
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Smart surface-enhanced Raman scattering traceable drug delivery systems†
Lei
Liu
a,
Yonghong
Tang
b,
Sheng
Dai
*a,
Freddy
Kleitz
c and
Shi Zhang
Qiao
*a
aSchool of Chemical Engineering, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: s.qiao@adelaide.edu.au; s.dai@adelaide.edu.au
bCentre for Nano Scale Science and Technology, School of Computer Science, Engineering, and Mathematics, Flinders University, Adelaide, SA 5042, Australia
cDepartment of Chemistry and Centre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, Quebec City, QC G1 V 0A6, Canada
First published on 30th May 2016
Abstract
A novel smart nanoparticle-based system has been developed for tracking intracellular drug delivery through surface-enhanced Raman scattering (SERS). This new drug delivery system (DDS) shows targeted cytotoxicity towards cancer cells via pH-cleavable covalent carboxylic hydrazone links and the SERS tracing capability based on gold@silica nanocarriers. Doxorubicin, as a model anticancer drug, was employed to compare SERS with conventional fluorescence tracing approaches. It is evident that SERS demonstrates higher sensitivity and resolution, revealing intracellular details, as the strengths of the original Raman signals can be amplified by SERS. Importantly, non-destructive SERS will provide the designed DDS with great autonomy and potential to study the dynamic procedures of non-fluorescent drug delivery into living cells.
Introduction
Most of the small-molecule anticancer drugs have potent chemotherapeutic characteristics, but their therapeutic efficacy is often limited by severe side effects caused by systemic toxicity and rapid resistance or aggregation issues caused by poor solubility, low stability and unsuitable pharmacokinetic profiles as well as the lack of selectivity.1,2 For the improvement of the practical and clinical performances of drugs to combat human cancer, nanoparticle-based drug delivery systems (DDSs) have, over the recent years, drawn worldwide attention in the field of anticancer research. For example, impressive efforts have been made to construct physical feature-controlled,3,4 highly biocompatible5 and multifunctional6 DDSs. Among those nanoparticles applied for drug delivery, silica nanoparticles (SNs) possess the excellent properties of high stability against chemical, mechanical and thermal changes, as well as high biocompatibility and straightforward surface functionalisation,7–9 thus they are considered as an ideal platform for functional DDSs. Unlike physical adsorption and diffusion, which may lead to premature, burst drug release and ultimately severe side effects, smart covalent linkages between the drug and carrier, especially carboxylic hydrazone links,10–12 could bring effective impacts on sustainable drug release and reduce side effects, making the development of such smart DDSs a high priority.When it comes to tracking the intracellular performance of DDSs, the conventional widely implemented approach is fluorescence evaluation. However, it has certain inevitable limitations. In general, DDSs require artificial labelling, which might change physicochemical properties, uptake pathways and pharmacological effects of the loaded drugs. Moreover, photo-bleaching fluorophores and the fluorescent background interruptions from cells tend to limit long-term tracing.13 As the characteristic peak in the fluorescence spectra is very broad, it raises the difficulty of distinguishing overlapping spectra from multi-fluorescence labelled tracing systems.14 Conversely, surface-enhanced Raman scattering (SERS) has been introduced into life science research,14–16 where these fluorescence barriers could be mitigated and crossed. The results of SERS tracing demonstrate high sensitivity and resolution features, as the strength of the original Raman peak can be theoretically enhanced to 8–11 orders of magnitude by SERS.17–19 Enhancement of fingerprint Raman signals (10–100 times narrower than fluorescence14) can materialise the navigation pathway of a single molecule from samples, cells, or even intracellular compartments. However, only limited work has been performed in the area of SERS-tracking drug delivery. For instance, single metal nanoparticles with a silica coating of size larger than 100 nm were reported,20 as this size of noble metal particles is acceptable for SERS tracing. However, clusters of small (5–10 nm) metal nanoparticles can also form hot spots in the junctions between them to enhance Raman signals,16 which could make them suitable to replace larger single nanoparticles as the most effective SERS traceable agents. However, to date, DDSs based on small metal nanoparticle-induced SERS have remained largely unexplored.
Herein, a smart SERS traceable drug delivery system (DDS) has been constructed, in which the drug-carrier linkages are pH-cleavable. Silica nanoparticles (SNs) are viewed as the framework, and small exposed gold particles with SERS reporters are settled on SNs. Owing to the covalent pH-cleavable drug-carrier links, the chosen anticancer drug (DOX) will remain conjugated into the DDS to avoid premature and uncontrolled drug leakage in healthy cell regions. As shown in Scheme 1, when delivered to cancer cells, the chemical links of the DDS will be cleaved by the more acidic surroundings of the cancer cells, and DOX will be dissociated and released into the cells to perform its pharmacological purpose. Therefore, this smart DDS design will offer targeted cytotoxicity while being efficiently tracked by SERS.
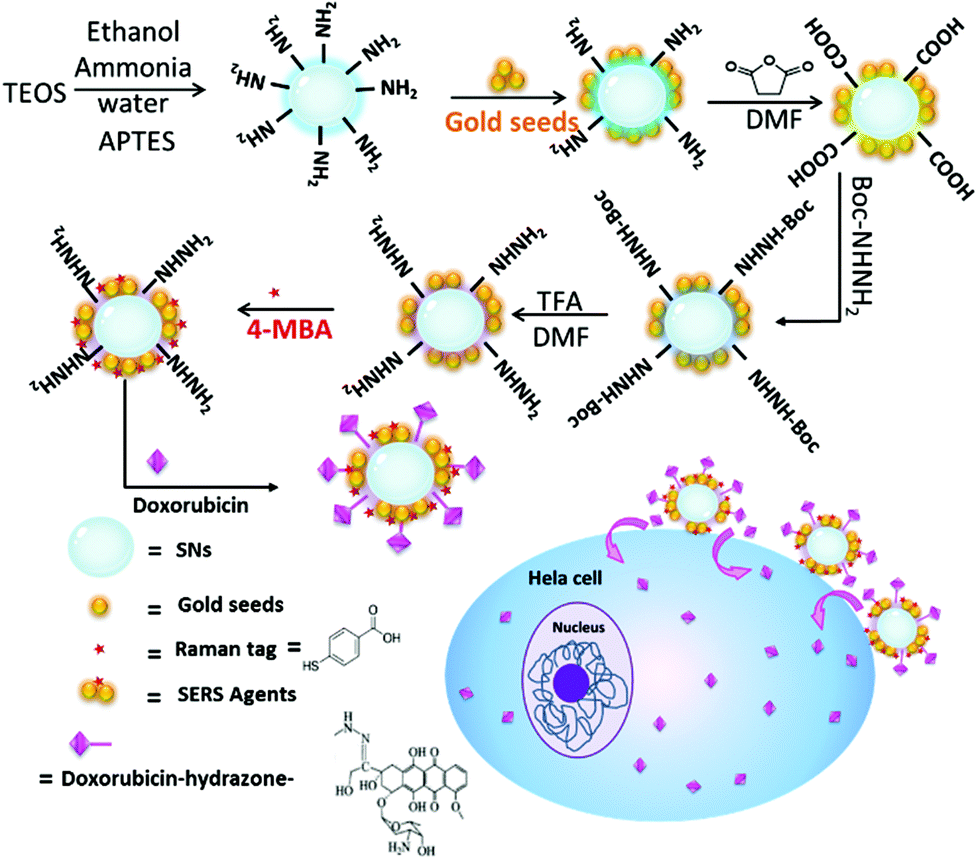 | ||
| Scheme 1 Schematic diagram representing the synthesis of doxorubicin-conjugated SERS nanocomposites and the pathway of intracellular drug release. | ||
Experimental
Chemicals
Fetal bovine serum (FBS), Dulbecco's modified eagle medium (DMEM), pH = 7.4 phosphate buffered saline (PBS), antibiotic-antimycotic (Anti-anti, 100×), 0.25% (w/v) trypsin–0.03% (w/v) ethylenediaminetetraacetic acid (EDTA) solution, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and LIVE/DEAD® viability/cytotoxicity kit (calcein-AM/ethidium homodimer-1) were purchased from Life Technologies Australia Pty Ltd. Doxorubicin hydrochloride (DOX), which is selected as a model drug for cancer therapy, was purchased from Beijing Huafeng United Technology Co Ltd. Gold(III) chloride trihydrate (HAuCl4·3H2O) and aqueous ammonia (NH4OH, 25%) were purchased from Chem-Supply Australia Pty Ltd. Sodium borohydride (NaBH4), N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 4-mercaptobenzoic acid (4-MBA), tetraethyl orthosilicate (TEOS, ≥98%), (3-aminopropyl)triethoxysilane (APTES, ≥98%), sodium citrate (Na3Ct), trifluoroacetic acid (TFA), N,N-dimethylformamide (DMF), tert-butyl carbazate (Boc-NHNH2), succinic anhydride, dimethyl sulfoxide (DMSO) and other chemicals were purchased from Sigma Aldrich. All materials were of analytical grade and used as received without further purification. Millipore water was obtained from a three-stage Millipore Mill-Q plus 185 purified cation system (Academic) with a resistivity higher than 18.2 MΩ cm.Characterisation
Scanning electron microscopy (SEM) observations were carried out on an FEI Quanta 450 FEG environmental emission scanning electron microscope operated at 10 kV. A 5 nm platinum layer coating by ion sputtering was required for each specimen before SEM observation. For obtaining transmission electron microscopy (TEM) images, an FEI Tecnai G2 Spirit transmission electron microscope at an acceleration voltage of 120 kV was employed. Fourier transform infrared (FTIR) spectra were recorded at room temperature on a Thermo Scientific NICOLET 6700 FTIR spectrometer. The hydrodynamic sizes and zeta-potentials of different products dispersed in deionized water were determined at room temperature by using a Malvern Zeta-sizer Nano ZS (Malvern Inst. Ltd, UK) equipment. The concentration of particles in water was kept at 0.1 ± 0.05 mg mL−1 for particle size analysis, while the zeta potential results were calculated based on the statistical analysis of 10 parallel runs. The pH values were measured by using a pH meter (EL20, METTLER TOLEDO). UV-Vis absorption spectra were recorded on a UV-2600 spectrophotometer (Shimadzu Corporation). X-ray diffraction (XRD) patterns were obtained on a powder X-ray diffractometer at 40 kV and 15 mA using Co-Kα radiation (Miniflex, Rigaku). In addition, an Axis Ultra (Kratos Analytical, UK) XPS spectrometer equipped with an Al Kα source (1486.6 eV) was used for X-ray photoelectron spectrum (XPS) collection. Fluorescence excitation and emission images were obtained on a ZEISS Axio Vert. A1 inverted microscope equipped with 488 nm and 530 nm lasers. A HORIBA Lab RAM HR Evolution equipped with a 785 nm laser was employed for obtaining Raman spectra and SERS mapping images.Synthetic procedures of SN-NH2
The SN-NH2 were fabricated by a modified direct Stöber approach.21 In a typical synthesis run, TEOS (2.3 mL) was quickly added to a mixed solution of 25% aqueous ammonia (3.0 mL), deionized water (1.0 mL), and ethanol (60 mL). The magnetic stirring rate is ca. 800–1000 rpm. The mixture was stirred at 40 °C for 4 h, and then 0.1 mL APTES was added into the system. After 800–1000 rpm stirring for another 2 h at 40 °C and washing with ethanol, the aminated silica nanospheres, labelled as SN-NH2, were obtained.Synthetic procedures of Au–SN-NH2
The gold nanoparticles were prepared by the NaBH4 and Na3Ct reduction method.22,23 1.0% HAuCl4·3H2O (0.1 mL) and deionized water (9 mL) were stirred for 1 min with 800–1000 rpm stirring rate at room temperature, and then 38.8 mM Na3Ct (0.2 mL) was added into the system. After adding 0.075% NaBH4 (0.1 mL), gold nanoparticles of ca. 5–10 nm were synthesised. Subsequently, these gold nanoparticles were attached to the SN-NH2 with a gold nanoparticle-to-SN molar ratio of 4, under 30 min sonication in deionized water. The resulting Au–SN-NH2 nanoparticles were centrifuged and then washed with ethanol, and the final precipitates were dried in an oven at 60 °C for 24 h.Synthetic procedures of 4-MBA–Au–SN-NHNH2
As illustrated in Scheme 1, 100 mg Au–SN-NH2 particles were dispersed in DMF (12 mL) by ultrasonic treatment. 900 mg succinic anhydride was dissolved in DMF and then added into the prepared Au–SN-NH2 suspension. After stirring for 6 h at room temperature, the products were centrifuged, washed with ethanol and water and dried to obtain Au–SN-COOH powder. Then, Au–SN-COOH particles were added in DMF (20 mL), and stirred with 54 mg EDC and 32 mg NHS for 1 h at room temperature, before 37 mg Boc-NHNH2 were finally added into the system. The mixture was further stirred at room temperature for another 24 h. The products were centrifuged, washed with ethanol and water and dried to yield Au–SN-NHNH-Boc. To remove the protective Boc group, Au–SN-NHNH-Boc particles were dispersed in a mixture of DMF (10 mL) and TFA (5 mL). The system was stirred at room temperature for 24 h, before the final products Au–SN-NHNH2 were centrifuged, washed with ethanol and dried in an oven at 60 °C. 1 mM 4-MBA was added to the Au–SN-NHNH2 aqueous suspensions with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. After stirring for 24 h at room temperature, the SERS tag 4-MBA was integrated onto the gold nanoparticles. The 4-MBA–Au–SN-NHNH2 nanocarriers were centrifuged and then washed several times with ethanol to remove any unreacted 4-MBA, and the final precipitates were dried in an oven at 60 °C for 24 h.
:100. After stirring for 24 h at room temperature, the SERS tag 4-MBA was integrated onto the gold nanoparticles. The 4-MBA–Au–SN-NHNH2 nanocarriers were centrifuged and then washed several times with ethanol to remove any unreacted 4-MBA, and the final precipitates were dried in an oven at 60 °C for 24 h.
DOX conjugation and in vitro release
Regarding doxorubicin conjugation (Scheme 1), 1.0 mg of dried 4-MBA–Au–SN-NHNH2 nanocarriers was added into ca. 0.5 mg mL−1 DOX/PBS solution, and stirred at 1000 rpm at room temperature for 24 h in a dark environment. After washing with ethanol 3 times, the final 4-MBA–Au–SN–hydrazone–DOX products were collected by centrifugation, washed with standard pH 7.4 PBS and dried in an oven at 60 °C for 24 h. The supernatant of the initial DOX/PBS solution and all the washing supernatants were collected for the evaluation of the DOX conjugation efficiency. The corresponding DOX doses were estimated by using 480 nm UV-vis adsorption values and a standard DOX calibration curve.The release profiles of DOX from 4-MBA–Au–SN–hydrazone–DOX were evaluated independently in three PBS media with different pH values 5.0, 6.0 and 7.4. The pH value of each system was adjusted using acetate buffer and PBS, and measured by using a pH meter. For each release investigation, 5 mg DOX-conjugated particles were added into the buffer solution (5 mL), and continuously stirred with a rate of 100 rpm at 37 °C under dark conditions. 1 mL of release medium was removed at selected time intervals. The supernatant was centrifuged for drug release analysis, and the residual parts together with an identical amount of fresh buffer solution were put back into the system. The released amounts of DOX were analysed by using 480 nm UV-vis adsorption values and a standard DOX calibration curve.
Cytotoxicity studies
The cell viability results obtained by MTT assays against human cervical cancer HeLa cells and human embryonic kidney HEK 293 cells were used to investigate the cytotoxicities of the fabricated nanocarriers 4-MBA–Au–SN-NHNH2 before and after DOX conjugation. Both HeLa and HEK293 cells were incubated in cell culture medium (DMEM, 10% FBS and 1× Anti-anti) at 37 °C in a 5% CO2 humid environment. For MTT assays, cells were seeded into a 96-well plate at a density of 5 × 104 cells per mL, and incubated for 24 h in cell culture medium (0.1 mL). Subsequently, an identical volume of cell culture medium (0.1 mL) containing different particles (nanocarriers, DOX-conjugated products and free DOX) was replaced by the previous medium and further incubated for 24 h. Then, 0.5 mg mL−1 MTT (0.01 mL) was added to each well, and after incubation for 4 h, DMSO (0.15 mL) was used to replace the medium and dissolve formazan crystals. The MTT assays were conducted in quadruplicate for each sample, and the final absorbance of formazan crystals was measured by using an ELx808 Absorbance Microplate Reader (Biotek, USA) at a wavelength of 595 nm.The cytotoxicity results of 4-MBA–Au–SN–hydrazone–DOX were further confirmed by using fluorescence LIVE/DEAD® viability/cytotoxicity studies. HEK293 and HeLa cells were added into a 24-well plate at 5 × 104 cells per mL seeding density, and incubated in cell culture medium (1 mL) for 24 h. Later, 0.2 mg mL−1 DOX-conjugated products in cell culture medium (1 mL) were replaced and incubated for another 24 h. After removal of the medium and PBS washing, the cells were harvested by using EDTA and resuspended in PBS solution. 0.4 mL LIVE/DEAD® viability/cytotoxicity kit (2 μM calcein-AM and 4 μM ethidium homodimer-1) was added in a dark environment, and then incubated for 30 min. The cells were gently washed with PBS to remove excessive dyes. The final cytotoxicity results were checked by using images obtained with a fluorescence microscope.
SERS tracking of 4-MBA–Au–SN–hydrazone–DOX
HeLa cells were seeded at a concentration of 2 × 105 cells per mL with 2 mL cell culture medium per well into a 6-well plate with 22 mm cover-glass slides inside, and incubated for 24 h at 37 °C under 5% CO2. The fresh medium (2 mL) containing 0.4 mg DOX-conjugated products was replaced and incubated for another 4 h. Subsequently, the glass substrate with cells and DOX conjugated products was collected after three times of gentle washing with PBS. The Raman study was conducted with a 785 nm laser and 100× objectives. The area is 30 × 30 μm with a resolution of 961 data points for mapping. The acquisition time for each data point is 1 s.Flow cytometry analysis of 4-MBA–Au–SN–hydrazone–DOX
In terms of flow cytometry analysis, 0.2 mg mL−1 DDSs were dispersed in cell culture medium (DMEM with 10% FBS and 1% PS). HeLa cells were added into a 24-well plate at 2 × 104 cells per mL seeding density, and incubated in 1 mL cell culture medium for 24 h at 37 °C under 5% CO2. Fresh media (1 mL) containing 0.2 mg mL−1 4-MBA–Au–SN–hydrazone–DOX was incubated for different time periods. The cells were harvested by the digestion of EDTA solution, and gently washed with PBS twice. With a 200–600 cells per second flow rate, ca. 1 × 104 cells were analysed by using a FACS Calibur flow cytometer (Becton Dickinson). The testing result of healthy HeLa cells was selected as a negative control, and the red fluorescence intensities of DOX were directly measured by cytometry. The data were analysed using Cell Quest 3.3 software, while one-color flow cytometry was used to evaluate cellular uptake.Results and discussion
Characterization of the smart drug delivery system
The 4-MBA–Au–SN–hydrazone–DOX smart DDS was prepared following the procedure illustrated in Scheme 1. The amino-functionalized silica nanoparticles (SN-NH2) were synthesized in one pot by a modified Stöber method.21 The scanning electron microscopy image (SEM, Fig. 1a) and transmission electron microscopy image (TEM, Fig. 1b) demonstrate that SNs are uniform nanospheres with an average particle diameter of ca. 80–100 nm. The size results are further verified from dynamic light scattering (DLS) measurements (Table S1† and Fig. 2a), where the average hydrodynamic diameter of SN-NH2 is 91 nm with an average zeta potential of +18.3 mV. In the FTIR spectra (Fig. S1†) of SN-NH2, the typical silica Si–O–Si asymmetric stretching, Si–OH and Si–O symmetric stretching vibrations can be observed at around 1066, 945 and 795 cm−1, respectively.24 The –OH stretching vibration at approximately 1635 cm−1 may be the deformation band of physically absorbed H2O. The 3357 cm−1 peak is the signature of N–H stretching vibrations, and N–H bending bands appear around 1500 cm−1. The existence of amino groups is crucial for further anchoring of gold nanoparticles. | ||
| Fig. 1 Electron microscopy images. (a) SEM, (b) TEM images of SN-NH2, (c) gold nanoparticles, and (d) Au–SN-NHNH2. | ||
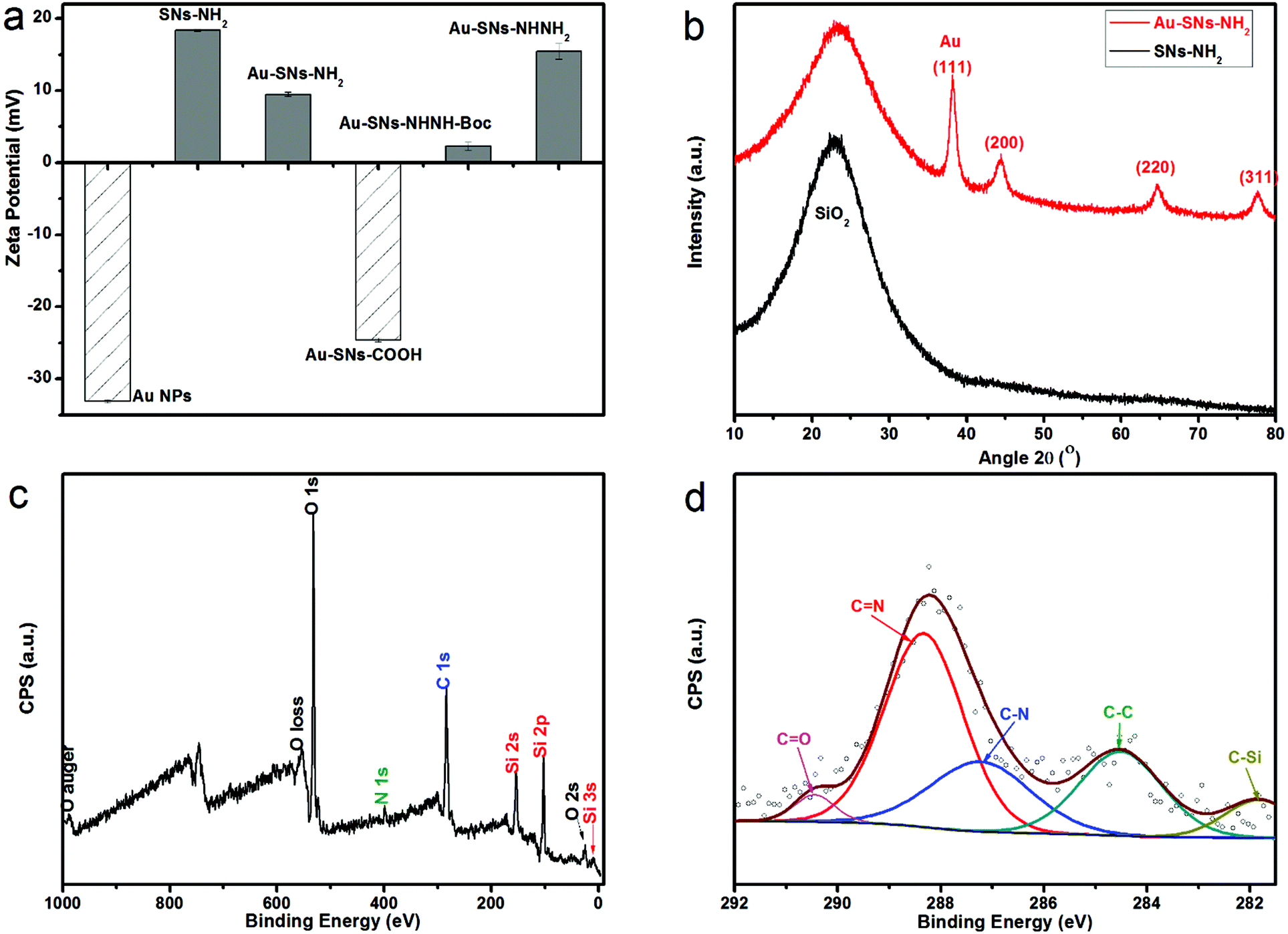 | ||
| Fig. 2 (a) Zeta potentials of different products at different synthesis stages, (b) XRD patterns of SN-NH2 and Au–SN-NH2, (c) XPS spectra of DOX-conjugated composites, and (d) the enlarged C 1s region. | ||
Gold nanoparticles (Au NPs) were synthesised by sodium borohydride reduction and sodium citrate stabilization.22,24 Sphere-like Au NPs with an average 5–10 nm diameter can be observed in the high resolution TEM images (Fig. 1c). The hydrodynamic size of Au NPs measured by DLS (Table S1† and Fig. 2a) is around 12 nm with a negative average zeta potential of −33.1 mV, which is in line with the TEM size results. As displayed in the enlarged image (inset of Fig. 1c), the average distance of the lattice fringes is approximately 0.23 nm, which is evidence of the presence of the (111) crystallographic planes.24,25 As shown in Fig. S2a,† the surface plasmon resonance (SPR) of the synthesised Au NPs, collected by UV-vis measurements, can be clearly visible as a peak at 520 nm in the UV-vis spectra. According to the reference experimental data,23,26 this SPR peak fits the behaviour of spherical Au NPs with the particle size range of 5–10 nm. In addition to the theoretical calculation, the measured ratio of the absorbance at the SPR peak to the absorbance at 450 nm is 1.20, and according to the size in dependence of the particle diameter data of spherical Au NPs,23 the particle diameter can be estimated as 4–5 nm.
Au–SN-NH2 was formed by attachment of Au NPs on SN-NH2 based on selective bonding between gold atoms and the amine groups.27 From the DLS data (Table S1† and Fig. 2a), the hydrodynamic diameter of Au–SN-NH2 is around 103 nm with an average zeta potential of +9.4 mV. The decrease of zeta potential from SN-NH2 to Au–SN-NH2 may be attributed to the presence of Au NPs linked to SN-NH2. Furthermore, in Fig. S2a,† the SPR peak shifts from 520 nm to 535 nm, which might be due to the Au NP aggregation on the silica particle surfaces. According to the wide-angle XRD pattern (Fig. 2b) and standard pattern JCPDS card no. 04-0784, the first broad diffraction pattern from the 2θ range 10 to 30° can be assigned to the amorphous silica component, while the peaks at 2θ = 38.18°, 44.39°, 64.58° and 77.55° correspond to the (111), (200), (220) and (311) planes of Au NPs, respectively. The estimated average size of Au NPs calculated using the Scherrer formula is about 5.5 nm, in accordance with the full width at half maximum of the intense (111) reflection of Au NPs. Furthermore, the FTIR spectroscopy result (Fig. S1†) of Au–SN-NH2 is almost identical to SN-NH2, with signals at 1066, 945 and 795 cm−1 confirming the presence of silica, while 3357 and 1500 cm−1 are evidence of the amino groups.
Towards surface modification, two intermediate products, Au–SN-COOH and Au–SN-NHNH-Boc, were synthesised. From the DLS data (Table S1† and Fig. 2a), the hydrodynamic size of Au–SN-COOH is about 115 nm with an average zeta potential of −24.6 mV, while Au–SNsNHNH-Boc with a hydrodynamic size of ca. 174 nm has an average zeta potential of +2.3 mV. The negative–positive change of the zeta potentials could be attributed to the conversion of surface groups from –COOH to –NHNH-Boc. From the FTIR spectra (Fig. S1†), the peak at 1720 cm−1 is attributed to the symmetric stretching of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which confirms the presence of carboxyl groups, while the 1657 cm−1 peak indicates the existence of –NH–CO stretching vibrations.
O, which confirms the presence of carboxyl groups, while the 1657 cm−1 peak indicates the existence of –NH–CO stretching vibrations.
After removing Boc groups using TFA,10 Au–SN-NHNH2 was obtained. From the TEM image (Fig. 1d), the average particle size is about 150–170 nm, combined with the ∼5–10 nm dark spots recognized as Au NPs. By further characterization by DLS (Table S1† and Fig. 2a), the hydrodynamic size of Au–SN-NHNH2 is about 167 nm with an average zeta potential of +15.4 mV. This size of nanocarriers is believed to be superior for drug delivery applications not only for favouring the enhanced permeability and retention effects (EPR), but also for limiting non-targeted cellular uptake and undesired cytotoxicity.28 The zeta potential increase from Au–SN-NHNH-Boc to Au–SN-NHNH2 is attributed to the surface hydrazine groups on Au–SN-NHNH2. The presence of hydrazine groups was also confirmed by FTIR. The 3357 cm−1 N–H stretching and 1500 cm−1 N–H bending vibration peaks can be clearly identified in Fig. S1.†
4-MBA–Au–SN–hydrazone–DOX DDSs were created by attaching 4-MBA as SERS tags,29,30 and DOX conjugation was realized via a pH-cleavable carboxylic hydrazone linkage.10,31
In the FTIR spectra (Fig. S1†), the peak at 1734 cm−1 is attributed to the –CO symmetric stretching vibrations from 4-MBA. The signal at 1657 cm−1 represents the hydrazone bonds, which arise from the reaction between hydrazine and the carbonyl groups from DOX. Characteristic peaks at 1540, 1467 and 1412 cm−1 belong to the skeleton vibrations of the ring of DOX,32 which proves the presence of DOX on the DDS particles. The 2850 and 2917 cm−1 bands are attributed to C–H vibrations.
The DOX conjugation was also verified by testing the fluorescence of the well-washed DDS composites. From Fig. S2b,† the 503 nm excitation and 557 nm emission peaks are evidence of DOX linked to DDSs. Moreover, the X-ray photoelectron spectroscopy results shown in Fig. 2c illustrate that DDSs contain C, O, N and Si elements. However, the presence of S from 4-MBA and Au element could not be detected plausibly because of their too low surface contents. The enlarged C 1s region (Fig. 2d) demonstrates the presence of C–Si, C–C, C–N, CN, and CO groups. These high resolution XPS results confirm the occurrence of hydrazone bonds, which is also proof of the DOX–hydrazone conjugations.
SERS effective enhancement factor
The Raman spectra of bulk 4-MBA and 4-MBA–Au–SN-NHNH2 (Fig. 3) are compared to estimate the SERS effective enhancement factor (EEF).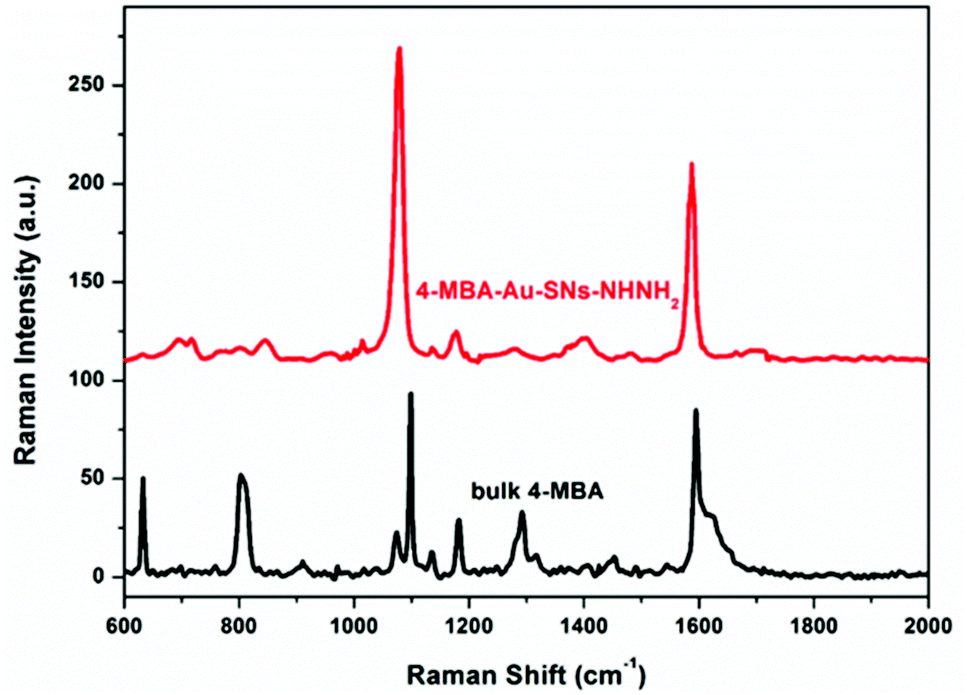 | ||
| Fig. 3 Raman spectra of bulk 4-MBA and the SERS spectra of 4-MBA–Au–SN-NHNH2. | ||
For 4-MBA, the Raman peaks at 1099 cm−1 and 1595 cm−1 are attributed to ν8a and ν12 aromatic ring vibrations of 4-MBA.29 However, for 4-MBA–Au–SN-NHNH2, these two peaks shift to 1078 cm−1 and 1580 cm−1, respectively. The occurrence of such blue shifts confirms the interaction between 4-MBA and Au NPs. The EEF can be estimated based on the calculation result using the 1078 cm−1 band with the following equation:
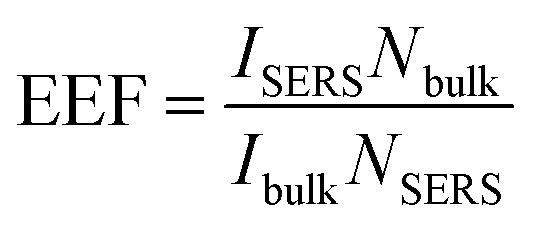
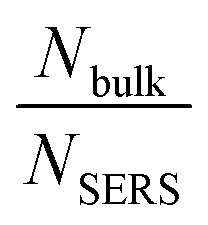 is the molar ratio between the bulk and SERS sample.29,33 Thus, the EEF of 4-MBA–Au–SN-NHNH2 is approximately 1.7 × 105.
is the molar ratio between the bulk and SERS sample.29,33 Thus, the EEF of 4-MBA–Au–SN-NHNH2 is approximately 1.7 × 105.
Biocompatibility and targeted cytotoxicity of DDSs
The biocompatibility of the nanocarriers (4-MBA–Au–SN-NHNH2) and the cytotoxicity of the DDS, and free DOX as a reference, were determined and compared for the HeLa (cancer) and HEK293 (healthy) cell lines. As shown in Fig. 4a, the nanocarriers demonstrate great biocompatibility in both cell lines. After 24 h of incubation at 37 °C under 5% CO2, cell viabilities remained above 85%, for particle concentrations below 200 μg ml−1.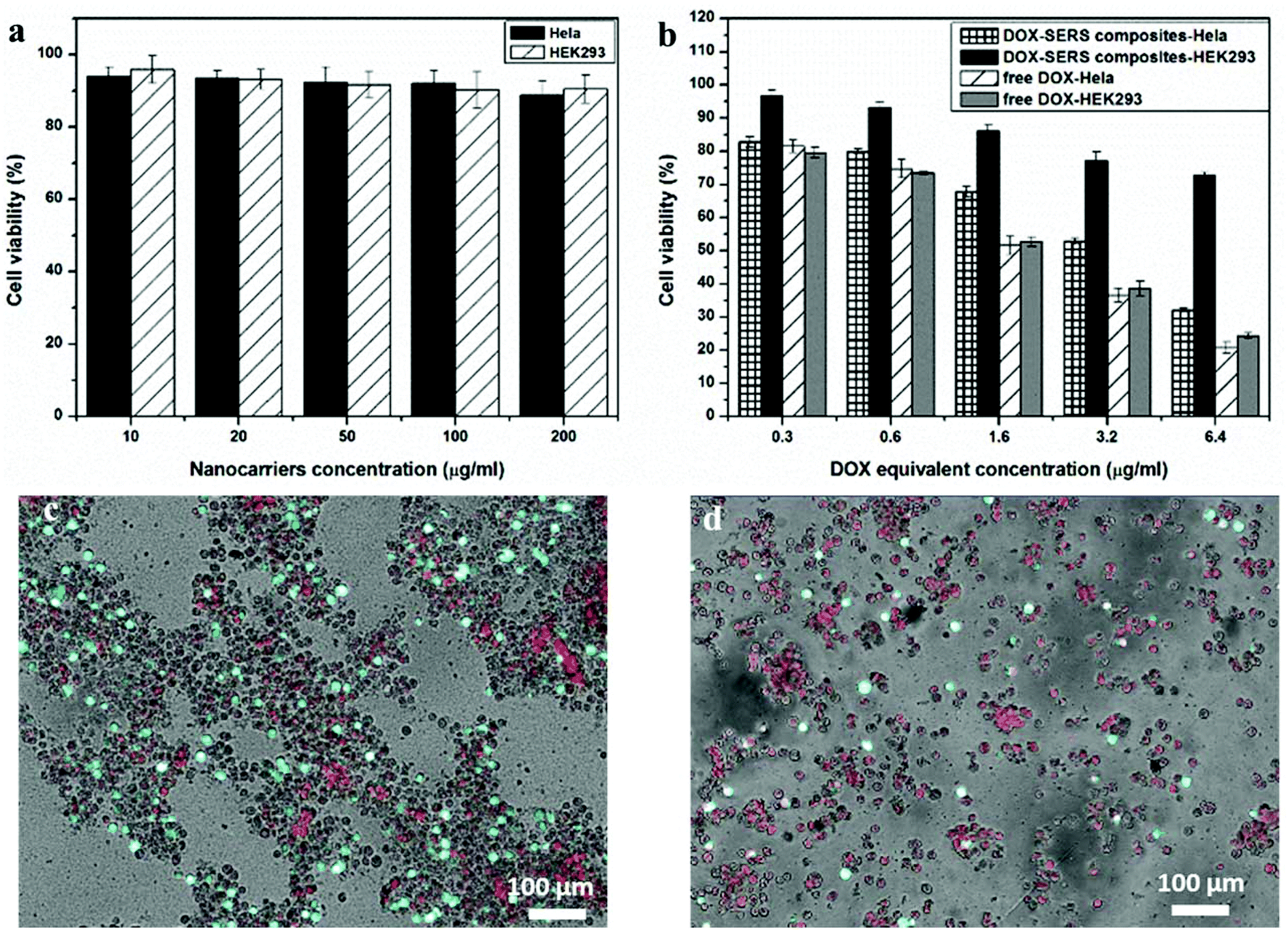 | ||
| Fig. 4 Cytotoxicity data of (a) SERS traceable nanocarriers, (b) DDSs and free DOX, and LIVE/DEAD® analysis of 24 h DDS incubation of (c) HEK293, and (d) HeLa cells. | ||
For the DDS particles and free DOX, the results, shown in Fig. 4b, reveal that cell viabilities are decreasing with the increasing DOX equivalent concentration in both cell lines. For free DOX, no distinct differences of cytotoxicity are observed between the cell lines. However, for DDSs, the tolerability of HEK 293 cells is much better than that of HeLa cells at high particle dosage. At 6.4 μg ml−1 of DOX equivalent concentration, the cell viability of HEK 293 is around 70%, which is higher than the 30% in HeLa. This difference could be attributed to the acidic-cleavage of the carboxylic hydrazone bond existing between DOX and nanocarriers, which accelerated the release of DOX from the nanocarriers in the cancer cells. The difference in DDS cytotoxicities between cancer and healthy cells evidences the feasibility of a pH-smart DDS. Furthermore, the SERS tracing agents seem to exert little influence on cellular selections. In addition, DDS cytotoxicities in both HEK 293 and HeLa cell lines were investigated by LIVE/DEAD® viability/cytotoxicity measurements. For this, 6.4 μg ml−1 DOX equivalent concentration of DDS particles was incubated for 24 h in both the cell lines. Living cells were stained fluorescent green with Calcein AM, while the dead cells were stained red with ethidium homodimer-1. Fig. 4c and d show the merged optical-fluorescence images of the HEK293 and HeLa cells. For HEK293, the amount of fluorescent green live cells is more than red ones with an estimated cell viability of 70%. While for the HeLa cells, the amount of green living cells is lower than that of the red dead cells, where the cell viability might be estimated to be about 25%.
Cellular uptake using SERS tracing
The cellular uptake performances of the DDS in the HeLa cell lines were analysed by SERS mapping and compared with the fluorescence method. The DDS-treated cells were placed on a microscopy glass substrate and directly observed by Raman microscopy.Fig. 5a shows the bright field image of the HeLa cells with a 30 × 30 μm mapping area (red square marked). This area demonstrates a single-cell, and its enlarged image with 2 selected positions is shown in the lower inset of Fig. 5a. These testing positions 1 and 2 are selected to represent the signal obtained from the substrate and that from the cell, respectively. Their corresponding SERS spectra are depicted in Fig. 5b. Although there are background fluorescence interruptions of the glass substrate around 400 and 1400 cm−1, the narrow 1078 and 1580 cm−1 SERS peaks are readily distinguished. Thus, the SERS approach offers a certain competitive advantage over the fluorescence method, such as a finger-print narrow band, high sensitivity and easily distinguished signals. Fig. 5c shows the tracing image by SERS mapping, in which the intensities of the 1078 cm−1 band are demonstrated by the brightness of green cursors. The possibility of obtaining an SERS tracing image of DDS intracellular performances validates the feasibility of the SERS tracing method. The fluorescence tracing image of DOX (Fig. 5d) was obtained from the same sample used in SERS mapping. Also, the DOX fluorescence tracing was further checked by in vitro flow cytometry analysis. DOX fluorescence is tracked from different incubation time points from 2 to 24 h. As demonstrated in Fig. 5f, the DOX fluorescence intensity is proportional to the incubation time, which is indicating the cellular uptake of DOX molecules.34 To compare the SERS and fluorescence tracing results, the fluorescence image was captured at the same position of the same sample tested, and the enlarged optical image obtained from fluorescence microscopy is shown in the upper inset of Fig. 5a. This single-cell image is identical to the optical image obtained from Raman microscopy shown in the lower inset of Fig. 5a. The merged image in Fig. 5e demonstrates the overlapping area of the optical, SERS and fluorescence images. As the fluorescence tracing result agrees well with the SERS one, efficient SERS tracing is further confirmed. Interestingly, the single-cell SERS tracing image exhibits high resolution and reveals more intracellular details than the red fluorescence one. The black area in the SERS image might demonstrate the position of the cell nucleus, where the DDS particles could not be delivered into.
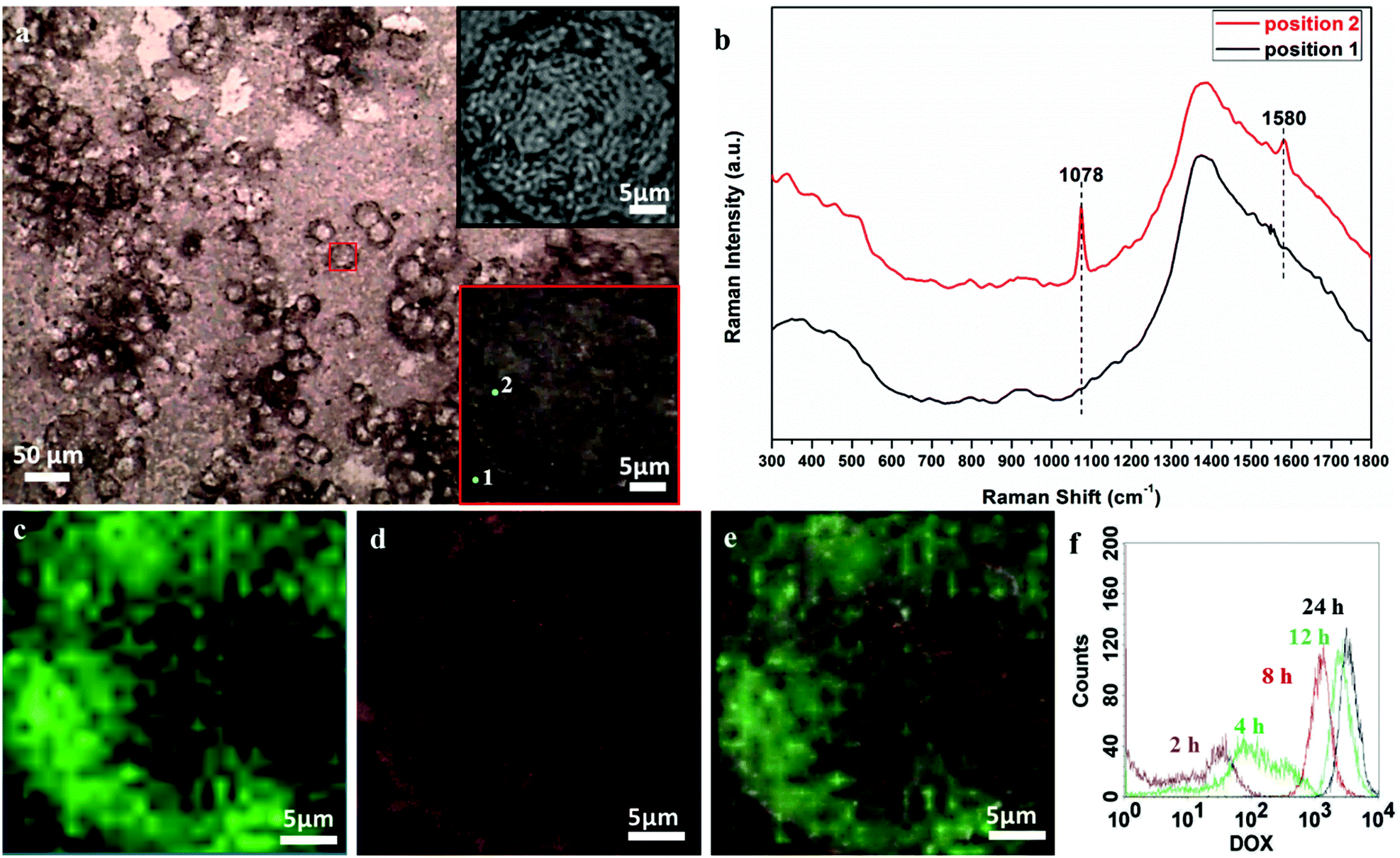 | ||
| Fig. 5 (a) Optical image of HeLa cells with a red rectangle mapping area, optical images of the enlarged mapping area from the upper inset of (a) fluorescence, and the lower inset of (a) Raman microscopy with two selected positions 1 and 2, (b) Raman spectra of selected positions, (c) SERS mapping image, (d) fluorescence image, (e) merged optical-fluorescence-SERS images, and (h) flow cytometry results with different DOX incubation times. | ||
Conclusions
A new smart DDS was designed through SERS-traceable nanocarriers bearing a carboxylic hydrazone-conjugated DOX anticancer agent. The SN platform contributes to the aggregation of small exposed gold nanoparticles on their surfaces, which benefits SERS tracing. The smart DDS is almost biocompatible in healthy cells, while exhibits targeted cytotoxicities in cancer cells. By choosing DOX as an anticancer model drug, intracellular uptake of DDS particles could be traced by comparing the SERS and fluorescence approaches. As SERS signals have higher sensitivities than the fluorescence ones, it could effectively replace the fluorescence method in tracing. With further consideration of the non-destructive features of SERS methods, these designed DDSs might be implemented to evaluate other general drugs without fluorescence analysis. This point is also valid for general drugs without inhibiting cell functions. In that case, we envision that these structured nanocarriers might have the capability to unveil dynamic processes of drug delivery in living cells.Acknowledgements
This work was financially supported by the Australian Research Council (ARC) Discovery Projects (DP140104062 and DP130104459). Lei Liu acknowledges a scholarship from the University of Adelaide. Freddy Kleitz acknowledges financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC).Notes and references
- X. Zhang, F. Li, S. Guo, X. Chen, X. Wang, J. Li and Y. Gan, Biomaterials, 2014, 35, 3650–3665 CrossRef CAS PubMed.
- F. Qiu, D. Wang, Q. Zhu, L. Zhu, G. Tong, Y. Lu, D. Yan and X. Zhu, Biomacromolecules, 2014, 15, 1355–1364 CrossRef CAS PubMed.
- K. C. W. Wu and Y. Yamauchi, J. Mater. Chem., 2012, 22, 1251–1256 RSC.
- V. Malgras, Q. Ji, Y. Kamachi, T. Mori, F.-K. Shieh, K. C. W. Wu, K. Ariga and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2015, 88, 1171–1200 CrossRef CAS.
- H.-Y. Lian, M. Hu, C.-H. Liu, Y. Yamauchi and K. C. W. Wu, Chem. Commun., 2012, 48, 5151–5153 RSC.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- X. Du, L. Xiong, S. Dai, F. Kleitz and S. Z. Qiao, Adv. Funct. Mater., 2014, 24, 7627–7637 CrossRef CAS.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- X. Fang, X. Zhao, W. Fang, C. Chen and N. Zheng, Nanoscale, 2013, 5, 2205–2218 RSC.
- J. Fan, G. Fang, X. Wang, F. Zeng, Y. Xiang and S. Wu, Nanotechnology, 2011, 22, 455102 CrossRef PubMed.
- Y. Zhang, C. Xiao, M. Li, J. Chen, J. Ding, C. He, X. Zhuang and X. Chen, Macromol. Biosci., 2013, 13, 584–594 CrossRef CAS PubMed.
- C. Liu, F. Liu, L. Feng, M. Li, J. Zhang and N. Zhang, Biomaterials, 2013, 34, 2547–2564 CrossRef CAS PubMed.
- A. Samanta, K. K. Maiti, K.-S. Soh, X. Liao, M. Vendrell, U. S. Dinish, S.-W. Yun, R. Bhuvaneswari, H. Kim, S. Rautela, J. Chung, M. Olivo and Y.-T. Chang, Angew. Chem., Int. Ed., 2011, 50, 6089–6092 CrossRef CAS PubMed.
- M. Vendrell, K. K. Maiti, K. Dhaliwal and Y.-T. Chang, Trends Biotechnol., 2013, 31, 249–257 CrossRef CAS PubMed.
- S. Zeng, D. Baillargeat, H.-P. Ho and K.-T. Yong, Chem. Soc. Rev., 2014, 43, 3426–3452 RSC.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- M. Fan, G. F. S. Andrade and A. G. Brolo, Anal. Chim. Acta, 2011, 693, 7–25 CrossRef CAS PubMed.
- X. Gong, Y. Bao, C. Qiu and C. Jiang, Chem. Commun., 2012, 48, 7003–7018 RSC.
- S. Zong, Z. Wang, H. Chen, J. Yang and Y. Cui, Anal. Chem., 2013, 85, 2223–2230 CrossRef CAS PubMed.
- Y. Takeda, Y. Komori and H. Yoshitake, Colloids Surf., A, 2013, 422, 68–74 CrossRef CAS.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- X. Du and J. He, Nanoscale, 2012, 4, 852–859 RSC.
- X. Li, H. Liu, J. Yang, S. Z. Qiao and X. W. Du, RSC Adv., 2014, 4, 1185–1188 RSC.
- G. A. López-Muñoz, J. A. Pescador-Rojas, J. Ortega-Lopez, J. S. Salazar and J. A. Balderas-López, Nanoscale Res. Lett., 2012, 7, 1–6 CrossRef PubMed.
- S. Y. Quek, L. Venkataraman, H. J. Choi, S. G. Louie, M. S. Hybertsen and J. B. Neaton, Nano Lett., 2007, 7, 3477–3482 CrossRef CAS PubMed.
- K. E. Schmalenberg, L. Frauchiger, L. Nikkhouy-Albers and K. E. Uhrich, Biomacromolecules, 2001, 2, 851–855 CrossRef CAS PubMed.
- L. Wei, B. Jin and S. Dai, J. Phys. Chem. C, 2012, 116, 17174–17181 CAS.
- A. Michota and J. Bukowska, J. Raman Spectrosc., 2003, 34, 21–25 CrossRef CAS.
- D. Li, J. Tang, C. Wei, J. Guo, S. Wang, D. Chaudhary and C. Wang, Small, 2012, 8, 2690–2697 CrossRef CAS PubMed.
- A. Popat, J. Liu, Q. Hu, M. Kennedy, B. Peters, G. Q. Lu and S. Z. Qiao, Nanoscale, 2012, 4, 970–975 RSC.
- P. H. C. Camargo, L. Au, M. Rycenga, W. Li and Y. Xia, Chem. Phys. Lett., 2010, 484, 304–308 CrossRef CAS PubMed.
- X. Du, L. Xiong, S. Dai and S. Z. Qiao, Adv. Healthcare Mater., 2015, 4, 771–781 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr03869g |
| This journal is © The Royal Society of Chemistry 2016 |