
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new 2D monolayer BiXene, M2C (M = Mo, Tc, Os)†
Weiwei
Sun
*a,
Yunguo
Li
 *b,
Baotian
Wang
cd,
Xue
Jiang
e,
Mikhail I.
Katsnelson
fg,
Pavel
Korzhavyi
hi,
Olle
Eriksson
a and
Igor
Di Marco
*a
*b,
Baotian
Wang
cd,
Xue
Jiang
e,
Mikhail I.
Katsnelson
fg,
Pavel
Korzhavyi
hi,
Olle
Eriksson
a and
Igor
Di Marco
*a
aDepartment of Physics and Astronomy, Materials Theory, Uppsala University, Box 516, SE-75120 Uppsala, Sweden. E-mail: sun.weiwei@physics.uu.se; igor.dimarco@physics.uu.se
bDepartment of Earth Sciences, University College London, London WC1E 6BT, UK. E-mail: yunguo.li@ucl.ac.uk
cChina Spallation Neutron Source (CSNS), Institute of High Energy Physics (IHEP), Chinese Academy of Sciences (CAS), Dongguan 523803, China
dDongguan Institute of Neutron Science (DINS), Dongguan 523808, China
eKey Laboratory of Materials Modification by Laser, Ion and Electron Beams (Dalian University of Technology), Ministry of Education, Dalian 116024, China
fRadboud University of Nijmegen, Institute for Molecules and Materials, Heijendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
gTheoretical Physics and Applied Mathematics Department, Ural Federal University, Mira Street 19, 620002 Ekaterinburg, Russia
hDepartment of Material Science and Engineering, KTH-Royal Institute of Technology, Stockholm SE-10044, Sweden
iInstitute of Metal Physics, Ural Division of the Russian Academy of Sciences, 620219 Ekaterinburg, Russia
First published on 2nd August 2016
Abstract
The existence of BiXenes, a new family of 2D monolayers, is hereby predicted. Theoretically, BiXenes have 1H symmetry (P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2) and can be formed from the 4d/5d binary carbides. As the name suggests, they are close relatives of MXenes, which instead have 1T symmetry (P
m2) and can be formed from the 4d/5d binary carbides. As the name suggests, they are close relatives of MXenes, which instead have 1T symmetry (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1). The newly found BiXenes, as well as some new MXenes, are shown to have formation energies close to that of germanene, which suggests that these materials should be possible to be synthesised. Among them, we illustrate that 1H-Tc2C and 1T-Mo2C are dynamically stable at 0 K, while 1H-Mo2C, 1T-Tc2C, 1H-Os2C, and 1T-Rh2C are likely to be stabilised via strain or temperature. In addition, the nature of the chemical bonding is analysed, emphasizing that the covalency between the transition metal ions and carbon is much stronger in BiXenes than in MXenes. The emergence of BiXenes can not only open up a new era of conducting 2D monolayers, but also provide good candidates for carrier materials aimed at energy storage and spintronic devices that have already been unveiled in MXenes.
m1). The newly found BiXenes, as well as some new MXenes, are shown to have formation energies close to that of germanene, which suggests that these materials should be possible to be synthesised. Among them, we illustrate that 1H-Tc2C and 1T-Mo2C are dynamically stable at 0 K, while 1H-Mo2C, 1T-Tc2C, 1H-Os2C, and 1T-Rh2C are likely to be stabilised via strain or temperature. In addition, the nature of the chemical bonding is analysed, emphasizing that the covalency between the transition metal ions and carbon is much stronger in BiXenes than in MXenes. The emergence of BiXenes can not only open up a new era of conducting 2D monolayers, but also provide good candidates for carrier materials aimed at energy storage and spintronic devices that have already been unveiled in MXenes.
Introduction
The emergence of graphene has promoted the discovery and investigation of novel two-dimensional (2D) materials,1,2 as for instance elementary silicene and germanene,3,4 functional graphene, binary single layer boron-nitride,5 as well as monolayer transition metal dichalcogenides.6 Their fascinating and exotic electronic, optical, mechanical, magnetic and thermal properties provide extensive possibilities to synthesise artificial materials for potential applications in catalysis, new energy areas, etc.7 Due to these high expectations, the 2D materials world is constantly enriched by the discovery of more and more emerging materials.8 Nevertheless very few conductors with pronounced metallic character have been found so far, which is a limit for technological applications. Among the best conductors one can find MXene films, which are a class of 2D early transition metal carbides (MCs) and carbonitrides with 1T symmetry.9 The metallic conductivity of MXenes is also accompanied by surface hydrophilicity, which means that they behave as “conductive clays”.10 Hence, MXenes have attracted more and more attention, because they have tremendous potential for applications in electrode materials, sensors, catalysis and electrochemical energy storage.11–14MXenes are produced by immersing selected MAX phase powders in hydrofluoric acid (HF). MAX phases include compounds of composition Mn+1AXn, where M is a transition metal element, A is an element mostly from IIIA and IVA columns, and X is carbon or nitrogen. The index n can be 1, 2 or 3, depending on the geometrical arrangement of atoms.15,16 In principle, many MCs can be intercalated by light ions to form 2D materials, providing several fascinating properties like superconductivity, good electrochemical performance and storage properties for hydrogen and lithium ions.13 Unfortunately, to date, existing 2D materials derived from MCs are limited to MXenes, and involve mostly a 3d M and a very limited number of heavier elements.17
We have recently reported18 that under ambient conditions bulk Ru2C can be stable in the space groups R3m and Rm. These structures are composed of layers that are ideal candidates as novel 2D systems. It is natural, therefore, to explore the structural and dynamical stability of 2D monolayers arising from Ru2C and neighbouring systems, Tc2C, Rh2C and Os2C, which are also likely to exist in a layered structure. To these compounds, one can add Mo2C, which was recently studied in a couple of experiments. First, a large surface of a superconducting Mo2C thin film of 10 Å thickness was synthesised at a high temperature.19 The preparation procedure was based on a modification of the method used for growing high-quality graphene, chemically-functionalized and defective reduced graphene oxide, or also for preparing MXenes. This approach may be used as a general strategy for fabricating monolayer carbides and other 2D crystals, including transition metal nitrides (MNs).20 Further, large-scale synthesis of 2D Mo2C/Mo2CTx flakes (where T is a surface termination group, e.g. –O –OH or –F) was also realized, via selective etching of Ga from Mo2Ga2C powders.17,21 Holding to these clues, it becomes crucial to understand the structural and dynamical stability of 2D monolayers arising from Mo, Tc, Ru, Rh, and Os binary carbides.
We hereby predict the existence of a new family of 2D materials having 1H symmetry and involving two layers of heavy M atoms sandwiching a graphene-like interlayer of C atoms. Due to the fact that they originate from binary carbides and have the same chemical composition of MXenes, we name these systems BiXenes. BiXenes differ from MXenes not only for their symmetry, but also for the nature of the M components. While MXenes are mostly composed of light M elements, e.g. 3d elements, BiXenes contain 4d/5d elements, which results in wider d bands. This is likely to bring out different functionalities like magnetic or superconducting properties.22 As for several known 2D monolayers, distinctive geometry and compositions are of great importance for improving mechanical stability and related properties.23 The metallic character makes BiXenes also potentially interesting as transparent conductors to be used in modern technology, e.g. in touch screens.24 The likely high thermal conductivity may be potentially interesting for electronics where the problem of heat elimination is one of the crucial obstacles for industrial exploitation. In any case, these are only a few exemplary features, since the emergence of BiXenes, analogously to that of any new class of 2D materials, offers a truly exciting opportunity for future discoveries.
Computational methods
In this work we performed ab initio electronic structure calculations based on density-functional theory (DFT). We used the Vienna ab initio Simulation Package (VASP),25,26 based on a projected-augmented wave (PAW) method.27 Within PAW, 4p, 4d, and 5s states were treated as valence states for Mo, Tc, Ru, and Rh; 5p, 5d, and 6s states were used for Os and 2s and 2p states for C. The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional in the generalized-gradient approximation (GGA)28 was used for all elements. An energy cutoff of 650 eV was employed to achieve the energetic convergence of 0.001 meV as well as the force convergence of 1 meV Å−1. The formation energies of the bulk were evaluated using the diamond structure as a reference state for C. Using diamond instead of graphite is computationally easier since one does not need to include van der Waals (vdW) interactions. The reference state of the M elements was instead obtained from their corresponding ground-state structure, i.e. bcc for Mo, hcp for Tc, Ru and Os, fcc for Rh. A more detailed analysis of the motivation and consequences of these choices can be found in our previous work on Ru2C.18 Finally, we sampled the Brillouin Zone (BZ) with a dense Monkhorst–Pack k-grid29 of 17 × 17 × 5 points.For 2D materials, a supercell was constructed including a vacuum layer with a thickness of at least 15 Å. Full optimisation was carried out in the in-plane directions. The formation energies Eform with respect to the bulk were evaluated as:
| Eform = E2D(M2C) − Ebulk(n[M2C])/n | (1) |
m1 for MXene, and the energetically favoured structure between R3m and P63/mmc for BiXene (see also Fig. 1). For the sake of completeness, for the 2D materials, another set of calculations was performed including vdW interactions by means of the optB86b-vdW functional.30,31 The BZ was sampled with a dense Monkhorst–Pack k-grid29 of 17 × 17 × 1 points. Note that for these open structures, an atomic centred basis set is much more precise. Therefore, we analysed the Fermi surfaces (FS) by means of the full-potential linear muffin-tin orbital (FP-LMTO) code by M. Van Schilfgaarde et al.32
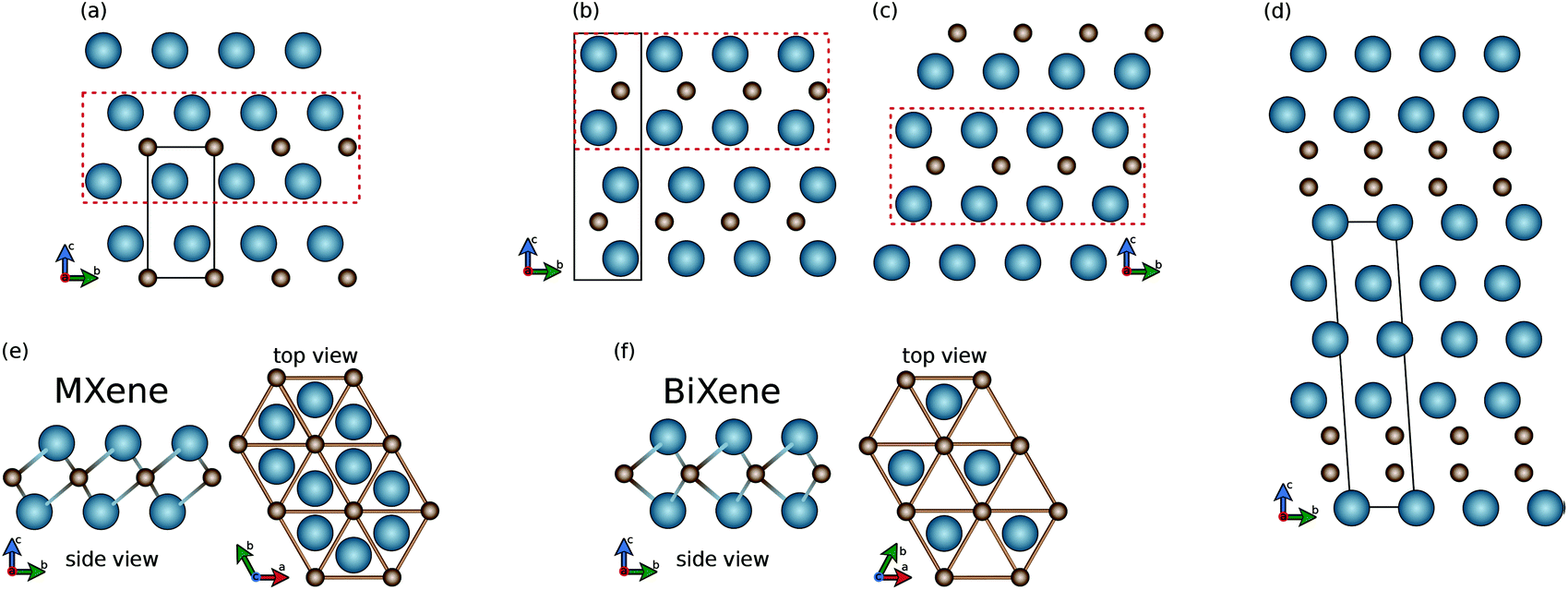 | ||
| Fig. 1 Crystal structures of the 3D and 2D materials studied in this work. For 3D materials: (a) Pm1; (b) P63/mmc; (c) R3m; (d) Rm. For 2D materials: (e) MXene 1T (Pm1); (f) BiXene 1H (Pm2). The blue spheres stand for M atoms and the brown spheres stand for C atoms. The dotted red rectangles in panels (a)–(d) represent the periodic block composing the 2D materials in panels (e) and (f). The black lines identify the unit cell. | ||
The density functional perturbation theory (DFPT) implemented in the PHONOPY code33 was employed to examine the dynamical stability. To ensure a reasonable convergence, we used an energy cutoff of 700 eV with a precision of 1 × 10−8 eV. The force constants in the bulk were calculated for an isotropic 5 × 5 × 5 supercell whose BZ was sampled with 5 × 5 × 5 k-points. Those in the 2D materials were instead obtained for an isotropic 5 × 5 × 1 supercell whose BZ was sampled with 5 × 5 × 1 k-points. For Mo2C, it was necessary to extend the 2D supercell to 6 × 6 × 1. The Fermi–Dirac (FD) broadening scheme34 for an electronic temperature σ was used in the self-consistency cycle to smear out the abrupt change of the Fermi–Dirac statistics in the ground state.
Results and discussion
From 3D to 2D M2Cs
We first investigate the bulk M2Cs in the Pm1, P63/mmc, R3m and Rm structures (see panels (a)–(d) of Fig. 1). In the ESI† we provide an overview of the ground-state structures of the bulk, formation energies and phonon spectra, as well as motivation to restrict our analysis to the four aforementioned structures.
The 3D structures are composed of layers of different symmetries as basic building blocks. As shown in panel (e), in the Pm1 bulk structure one can identify layers with 1T symmetry with inversion (point group D3d). This corresponds to the 2D structure of MXenes, mentioned above. As shown in panel (f), instead, from the P63/mmc and R3m bulk structures, one can extract 2D systems with the 1H symmetry without inversion (point group D3h). This corresponds to the 2D structure of MoS2 but has never been found before for carbides or nitrides.8 Moreover, there is a fundamental feature which makes BiXene different from the transition metal dichalcogenides MX2 (X = S, Se, Te). This feature is that metal and non-metal atoms are swapped. The different arrangement leads to very different electronic structures and related properties, as we will see in section 2. Finally, it is worth stressing that both the 1H and 1T structures exhibit hexagonal lattices, while their main geometrical difference concerns the M layers, which are overlapping in the top view in BiXene (see Fig. 1).
The relative energies shown in the top panel in Table 1 indicate that either 1H or 1T is more energetically favoured. In the framework of PBE, all 1H-M2Cs are favoured, with the exception of 1T-Rh2C. Inclusion of vdW interactions makes also 1T-Os2C more favourable than the corresponding 1H structure. We notice that for the early elements Tc and Mo, results are only barely affected by including vdW interactions, while for the other systems the corrections are larger. We are not sure that this trend is physical, since it may be related to the particular vdW implementation used in VASP, whose accuracy increases with the number of valence electrons.35
| PBE | vdW | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mo2C | Tc2C | Ru2C | Rh2C | Os2C | Mo2C | Tc2C | Ru2C | Rh2C | Os2C | |
| E Rtot (1H) | 0 | 0 | 0 | 0.25 | 0 | 0 | 0 | 0 | 0.24 | 0.01 |
| E Rtot (1T) | 0.19 | 0.25 | 0.08 | 0 | 0.11 | 0.24 | 0.27 | 0.49 | 0 | 0 |
| E form (1H) | 2.06 | 2.82 | 2.19 | 1.58 | 2.57 | 2.39 | 2.91 | 2.57 | 1.90 | 2.92 |
| E form (1T) | 2.69 | 2.17 | 1.81 | 1.38 | 1.81 | 3.03 | 2.52 | 2.56 | 1.66 | 2.06 |
Most of the 2D materials are exfoliated slices of powders (bulk)/thin films or can be prepared by designed methods such as chemical vapour deposition (CVD).19 Their feasibility can first be evaluated through their formation energies with respect to the bulk, i.e. Eform from eqn (1). The values for the 2D materials addressed in this work are reported in the bottom panel of Table 1. In general, most of the formation energies appearing in Table 1 are smaller than the values of the primary (intra-layer) chemical bonds, suggesting that the interaction between the M layers is sufficiently weak for possible exfoliation. In fact, we emphasise that, if normalised per atom, the values reported in Table 1 are comparable to (or smaller than) the formation energies of germanene and silicene.36 This trend is confirmed by the inclusion of vdW interactions. As expected, the latter make the bulk energetically more convenient, increasing the formation energies of the monolayers. Despite this small increase, the formation energies are still below 1 eV per atom. Overall, all these 2D compounds seem possible to be realised, provided that they are dynamically stable (see below). Mo2C stands out as the only material for which the 1H structure seems easier to realise than the 1T structure, with a difference in the formation energies of about 0.6 eV. Finally, an interesting speculation based on Table 1 concerns the dependence of Eform on the atomic number. For M-ions heavier than Tc, the 1T structure seems more favourable, while the opposite holds for lighter elements. The Tc ion in Tc2C, corresponding to a half-filled 4d-shell, seems in fact to act as a transition element, although more compounds should be investigated to support this analysis.
Dynamical stability and vibrational properties
Realising the monolayers in experiments requires also the dynamical stability with respect to the lattice vibrations. This can be analysed by means of the phonon spectra, which are reported in the left panel of Fig. 2. In general, one cannot identify a trend where 1H or a 1T polymorph seems to have stable 2D compounds. Moreover, one can notice a general softening for compounds in the 1H structure, which is also seen in the 2D M-dichalcogenides.37,38 Among all investigated systems, only 1T-Mo2C and 1H-Tc2C satisfy the strictest criterion for dynamical stability, i.e. the absence of imaginary branches. Nevertheless, 1T-Tc2C, 1T-Rh2C, 1H-Mo2C and 1H-Os2C are characterized by imaginary modes of a very small size, which makes them interesting for our purposes. In fact, these small pockets of imaginary modes may be the result of numerical truncation in our theory, associated with the finite size of the supercells (see also the ESI†).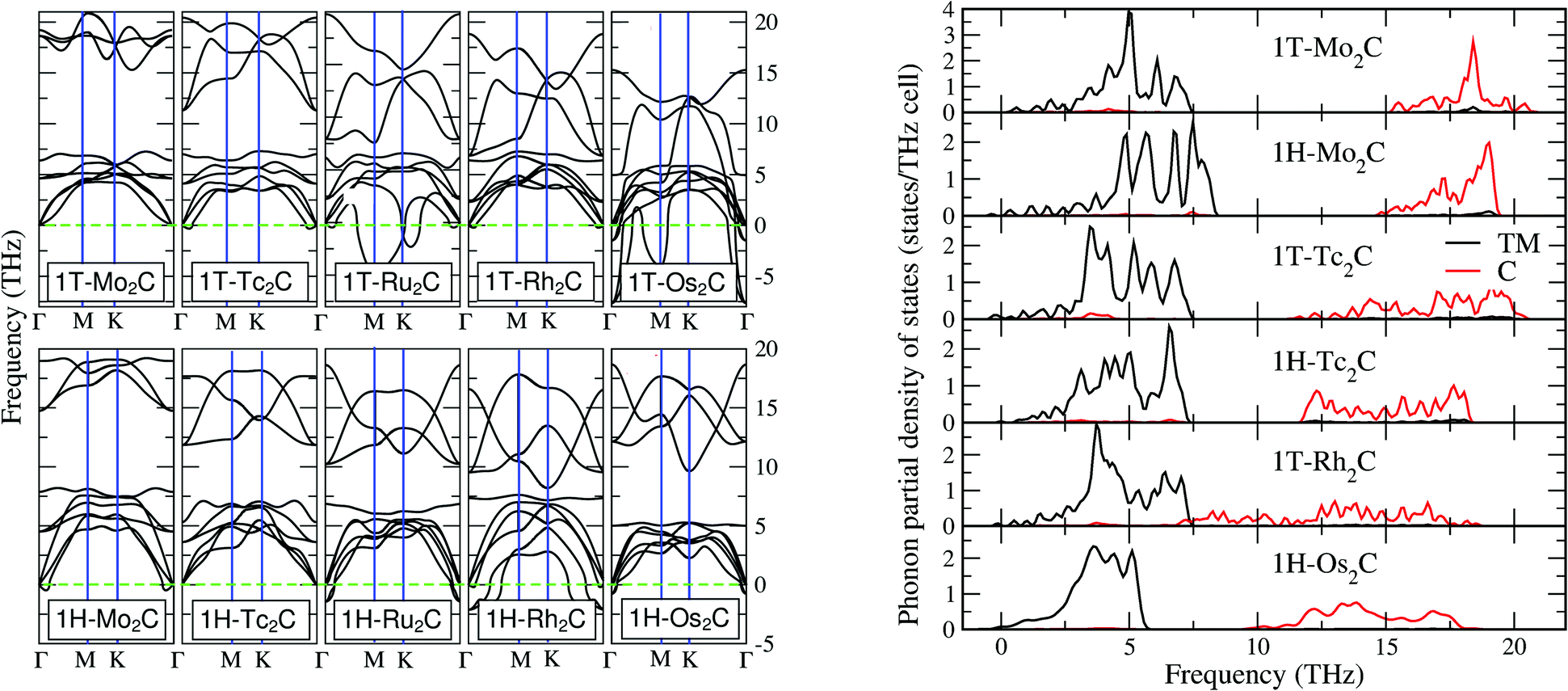 | ||
| Fig. 2 Left panel: The phonon dispersion curves for the single layer of M2C M = Mo, Tc, Ru, Rh, Os with both 1T and 1H symmetries. Right panel: The phonon site-projected density of states (PSPDOS) of 1T-Mo2C, 1H-Mo2C, 1T-Tc2C, 1H-Tc2C, and 1T-Rh2C and 1H-Os2C (from top to bottom). | ||
Furthermore one has to consider that under experimental conditions 2D materials are often grown on top of a substrate, such as Si or SiO2. The substrate may induce an effective strain on the monolayer, which can in turn modify its dynamical stability. Eventually, an appropriate substrate can be selected to remove small pockets of imaginary modes. In addition to the role of the substrate, one has to consider that effects due to a finite temperature can further improve the dynamical stability with respect to our zero temperature calculations (see below for a more extensive discussion). Therefore, it is worth considering not only the fully stable monolayers but also those monolayers characterised by small imaginary pockets, which are likely to be removed by strain and temperature. In total, we can thus identify 6 (quasi) stable monolayers, including 1T-Mo2C, 1T-Tc2C and 1T-Rh2C for the MXene structure, and 1H-Mo2C, 1H-Tc2C and 1H-Os2C for the BiXene structure.
The vibrational properties of these 6 structures are discussed by means of the phonon site-projected density of state (PSPDOS), reported in the right panel of Fig. 2. A general feature for all materials except 1T-Rh2C is the presence of a big gap between modes projected over M and C species, which is related to their large mass ratio. For 1T-Rh2C, the unusually wide phonon band of the C atoms extends from 18 THz to 8 THz, where it overlaps with the M band. The absence of a gap will be further discussed in the next subsection. In the low frequency region of all materials one can also notice an overlap of M and C modes, corresponding to co-vibrations. This overlap is weak but noticeable, especially for 1T-Tc2C, at around 4 THz. Concerning the general differences between the 1T and 1H structures (to be inferred from both Mo2C and Tc2C), they appear to be not too marked. Once can notice that in 1H a marked peak characterises the top of the M band, and also that the end of the C band is not as high in energy as in 1T. Finally, we note that the phonon spectrum of Os2C is slightly different from the other ones, as the M band is narrower and no multiple peaks can be resolved. This may be a consequence of the fact that Os is a 5d element, i.e. much heavier than the 4d elements involved in the other materials. However, it may also be connected to a chemical bonding of a different nature, as arising from the more delocalised 5d electrons. This issue will be addressed more in detail in the next subsection.
As we stressed previously, the quasi-stable structures can in principle be stabilised by the presence of a substrate or by effects due to a finite temperature. In the perspective of practical applications, it is important to analyse these factors more in detail. Among the quasi-stable structures, 1H-Mo2C is the most interesting system to address, since it offers the lowest formation energy compatible with the 1H symmetry (see Table 1). Moreover, 1T-Mo2C has already been identified as a very promising candidate for a high-performance thermoelectric material, and its functionalized 2D layer could find many applications within power generation, energy storage devices, and catalysis.39,40 This suggests that its counterpart 1H-Mo2C may have equally interesting properties. Therefore, we choose 1H-Mo2C as a test example to investigate the effects of strain and temperature on the dynamical stability.
In Fig. 3 we report the phonon spectra of 1H-Mo2C under various conditions. For practical purposes, strain is considered within a range of 3%, compatible with recent work on analogous systems.41,42 Given that GGA often tends to underestimate the binding, the exchange–correlation functional was switched from GGA to LDA for these calculations. The phonon spectrum with no strain at zero temperature, shown in panel (a) of Fig. 3, is very similar to the corresponding spectrum of Fig. 2. As expected, the imaginary modes are slightly enhanced and also the upper phonon branches (mainly due to C) are upshifted, revealing that LDA strengthens the Mo–C bonds. These effects are further emphasized if a biaxial compressive strain (3%) is applied, as illustrated in panel (b) of Fig. 3. Instead, applying a tensile strain (3%), leads to a strong downward shift of the upper phonon branches, as illustrated in panel (c) of Fig. 3. Moreover, the imaginary modes are strongly enhanced with respect to both the unstrained system and the system with a compressive strain. So, a tensile strain (3%) is the worst case scenario that will be considered below for investigating the effects due to a finite temperature.
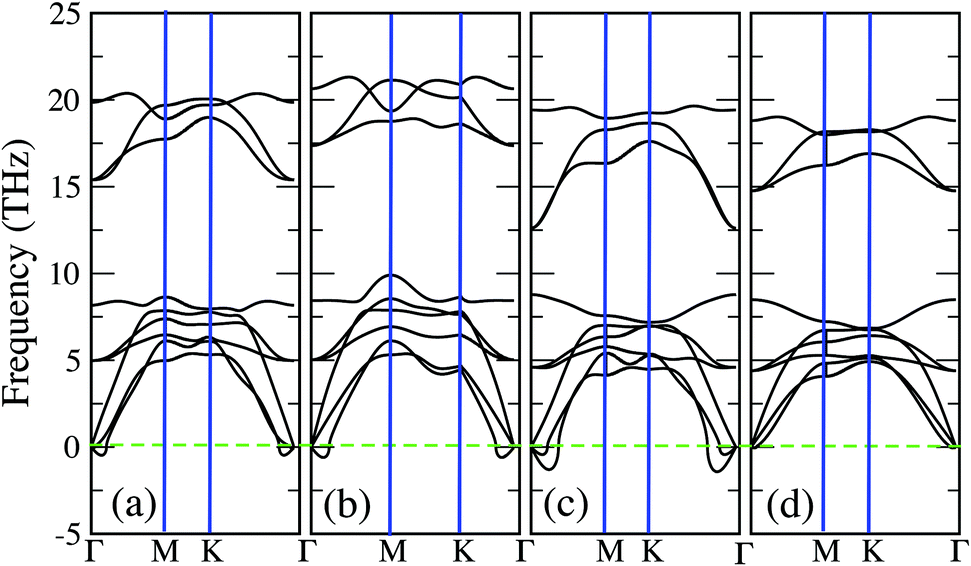 | ||
| Fig. 3 The phonon dispersion curves of the 1H-Mo2C monolayer in LDA and under different conditions: (a) free strain, zero temperature; (b) biaxial compressive strain of 3% in the basal plane; (d) biaxial tensile strain of 3% in the basal plane; (d) biaxial tensile strain of 3% in the basal plane at an effective temperature (Fermi–Dirac smearing) of σ = 0.4 eV. | ||
Our calculations provide a model of the monolayers at zero temperature, while real life experiments are performed at a finite temperature. Ideally, a finite temperature should affect the lattice system both directly, through phonon–phonon interactions, and indirectly, through the electronic system. The latter can in principle be handled in Kohn–Sham DFT by considering a Fermi–Dirac distribution of the Kohn–Sham eigenvalues as well as a temperature dependent exchange–correlation functional. Unfortunately, there are no widespread functionals with this capability,43 so we have to limit our analysis for considering thermal smearing in a Fermi–Dirac distribution, as routinely discussed in the literature, e.g. for Al hydrides44 or Nb/Mo nitrides.45,46 The phonon spectrum of 1H-Mo2C under a 3% tensile strain and for an effective smearing (temperature) of σ = 0.4 eV is reported in panel (d) of Fig. 3. There are no signs of negative phonon branches, so under these conditions the monolayer can be considered as dynamically stable. The value σ = 0.4 eV should not be compared to experimental temperatures directly, since we are still neglecting the temperature dependence of the exchange–correlation functional as well as the coupling with the lattice.44 Instead, it is much more relevant to compare our σ to values obtained for other systems that have already been synthesised. For example, the monolayer of 2H-TaSe2 exists in the experiment at room temperature,47 despite being predicted in theory to be dynamically stable for a thermal smearing of at least σ = 0.68 eV.48 Our σ, for the worst case scenario, is well below 0.68 eV, and therefore we can expect that the predicted monolayers are likely to be stable under experimental conditions. This conclusion is even stronger if we consider the less unstable cases, corresponding to panels (a) and (b) of Fig. 3, which are expected to become stable for a σ as small as 0.1 or 0.2 eV.
The temperature effects on the electronic system alone seem sufficient to stabilize the predicted monolayers, but we should not forget that the phonon spectra are also directly affected by the temperature in the form of phonon–phonon interactions. These are not included in our calculations, since they are beyond the harmonic approximation. Anharmonic effects are often responsible for stabilizing some phases at high temperature and can be taken into account by more sophisticated techniques such as the self-consistent ab initio lattice dynamics method (SCAILD).49–52 While this analysis is too demanding to be performed now, we wish to address this issue in future studies.
Chemical bonding and electronic structure
The next point of analysis concerns the chemical bonding and the electronic structure of the identified (quasi) stable monolayers. This will also help explain the differences observed in the vibrational properties. In the bottom panel of Fig. 4 the electronic density of states (DOS) of the investigated structures is reported. When going from Mo to Tc and Rh, the inclusion of more and more d electrons pushes the Fermi level Ef upwards, as shown by the green dashed lines. In contrast to the bulk, no clear formation of “valley states” dividing bonding from antibonding states53,54 is observed, except for 1H-Mo2C. This is a very interesting feature associated with the lower dimensionality and arises from quantum confinement effects (periodicity broken along the c axis). Most importantly, all the investigated systems have a strong metallic character, which is a rather rare feature of 2D materials. For example, among the transition metal dichalcogenides MX2, the tellurides show a semi-metallic character, while sulfides and selenides are semiconductors.55,56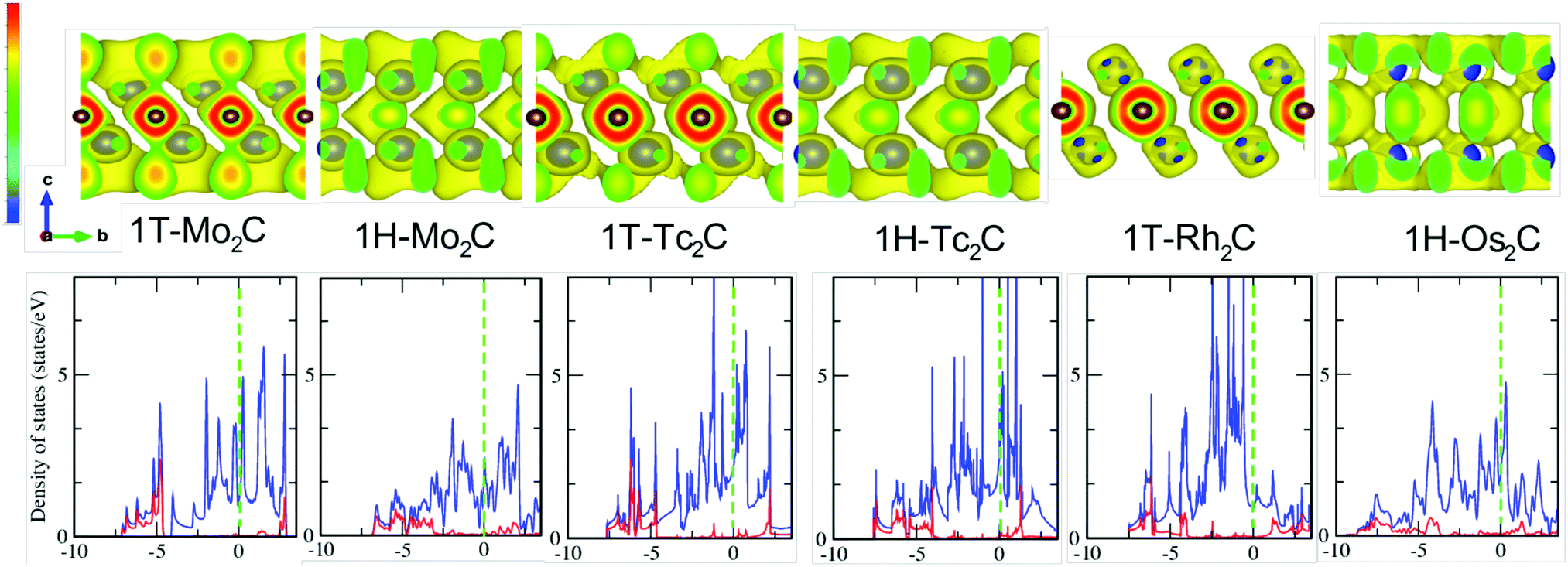 | ||
| Fig. 4 Top panel: The electron localisation function (ELF) of the possible (quasi) stable 2D materials: 1T-Mo2C, 1H-Mo2C, 1T-Tc2C, 1H-Tc2C, 1T-Rh2C, and 1H-Os2C. The level of the isosurface is set to be 0.38 of the local maximum value. The blue (grey) spheres represent the M (C). Bottom panel: Projected density of states of the M-d (in blue) and C-2p (in red) electrons. The dashed green line indicates the Fermi energy. | ||
The two structures of Tc2C show the highest number of states at Ef, which is related to four bands crossing the Fermi energy in this case, while there are only three for the other materials (see also next subsection). Another major feature noticeable in the DOS is that the states around Ef have mainly M character, and there is no significant hybridization with the C derived states. The most relevant hybridized M–C states lay instead at high binding energy, several eV below the Fermi energy. Moreover, for 1T-Rh2C and 1H-Os2C, no pronounced antibonding peak can be observed, due to the shift of Ef induced by filling the d shell. In 1H-Os2C, hybridized Os–C states spread over a wider range, which is partially due to the broader 5d band with respect to the 4d band. This is also noticeable through the high energy tail of the DOS, which in 1H-Os2C goes down to −8.5 eV, i.e. about 1 eV more than for the other systems. Moreover, although it is not visible from the bottom panel of Fig. 4, the tail does not suddenly stop at −8.5 eV but slowly decays at higher binding energies. This does not happen for the other systems. Finally, there seem to be no obvious differences between the DOS in the 1T and 1H structures, except that the latter appears to be characterised by slightly broader peaks.
Another interesting feature noticeable from the bottom panel of Fig. 4 is that occasionally a pseudo-gap is formed at about −4 eV. In one case, i.e. for 1T-Mo2C, this pseudo-gap becomes a real gap in the spectrum. This transition from a pseudo-gap to a full gap is related to the formation of unique inter-layered electrostatic interactions, and can be better analysed through the electronic localisation function (ELF), shown in the top panel of Fig. 4.
Before discussing the ELF of 1T-Mo2C, we first notice that the ELF of the 1H structures shows a chemical bonding featured with some covalency. The orientations of the ellipsoids of the electron clouds of the C layer are noticeably different. In 1H-Os2C they prefer to align parallel to the C axis, while in 1H-Mo2C and 1H-Tc2C the favoured direction is perpendicular to the C axis. Although the overlap of the electron clouds belonging to the M and C layers can be observed in all three cases, it is particularly strong for 1H-Os2C, where bridge-like channels emerge. It can also be inferred that the in-plane C–C π bonds are weaker in the case of 1H-Os2C, and this is different from the other 2D materials.
The 1T structures show conversely more localised charges around the C atoms, which points to the fact that they are more ionic than their 1H counterparts. The ELF of 1T-Mo2C stands out as significantly different from the others, due to the presence of the point charges at the surface (M layers). These point charges produce a unique inter-layered electrostatic interaction with the C layers, which induces the formation of the gap at −4 eV observed in the bottom panel of Fig. 4. Another interesting feature is that the ELF of 1T-Rh2C shows a strongly ionic Rh–C bond, where the states at the surface are almost empty. In general, the stronger the inter-layer bonds, the higher the frequency of the C modes in the phonon spectra. Therefore, the ELF data explain the extension of the C bands in the phonon spectra of Fig. 2.
Fermi surface and band structure
After having analysed the basic features of the electronic structure and the nature of the chemical bonding, we move to investigate the band structure and the Fermi surface (FS). These are shown in Fig. 5 for the 6 (quasi) stable structures identified above. The band structure of Tc2C, for both symmetries, stands out as it is the only material where four bands cross the Fermi energy. In the FS of 1H-Tc2C the 1st band forms the small hole pockets, the 2nd band forms the “dog-bone” sheets, the 3rd band forms holes, and the 4th band forms the inner smaller sheets inside the sheet formed by the 3rd band. For 1T-Tc2C, the components of FS are very similar to the 1H structure but the “dog-bone” sheets are replaced by a flower-like sheet, as shown in Fig. 5. As expected, filling the 4d shell, as in 1T-Rh2C or 1H-Os2C, leads to a simpler FS. The hole pockets become smaller, and even disappear in the case of 1T-Rh2C. The most noticeable features are very closely parallel sheets formed by the 1st and 2nd bands, which are evident for both 1T-Rh2C or 1H-Os2C. In the case of 1T-Rh2C parallel sheets can be found, whose nesting vector q has been defined in Fig. 5. This suggests that a Kohn anomaly57 may potentially occur at this vector q, e.g. in the phonon spectrum, as it happens for e.g. graphene58 or silicene.59 Graphene and silicene are in fact the prototypes of 2D monolayers exhibiting Kohn anomalies, which also lead to the formation of charge density waves.60 Hajiyev et al.47 have also revealed the existence of an incommensurate charge density wave phase transition in single- and double-layered 2H-TaSe, identified by sudden softening of a Raman mode resulting from the Kohn anomaly through strong electron–phonon coupling. Anyway, one should keep in mind that all these systems are insulating, and can therefore be very different for the present monolayers. Since this nesting direction is not along the high symmetry points, no anomalies have been observed in Fig. 2. Instead, 1H-Os2C differs more significantly from the other systems, as the FS sheets constructed by the 3rd band are no longer two-dimensional due to the complexity of the wiggled red bands shown in Fig. 5. This complexity can possibly be explained by the fact that each 5d orbital in Os has 12 lobes, while only 8 lobes are found for the 4d orbitals of the other systems.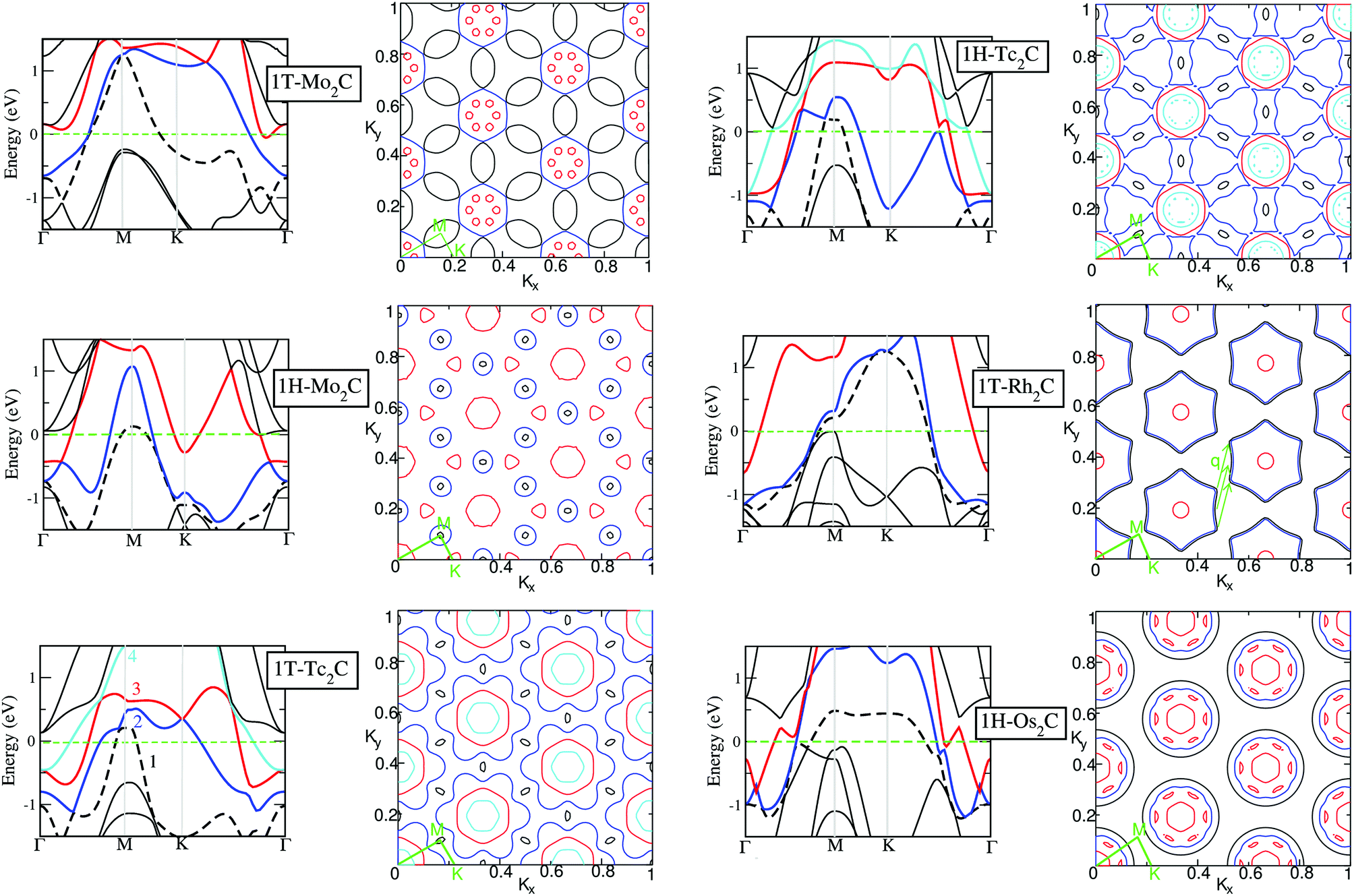 | ||
Fig. 5 Left side: Band structure along high symmetry directions for the (quasi) stable 2D materials 1T-Mo2C, 1H-Mo2C, 1T-Tc2C, 1H-Tc2C, 1T-Rh2C, and 1H-Os2C. The bands crossing Ef are labelled by counting their energetic order at the Γ point. The first band is depicted with dashed black lines, the second one with blue lines, the third one with red lines, and the fourth one (when present) with cyan lines. Right side: Corresponding Fermi surface (FS) in the plane of kx − ky in the reciprocal space. The unitary length along both directions corresponds to 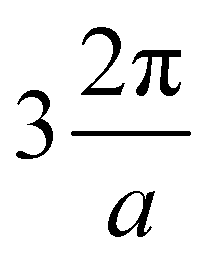 . The bands are labelled as in the band structures. The high symmetry points Γ, M and K, and the paths between them are shown in green. . The bands are labelled as in the band structures. The high symmetry points Γ, M and K, and the paths between them are shown in green. | ||
We can then analyse what happens by removing an electron from the half-filled 4d shell. This situation corresponds to a transition from Tc2C to Mo2C. In 1T-Mo2C we note that the 1st band forms the overlapping “saddles”, while the 2nd band forms hexagons with the holes from the 3rd band. These features are consistent with a simple evolution of the FS of 1T-Tc2C, which has one electron more. Instead, the evolution of the two 1H structures is not so straightforward. In the FS of 1H-Mo2C, there is one less band than in 1H-Tc2C, and the “dog-bone” sheets evolve into smaller holes. Moreover, for 1H-Mo2C, in contrast to 1H-Tc2C, the FS sheets formed by the 2nd band shrink, and additionally there is one less band missing in the FS, also mentioned above. Finally, notice that only 1H-Mo2C shows the coexistence of well-defined hole and electron pockets (see the 1st and 2nd bands in the corresponding electronic structure of Fig. 5). 1T-Mo2C and 1H-Os2C also show pockets of both characters, but the electronic ones are extremely small.
Experimental realisation
So far we have identified 6 (quasi) stable monolayers and investigated their electronic and vibrational properties. We can now analyse more in detail if and how they can be realised in experiment. We already mentioned that one possibility is to obtain a monolayer via mechanical exfoliation. This requires that the monolayer can be identified as a component of a bulk structure that is stable under ambient conditions. Bulk formation energies and phonon spectra are reported in the ESI.† 1H-Tc2C can be exfoliated from R3m and P63/mmc bulk structures, which have negative formation energies and stable phonon spectra. Also 1T-Mo2C can in principle be exfoliated from the stable bulk Mo2C in the Pm1 structure (also experimentally reported in the sixties61). All other monolayers are associated with their corresponding bulk structures with stable phonon spectra but positive formation energies. This last feature does not necessarily mean that these structures do not exist. For example, Mo2C in the P63/mmc structure is predicted in our theory to have a small but positive formation energy of 0.097 eV f.u.−1. Nevertheless, thin films of P63/mmc Mo2C were recently grown on top of graphene62 and could be used to exfoliate 1H-Mo2C. The realisation of bulk metastable structures is not limited to very small formation energies, but is indeed rather common for transition metal carbides, and can be achieved by various techniques.63 For example, Ru2C has positive formation energies but can be formed in the Pm1 structure at high pressure and high temperature and then quenched under ambient conditions.64 Bulk Os2C in the P63/mmc structure, Rh2C in the Pm1 structure, and Tc2C in the Pm1 structure have all formation energies smaller than that for the aforementioned Ru2C. Therefore, we believe that they can possibly be stabilised and then used to exfoliate 1H-Os2C, 1T-Rh2C and 1T-Tc2C. Concerning Os2C, we should stress that it has been recently claimed that the P63/mmc structure is likely to form in a reaction in a carbon-saturated environment.65
After this analysis, we can investigate the cleavage energy Ecl due to the interlayer coupling strength, which is a way to explore how feasible the exfoliation process is.66 The cleavage energies of the predicted monolayers with respect to the separation distance of a fracture in the bulk material are reported in Fig. 6. 1H-Os2C stands out as the monolayer having the highest cleavage energy and can be achieved by overcoming a barrier of at least 0.81 J m−2. This value has to be compared with analogous values found in the literature. Theoretical calculations show67 that the cleavage energy of ReSe2 is around 1.12 J m−2, and the corresponding monolayer has been successfully exfoliated in experiment.68 Another relevant term of comparison is the experimentally estimated Ecl of graphite, which is around 0.37 J m−2.69 The cleavage energies of 1T-Mo2C and 1T-Rh2C are comparable to this value, while those of 1H-Mo2C and 1T-Tc2C are slightly larger, but still smaller than 0.6 J m−2. Among these monolayers, 1H-Tc2C is the easiest to exfoliate, with an Ecl of about 0.2 J m−2. Overall, the values reported in Fig. 6 are much smaller than those obtained for other materials that are considered suitable for mechanical exfoliation, such as Ca2N, TcSe2 or TcS2.67,70
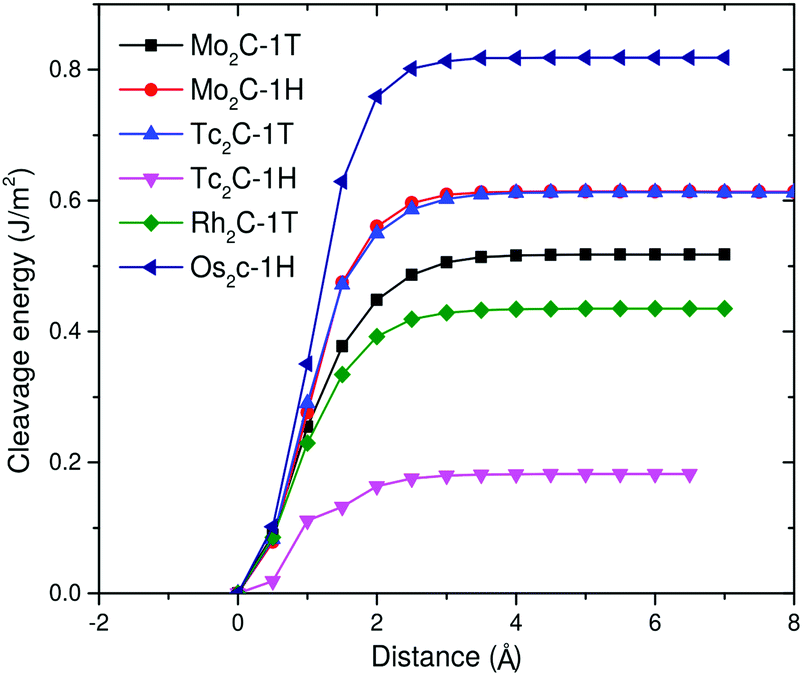 | ||
| Fig. 6 Cleavage energy Ecl in J m−2 as a function of the separation distance d for a fracture in bulk materials for the six (quasi) stable monolayers. | ||
An alternative process to synthesise the three predicted monolayers of the MXene family is via etching from related MAX phases. In fact, 1T-Mo2C has been already realised experimentally from the non-standard MAX phase Mo2Ga2C.17,21 Electronic structure calculations of the MAX phases M2XC, where M = Tc, Rh and X = Al, Si are reported in the ESI.† Among those, Tc2AlC and Rh2AlC have negative formation energies, but only the latter is dynamically stable and can therefore be used to create 1T-Rh2C. In order to find a corresponding MAX phase for 1T-Tc2C, we also performed calculations for Tc2Ga2C but this structure was also found to be unstable.
Conclusions and outlook
A new class of conductive transition metal carbide monolayers, BiXene, is here theoretically predicted. The BiXene family is reminiscent of the well-known MXenes, but is characterized by 1H symmetry, instead of 1T. The investigation of both BiXenes and MXenes arising from transition metal carbides TM2C, where TM = Mo, Tc, Ru, Rh, and Os, is motivated by their particularly layered structure, composed of a basic building block made by a sandwich of transition metal bilayers along with a graphene-like C layer in the middle. Among the systems studied here, we identify 1H-Mo2C, 1H-Tc2C and 1H-Os2C as possible BiXenes to synthesize in future experiments. The stronger inter-layered interactions of BiXene provide more stable 2D monolayers than MXene, which can be suitable for manipulating carriers in applications and devices. One can also expect larger and more dramatic effects induced by surface modifications and adsorption, which will be the object of our future studies. Also, the Fermi surface nesting found in 1T-Rh2C opens a link with the possible formation of charge-density waves. This, as well as exploring possible structural phase transitions induced by strain or temperature, will be addressed in future studies. Briefly, the emergence of BiXene will stimulate extensive studies on 2D metallic monolayers, and also supply another candidate material for nano-scale devices.In addition to the discovery of BiXene, in this work we also identify three systems in the MXene family, i.e. 1T-Tc2C, 1T-Rh2C, and 1T-Mo2C, which was also recently synthesised.17,21 These systems enrich the small group of MXenes originating from heavy transition metal carbides and are likely to lead to several interesting properties, which only future theoretical and experimental analyses will unveil. Moreover, the discovery of new pathways to MXenes through binary MCs or MNs will be very useful to overcome the limitation that most MXenes so far required to be produced from MAX phases including Al as an A element, which made many predicted monolayers not accessible experimentally.17
In practice, we suggest that these materials may be realised via mechanical exfoliation from their corresponding bulk structures. This seems immediately doable for 1T-Mo2C, 1H-Mo2C, and 1H-Tc2C, while 1H-Os2C, 1T-Rh2C and 1T-Tc2C require first that a suitable metastable bulk-like structure is realised in experiments. In contrast, 1T-Rh2C seems also possible to be synthesised from the MAX phase Rh2AlC.
Finally, the new M2C monolayers are analogous to MX2 in terms of structures (1T and 1H), but differ from the arrangement of the metal and non-metal sites, which is likely to lead to different chemical and physical properties. Our future research will focus on exploring the formation of BiXenes from other carbides and nitrides, possibly exploring all the transition metal elements that can form MAX phases. Furthermore, we will investigate in more detail the role of the temperature in stabilising the group of quasi-stable M2C monolayers, by means of more sophisticated techniques to describe thermally induced anharmonic effects.49–52
Acknowledgements
The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N) and at the National Supercomputer Cluster (NSC) in Sweden. The financial support from the Knut and Alice Wallenberg Foundation (KAW), the Swedish Research Council (VR), and eSSENCE is acknowledged. The authors appreciate the fruitful discussion with Prof. Shi-Li Zhang, Prof. Ulf Jansson and Ms Iulia Brumboiu.References
- K. Novoselov, A. K. Geim, S. Morozov, D. Jiang, M. I. Katsnelson, I. Grigorieva, S. Dubonos and A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- A. C. Neto, F. Guinea, N. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Tech., 2013, 3, 1782–1789 RSC.
- A. Acun, L. Zhang, P. Bampoulis, M. Farmanbar, A. van Houselt, A. N. Rudenko, M. Lingenfelder, G. Brocks, B. Poelsema, M. I. Katsnelson and H. J. W. Zandvliet, J. Phys.: Condens. Matter, 2015, 27, 443002 CrossRef CAS PubMed.
- N. Roome and J. D. Carey, ACS Appl. Mater. Interfaces, 2014, 6, 7743–7750 CAS.
- Y. Ma, Y. Dai, M. Guo, C. Niu, J. Lu and B. Huang, Phys. Chem. Chem. Phys., 2011, 13, 15546–15553 RSC.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- P. Miro, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537–6554 RSC.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6(2), 1322–1331 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- Z. Ling, C. E. Ren, M. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- C. Eames and M. S. Saiful, J. Am. Chem. Soc., 2014, 136, 16270–16276 CrossRef CAS PubMed.
- Y. Xie, Y. Dall'Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. C. Kent, ACS Nano, 2014, 8(9), 9606–9615 CrossRef CAS PubMed.
- M. W. Barsoum and M. Radovic, Annu. Rev. Mater. Res., 2011, 41, 159–227 CrossRef.
- M. W. Barsoum, T. Zhen, S. R. Kalidindi, M. Radovic and A. Murugaiak, Nat. Mater., 2003, 2, 107 CrossRef CAS PubMed.
- R. Meshkian, L.-A. Näslund, J. Halim, J. Lu, M. W. Barsoum and J. Rosen, Scr. Mater., 2015, 108, 147–150 CrossRef CAS.
- W. Sun, Y. Li, L. Zhu, Y. Ma, I. Di Marco, B. Johansson and P. Korzhavyi, Phys. Chem. Chem. Phys., 2015, 17, 9730 RSC.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X.-L. Ma, H.-M. Cheng and W. Ren, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- Y. Gogotsi, Nat. Mater., 2015, 14, 1079–1080 CrossRef CAS PubMed.
- J. Halim, S. Kota, M. R. Lukatskaya, M. Naguib, M.-Q. Zhao, E. J. Moon, J. Pitock, J. Nanda, S. J. May, Y. Gogotsi and M. W. Barsoum, Adv. Funct. Mater., 2016, 26, 3118–3127 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248 CrossRef CAS PubMed.
- R. B. Kaner, J. J. Gilman and S. H. Tolbert, Science, 2005, 308, 1269 CrossRef PubMed.
- J. K. Wassei and R. B. Kaner, Mater. Today, 2010, 13, 52–59 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 48, 13115 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. Klimes˘, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter, 2010, 82, 081101 CrossRef.
- M. Methfessel, M. van Schilfgaarde and R. A. Casali, A full-potential LMTO method based on smooth Hankel functions, Springer-Verlag, Berlin, 2000 Search PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter, 2008, 78, 134106 CrossRef.
- N. Mermin, Phys. Rev., 1965, 137, A1441 CrossRef.
- http://cms.mpi.univie.ac.at/vasp/vasp/vdW_DF_functional_Langreth_Lundqvis%t_et_al.html .
- S. Cahangirov, M. Topsakal, E. Aktürkürk, H. Şahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS PubMed.
- T. Li, Phys. Rev. B: Condens. Matter, 2012, 85, 235407 CrossRef.
- Y. Li, Y.-L. Li, W. Sun and R. Ahuja, Comput. Mater. Sci., 2014, 92, 206 CrossRef CAS.
- M. Khazaei, M. Arai, M. Sasaki, T. Estili, M. Estili and Y. Sakka, Phys. Chem. Chem. Phys., 2014, 16, 7841–7849 RSC.
- J. R. Sootsman, D. Y. Chung and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2009, 48, 8616–8639 CrossRef CAS PubMed.
- Z. Guo, J. Zhou, C. Si and Z. Sun, Phys. Chem. Chem. Phys., 2015, 17, 15348–15354 RSC.
- G. Gao, G. Ding, J. Li, K. Yao, M. Wu and M. Qian, Nanoscale, 2016, 8, 8986–8994 RSC.
- R. M. Martin, Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press, 2004 Search PubMed.
- D. Y. Kim, R. H. Scheicher and R. Ahuja, Phys. Rev. B: Condens. Matter, 2008, 78, 100102 CrossRef.
- V. I. Ivashchenko, P. E. A. Turchi and E. I. Olifan, Phys. Rev. B: Condens. Matter, 2010, 82, 054109 CrossRef.
- S. Gupta and P. K. Jha, Physica B, 2012, 407, 1978 CrossRef CAS.
- P. Hajiyev, C. Cong, C. Qiu and T. Yu, Sci. Rep., 2013, 3, 2593 Search PubMed.
- J.-A. Yan, M. A. D. Cruz, B. Cook and K. Varga, Sci. Rep., 2015, 5, 16646 CrossRef CAS PubMed.
- P. Souvatzis, O. Eriksson, M. I. Katsnelson and S. P. Rudin, Phys. Rev. Lett., 2008, 100, 095901 CrossRef CAS PubMed.
- P. Souvatzis, O. Eriksson, M. I. Katsnelson and S. P. Rudin, Comput. Mater. Sci., 2009, 44, 888–894 CrossRef CAS.
- P. Souvatzis and S. P. Rudin, Phys. Rev. B: Condens. Matter, 2008, 78, 184304 CrossRef.
- P. Souvatzis, T. Bjorkman, O. Eriksson, P. Andersson, M. I. Katsnelson and S. P. Rudin, J. Phys.: Condens. Matter, 2009, 21, 175402 CrossRef CAS PubMed.
- O. Pierson, Handbook of Refractory Carbides and Nitrides: Properties, Characteristics and Applications, Noyes Publications, Westwood, New Jersey, 1996 Search PubMed.
- W. Sun, H. Ehteshami and P. Korzhavyi, Phys. Rev. B: Condens. Matter, 2015, 91, 134111 CrossRef.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- J. A. Wilson and A. D. Yoffe, Adv. Phys., 1969, 18, 193–335 CrossRef CAS.
- W. Kohn, Phys. Rev. Lett., 1959, 9, 393 CrossRef.
- S. Piscanec, M. Lazzeri, F. Mauri, A. Ferrari and J. Robertson, Phys. Rev. Lett., 2004, 93, 185503 CrossRef CAS PubMed.
- J.-A. Yan, R. Stein, D. M. Schaefer, X.-Q. Wang and M. Y. Chou, Phys. Rev. B: Condens. Matter, 2013, 88, 121403 CrossRef.
- M. D. Johannes and I. I. Mazin, Phys. Rev. B: Condens. Matter, 2008, 77, 165135 CrossRef.
- E. Parthé and V. Sadagopan, Acta Crystallogr., 1963, 16, 202–205 CrossRef.
- C. He and J. Tao, Chem. Commun., 2015, 16, 8323–8325 RSC.
- S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Springer, Dordrecht, the Netherlands, 1996 Search PubMed.
- N. R. S. Kumar, N. V. C. Shekar, S. Chandra, J. Basu, R. Divakar and P. C. Sahu, J. Phys.: Condens. Matter, 2012, 24, 362202 CrossRef PubMed.
- E. A. Juarez-Arellano, B. Winkler, A. Friedrich, L. Bayarjargal, V. Milman, J. Yan and S. M. Clark, J. Alloys Compd., 2009, 481, 577–581 CrossRef CAS.
- N. I. Medvedeva, O. N. Mryasov, Y. N. Gornostyrev, D. L. Novikov and A. J. Freeman, Phys. Rev. B: Condens. Matter, 1996, 54, 13506–13514 CrossRef.
- Y. Jiao, L. Zhou, F. Ma, G. Gao, L. Kou, J. Bell, S. Sanvito and A. Du, ACS Appl. Mater. Interfaces, 2016, 8, 5385–5392 CAS.
- E. Liu, Y. Fu, Y. Wang, Y. Feng, H. Liu, X. Wan, W. Zhou, B. Wang, L. Shao, C.-H. Ho, Y.-S. Huang, Z. Cao, L. Wang, A. Li, J. Zeng, F. Song, X. Wang, Y. Shi, H. Yuan, H. Y. Hwang, Y. Cui, F. Miao and D. Xing, Nat. Commun., 2015, 6, 6991 CrossRef CAS PubMed.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter, 2004, 69, 155406 CrossRef.
- S. Zhao, Z. Li and J. Yang, J. Am. Chem. Soc., 2014, 136, 13313–13318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C6NR03602C |
| This journal is © The Royal Society of Chemistry 2016 |