–41 For their fabrication, first, a carbon–graphite ink formulation (product code C2000802P2; Gwent Electronic Materials Ltd, UK) was screen-printed onto a polyester (Autostat, 250 μm thickness) flexible film (denoted throughout as standard-SPE); this layer was cured in a fan oven at 60 °C for 30 minutes. Next, a silver/silver chloride reference electrode was included by screen-printing Ag/AgCl paste (product code C2040308D2; Gwent Electronic Materials Ltd, UK) onto the polyester substrates and a second curing step was undertaken where the electrodes were heated at 60 °C for 30 minutes. Finally, a dielectric paste (product code D2070423D5; Gwent Electronic Materials Ltd, UK) was then printed onto the polyester substrate to cover the connections. After a final curing at 60 °C for 30 minutes these SPEs are ready to be used. These SPEs have been reported previously and shown to exhibit a heterogeneous electron transfer (HET) rate constant, ko, of ca. 10−3 cm s−1, as measured using the [Ru(NH3)6]3+/2+ redox probe.40,42–45 For the purpose of this work, electrochemical experiments were performed using the working electrode of the SPEs (only) and external reference and counter electrodes were implemented as detailed earlier to allow a direct comparison between all the utilised electrodes as well as with academic literature.
The 2D-MoS2 was commercially procured from ‘Graphene Supermarket’ (Reading, MA, USA).46 The 2D-MoS2 nanosheets have a reported purity of >99% and are dispersed in ethanol at a concentration of 18 mg L−1.46 Our previous work has implemented extinction spectroscopy (ESI Fig. 1†) to determine the lateral length and number of 2D-MoS2 nanosheets in our commercially sourced sample which are found to correspond to 61.5 nm and an average of 3 (2.89) monolayers per nanosheet, respectively.1,46 The modification of each electrode was carried out using a drop casting approach, where an aliquot of the 2D-MoS2 suspension was deposited onto the desired supporting electrode surface using a micropipette.42 This deposition was allowed to dry for 5 minutes (at 35 °C) to ensure complete ethanol evaporation. Finally, the electrode was allowed to cool to ambient temperature, after which the process was repeated until the desired mass was deposited onto the surface, at which point the electrode was ready to be used.
Where specific masses of modification are donated within the paper (i.e. ng cm−2), note that this value represents the quantity/mass of 2D-MoS2 that will be present over the averaged area specified and this does not stipulate that an even spread/distribution of monolayer 2D-MoS2 is present. Rather, the reader should be aware that in reality it is likely that there are areas of multilayer, bilayer and indeed monolayer 2D-MoS2 randomly distributed across the electrode surface.47 Interested readers are directed to ESI Fig. 2,† which shows how different masses of 2D-MoS2 distribute across the surface of a SPE. Essentially, the values reported represent the mass of 2D-MoS2 deposited respective to the area of the electrode utilised.
An Agilent 8453 UV-visible Spectroscopy System (equipped with a tungsten lamp assembly, G1315A, 8453 for absorption between 250 nm and 1500 nm and a deuterium lamp, 2140-0605 for absorption between 200 nm and 400 nm) was used to obtain the absorption spectroscopy. The absorption spectrum was analysed using UV-Visible ChemStation software. Scanning electron microscope (SEM) images and surface element analysis were obtained using a JEOL JSM-5600LV model SEM equipped with an energy-dispersive X-ray microanalysis (EDS) package. Transmission electron microscopy (TEM) images were obtained using a 200 kV primary beam under conventional bright-field conditions. The 2D-hBN sample was dispersed onto a holey-carbon film supported on a 300 mesh Cu TEM grid. Raman Spectroscopy was performed using a ‘Renishaw InVia’ spectrometer equipped with a confocal microscope (×50 objective) and an argon laser (514.3 nm excitation). Measurements were performed at a very low laser power level (0.8 mW) to avoid any heating effects. X-ray diffraction (XRD) was performed using an “X'pert powder PANalytical model” with a copper source of Kα radiation (of 1.54 Å) and Kβ radiation (of 1.39 Å), using a thin sheet of nickel with an absorption edge of 1.49 Å to absorb Kβ radiation. The omega was set to 3.00 and the 2θ range was set between 10 and 100 2θ in correspondence with literature.48 Additionally, to ensure well defined peaks, an exposure of 100 seconds per 2θ step was implemented for all the above analysis. 2D-MoS2 was utilised after deposition onto a sterilised glass slide (coated with excess 2D-MoS2 in ethanol then allowed to dry) or a silicon wafer where appropriate. The X-ray photoelectron spectroscopy (XPS) data was acquired using a bespoke ultra-high vacuum system fitted with a Specs GmbH Focus 500 monochromated Al Kα X-ray source, Specs GmbH Phoibos 150 mm mean radius hemispherical analyser with 9-channeltron detection, and a Specs GmbH FG20 charge neutralising electron gun.49 Survey spectra were acquired over the binding energy range 1100–0 eV using a pass energy of 50 eV and high resolution scans were made over the C 1s and O 1s lines using a pass energy of 20 eV. Under these conditions the full width at half maximum of the Ag 3d5/2 reference line is ca. 0.7 eV. In each case, the analysis was an area-average over a region approximately 1.4 mm in diameter on the sample surface, using the 7 mm diameter aperture and lens magnification of ×5. The energy scale of the instrument is calibrated according to ISO 15472, and the intensity scale is calibrated using an in-house method traceable to the UK National Physical Laboratory.50 Data were quantified using Scofield cross sections corrected for the energy dependencies of the electron attenuation lengths and the instrument transmission.51 Data interpretation was carried out using CasaXPS software v2.3.16.52
3. Results and discussion
3.1 Characterisation of the commercially obtained 2D-MoS2
Extensive physiochemical characterisation of the 2D-MoS2 has been previously conducted and reported,1 including: Raman spectroscopy, EDS, SEM, TEM, UV-Vis spectroscopy, XRD and XPS. Full characterisation is presented in the ESI† and is summarised below for convenience. Despite some aggregation, which is the case for all 2D materials, upon close inspection a lateral grain size of ca. 100–400 nm is evident. UV-Vis (ESI Fig. 1†) indicates that the lateral length and stacking number of the 2D-MoS2 corresponds to 61.5 nm and 3 (2.89) respectively when dispersed in ethanol pre-deposition onto the surface of the electrode supporting material utilised. SEM and TEM images of the commercially sourced 2D-MoS2 are shown in ESI Fig. 3 and 4.† EDS (ESI Fig. 3†) and XPS (ESI Fig. 5 and 6†) confirm the presence of Mo and S at the expected ratios (0.55 Mo at% to 1.35 S and Mo to S at% concentrations at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 ratio respectively) thus indicating the presence of 2D-MoS2. This was supported by XRD analysis (ESI Fig. 7†), which shows a diffraction peak for 2D-MoS2 with a 2θ corresponding to 14.2°.33,48 Last, Raman spectroscopy (ESI Fig. 8 and 9†) indicates that the separation between the A1g and E12g vibrational bands give a consistent value of 24.7 cm−1, which corresponds with literature to bulk MoS2.53 This implies that upon deposition of the 2D-MoS2 utilised herein onto the supporting electrode materials, the structural model is likely that of re-assembly, with few-layer nanosheets forming as bulk.
:2.2 ratio respectively) thus indicating the presence of 2D-MoS2. This was supported by XRD analysis (ESI Fig. 7†), which shows a diffraction peak for 2D-MoS2 with a 2θ corresponding to 14.2°.33,48 Last, Raman spectroscopy (ESI Fig. 8 and 9†) indicates that the separation between the A1g and E12g vibrational bands give a consistent value of 24.7 cm−1, which corresponds with literature to bulk MoS2.53 This implies that upon deposition of the 2D-MoS2 utilised herein onto the supporting electrode materials, the structural model is likely that of re-assembly, with few-layer nanosheets forming as bulk.
Thus the 2D-MoS2 utilised in this work has been fully characterised and revealed as high quality, few layer sheets of MoS2, which are next implemented towards the ORR.
3.2 Catalytic activity of 2D-MoS2 towards the ORR at an assigned coverage
Previous work focused on using 2D-MoS2 as an electrocatalyst for the HER and showed 2D-MoS2 to be electroactive when immobilised on carbon based electrode substrates.1 It was therefore essential to benchmark the electrochemical activity of the 2D-MoS2 when electrically wired using BDD, EPPG, GC and SPEs and explored in degassed 0.1 M H2SO4. This was to ensure that no electroactivity was observed in the region of a linear sweep voltammogram (LSV) where the ORR is expected to occur, as this would convolute the interpretation of the ORR, the results of which can be observed in ESI Fig. 10.†Fig. 1(A) shows LSVs for EPPG, GC and SPEs in a 0.1 M H2SO4 solution which was oxygenated for 1 hour giving a 0.9 mM concentration of oxygen.22,23 Through inspection of these figures, a clear peak is observed for the ORR. An onset potential of ca. −0.22, −0.30 and −0.39 and an oxygen reduction peak maxima at ca. −0.51, −0.85 and −1.00 V is observed for EPPG, GC and SPE respectively. All of which are significantly more electronegative than that of the Pt's ORR peak and onset potential of +0.46 and +0.13 V respectively. The lack of an observable oxygen reduction peak for the BDD electrode (whilst using an acidic electrolyte) corresponds with previous literature.22 Yano et al.54 suggest that for the ORR to be initiated at a BDD electrode it must first undergo a pre-treatment step at +1.4 V vs. (Ag/AgCl).22 This pre-treatment step serves to oxidise the sp2 hybridised carbon species, the likely location for the sp2 species being the grain boundaries of the sp3 diamond structure.19 The oxidised sp2 species subsequently mediate the ORR.
 | ||
| Fig. 1 (A) LSVs of bare/unmodified EPPG, GC, SPE, BDD and Pt electrodes showing signals corresponding to the ORR. (B) LSVs recorded using 1524 ng cm−2 2D-MoS2 modified EPPG, GC, SPE, BDD and Pt electrodes showing the position of ORR peaks. In all cases; scan rate: 25 mV−1 s−1 (vs. SCE) and a solution composition of 0.1 M H2SO4 which is oxygen saturated. | ||
Fig. 1(B) shows LSV's of BDD, EPPG, GC, SPE and Pt (all of which had been modified with 1524 ng cm−2 of 2D-MoS2). Inspection of this figure reveals that there is a significant positive shift in the ORR onset to ca. +0.1 V for all of the carbon electrodes utilised. There is a corresponding decrease in the observed oxygen reduction peak potentials by ca. 0.25, 0.39 and 0.82 V for EPPG, GC and SPEs respectively compared to their bare/unmodified counterparts. For the case of the BDD, this is now able to reduce oxygen at −0.29 V, which is comparable with the three other carbon based electrodes utilised. The SPEs exhibit the least electronegative ORR peak potential of −0.16 V. Clearly, the immobilisation of 2D-MoS2 onto the chosen carbon based electrodes significantly reduces the overpotential for the ORR to occur when compared against the bare/unmodified electrodes. Thus, there has been a reduction in the reaction's activation energy to a potential that is closer to the value obtained at the unmodified Pt electrode (ca. +0.46 V). The above data implies that 2D-MoS2 is an effective electrocatalyst for the ORR when electrically wired with various carbon based electrodes.
3.3 Electrocatalytic activity of 2D-MoS2 towards the ORR at differing coverages
Previous work utilising 2D-MoS2 as an electrocatalyst for the HER revealed that there is an optimal immobilised mass, where the structure of said material has the highest ratio of active edge planes to comparatively inert basal planes.1 We therefore investigate the effect of altering the immobilised mass of 2D-MoS2 onto the carbon based electrodes towards the ORR. Fig. 2 shows the peak positions of the ORR (black circles) using LSV (25 mV−1 s−1vs. SCE) in 0.1 M H2SO4 for BDD, EPPG, GC and SPEs following modification with 0, 252, 504, 762, 1009, 1267, 1524, 1771, 2009, 2261 and 2533 ng cm−2 of 2D-MoS2. It is evident from inspection of Fig. 2 that there is a trend of a decreasing ORR reduction peak position associated with an increase in the mass of 2D-MoS2 immobilised onto each of the electrode surfaces utilised. The EPPGs, GCs and SPEs modified with 256 and 504 ng cm−2 of 2D-MoS2 experienced a dramatic decrease in the ORR peak potential from ca. −0.46, −0.59 and −0.85 V for the bare/unmodified to ca. −0.23, −0.25 and −0.32 V for the modified (with 504 ng cm−2 2D-MoS2) electrodes respectively. Subsequent increases in the mass of 2D-MoS2 immobilisation resulted in minor reductions of the ORR peak position, which was incrementally reduced to −0.16, −0.15 and −0.2 V by 2533 ng cm−2 of 2D-MoS2 for the EPPG, GC and SPE respectively. Interestingly, no ORR peak was observable for modifications less than 1009 ng cm−2 on the BDD electrode, whilst a BDD modified with 1009 ng cm−2 of 2D-MoS2 had a ORR peak potential of ca. −0.37 V. As with the other carbon based electrodes, the ORR peak potential was incrementally reduced to −0.23 V by 2533 ng cm−2 of 2D-MoS2 modification on BDD. | ||
| Fig. 2 ORR peak positions (black circles, left Y axis) taken from LSV, the ORR onset potential (blue triangles, left Y axis) and the number of electrons involved in the reaction mechanism (red squares, right Y axis) for 0, 252, 504, 762, 1009, 1267, 1524, 1771, 2018, 2261 and 2533 ng cm−2 of 2D-MoS2 deposited onto the following electrodes: (A) BDD, (B) EPPG, (C) GC and (D) SPE. Error bars are the standard deviation of 3 replicates. In all cases; scan rate: 25 mV−1 s−1 (vs. SCE) and a solution composition of 0.1 M H2SO4 which is oxygen saturated. | ||
With respect to the ORR onset, Fig. 2 (blue triangles) implies that for the bare/unmodified electrodes, the SPE has the most electronegative ORR onset potential at −0.54 V, closely followed by GC at −0.4 V. EPPG has the least negative thus the most favourable ORR onset potential at −0.1 V whilst (as mentioned previously) the ORR does not occur at a bare/unmodified BDD. EPPG, GC and SPE electrodes all have a positive shift in their ORR potential with increased mass deposition of 2D-MoS2 until at 1009 ng cm−2, where the ORR onset potential is +0.1 V in all cases: after which the onset potential remains unchanged until the final mass of modification of 2533 ng cm−2. This demonstrates that after 2D-MoS2 has been electrically wired via immobilisation onto a carbon based electrode, the kinetics of the supporting electrode itself has little effect upon the ORR onset potential, particularly after complete coverage of the surface at 1009 ng cm−2. This work suggests that the ORR onset potential is solely determined via the mass of 2D-MoS2 deposited until complete coverage, thus the response of +0.1 V which is likely that solely of 2D-MoS2.
It is apparent from the above discussion (and inspection of Fig. 2) that the observed increase in the catalytic performance of a given modified electrode material (which corresponds to the addition of 2D-MoS2) begins to plateau, after which further additions of our target material result in increasingly smaller improvements to the electrochemical performance. This ‘critical mass’ of modification is likely due to either achieving complete coverage of the given underlying electrode material or that the structural model of the 2D-MoS2 immobilised upon the electrode/support surface is that of reassembly, whereby few layer MoS2 alters to a bulk morphology. Forming bulk MoS2 would result in the exposure of less edge planes in proportion to basal planes and consequently mitigate the beneficial electrochemical properties of single-, few-, quasi-2D-MoS2 nanosheets. Alternatively, this plateau could signify the mass (a critical mass of ca. 504 ng cm−2 of 2D-MoS2 for the ORR peak potential and 1009 ng cm−2 for the ORR onset potential)1 at which the structure of MoS2 can no longer structurally support itself upon the electrode surface (becoming unstable due to the quantity/mass present) and delaminates thereby eliminating the catalytic benefits of additional 2D-MoS2 immobilisation, which does not adhere to the electrode's surface throughout the course of the experiment. Similar observations have been reported for the case of graphene.47,55–57 This is likely not the case here however as we do not see a reduction in the performance of the modified electrodes. Trying to visually assess the extent of 2D-MoS2 coverage on the surface of an SPE and any subsequent possible SEM analysis was found to be inconclusive as the 2D-MoS2 proved to be indistinguishable from the SPE surface.
The intra-repeatability of the modified and bare/unmodified SPEs was tested (N = 3) and can be observed in Fig. 2(D). The % Relative Standard Deviation (% RSD) in the ORR peak position was found to diminish with a greater mass of 2D-MoS2 immobilised onto the SPE's surface. The % RSD decrease with greater modifications of 2D-MoS2 confirms that the observed plateauing is not due to the delamination of the 2D-MoS2 from the electrode's surface, as this would likely result in increasing % RSDs with increased mass of modification. The observed plateauing effect is therefore likely a result of the 2D-MoS2 reassembling to a stable bulk structure and in doing so exposing less reactive edge planes (or this quantity remaining constant).
The coverage effect reported above is interesting, as such we next consider whether the responses observed are strictly due to the electronic properties of the 2D-MoS2 (and are solely diffusional in nature) or if thin-layer effects are present and complicating the interpretation. The diffusion layer thickness, δ, can be estimated using  where D is the diffusional coefficient (2.0 × 10−5 cm2 s−1), ΔE is the potential width and v is scan rate.58 At a scan rate of 25 mV−1 s−1 the diffusion layer thickness is ∼44 microns, which is significantly larger than that of the 2D-MoS2 surface roughness. Additionally, scan rate studies were performed on the full range of herein utilised 2D-MoS2 modified electrodes, where the voltammetric peak height (Ip) was monitored as a function of scan rate (v), with a plot of peak height versus square-root of the scan rate revealing clear linear trends and resultantly indicating diffusional processes. Furthermore, as is expected for the case of the semi-infinite diffusion model as governed by the Randles–Ševćik equation, analysis of logIpversus logv revealed gradients of no greater than ca. 0.52 in all cases, indicating the absence of thin-layer effects (such that the analyte is not trapped within the mesh/framework of the modified electrode) and representing a response that is purely diffusional in each case.47,57 Overall, based on the above data, the voltammetry is not dominated by the 2D-MoS2 layer and rather semi-infinite diffusion is in operation.
where D is the diffusional coefficient (2.0 × 10−5 cm2 s−1), ΔE is the potential width and v is scan rate.58 At a scan rate of 25 mV−1 s−1 the diffusion layer thickness is ∼44 microns, which is significantly larger than that of the 2D-MoS2 surface roughness. Additionally, scan rate studies were performed on the full range of herein utilised 2D-MoS2 modified electrodes, where the voltammetric peak height (Ip) was monitored as a function of scan rate (v), with a plot of peak height versus square-root of the scan rate revealing clear linear trends and resultantly indicating diffusional processes. Furthermore, as is expected for the case of the semi-infinite diffusion model as governed by the Randles–Ševćik equation, analysis of logIpversus logv revealed gradients of no greater than ca. 0.52 in all cases, indicating the absence of thin-layer effects (such that the analyte is not trapped within the mesh/framework of the modified electrode) and representing a response that is purely diffusional in each case.47,57 Overall, based on the above data, the voltammetry is not dominated by the 2D-MoS2 layer and rather semi-infinite diffusion is in operation.
The “critical mass” of 2D-MoS2 describes the mass of 2D-MoS2 immobilisation on a carbon based electrode's surface where optimal catalytic activity is observed and after which the catalytic benefits plateau or diminish with additional masses of 2D-MoS2 immobilisation. The findings presented herein are strongly supported by the results of a previous study which observed a similar correlation between the mass of 2D-MoS2 immobilised onto a carbon electrode substrate and its catalytic activity towards the HER. In this study the critical mass was observed to be ca. 1267 ng cm−2 on SPEs, at which point the HER onset was lowered by 0.29 V.1 The combination of the results presented herein and those of the aforementioned HER study confirms that the electrocatalytic activity of 2D-MoS2 is mass and therefore structure dependent. Future studies reported in literature involving 2D-MoS2 based materials should endeavour to vary the mass utilised in order to deconvolute its optimum electrocatalytic activity. It also proves that 2D-MoS2 is a promising catalyst that could be utilised to increase the efficiency and energy output of hydrogen fuel cells, thereby making them a more viable alternative to FF combustion as a method of energy generation.
3.4 Tafel assessment of the reaction pathway mechanism
It is evident from above that immobilisation of 2D-MoS2 onto a carbon based electrode substrate reduces the ORR onset and peak potential. Next, consideration was given to the question of whether 2D-MoS2, once immobilised onto the carbon based electrodes, demonstrated preferential selectivity for the ORR to occur via the desirable 4 electron pathway (producing H2O) or the 2 electron pathway (producing H2O2, which is detrimental to PEM fuel cells).19 Tafel analysis is a common approach employed within the literature to deduce the number of electrons involved in the ORR electrochemical mechanism.59Initially, a plot of ln(I) vs. Ep (V) was considered for each of the four carbon based electrodes (see ESI Table 3 and Fig. 11†) and for each mass of 2D-MoS2 modification. This was performed via analysis of the voltammograms depicting the ORR (which were utilised to produce Fig. 2) and using the following equation:59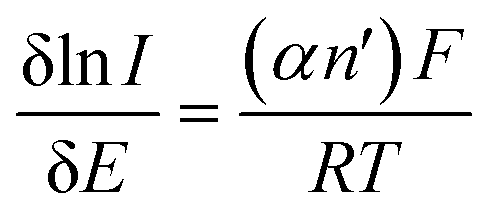 . The slope of the ln(I) vs. Ep (V) plot mentioned above corresponds to δlnI/δEp, where α is the electron transfer coefficient, F is the Faraday constant, n′ is the number of electrons transferred in the rate determining step, R is the gas constant and T is the temperature of the solution temperature in kelvin. Literature has previously suggested that the rate determining step involving the transfer of the first electron is electrochemically irreversible resulting in n′ being 1,60 with αn′ values for SPEs across all masses of modification were deduced. Using these values, the number of electrons involved in the ORR reaction mechanism, n, was deduced using the αn′ calculated from the Tafel equation (see above) and the Randles–Ševćik equation for an irreversible electrochemical process, seen below:61
. The slope of the ln(I) vs. Ep (V) plot mentioned above corresponds to δlnI/δEp, where α is the electron transfer coefficient, F is the Faraday constant, n′ is the number of electrons transferred in the rate determining step, R is the gas constant and T is the temperature of the solution temperature in kelvin. Literature has previously suggested that the rate determining step involving the transfer of the first electron is electrochemically irreversible resulting in n′ being 1,60 with αn′ values for SPEs across all masses of modification were deduced. Using these values, the number of electrons involved in the ORR reaction mechanism, n, was deduced using the αn′ calculated from the Tafel equation (see above) and the Randles–Ševćik equation for an irreversible electrochemical process, seen below:61
| IIrrevP = ±0.496(αn′)1/2nFAC(FDv/RT)1/2 |
Note, of the carbon based electrodes utilised within this study, GC had the lowest number of electrons involved in its ORR reaction mechanism, this raises the question of why it is the commonly used electrode within the literature as it is clearly the least effective at enabling the desirable 4 electron ORR mechanism. Future studies should use a range of bare/unmodified carbon based electrodes, which exhibit different HET kinetics resulting in unique interactions between the supporting carbon based electrode and any deposited material, breaking from the convention of solely using GC; this will help establish the true electrocatalytic activity of a given material.
SPEs show the highest initial n involved in the ORR reaction mechanism at 2.67 for a bare/unmodified electrode (this corresponds to the literature).60 From 256 to 1009 ng cm−2 of 2D-MoS2 immobilised on a SPE's surface there is an increase in the n involved in the ORR mechanism to 3.96. Greater than 1009 ng cm−2 masses of 2D-MoS2 modification result in a decrease in the n involved until n is 2.64 at 2533 ng cm−2. Unlike BDD, EPPG and GC electrodes, it is clear that 2D-MoS2, once deposited onto a SPE, not only results in a significant decrease in the ORR onset and peak position but also in a beneficial change in the ORR reaction mechanism from ca. 2 to a 4 electron process. Indicating that the major product of the ORR is the desired H2O and not the detrimental H2O2. The reason for 2D-MoS2 altering the n involved for SPE and not for BDD, EPPG and GC is likely due to the SPEs having “rougher” surfaces, resulting in the 2D-MoS2 (once deposited) exhibiting structural/electronic orientations not capable on the “smoother” surface of BDD, EPPG and GC.60
A comparison was made between the surface topography of BDD, EPPG, GC and SPE using white light profilometry (a ZeGage 3D Optical Surface Profiler, produced by Zygo, was utilised for this). The surface of a SPE was observed to be significantly rougher, with a root mean squared value of the heights over the whole surface (SQ) of 1904.9 nm, than that of BDD, EPPG and GC which had values of 7.5, 26.1 and 15.9 nm respectively (see ESI Fig. 12†). Next, it was necessary to determine whether the SPEs greater roughness resulted in a greater exposure of 2D-MoS2. This was determined via an evaluation of the roughness factors (RF), for BDD, EPPG, GC and SPE modified with 0, 256, 1009 and 2018 ng cm−2 of 2D-MoS2. In order to deduce RF values which are representative of the true electrochemical area of an electrode, a double layer capacitance technique can be employed (the methodology of which can be seen in ESI† section 1.3 “Roughness factor calculations”). ESI Table 2† clearly shows that SPEs have significantly larger RF values at every mass of 2D-MoS2 modification; for example at 2D-MoS2 2018 ng cm−2 the RF for SPE is 37 whereas the RF value for BDD, EPPG and GC is 13.5, 2 and 6.4 respectively. Given the topographic roughness and the RF values determined above we suggest that the correlation between an underlying substrate's roughness and the ability of immobilised 2D-MoS2 to electrocatalyse the ORR via a 4 electron process is likely a result of the structural/electronic orientations which occur for 2D-MoS2 when it is immobilised on a rough surface. This is further supported by ESI Fig. 13 and 14.† ESI Fig. 13† shows SEM images of; (A) the surface of a typical SPE and (B) the surface of an SPE which has been polished. ESI Fig. 13(B)† can visually be seen to be smoother than that of ESI Fig. 13(A).†60,64 ESI Fig. 14† shows that an SPE following being polished has a significantly smaller SQ value of 593 nm compared to that of 1905 nm for an unpolished SPE, indicating post polishing, the SPE's surface is considerably smoother. ESI Fig. 14(B) and (D)† show that the surface of an SPE becomes smoother post 1009 ng cm−2 of 2D-MoS2 immobilisation. When 1009 ng cm−2 of 2D-MoS2 is deposited upon the polished SPE's surface the RF value obtained is 13.5 (see ESI Table 2†) and allowed the ORR to occur via a 3.4 electron pathway; both of which are significantly less than that of the unpolished (rougher) alternative. We infer that the increased catalytic behaviours, observed for a rougher surface electrode are due to the unique structural/electronic orientations which are formed once 2D-MoS2 is immobilised onto an SPE. Resulting in an exposure of larger numbers of active edge plane sites/edge plane-like defects than their BDD, EPPG and GC counterparts, thereby, offering a greater catalytic prospective. Future studies should consider which supporting material they employ as the results observed above show that this has a significant effect upon the deposited material's structure and electron transfer kinetics.
Whilst other studies have managed to produce a 4 electron pathway using alkaline conditions, (such as Suresh et al.35) we believe, given that all previous studies utilising MoS2 materials towards the ORR are shown in Table 1, that this report is the first to observe the ORR occur via the 4 electron pathway (thus producing H2O rather than H2O2) in acidic conditions using an 2D-MoS2 based electrocatalytic material on a carbon based substrate (SPEs). Clearly, these results are of significant importance as it is acidic conditions found within a PEM fuel cell, thusly making the results of this study highly applicable to real world industry.
This work clearly indicates that there is an optimal/critical mass, which we determine to be ca. 1009 ng cm−2 for SPEs, whereby there is the largest average n (4) involved in the ORR reaction mechanism as well as a significant improvement in the ORR onset and peak potential. Subsequent studies within the literature which use 2D-MoS2 should consider using a range of differing loadings/modifications in order to deconvolute the true/optimal electrocatalytic performance of a given electrocatalyst. The findings of this study have clear implications that are applicable when using any 2D material.
4. Conclusions
This study sought to break from the conventions found within the literature when 2D-MoS2 materials are explored towards the ORR; of solely using GC as a supporting electrode, using only one mass of the electrocatalytic material to modify the supporting electrode and using KOH as the electrolyte.Our investigations implemented a range of diligent control experiments. Rather than solely using GC as a supporting electrode we employed BDD, EPPG, GC and SPE's. The ORR onset was reduced to ca. +0.1 V for EPPG, GC and SPEs at a 2D-MoS2 1524 ng cm−2 modification, which is far closer to Pt at +0.46 V compared to the bare/unmodified EPPG, GC and SPE counterparts. BDD was observed to have an ORR onset potential of −0.03 V at 2D-MoS2 1524 ng cm−2 modification. Using a range of 2D-MoS2 modification masses (rather than one set mass) allowed us to observe that a critical/optimal mass of 2D-MoS2 existed (in this case ca. 1009 ng cm2). At this critical mass, there is optimal catalytic activity, after which the catalytic benefits plateau with additional masses of 2D-MoS2 immobilisation. This is likely a result of the structure of 2D-MoS2 at the critical mass exposing the largest ratio of electroactive edge planes, after which the structure is that of bulk MoS2. 0.1 M H2SO4 was utilised as an electrolyte for all the experiments described herein, unlike previous studies which used KOH. Performing the experiments in an acidic electrolyte resembles the conditions that PEM fuel cells operate, making the observations presented herein highly applicable to industry.
SPEs were the only carbon based electrode found to allow the ORR to occur via the desirable 4 electron pathway (producing H2O rather than H2O2) at 2D-MoS2 (ca. 1009 ng cm2). This is likely as a result of the structurally rougher SPE surfaces allowing for unique 2D-MoS2 structural/electronic orientations, where larger numbers of active edge planes are exposed, which are not possible on the “smoother” BDD, EPPG and GC electrodes. Whilst other reports have managed to produce a 4 electron process, we believe that this report is the first to observe the ORR to occur via a 4 electron process in acidic conditions using a 2D-MoS2 based electrocatalyst material on a carbon based substrate. There is no reason why the findings of this study would not be applicable to other 2D materials, this opens up new avenues of research where the surface roughness of a supporting electrode could be altered, allowing 2D materials to exhibit unique and unreported structural/electronic orientations and electrochemical behaviours.
By straying from these literature conventions we de-convoluted the true electrochemical behaviour of 2D-MoS2, which was shown to be an effective electrocatalyst towards the ORR. We also revealed modified SPEs as a valid alternative to GC for research purposes and for Pt in real world fuel cell applications. SPEs are significantly cheaper, adaptable and mass producible when compared to Pt and other carbon based electrodes examined herein, whilst upon modification with an optimal mass of 2D-MoS2, exhibit preferential electrocatalytic activity towards the ORR.
Acknowledgements
The authors acknowledge a British Council Institutional Link grant (no. 172726574) for the support of this research. D. A. C. Brownson acknowledges funding from the Ramsay Memorial Fellowships Trust.References
- S. J. Rowley-Neale, D. A. C. Brownson, G. C. Smith, D. A. G. Sawtell, P. J. Kelly and C. E. Banks, Nanoscale, 2015, 7, 18152–18168 RSC.
- A. J. López-Menéndez, R. Pérez and B. Moreno, J. Environ. Manage., 2014, 145, 368–373 CrossRef PubMed.
- Y. Zhao, Y. Zhang, Z. Yang, Y. Yan and K. Sun, Sci. Technol. Adv. Mater., 2013, 14 Search PubMed.
- T. Wang, L. Liu, Z. Zhu, P. Papakonstantinou, J. Hu, H. Liu and M. Li, Energy Environ. Sci., 2013, 6, 625–633 CAS.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed.
- J. P. Barton and D. G. Infield, IEEE Trans. Energy Convers., 2004, 19, 441–448 CrossRef.
- F. Blaabjerg, Z. Chen and S. B. Kjaer, IEEE Trans. Power Electron., 2004, 19, 1184–1194 CrossRef.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. D. M. G. Schultz, G. P. Brasseur and W. Zittel, Science, 2003, 302, 624–627 CrossRef PubMed.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- M. Jouin, R. Gouriveau, D. Hissel, M. C. Péra and N. Zerhouni, IFAC-PapersOnLine, 2015, 48, 26–31 CrossRef.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- M.-R. Gao, J.-X. Liang, Y.-R. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed.
- Y. Liu, X. Yue, K. Li, J. Qiao, D. P. Wilkinson and J. Zhang, Coord. Chem. Rev., 2016, 315, 153–177 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang, Y.-R. Zheng and S.-H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- M. Gara and R. C. Compton, New J. Chem., 2011, 35, 2647–2652 RSC.
- K. Uosaki, G. Elumalai, H. Noguchi, T. Masuda, A. Lyalin, A. Nakayama and T. Taketsugu, J. Am. Chem. Soc., 2014, 136, 6542–6545 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755–B769 CrossRef CAS.
- M. Gara and R. G. Compton, New J. Chem., 2011, 35, 2647–2652 RSC.
- E. P. Randviir and C. E. Banks, Electroanalysis, 2014, 26, 76–83 CrossRef CAS.
- K. K. Ravindra, L. L. Bencs and R. V. Grieken, Sci. Total Environ., 2004, 318, 1–43 CrossRef CAS PubMed.
- M.-R. Gao, S. Liu, J. Jiang, C.-H. Cui, W.-T. Yao and S.-T. Yu, A, 2010, 20, 9355–9361 CAS.
- E. P. Randviir, D. A. C. Brownson, M. Gomez-Mingot, D. K. Kampouris, J. Iniesta and C. E. Banks, Nanoscale, 2012, 4, 6470–6480 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- C. Ataca, M. Topsakal, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2011, 115, 16354–16361 CAS.
- N. A. Dhas and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 2368–2369 CrossRef CAS PubMed.
- F. Cesano, S. Bertarione, A. Piovano, G. Agostini, M. M. Rahman, E. Groppo, F. Bonino, D. Scarano, C. Lamberti, S. Bordiga, L. Montanari, L. Bonoldi, R. Millini and A. Zecchina, Catal. Sci. Technol., 2011, 1, 123–136 CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- S. Ji, Z. Yang, C. Zhang, Z. Liu, W. W. Tjiu, I. Y. Phang, Z. Zhang, J. Pan and T. Liu, Electrochim. Acta, 2013, 109, 269–275 CrossRef CAS.
- S. S. Chou, M. De, J. Kim, S. Byun, C. Dykstra, J. Yu, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2013, 135, 4584–4587 CrossRef CAS PubMed.
- H. Huang, X. Feng, C. Du, S. Wu and W. Song, J. Mater. Chem. A, 2015, 3, 16050–16056 CAS.
- C. Suresh, S. Mutyala and J. Mathiyarasu, Mater. Lett., 2016, 164, 417–420 CrossRef CAS.
- C. Médard, M. Lefèvre, J. P. Dodelet, F. Jaouen and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202–3213 CrossRef.
- N. A. Choudry, D. K. Kampouris, R. O. Kadara and C. E. Banks, Electrochem. Commun., 2010, 12, 6–9 CrossRef CAS.
- L. R. Cumba, J. P. Smith, D. A. C. Brownson, J. Iniesta, J. P. Metters, D. R. D. Carmo and C. E. Banks, Analyst, 2015, 140, 1543–1550 RSC.
- C. W. Foster, J. Pillay, J. P. Metters and C. E. Banks, Sensors, 2014, 14, 21905–21922 CrossRef CAS PubMed.
- C. W. Foster, J. P. Metters and C. E. Banks, Electroanalysis, 2013, 25, 2275–2282 CAS.
- J. P. Metters, M. Gomez-Mingot, J. Iniesta, R. O. Kadara and C. E. Banks, Sens. Actuators, B, 2013, 177, 1043–1052 CrossRef CAS.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- J. P. Smith, J. P. Metters, O. I. G. Khreit, O. B. Sutcliffe and C. E. Banks, Anal. Chem., 2014, 86, 9985–9992 CrossRef CAS PubMed.
- O. Ramdani, J. P. Metters, L. C. S. Figueiredo-Filho, O. Fatibello-Filho and C. E. Banks, Analyst, 2013, 138, 1053–1059 RSC.
- J. P. Smith, J. P. Metters, D. K. Kampouris, C. Lledo-Fernandez, O. B. Sutcliffe and C. E. Banks, Analyst, 2013, 138, 6185–6191 RSC.
- Graphene Supermarket, https://graphene-supermarket.com/MoS2-Pristine-Flakes-in-Solution.html, accessed 24/03/2016.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944–6976 RSC.
- P. C. Joensen, E. D. Crozier, N. Alberding and R. F. Frindt, J. Phys. C: Solid State Phys., 1987, 20, 4043–4053 CrossRef CAS.
- Innovation in Surface Spectroscopy and Microscopy Systems, http://www.specs.de/cms/front_content.php?idcat=209, accessed 20/03/2016.
- M. P. Seah and S. J. Spencer, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 178–181 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- CasaXPS, http://www.casaxps.com/, accessed 2/02/2016.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- T. Yano, E. Popa, D. A. Tryk, K. Hashimoto and A. Fujishima, J. Electrochem. Soc., 1999, 146, 1081–1087 CrossRef CAS.
- D. A. C. Brownson, A. C. Lacombe, D. K. Kampouris and C. E. Banks, Analyst, 2012, 137, 420–423 RSC.
- D. A. C. Brownson, L. C. S. Figueiredo-Filho, X. Ji, M. Gomez-Mingot, J. Iniesta, O. Fatibello-Filho, D. K. Kampouris and C. E. Banks, J. Mater. Chem. A, 2013, 1, 5962–5972 CAS.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- C. E. Banks and R. G. Compton, Understanding voltammetry, Imperial College Press, London, 2007 Search PubMed.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, V. Radmilovic, N. M. Markovic and P. N. Ross, J. Phys. Chem. B, 2002, 106, 4181–4191 CrossRef CAS.
- E. P. Randviir and C. E. Banks, Electroanalysis, 2014, 26, 76–83 CrossRef CAS.
- D. A. C. Brownson and C. E. Banks, The Handbook of Graphene Electrochemistry, Springer, London, 2014 Search PubMed.
- T. Kaskiala, Miner. Eng., 2002, 15, 853–857 CrossRef CAS.
- P. Han and D. M. Bartels, J. Phys. Chem., 1996, 100, 5597–5602 CrossRef CAS.
- L. R. Cumba, C. W. Foster, D. A. C. Brownson, J. P. Smith, J. Iniesta, B. Thakur, D. R. D. Carmo and C. E. Banks, Analyst, 2016, 141, 2791–2799 RSC.
- R. Illathvalappil, S. M. Unni and S. Kurungot, Nanoscale, 2015, 7, 16729–16736 RSC.
- J. Zhou, H. Xiao, B. Zhou, F. Huang, S. Zhou, W. Xiao and D. Wang, Appl. Surf. Sci., 2015, 358(Part A), 152–158 CrossRef CAS.
- T. Wang, J. Zhuo, Y. Chen, K. Du, P. Papakonstantinou, Z. Zhu, Y. Shao and M. Li, ChemCatChem, 2014, 6, 1877–1881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr04073j |
| This journal is © The Royal Society of Chemistry 2016 |