
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical reactivity of graphene oxide towards amines elucidated by solid-state NMR†
Isabella A.
Vacchi
a,
Cinzia
Spinato
a,
Jésus
Raya
b,
Alberto
Bianco
*a and
Cécilia
Ménard-Moyon
*a
aCNRS, Institut de Biologie Moléculaire et Cellulaire, Immunopathologie et Chimie Thérapeutique, 67000 Strasbourg, France. E-mail: a.bianco@ibmc-cnrs.unistra.fr; c.menard@ibmc-cnrs.unistra.fr
bMembrane Biophysics and NMR, Institute of Chemistry, UMR 7177, University of Strasbourg, Strasbourg, France
First published on 22nd June 2016
Abstract
Graphene oxide (GO) is an attractive nanomaterial for many applications. Controlling the functionalization of GO is essential for the design of graphene-based conjugates with novel properties. But, the chemical composition of GO has not been fully elucidated yet. Due to the high reactivity of the oxygenated moieties, mainly epoxy, hydroxyl and carboxyl groups, several derivatization reactions may occur concomitantly. The reactivity of GO with amine derivatives has been exploited in the literature to design graphene-based conjugates, mainly through amidation. However, in this study we undoubtedly demonstrate using magic angle spinning (MAS) solid-state NMR that the reaction between GO and amine functions occurs via ring opening of the epoxides, and not by amidation. We also prove that there is a negligible amount of carboxylic acid groups in two GO samples obtained by a different synthesis process, hence eliminating the possibility of amidation reactions with amine derivatives. This work brings additional insights into the chemical reactivity of GO, which is fundamental to control its functionalization, and highlights the major role of MAS NMR spectroscopy for a comprehensive characterization of derivatized GO.
Introduction
Since its isolation in 2004, graphene has emerged as a fascinating nanomaterial with unique physical properties.1 Graphene is a one-atom-thick two-dimensional sheet constituted of hexagonally arrayed sp2-bonded carbon atoms. This material has attracted attention due to its unique structural, thermal, mechanical, optical, and electrical properties.2 Intensive research is ongoing to investigate the applications of graphene and graphene oxide (GO)3 in many fields,4 including the development of nanoelectronic devices,5 nanocomposite materials,6 as well as in biotechnology7 and nanomedicine.8 Nevertheless, despite its high potential in various fields, major challenges still remain. Indeed, the low dispersibility of graphene hampers full exploitation of some of its properties.9 To overcome these issues, rational functionalization chemistry is needed to improve the processability and impart graphene with novel properties. In this context, GO is a useful platform for the design of graphene-based hybrid materials.10 GO consists of a single-layer of graphite oxide and it is usually produced by oxidation of graphite using strong acids. GO can be transformed to graphene by thermal or chemical reduction.11 The polar oxygen-containing functional groups of GO render it highly hydrophilic, leading to good dispersibility in water and many other solvents. In addition, the derivatization of these oxygenated functions is a versatile and effective method to prepare chemically functionalized graphene for a wide range of applications.10,12 However, the precise atomic structure of GO remains uncertain and needs to be fully elucidated. Investigations of the chemical structure of GO are carried out through the structural analysis of graphite oxide. But, the composition of graphite oxide is also still under considerable debate. Many models of the chemical composition and structure of graphite oxide and GO have been reported over the years, but currently no unambiguous chemical structure exists.12c Several conflicting models to assess the nature, stoichiometry and distribution of the oxygen-containing functional groups have been proposed successively. These models are still heavily discussed as they do not converge towards a common structure.13 There are many reasons that could explain these discrepancies, including the complexity of the starting material due to the amorphous and berthollide character (i.e., non-stoichiometric atomic composition) of GO, as well as the possible batch-to-batch variability of graphite sources.14 Variations of the protocols used to prepare GO resulting in different extents of oxidation could also explain the inhomogeneity between the samples.12c,15 In addition, even if graphite and graphene oxide have been thoroughly characterized by several spectroscopic techniques, there is a lack of sufficiently sensitive analytical techniques. The most well-known and currently accepted model is the Lerf–Klinowski model,13a where the oxygen-containing functions are mostly in the form of epoxy and hydroxyl groups on the basal plane, with a very low amount of carboxylic acid, carbonyl, quinone, phenol, and lactone functions at the edges of the GO sheet.16 There is still uncertainty and ambiguity regarding the presence or absence of the carboxyl groups on the periphery of the basal plane. Indeed, the Dékány model, proposed as an alternative to the Lerf–Klinowski model, states that the edges of graphite oxide do not possess carboxylic acid groups.13b Klinowski and coworkers did pioneering work on the reactivity of graphite oxide towards different reagents, leading to the assessment of the structure of graphite oxide based on NMR spectroscopy.13a Due to the presence of many oxygenated functional groups and their high chemical reactivity, multiple derivatization reactions of these functions may occur simultaneously. On the other hand, a precise understanding of the reactivity of the different groups and of their possible derivatization is necessary for applications in nanomedicine as it will open the route to use GO as a suitable platform for the self-assembly and conjugation of biomolecules aiming to create innovative hybrids able to affect biological processes related to specific diseases.7,8,17 Therefore, a more detailed study differentiating these reactions is necessary, but it remains challenging. In most studies reported in the literature, amidation of the carboxylic groups of GO with amine derivatives has been described as one of the main strategies for the functionalization of GO.18 However, as underlined above, multiple reactions may occur simultaneously. Indeed, it is worth pointing out that epoxy ring opening can take place under the conditions used to perform the amidation as epoxides are highly reactive towards nucleophilic attack reactions,13a,19 in particular involving amine functions.20 Unfortunately, many articles in the literature reported the amidation of GO without taking into account the possibility of concomitant epoxy ring opening. Therefore, to clarify the situation of the chemistry performed on GO, this work is aimed at investigating the reactivity of GO towards amine derivatives and characterizing the derivatized GO by magic angle spinning (MAS) NMR, X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), FT-IR spectroscopy, Raman spectroscopy, and transmission electron microscopy (TEM). As the ratio between the different oxygenated groups may vary from one batch to another, we used GO samples from two different commercial sources. This study points out the absence of reactive COOH groups, thus eliminating the possibility of the amidation reaction with amine derivatives, and gives further insights into the reactivity of GO with amino compounds, resulting in epoxide opening, and not to amidation even in the presence of coupling reagents. Overall, this work leads to a better understanding of the reactivity of GO, which is essential for controlled derivatization.Results and discussion
In our study we have decided to focus on GO samples from two different commercial sources (NanoInnova and Grupo Antolin). GO from NanoInnova (named GO-N) is prepared by the Hummers’ method. GO from Antolin (named GO-A) is synthesized by a different method. It is derived from carbon nanofibers that consist of ribbons of about five stacked graphene layers rolled up along the fiber axis and developing a continuous spiral. These rolled carbon nanofibers are cut and exfoliated into GO. As the two samples are prepared by a different process, they differentiate in their physicochemical characteristics and may display differences regarding the amount of functional groups.21 Indeed, the thickness of the GO-N layers reaches 20 nm, indicating the presence of aggregated GO sheets. Differently, GO-A contains monolayer sheets of ∼1 nm thickness, along with many irregular particles with a thickness from 3 to 10 nm.22 As nucleophiles react with epoxides in an SN2 type of reaction at 180° to the C–O bond that breaks, the reactivity of the two GO samples may be influenced by their aggregation state. In addition to the differences in their dimension, the water dispersibility of GO-A was higher in comparison with GO-N, which is correlated with the higher aggregation state of GO-N. We recently studied the biodegradability of three GO samples by myeloperoxidase derived from human neutrophils and we observed that the degradation of the graphene sheets was strongly dependent on the water dispersibility of the samples.22 XPS analysis of both GO samples revealed that the C/O ratio was rather similar: 2.24 (69.1% C and 30.9% O) and 2.21 (68.8% C and 31.2% O) for GO-N and GO-A, respectively (Fig. S1, ESI†).Because of the discrepancy between the different GO structural models and the debate on the presence or absence of carboxylic groups, the percentage of COOH functions was assessed by detailed deconvolution of the C (1s) peak. We found that the amount of carboxylic groups was 1.06% and 1.52% for GO-N and GO-A, respectively.22 We further investigated the structure of both GO samples using MAS NMR spectroscopy. The 13C NMR spectrum of the starting GO-N is presented in Fig. 1a. In good agreement with the data reported in the literature, the peaks at 60.9 ppm and 70.8 ppm are attributed to the epoxide and C–OH groups, respectively.13a,23 The peak centered at 129.4 ppm corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conjugated double bonds, while the weak broad peak around 90–100 ppm is still unassigned.13b We performed a quantitative 13C NMR analysis and by integrating each peak after fitting (Fig. S2a†), we were able to calculate the percentage of all peaks (Table S1a†). The amount of carbonyl groups (e.g., carboxylic acids, ketones) was very low (3.5%). In order to have additional information and try to elucidate the chemical structure of GO-N, we performed an advanced solid-state NMR 2D 1H–13C correlation experiment, namely a 1H–13C Frequency-Switched Lee-Goldburg HETeronuclear CORrelation (FSLG-HETCOR) at high magnetic field and spinning speed.24Fig. 1b clearly shows the benefit of using very high fields (17.6 T) in 2D 1H–13C HETCOR experiments with FSLG irradiation during the evolution time, leading to unattained resolution until now for proton in the solid state for such materials. Indeed, a 0.05 ppm resolution was reached; thanks to the combination of the high field, the 30 kHz MAS spinning speed and the carefully set FSLG proton-proton decoupling (see the ESI†).
C conjugated double bonds, while the weak broad peak around 90–100 ppm is still unassigned.13b We performed a quantitative 13C NMR analysis and by integrating each peak after fitting (Fig. S2a†), we were able to calculate the percentage of all peaks (Table S1a†). The amount of carbonyl groups (e.g., carboxylic acids, ketones) was very low (3.5%). In order to have additional information and try to elucidate the chemical structure of GO-N, we performed an advanced solid-state NMR 2D 1H–13C correlation experiment, namely a 1H–13C Frequency-Switched Lee-Goldburg HETeronuclear CORrelation (FSLG-HETCOR) at high magnetic field and spinning speed.24Fig. 1b clearly shows the benefit of using very high fields (17.6 T) in 2D 1H–13C HETCOR experiments with FSLG irradiation during the evolution time, leading to unattained resolution until now for proton in the solid state for such materials. Indeed, a 0.05 ppm resolution was reached; thanks to the combination of the high field, the 30 kHz MAS spinning speed and the carefully set FSLG proton-proton decoupling (see the ESI†).
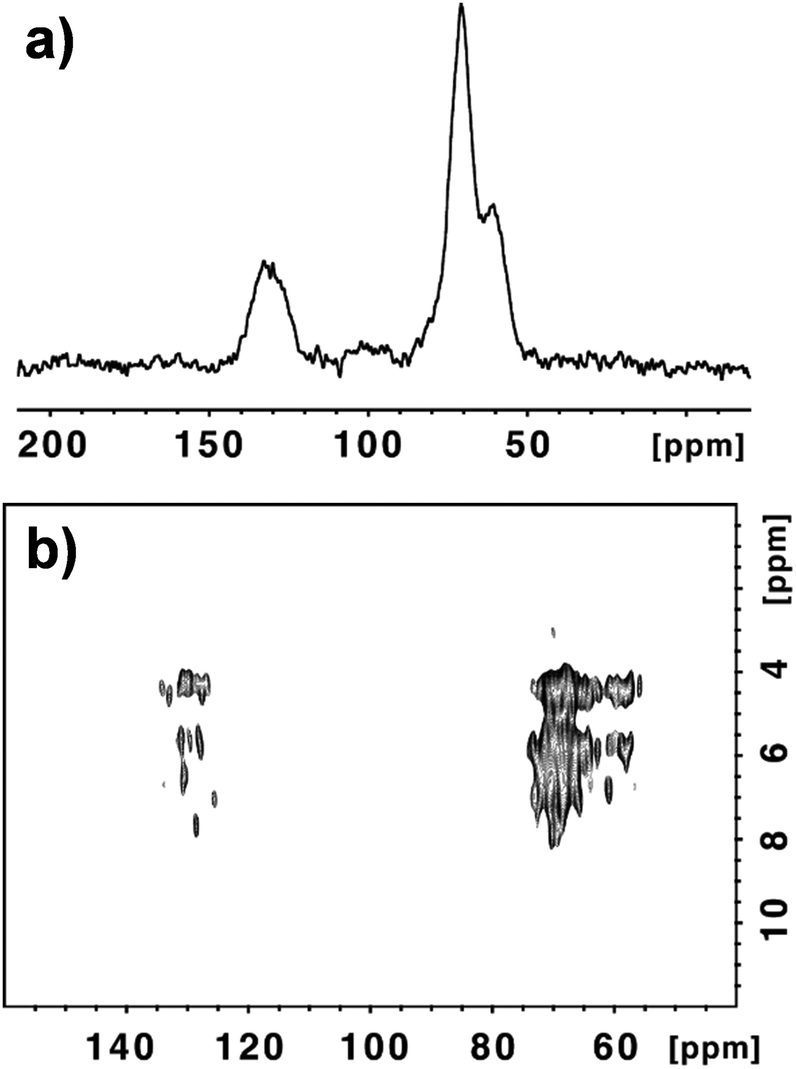 | ||
| Fig. 1 13C NMR (a) and 1H–13C correlation (b) spectra of GO-N. | ||
In order to sort out most of the carbons (that are unprotonated) we found that a Cross-Polarization (CP) contact time of 2 ms was optimal. Under these conditions even the weakly proton dipolar coupled rare spins like carbonyls give rise to rather well detectable cross-peaks. The FSLG-HETCOR spectrum illustrates the complexity of the chemical environment of the carbon and hydrogen atoms present onto GO-N (Fig. 1b and S3†). In the region corresponding to the aliphatic carbons between 55 and 75 ppm, we can count around 25 cross-peaks that are associated with the spatial proximity and close contact between protons and carbons in C–OH and C–O–C groups. Similarly, in the aromatic region between 125 and 135 ppm we could count 13 cross peaks. They are generated by the spatial contact between the protons, most probably on the hydroxyl functional groups and the CC aromatic bonds. Some of the cross-peaks in the carbon dimension (F2) have the same proton chemical shifts (F1). For example, the protons at 4.38 ppm correlate with carbons at 57.42, 65.55 and 134.11 ppm; the protons at 4.46 ppm cross-correlate with carbons at 62.60, 68.96, 70.04 and 127.64 ppm; and the protons at 5.72 ppm are in the proximity of carbons at 60.76, 67.99, 69.50, 73.27 and 127.96 ppm (Fig. S3†).
Fig. S4† illustrates a possible chemical structure that could explain the vicinity between the sp2 and sp3 carbons and the protons of the hydroxyl groups. The presence of adsorbed water molecules certainly contributes to a certain number of peaks in both regions rendering a thorough description of the chemical structure difficult. However, most importantly, the MAS NMR experiments confirm that the presence of the carboxylic acid functions is negligible as only extremely weak broad peaks above 140 ppm (centered around 160 and 190 ppm) can be observed. This confirms the absence or very little amount of COOH groups, as highlighted by the Dékány model proposed for GO.13b
We performed similar MAS NMR experiments with GO-A (Fig. S5†). Similarly, the amount of carboxyl groups was very low (2.5%) (Fig. S2b and Table S1b†). The FSLG-HETCOR spectrum also shows the complex chemical structure of GO-A (Fig. S5b†).
To assess the reactivity of the functional groups of GO, we performed a series of reactions with different amino derivatives. We first investigated the opening of the epoxide rings in the presence of Boc mono-protected triethylene glycol (TEG) diamine 1 (Scheme 1). The reaction was performed under mild conditions, at room temperature in DMF. No catalyst was necessary as epoxides are highly reactive towards amine derivatives.13a,19,20 The ring-opening amination of epoxides yields 1,2-amino alcohols on the GO sheets, leading to the formation of GO 2.
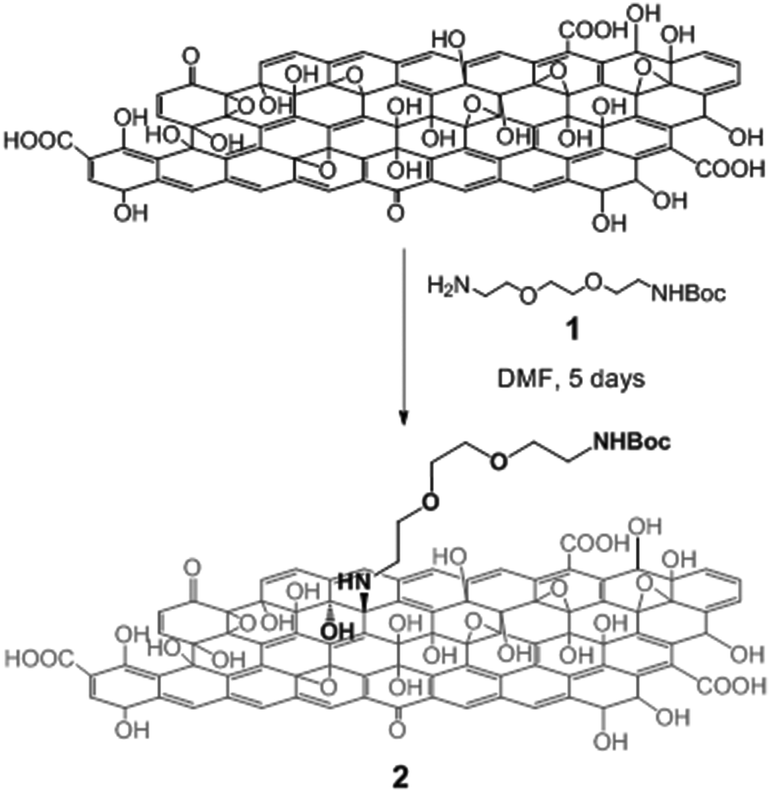 | ||
| Scheme 1 Derivatization of GO via nucleophilic epoxy ring opening by the TEG diamine derivative 1. For the sake of clarity, only one epoxide group is derivatized. | ||
We then examined the amidation reaction of the carboxylic acids of GO with the same diamine derivative (Scheme S1†). For this purpose, GO was reacted with Boc mono-protected TEG diamine 1 in the presence of N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide (EDC) and 1-hydroxybenzotriazole hydrate (HOBt) as coupling reagents, which are commonly used to perform amidation reactions. Under these conditions, in addition to the formation of amide bonds, the amine derivative simultaneously reacts with the epoxides, leading to GO 3 (Scheme S1†). We did not use thionyl chloride or oxalyl chloride to activate the COOH groups as these reagents can also react with hydroxyl functional groups.
Different analytical techniques were used to characterize GO 2 and 3 prepared from GO-N and GO-A, such as MAS NMR spectroscopy, XPS, TGA, FT-IR, Raman spectroscopy and TEM.
MAS NMR spectroscopy of GO-N 2 showed drastic changes in comparison with the starting GO-N (Fig. 2). Indeed, in the 13C spectrum of GO-N 2 the relative intensity of the peak at ∼60 ppm assigned to the epoxide groups is considerably lower, which supports that the epoxides were opened by the amine derivative (Fig. 2a). In addition, new peaks appeared at ∼30 and 40 ppm, which can be attributed to the methylene groups alpha to the nitrogen atoms and to the methyls of the Boc group. The FSLG-HETCOR spectrum confirmed the appearance of these new peaks (Fig. 2b). Likewise, the MAS NMR experiments performed with the GO-A 2 sample led to similar results, demonstrating that the reaction between GO and amine functions occurred via ring opening of the epoxides, and not through amidation (Fig. 2c and d).
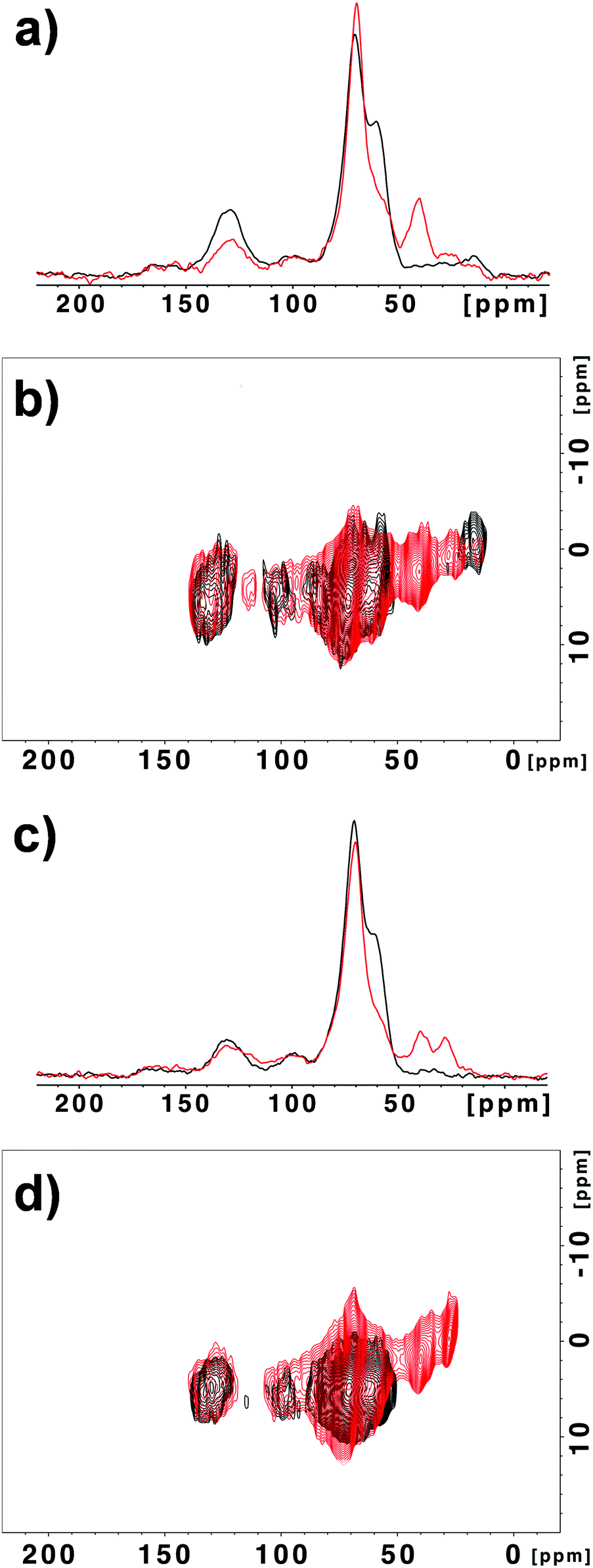 | ||
| Fig. 2 13C NMR (a) and 1H–13C correlation (b) spectra of GO-N (black) and GO-N 2 (red). 13C NMR (c) and 1H–13C correlation (d) spectra of GO-A (black) and GO-A 2 (red). | ||
XPS of GO after derivatization by amidation and/or epoxide opening showed the appearance of a peak corresponding to the N (1s) envelope at a value close to 400 eV in both cases, confirming the incorporation of nitrogen (Fig. 3a, b and S6a, b†). In the case of GO from NanoInnova, GO-N 2 and 3 displayed 3.1 and 2.3% N, respectively. With regard to GO from Antolin (GO-A 2 and 3), the amount of nitrogen was 1.9 and 3.0%, respectively. In addition, the C/O ratio increased from 2.24 to 2.62 and 2.53 for GO-N 2 and 3, respectively, which was consistent with the introduction of the TEG chain. It also demonstrated that GO was not reduced under these conditions. We observed a similar trend for the Antolin conjugates. Indeed, C/O increased from 2.21 to 2.43 and 2.73 for GO-A 2 and 3, respectively. The introduction of nitrogen after amino-functionalization of GO was also confirmed by elemental analysis. The percentage of N was 2.8% and 1.6% for GO-N 2 and GO-A 2, respectively. These values are consistent with the XPS data.
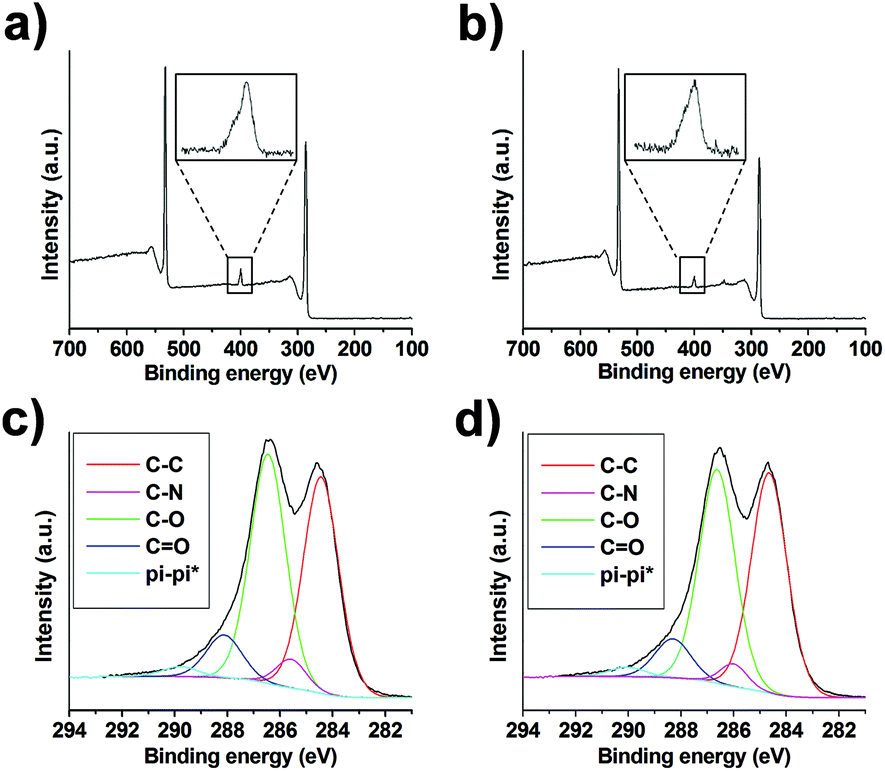 | ||
| Fig. 3 XPS of GO-N 2 (a) and GO-N 3 (b) with a zoom of the N (1s) peak. Deconvolution of the C (1s) peak for GO-N 2 (c) and GO-N 3 (d). | ||
We prepared control samples by treating GO-N and GO-A under the same conditions of the epoxide opening, but without adding amino compound 1 (these control samples are named “GO-N CONT” and “GO-A CONT”). The XPS analysis of the control samples revealed no trace of nitrogen, showing that there is no residual DMF in the samples (Fig. S7†). Deconvolution of the high resolution C (1s) peak allowed further investigation of the reaction outcome (Fig. 3c, d, S6c, d and S8†). The incorporation of nitrogen was confirmed by the appearance of the C–N peak at 286 eV for GO 2 and 3 samples, using both NanoInnova and Antolin materials.
The thermal profile of GO before and after functionalization was assessed by TGA. The analyses were performed under an inert atmosphere at a heating rate of 10 °C min−1. TGA is commonly used to assess the level of functionalization of carbon-based nanomaterials.25 But, in the case of GO, the interpretation of the data is rather difficult as GO is thermally unstable.26 GO displays a typical thermogram with degradation in three steps (Fig. 4).27,28 The significant weight loss below 100 °C is caused by the desorption of adsorbed water and some unstable oxygen-containing functional groups. The main weight loss takes place at around 200 °C and is ascribed to the decomposition of other labile oxygen functionalities. The smaller weight loss occurring over the whole temperature range above 250 °C can be attributed to the removal of more stable oxygenated functional groups. The thermogravimetric profile of Boc-deprotected GO 2 and 3 was different compared to starting GO, revealing higher thermal stability.29 We observed that the control treated GO samples also displayed higher thermal stability. Even though conditions used for amidation and/or epoxide opening are mild, some labile functionalities have been removed from GO.30
 | ||
| Fig. 4 TGA of GO, GO 2 (after Boc deprotection), GO 3 (after Boc deprotection), and the control treated sample (GO CONT) for the NanoInnova (a) and Antolin (b) materials. | ||
In the case of GO from NanoInnova, the weight loss observed for deprotected GO-N 2 and 3 between 150 and 400 °C can be assigned to the thermal decomposition of both the oxygenated moieties and the organic functional groups arising from the newly formed carbon-bound TEG (Fig. 4a). More precisely, the weight loss difference between GO-N 2 and the control treated GO-N represents the amount of TEG diamine introduced onto GO. As the thermal decomposition of the TEG diamine derivative 1 takes place at a temperature below 200 °C, the higher degradation temperature of deprotected GO-N 2 and 3 is a proof of covalent bonding of the TEG diamine onto GO. In addition, the main weight loss (at 210 °C) arises at a lower temperature compared to GO (230 °C), likely due to the presence of the TEG diamine moiety.
The thermogravimetric curves of deprotected GO-N 2 and 3 are rather similar, indicating that, if amidation would have occurred, the amount of the TEG diamine derivative grafted through amide bonds would be very low. This observation supports the conclusions resulting from the characterization by MAS NMR spectroscopy. In the case of GO-A, the interpretation of the TGA data is instead not clear (Fig. 4b). Whereas the GO-A control sample displays higher thermal stability compared to starting GO-A, the weight loss difference between the deprotected GO-A 2 and 3 and the control sample is not really significant. Overall, as GO is thermally unstable, it is quite complicated to interpret the weight loss observed for the different samples.26 Thus, it seems that TGA is not an appropriate technique to quantitatively assess the level of functionalization for GO-based samples. It should be used in combination with other techniques like FT-IR and mass spectroscopy to make the interpretation easier.
TGA is certainly more useful when molecules with a high molecular weight are used to derivatize GO as the weight loss difference with the control sample in this case should be more pronounced. Hence, to further prove the covalent grafting of molecules onto GO by epoxide ring opening, we derivatized GO-N with aminopolyethylene glycol monomethyl ether (average MW 1000), resulting in GO-N 4 (Scheme S2†). As polyethylene glycol (PEG) chains have the propensity to adsorb on GO,31 we performed a control reaction by mixing GO-N with poly(ethylene glycol) dimethyl ether (MW 1000) under similar conditions to assess the amount of the adsorbed PEG derivative, leading to a GO-N 5 sample (Scheme S3†). Both samples were characterized by TGA under an inert atmosphere and the curves were compared with the control sample GO-N CONT (Fig. S9†). The adsorbed sample GO-N 5 displayed a similar weight loss compared to GO-N CONT, demonstrating that the PEG chains do not adsorb on GO-N under the conditions we used for the reaction and purification of the samples. However, the thermal profile of GO-N 4 is different, as expected. Indeed, it presents a slighter weight loss below 100 °C compared to GO-N and GO-N CONT, which is indicative of a change of the surface of the GO layers. This could be explained by a lower amount of adsorbed water due to the presence of PEG molecules on GO. In addition, we observed a significant weight loss in the temperature range 250–400 °C, which can be attributed to the removal of a covalently grafted PEG derivative, as this slope was absent in non-derivatized GO-N 5 and in the control sample. Therefore, we showed that TGA can be a useful technique to characterize GO derivatized with high molecular weight molecules. The dispersibility of GO-N 4 in water was found to be better in comparison with GO-N 5, which is a further indication of the successful covalent functionalization.
FT-IR spectroscopy was also used to characterize the GO samples. It is rather difficult to monitor the derivatization of the epoxy rings as the band that could be assigned to the C–O–C vibration band of epoxide (at ∼1225 cm−1) is small and covered by other unassigned bands (Fig. S10†).32 However, a change could be observed after reaction with the TEG diamine derivative (Fig. S11†). In addition, new bands appear at ∼2918 and 2850 cm−1 which can be attributed to the stretching vibrational bands of the methylene groups of the TEG chain. The band at ∼1720 cm−1, which has been attributed mainly to ketones and quinones,32 is visible in the FT-IR spectra of the starting GO samples, but it disappears after the reaction and also in the FT-IR spectra of the control treated GO samples (Fig. S12†). This is an indication of the labile character of these carbonyl groups.
The characterization of the GO samples by TEM showed that the morphology of the graphene sheets was preserved after derivatization (Fig. S13†). The GO sheets have a wavy shape with the edges folding back and a lateral dimension of a few micrometers.
Finally, GO 2 and 3 samples were analyzed by Raman spectroscopy. The intensity ratio between the characteristic D band (∼1350 cm−1) and G band (∼1600 cm−1) (ID/IG) gives information about the degree of disorder in the graphene lattice.33 The ID/IG ratio is already high in the starting materials for GO-N and GO-A due to the drastic treatments needed to obtain GO sheets (Fig. S14†). After derivatization, there are no clear changes in the ID/IG ratio, meaning that the functionalization conditions do not introduce many defects in the graphenic lattice. The Raman spectra of GO 2 and 3 are similar for the NanoInnova and Antolin materials. Overall, the detailed characterization of GO 3 samples shows that there is no significant difference with GO 2, demonstrating that the reaction between GO and amine derivatives leads mainly to epoxide ring opening.
Taken altogether, among all the analytical techniques used to characterize the GO samples, MAS NMR spectroscopy appears to be the most appropriate technique to discriminate between two potential reactions involving amine derivatives: epoxide ring opening and amidation. The latter can be excluded because of the absence of a significant proportion of carboxylic acids among the different oxygenated moieties present on GO.
Conclusions
We explored the chemical reactivity of two different GO samples. By investigating the reaction of GO with amine derivatives, we demonstrated the negligible presence of reactive carboxylic acid groups in both GO samples by MAS NMR experiments. We evidenced that the reaction between GO and amine functions involves mostly the epoxy groups through ring opening, and not the carboxylic acids that are present in a small amount. The reaction takes place under mild conditions at room temperature. This strategy was applied to two different amine derivatives and could be extended to more complex amino-containing molecules, like peptides and small proteins. This study helps to clarify the current situation of GO functionalization as many articles have reported an incorrect and/or incomplete structure of GO functionalized with amine derivatives. Indeed, the reactivity of GO with amine derivatives is reported in the literature to lead mainly to amidation. Our study brings additional information on the chemical reactivity of GO, which is essential to control its derivatization, and points out the key role of MAS NMR spectroscopy for a comprehensive characterization of functionalized GO. Due to the complexity of the GO chemical structure, much work remains to be done for developing reliable characterization methods that will help to obtain an unambiguous structural identification as well as synthetic procedures that lead to relatively uniform products. Finally, this work opens novel opportunities to control the derivatization of GO and brings an unprecedented knowledge of the GO chemistry, providing valuable insight into its reactivity and eventually its properties.Experimental
Materials and methods are detailed in the ESI.†Preparation of GO 2
To a suspension of GO (20 mg) in dry DMF (20 mL) sonicated in a water bath for 10 min, Boc mono-protected triethylene glycol (TEG) diamine 1 (see the ESI†) (80 mg, 0.32 mmol) was added. The reaction mixture was stirred for 5 days. GO was separated from the solvent through centrifugation (4500 rpm, 5 min). The precipitate was dispersed in DMF, sonicated in a water bath for a few minutes and centrifuged. This work-up sequence was repeated twice with DMF, twice with MeOH, and twice with DCM. The precipitate was dried under vacuum to obtain GO 2.Preparation of GO 3
To a suspension of GO (20 mg) in dry DMF (20 mL) sonicated in a water bath for 10 min and cooled at 0 °C, were added Boc mono-protected TEG diamine 1 (80 mg, 0.32 mmol), HOBt (20 mg, 0.15 mmol) and EDC (30 mg, 0.19 mmol). The reaction mixture was stirred for 5 days at room temperature. GO was separated from the solvent through centrifugation (4500 rpm, 5 min). The precipitate was dispersed in DMF, sonicated in a water bath for a few minutes and centrifuged. This work-up sequence was repeated twice with DMF, twice with MeOH, and twice with DCM. The precipitate was dried under vacuum to obtain GO 3.Preparation of GO CONT
A suspension of GO (20 mg) in dry DMF (20 mL) was sonicated in a water bath for 10 min and stirred for 5 days. GO was separated from the solvent through centrifugation (4500 rpm, 5 min). The precipitate was dispersed in DMF, sonicated in a water bath for a few minutes and centrifuged. This work-up sequence was repeated twice with DMF, twice with MeOH, and twice with DCM. The precipitate was dried under vacuum to obtain GO CONT.Boc deprotection of GO 2, GO 3, and GO CONT
To a suspension of GO 2, GO 3, or GO CONT (20 mg) in 1,4-dioxane (10 mL) sonicated in a water bath for 10 min, was added a solution of 4 M HCl in 1,4-dioxane (10 mL). The reaction mixture was stirred for 15 h. After centrifugation (4500 rpm, 5 min), the precipitate was dispersed in DMF, sonicated in a water bath for a few minutes and centrifuged. This work-up sequence was repeated twice with DMF, twice with MeOH and twice with DCM. The precipitate was dried under vacuum to obtain Boc-deprotected GO 2, GO 3, and GO CONT.The synthesis and characterization of Boc mono-protected TEG diamine 1 are reported in the ESI.†
Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS) and by the International Center for Frontier Research in Chemistry (icFRC). The authors gratefully acknowledge financial support from the EU FP7-ICT-2013-FET-F GRAPHENE Flagship project (no. 604391) and from the Agence Nationale de la Recherche (ANR) through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC). Dominique Bégin and Caroline Hadad are gratefully acknowledged for preliminary studies by XPS and TGA, respectively. TEM images were recorded at the “Plateforme Imagerie In Vitro” at the Center of Neurochemistry and at the RIO Microscopy Facility Platform of Esplanade Campus (Strasbourg, France). We wish to acknowledge Petra Hellwig and Frédéric Melin for giving access to Raman spectrometer.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- (a) R. S. Edwards and K. S. Coleman, Nanoscale, 2012, 5, 38–51 RSC; (b) J. K. Wassei and R. B. Kaner, Acc. Chem. Res., 2013, 46, 2244–2253 CrossRef CAS PubMed; (c) H. Chang and H. Wu, Adv. Funct. Mater., 2013, 23, 1984–1997 CrossRef CAS; (d) X. Wan, Y. Huang and Y. Chen, Acc. Chem. Res., 2012, 45, 598–607 CrossRef CAS PubMed; (e) Y. Zhu, D. K. James and J. M. Tour, Adv. Mater., 2012, 24, 4924–4955 CrossRef CAS PubMed.
- (a) A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS; (b) P. Wick, A. E. Louw-Gaume, M. Kucki, H. F. Krug, K. Kostarelos, B. Fadeel, K. A. Dawson, A. Salvati, E. Vázquez, L. Ballerini, M. Tretiach, F. Benfenati, E. Flahaut, L. Gauthier, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2014, 53, 7714–7718 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- N. O. Weiss, H. Zhou, L. Liao, Y. Liu, S. Jiang, Y. Huang and X. Duan, Adv. Mater., 2012, 24, 5782–5825 CrossRef CAS PubMed.
- (a) X. Qi, C. Tan, J. Wei and H. Zhang, Nanoscale, 2013, 5, 1440–1451 RSC; (b) X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC; (c) I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2013, 46, 2235–2243 CrossRef CAS PubMed.
- (a) K. V. Krishna, C. Ménard-Moyon, S. Verma and A. Bianco, Nanomedicine, 2013, 8, 1669–1688 CrossRef CAS PubMed; (b) Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS PubMed; (c) J. Yao, Y. Sun, M. Yang and Y. Duan, J. Mater. Chem., 2012, 22, 14313–14329 RSC; (d) Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283–2307 RSC.
- (a) K. Kostarelos and K. S. Novoselov, Science, 2014, 344, 261–263 CrossRef CAS PubMed; (b) K. Kostarelos and K. S. Novoselov, Nat. Nanotechnol., 2014, 9, 744–745 CrossRef CAS PubMed; (c) C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed; (d) H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407–3424 CrossRef CAS PubMed; (e) D. A. Jasim, C. Ménard-Moyon, D. Bégin, A. Bianco and K. Kostarelos, Chem. Sci., 2015, 6, 3952–3964 RSC.
- (a) Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC; (b) F. M. Koehler and W. J. Stark, Acc. Chem. Res., 2013, 46, 2297–2306 CrossRef CAS PubMed; (c) L. Rodríguez-Pérez, M. Á. Herranz and N. Martín, Chem. Commun., 2013, 49, 3721–3735 RSC; (d) V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed; (e) J. Park and M. Yan, Acc. Chem. Res., 2013, 46, 181–189 CrossRef PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- (a) S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS; (b) C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC; (c) S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen, J. Oh, S. O. Kim, Y.-H. Kim, Y. Ishii and R. S. Ruoff, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- (a) Y. Pan, N. G. Sahoo and L. Li, Expert Opin. Drug Delivery, 2012, 9, 1365–1376 CrossRef CAS PubMed; (b) K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed; (c) D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC; (d) D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC; (e) S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- (a) A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS; (b) T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef; (c) W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed; (d) A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- Z.-L. Chen, F.-Y. Kam, R. G.-S. Goh, J. Song, G.-K. Lim and L.-L. Chua, Chem. Mater., 2013, 25, 2944–2949 CrossRef CAS.
- (a) A. Y. S. Eng, C. K. Chua and M. Pumera, Nanoscale, 2015, 7, 20256–20266 RSC; (b) C. K. Chua, Z. Sofer and M. Pumera, Chem. – Eur. J., 2012, 18, 13453–13459 CrossRef CAS PubMed.
- L. R. De Jesus, R. V. Dennis, S. W. Depner, C. Jaye, D. A. Fischer and S. Banerjee, J. Phys. Chem. Lett., 2013, 4, 3144–3151 CrossRef CAS PubMed.
- J. Russier, E. Treossi, A. Scarsi, F. Perrozzi, H. Dumortier, L. Ottaviano, M. Meneghetti, V. Palermo and A. Bianco, Nanoscale, 2013, 5, 11234–11247 RSC.
- (a) J. Wang, H.-Z. Geng, Z.-J. Luo, S. Zhang, J. Zhang, J. Liu, H.-J. Yang, S. Ma, B. Sun, Y. Wang, S.-X. Da and Y.-Q. Fu, RSC Adv., 2015, 5, 105393–105399 RSC; (b) H. Qin, T. Gong, Y. Cho, C. Lee and T. Kim, Polym. Chem., 2014, 5, 4466–4473 RSC; (c) H. Wu, H. Shi, Y. Wang, X. Jia, C. Tang, J. Zhang and S. Yang, Carbon, 2014, 69, 379–389 CrossRef CAS; (d) H. Bao, Y. Pan, Y. Ping, N. G. Sahoo, T. Wu, L. Li, J. Li and L. H. Gan, Small, 2011, 7, 1569–1578 CrossRef CAS PubMed.
- (a) S. Eigler, Y. Hu, Y. Ishii and A. Hirsch, Nanoscale, 2013, 5, 12136–12139 RSC; (b) H. R. Thomas, A. J. Marsden, M. Walker, N. R. Wilson and J. P. Rourke, Angew. Chem., Int. Ed., 2014, 53, 7613–7618 CrossRef CAS PubMed; (c) T. Taniguchi, S. Kurihara, H. Tateishi, K. Hatakeyama, M. Koinuma, H. Yokoi, M. Hara, H. Ishikawa and Y. Matsumoto, Carbon, 2015, 84, 560–566 CrossRef CAS.
- (a) N. Azizi and M. R. Saidi, Org. Lett., 2005, 7, 3649–3651 CrossRef CAS PubMed; (b) S. Bonollo, D. Lanari and L. Vaccaro, Eur. J. Org. Chem., 2011, 2587–2598 CrossRef CAS; (c) S. Bonollo, D. Lanari, A. Marrocchi and L. Vaccaro, Curr. Org. Synth., 2011, 8, 319–329 CrossRef CAS; (d) A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabó, A. Szeri and I. Dékány, Langmuir, 2003, 19, 6050–6055 CrossRef CAS; (e) H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC; (f) S. Wang, P.-J. Chia, L.-L. Chua, L.-H. Zhao, R.-Q. Png, S. Sivaramakrishnan, M. Zhou, R. G.-S. Goh, R. H. Friend, A. T.-S. Wee and P. K.-H. Ho, Adv. Mater., 2008, 20, 3440–3446 CrossRef CAS; (g) Q. Mei, K. Zhang, G. Guan, B. Liu, S. Wang and Z. Zhang, Chem. Commun., 2010, 46, 7319–7321 RSC; (h) S. H. Ryu and A. M. Shanmugharaj, Chem. Eng. J., 2014, 244, 552–560 CrossRef CAS; (i) J. U. Lee, W. Lee, J. W. Yi, S. S. Yoon, S. B. Lee, B. M. Jung, B. S. Kim and J. H. Byun, J. Mater. Chem. A, 2013, 1, 12893–12899 RSC; (j) B. Xue, J. Zhu, N. Liu and Y. Li, Catal. Commun., 2015, 64, 105–109 CrossRef CAS.
- N. Morimoto, T. Kubo and Y. Nishina, Sci. Rep., 2016, 6, 21715 CrossRef CAS PubMed.
- R. Kurapati, J. Russier, M. A. Squillaci, E. Treossi, C. Ménard-Moyon, A. E. Del Rio-Castillo, E. Vazquez, P. Samorì, V. Palermo and A. Bianco, Small, 2015, 11, 3985–3994 CrossRef CAS PubMed.
- (a) H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS; (b) W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- B.-J. van Rossum, H. Förster and H. J. M. de Groot, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214–2230 RSC.
- S. Eigler, C. Dotzer, A. Hirsch, M. Enzelberger and P. Müller, Chem. Mater., 2012, 24, 1276–1282 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- J. Chu, X. Li and P. Xu, J. Mater. Chem., 2011, 21, 11283–11287 RSC.
- (a) S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. de Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed; (b) C. K. Chua and M. Pumera, Small, 2015, 11, 1266–1272 CrossRef CAS PubMed; (c) P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher and J. C. Grossman, Nat. Chem., 2014, 6, 151–158 CrossRef CAS PubMed; (d) Z. Lin, Y. Yao, Z. Li, Y. Liu, Z. Li and C.-P. Wong, J. Phys. Chem. C, 2010, 114, 14819–14825 CrossRef CAS; (e) J. T. Paci, T. Belytschko and G. C. Schatz, J. Phys. Chem. C, 2007, 111, 18099–18111 CrossRef CAS.
- F. Barroso-Bujans, F. Fernandez-Alonso, J. A. Pomposo, S. Cerveny, A. Alegría and J. Colmenero, ACS Macro Lett., 2012, 1, 550–554 CrossRef CAS.
- T. Szabó, O. Berkesi and I. Dékány, Carbon, 2005, 43, 3186–3189 CrossRef.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr03846h |
| This journal is © The Royal Society of Chemistry 2016 |