
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaked-eye fingerprinting of single nucleotide polymorphisms on psoriasis patients†
Paola
Valentini‡
a,
Alessandra
Marsella‡
ab,
Paolo
Tarantino
c,
Salvatore
Mauro
c,
Silvia
Baglietto
d,
Maurizio
Congedo
e and
Pier
Paolo Pompa
*a
aNanobiointeractions & Nanodiagnostics, Istituto Italiano di Tecnologia (IIT), Via Morego, 30-16163 – Genova, Italy. E-mail: pierpaolo.pompa@iit.it
bDepartment of Engineering for Innovation, University of Salento, Via per Monteroni, 73100 – Lecce, Italy
cLaboratory of Medical Genetics, Vito Fazzi Hospital, Piazzetta Muratore, 73100 – Lecce, Italy
dPattern Analysis and Computer Vision (PAVIS), Istituto Italiano di Tecnologia (IIT), Via Morego, 30-16163 – Genova, Italy
eDepartment of Dermatology, Vito Fazzi Hospital, Piazzetta Muratore, 73100 – Lecce, Italy
First published on 28th April 2016
Abstract
We report a low-cost test, based on gold nanoparticles, for the colorimetric (naked-eye) fingerprinting of a panel of single nucleotide polymorphisms (SNPs), relevant for the personalized therapy of psoriasis. Such pharmacogenomic tests are not routinely performed on psoriasis patients, due to the high cost of standard technologies. We demonstrated high sensitivity and specificity of our colorimetric test by validating it on a cohort of 30 patients, through a double-blind comparison with two state-of-the-art instrumental techniques, namely reverse dot blotting and sequencing, finding 100% agreement. This test offers high parallelization capabilities and can be easily generalized to other SNPs of clinical relevance, finding broad utility in diagnostics and pharmacogenomics.
Sequencing of the human genome has shown that a significant part of the genetic variability among individuals is due to single nucleotide polymorphisms (SNPs).1 Nowadays, a bulk of knowledge is being accumulated on the involvement of an increasing number of SNPs in constitutional susceptibility to diseases and predictability of the response to drugs.2,3 These relationships are the focus of pharmacogenomics, the genetic pillar at the basis of personalized medicine. Current technologies for SNP diagnostics are mainly based on sequencing, real-time PCR, and hybridization assays, which however have limited clinical applicability, owing to their significant costs and instrument requirements, thus hampering the implementation of personalized therapies.4
Psoriasis is a chronic inflammatory skin disease affecting 2% of the world population.5 It is a heterogeneous disease, both in its clinical symptoms and in the underlying genetics. In recent years, Genome-Wide Association Studies (GWAS) have demonstrated the role of chromosomal regions, called PSORiasis Susceptibility (PSORS), in the risk of developing psoriasis. Moreover, several lines of evidence support the hypothesis that patients having a particular psoriasis-associated SNP can respond to treatment better than others.6 In particular, the HLA-C*06:02 (Human Leucocyte Antigen-C*06:02), located in the PSORS1 region on chromosome 6, was the first identified allele associated with psoriasis susceptibility7,8 and correlates with disease severity.9 HLA-C*06:02 positive patients showed a better response to the biological drug ustekinumab.10 Moreover, several SNPs in many other genes, mainly involved in the immune response, are associated with psoriasis.11–13 Notably, patients harboring certain mutations in the TNFAIP3 (Tumor Necrosis Factor-Alpha-Induced Protein 3) gene, respond better to anti-TNFα biological therapy.14,15 In particular, rs610604 G allele in the TNFAIP3 gene16 has been shown to be associated with a better response to several anti-TNFα drugs (etanercept, infliximab, and adalimumab).15 Currently, HLA genotype and TNFAIP3 SNPs are not routinely analyzed in psoriasis patients, due to economic constraints and relative novelty of the understanding of their pharmacogenomics implications. However, recent reports demonstrated that a four SNPs tagging is sufficient to identify both the presence and zygosity of the psoriasis susceptibility allele HLA-C*06:02, thus possibly substituting the more demanding whole gene analysis.17,18
Results
In this study, we show the low-cost detection of SNPs in the HLA locus and in the TNFAIP3 gene, aimed at identifying patients carrying the HLA-C*06:02 allele and the rs610604 G allele of TNFAIP3. We exploited a rapid and instrument-free colorimetric method for SNP detection, recently developed in combination with isothermal DNA amplification.19 We achieved the colorimetric genotypization of 30 real patients’ samples, and validated the results through double blind comparison with two standard techniques, namely reverse dot blotting and sequencing, demonstrating 100% accuracy.In order to identify the C*06:02 allele, we exploited the 4 tagging SNPs typing (rs3130457, rs6904246, rs7745906 and rs10484554)12,17 recently reported by Nikamo et al.18 The fifth SNP analyzed in this test was the above mentioned rs610604, falling in the TNFAIP3 gene. A total of 30 psoriasis patients were included in the study. 28 patients had been diagnosed with psoriasis vulgaris and 2 patients with palmoplantar psoriasis. Genomic DNA from patients’ blood samples was amplified by PCR with primers specific for the regions of the HLA locus and TNFAIP3 gene harboring the SNPs of interest (Table S1†).
Raw PCR samples were directly used for the colorimetric detection, according to the scheme depicted in Fig. 1. Briefly, streptavidinated paramagnetic microparticles were used to capture biotinylated PCR amplicons, which include the target SNPs to be tested. The PCR amplicons were denatured and subsequently hybridized with two different probes, the first one specific for the SNP (discriminating probe) and the second one, linked to AuNPs, specific for a fixed portion of the discriminating probe, providing the colorimetric detection (detection probe). After incubation with the streptavidinated magnetic microparticles, magnetic washing was performed to denature the amplicons and obtain single stranded DNA targets for the subsequent hybridization with the probes. For each SNP, two aliquots of the amplicon were hybridized with the discriminating probes specific for either allele. The universal AuNP probes were then added to the reactions. These consist of 40 nm AuNPs, functionalized with polyT oligonucleotides, which recognize a polyA tail in the discriminating probes.
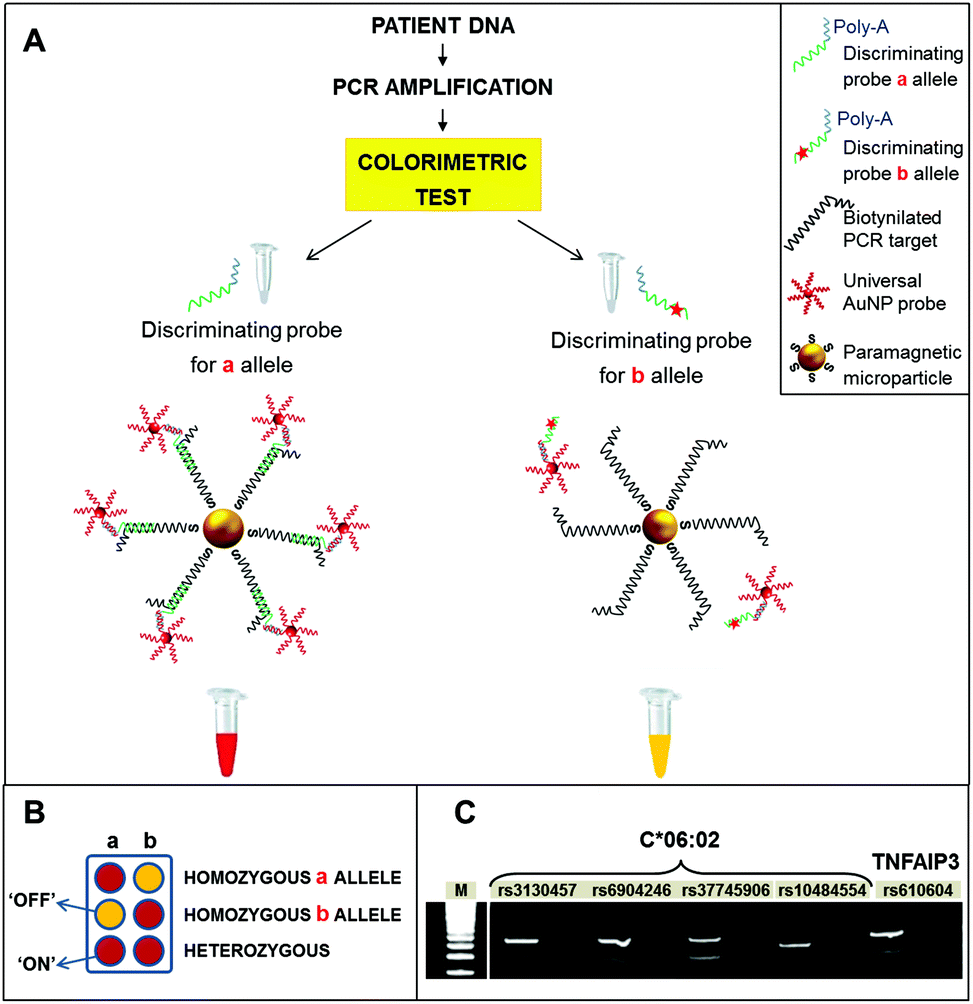 | ||
| Fig. 1 Strategy for the colorimetric SNP fingerprinting. (A) PCR reactions were directly analyzed by the colorimetric assay, using two discriminating probes, each specific for an allele variant (“a” or “b”). The PCR product hybridizes with the matching probe only (in this example, the one specific for allele a), capturing the AuNP probes on the microparticle surface, giving the sample a red color (left). In the case of mismatch (allele b is not present in this example), the sample remained yellow (right). The PCR negative control was analyzed with a mixture of the two discriminating probes and the resulting yellow color confirmed the absence of any amplified product (not shown in the scheme). (B) Legend for the interpretation of the results. Red color indicates “ON” result, i.e., the presence of that particular SNP variant. Yellow color indicates “OFF”. If both the samples containing the two discriminating probes turn red, the patient is heterozygous for that particular SNP. (C) Spot control of PCR specificity by polyacrylamide gel electrophoresis. | ||
Details about the synthesis, functionalization, and characterization of AuNP probes are provided in Materials and methods. Finally, samples were subjected to magnetic washing and the colorimetric result was assessed visually. In particular, red color (“ON” result), due to hybridized AuNPs, corresponds to a perfect match between the discriminating probe and the sample, indicating the detection of the specific allele. Conversely, yellow color (“OFF” result), due to pristine magnetic microparticles alone, indicated mismatch among the sample and probe, i.e., the absence of the allele tested for (Fig. 1B). Finally, as a further control, the negative PCR sample was hybridized with an equimolar mixture of the two discriminating probes. The resulting yellow color confirmed the absence of PCR contaminations, without the need for electrophoretic analysis. For TNFAIP3, homozygous GG, homozygous TT and heterozygous GT patients could be directly discerned from the colorimetric results. For HLA locus, the presence or absence of the C*06:02 allele was assessed by interpreting the results shown in Fig. 2. Indeed, the presence of a particular HLA allele can be identified by different combinations of SNPs, or haplotypes.17
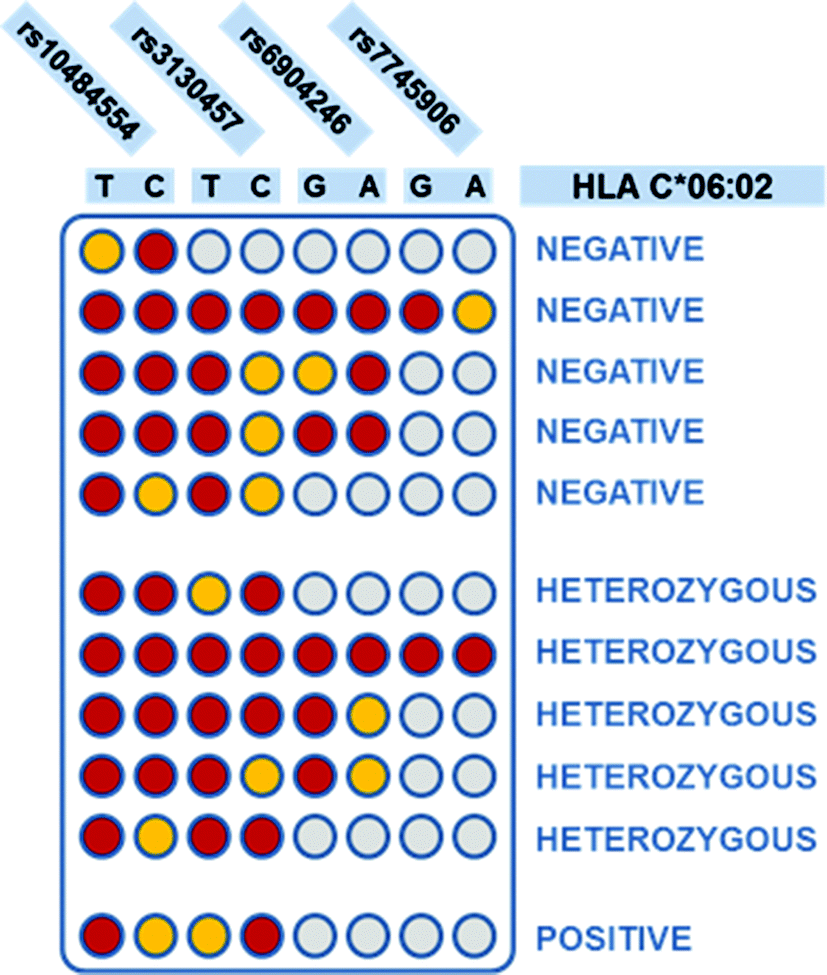 | ||
| Fig. 2 Interpretation of the four SNPs tagging,18 to determine C*06:02 presence and zygosity. Each column corresponds to a SNP variant, as indicated on top of the scheme. Each row corresponds to a possible combination of SNPs that can be found in a patient. Red color: “ON” result; yellow: “OFF”; grey: indifferent result (both presence and absence of the SNP variant return the same interpretation of the HLA-C*06:02 status). On the right, the interpretation of the HLA-C*06:02 status, for each SNP combination, is shown. | ||
Colorimetric test results are shown in Fig. 3. Each line of the plate corresponds to a patient, while each column corresponds to a discriminating probe, i.e., to an allele variant. The red/yellow experimental results corresponded well with the ON/OFF response of the test, and can be interpreted clearly and without bias by the naked-eye. Furthermore, the double test performed on each SNP allows distinguishing homozygous and heterozygous samples. For instance, according to the legend shown in Fig. 1B, the first patient is heterozygous for the first SNP (rs10484554), T allele homozygous for the second SNP (rs3130457), A allele homozygous for the third SNP (rs6904246), and so forth. The attribution of the C*06:02 allele could be easily inferred by visual comparison with a printed legend (Fig. 2). This latter describes all the possible combinations of the SNP status, corresponding to the absence or presence of the C*06:02 allele, or to heterozygosity for C*06:02. For each SNP, each of the two possible alleles may be present (red), absent (yellow) or either of the two (grey, indifferent). For instance, the first patient is C*06:02 negative (because it matches the status of the 3rd row in Fig. 2). Moreover, several different combinations of SNPs identify the presence of the C*06:02 allele in homozygosity (Fig. 2, last line). Indeed, any combination of SNPs rs6904246 and rs7745906 (homozygous for any of the alleles, or either one or both heterozygous) is compatible with C*06:02 homozygosity, while only SNP rs10484554 and SNP rs3130457 should necessarily be T and C, respectively.18 Similarly, other combinations of SNPs indicate heterozygosity or the absence of C*06:02.
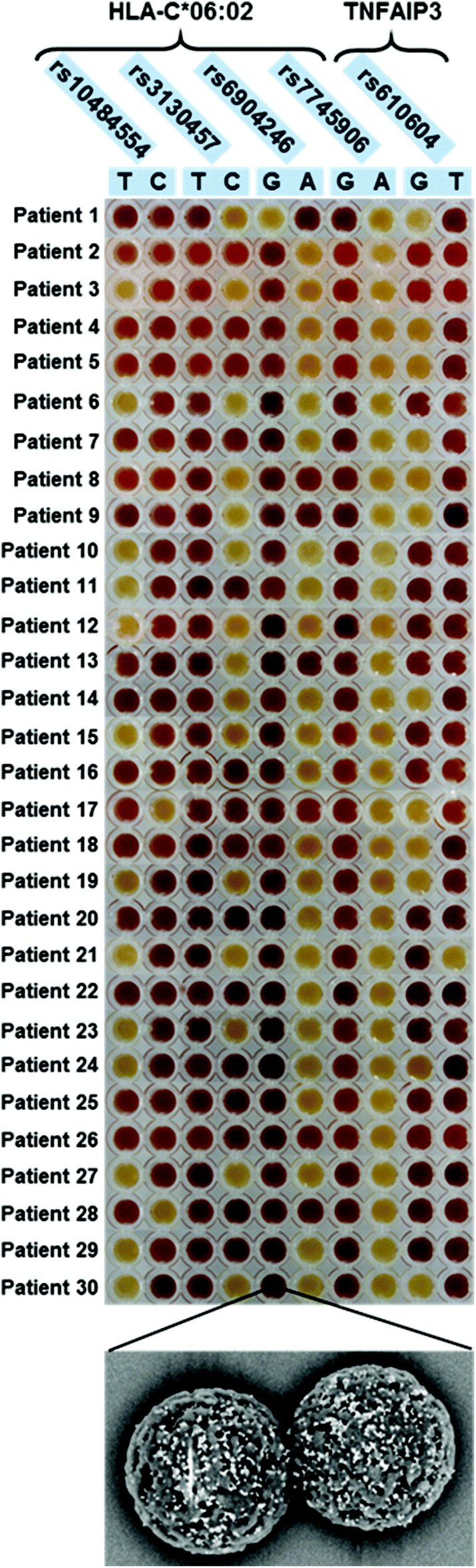 | ||
| Fig. 3 Results of the colorimetric test applied on 30 patients. Each row corresponds to a different patient, while each column to a different discriminating probe. For clarity, the negative control of the PCR reaction (always yellow), was not included in the picture (each patient's data row has been taken as a separate picture; pictures were then collated together for clarity of presentation). Bottom panel: representative SEM image of magnetic microparticles decorated with hybridized AuNPs (white dots). | ||
Notably, after standard PCR amplification, the duration of the colorimetric assay is ca. 30 min and requires no instrumentation. Moreover, all reagents, as well as the final colored samples, resulting from the presence or absence of hybridized AuNPs (Fig. 3, lower panel), are stable for months, allowing easy recording/archiving with no time-constraints.
All samples were also analyzed with two different state-of-the-art technologies, i.e. direct sequencing and reverse dot blotting (Fig. S2†). The primers used for direct sequencing are listed in Table S2.† All these studies were performed in double-blind. Interestingly, a comparison of our colorimetric test with standard techniques showed 100% agreement (Table 1), both when using the predictive method based on reverse dot blotting and when using direct sequencing, demonstrating an insignificant risk of producing erroneous results (both false positives and false negatives). The detailed results of the SNP genotypization by colorimetric test, sequencing, and reverse dot blot test, for each patient, are provided in Table S3.†
Discussion
The availability of a robust, rapid, and cost-effective tool for SNP fingerprinting can enhance pre-treatment screening and promote personalized medicine.4 Recently developed biological drugs for psoriasis have a better safety profile compared to conventional systemic drugs, but a cost up to ten times higher.11 Given the high variability of the therapeutic response among different patients,11 it would be crucial to predict the efficacy of a drug before treatment, as this could reduce unnecessary treatments and shrink the treatment-to-cure window, with great benefit to patients. However, treatments are most often administered on a trial-and-error basis, exposing the patient to avoidable side effects from ineffective drugs and delays in symptoms relief, which can be frustrating, especially for the severe forms of the disease, associated with serious and even potentially life-threatening comorbidities.Gold standard methods to genotype HLA alleles rely either on sequencing, or on PCR with sequence-specific-primers (SSP-PCR),20–22 or on array hybridization tests. In particular, a recently developed method, called the INNO-LiPA HLA-C kit, can detect human leukocyte antigen C alleles at the allele group level (first two digits of the HLA nomenclature,23e.g. HLA-C*06:xx), in a strip format, with one amplification and one detection line. This method does not directly detect the specific allele (second two digits in HLA nomenclature, e.g. HLA-C*06:02), but it can provide an estimate of it, based on statistical data, through the dedicated LiRAS software. Moreover, Olerup et al. have developed a SSP-PCR procedure,20,21 which requires two steps (and two different kits) to identify first the serological group, then the allele within that group. The PCR SSP results carry a large amount of information, as several allelic variants can be identified with the same test, and have to be carefully interpreted according to reference tables provided by the manufacturer. On the other hand, typing of tagging SNPs17 allows for simple interpretation of the results. The HLA-C*06:02 status can be assessed easily, based on a combination of four tagging SNPs18 (Fig. 2). Our test offers a low-cost and instrument-free alternative to standard techniques for SNP genotyping. Several AuNP-based colorimetric methods have been reported in the literature for the detection of nucleic acid targets, and also for single nucleotide polymorphisms (reviewed, for instance, in ref. 24, 25). Many of them exploit AuNPs aggregation and their consequent color-shift.26–29 When comparing simplified, colorimetric SNP detection assays with state-of-the-art instrumental techniques, the main issues are specificity and sensitivity.24 Although several reports have developed different strategies to deal with these issues, as a matter of fact, a great majority of commercial diagnostic tests for SNP genotyping currently rely on instrumental assays, mirroring the drawbacks that molecular strategies face when transferred to real-world samples. Compared to other hybridization-based methods, our test is very robust and simple to operate, and, importantly, displays zero background. This is due to a smart design that combines the use of AuNPs as a signal transducer and cooperative hybridization of probe oligonucleotides.30 Indeed, beyond the sensitive colorimetric detection, AuNPs provide enhanced specificity in mismatch discrimination, through their direct effect on the melting properties of DNA.31 Cooperative hybridization of adjacent probes, in turn, further enhances the stability of the binding of perfectly matched targets, over that of mismatched targets. In addition, the yellow-to-red colorimetric result proved to be bias-free, making the test appropriate for the development of an integrated point-of-care device, which can be operated and read even by untrained users, or by automated image analysis software (as shown, for instance, in Fig. S3†).
In this work, we have reported and validated a novel colorimetric test for the genotypization of a panel of SNPs, relevant in the pharmacogenomics of psoriasis. Given its diagnostic accuracy, simplicity and affordability, this test may find great utility in promoting personalized medicine of psoriasis patients. Furthermore, the use of universal AuNP detection probes, together with the great parallelization potential given by the reduced number of processing steps, simplifies the application of this assay to the colorimetric fingerprinting of other clinically relevant SNP panels.
Materials and methods
Synthesis and functionalization of 40 nm gold nanoparticles
40 nm citrate capped gold nanoparticles were synthesized as previously reported.32 AuNPs were conjugated with thiolated DNA probes consisting of a sequence of thymines (5′ T(30)–(O–CH2–CH2)3–SH 3′), purchased from IDT DNA, by standard gold–thiol chemistry. Functionalized AuNPs were centrifuged and washed four times with 0.3 M NaCl in 10 mM phosphate buffer, 0.01% SDS, in order to remove the excess unbound oligonucleotides. These conjugates DNA–AuNPs were stored at 4 °C until their use. Their concentration was determined by UV-vis spectrophotometry. In order to quantify the density of functionalization, functionalized AuNPs were digested overnight at 40 °C with 10 mM dithiothreitol (DTT) in order to displace the DNA from the AuNP surface. The digested suspension was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 rpm for 15 min and the supernatant was analyzed using the Quant-iT OliGreen ssDNA kit (Invitrogen). The number of oligonucleotides bound on the surface of a single AuNP was 430 ± 60. Both the as-synthesized AuNPs and AuNP probes were thoroughly characterized by UV-vis, Dynamic Light Scattering (DLS), and Transmission Electron Microscopy (TEM) (Fig. S1†).
400 rpm for 15 min and the supernatant was analyzed using the Quant-iT OliGreen ssDNA kit (Invitrogen). The number of oligonucleotides bound on the surface of a single AuNP was 430 ± 60. Both the as-synthesized AuNPs and AuNP probes were thoroughly characterized by UV-vis, Dynamic Light Scattering (DLS), and Transmission Electron Microscopy (TEM) (Fig. S1†).
Clinical samples and extraction of genomic DNA
A total of 30 psoriasis patients (21 females and 9 males, average age 47 years), under treatment at the department of Dermatology of Vito Fazzi Hospital in Lecce (Italy), were included in the study. Of these, 28 patients were affected by psoriasis vulgaris, with psoriatic lesions in specified body regions (upper and lower extremities, trunk, scalp, and nails), 2 patients by palmoplantar psoriasis. All patients provided written informed consent prior to any study-related procedures. In 4 patients the skin disorder was associated with psoriatic arthropathy (according to Moll and Wright criteria for PsA), diagnosed by imaging studies (enthesitis and tenosynovitis). PASI (Psoriasis Area and Severity Index) was used to assess psoriasis disease severity and the clinical efficacy of treatment. DNA samples for the genetic study were extracted from blood samples from all patients by using a NucliSENS® easyMAG® automated nucleic acid extraction system (BioMérieux, Durham, NC). The samples were then anonymized prior to testing. The concentration and purity of the extracted DNA were determined by measuring the absorbance at 260 nm by using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Inc.). The average concentration of genomic DNA in extracted samples was ≤5 ng μl−1. Only 1 μl of this material was used for the amplification and yielded enough material for the subsequent hybridization step.Live subject statement
All experiments were performed in compliance with the relevant laws and institutional guidelines. The study was approved by the ethical committee of the Vito Fazzi Hospital. Written informed consent was obtained from patients for DNA testing.Polymerase chain reaction (PCR) amplification and gel electrophoresis
Genomic DNA was amplified by traditional PCR to obtain approximately 100 bp fragments containing SNPs rs3130457, rs6904246, and rs7745906 for the determination of HLA C*06:02 allele and SNP rs610604 in the TNFAIP3 gene. The PCR reaction was set up in a 50 μL mixture containing 100 pg μL−1 of genomic DNA, 100 nM FW primer, 100 nM biotinylated REV primer, 2.5 mM MgCl2, 1× PCR reaction buffer, 200 μM dNTPs (Sigma-Aldrich) and 0.05 units per μL of Taq DNA Polymerase (Sigma-Aldrich). Sequences of all primers used are listed in Table S1.† DNA amplification was performed using the following thermal cycling conditions: 95 °C for 5 min, followed by 34 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min, with a final extension at 72 °C for 5 min. For the amplification of the region of the genome containing SNP rs10484554, which is a GC-rich region, the reaction mixture contained 200 pg μL−1 of genomic DNA, 1.875 mM MgCl2, 200 μM CleanAmp™ dNTP (Sigma-Aldrich) and 3% dimethyl sulfoxide (DMSO). In this latter case, thermal cycling conditions were: 95 °C for 5 min, followed by 38 cycles of 94 °C for 1 min, 59 °C for 30 s, 72 °C for 1 min, with a final extension at 72 °C for 5 min. PCR products from the first sample were checked by polyacrylamide gel electrophoresis, in order to assess the specificity of the primer sets and exclude off-target amplifications. Notably, the step of gel electrophoresis was not needed for subsequent samples, as these were directly analyzed with the colorimetric method, which also allows checking for possible interfering cross-contaminations of PCR reactions. The PCR products were separated on an 18% denaturing polyacrylamide gel, run in Tris-Borate-EDTA (TBE) buffer, they were then stained with SYBR® Gold (Thermo Fisher Scientific) and imaged on a Typhoon scanner (GE Healthcare).Colorimetric discrimination of single nucleotide polymorphisms (SNPs)
Colorimetric discrimination of the SNPs was performed according to a previously reported protocol19 with some optimizations. Briefly, each amplification product related to each SNP was analyzed in duplicate, in order to have a naked-eye discrimination of the results after hybridization with the two discriminating probes (Integrated DNA Technologies, Inc.) matching the two possible allele variants (Table S1†). For each duplicate, a quantity of 15 μL of the biotinylated PCR product was incubated for 5 min at room temperature with 5 μL of Dynabeads M-280 Streptavidin (Invitrogen) paramagnetic microparticles, preventively washed twice with hybridization buffer (HB) (1× PBS pH 7.4, 5% w/v PEG 600) and resuspended in an equal volume of HB. The microparticles coated with biotinylated PCR products were incubated with 50 μL of 0.15 M NaOH for 5 min to denature the double-stranded amplification product and magnetically separate the interfering non-biotinylated strand. The suspension was washed with 100 μL of 0.15 M NaOH and subsequently with 100 μL of HB, and finally it was resuspended in 20 μL of HB. The two samples were incubated for 5 min at room temperature with 10 μL of a 10 μM solution of one of the two discriminating probes. Then, 300 fmol of AuNPs conjugated with detection probes was added and kept at room temperature for 10 min. After a magnetic wash with 100 μL of HB at room temperature, the mixtures were incubated at 52 °C for 8 min and then resuspended in 25 μL of HB for the readout of the colorimetric result.Determination of the HLA C*06:02 allele
HLA-C*06:02 genotyping was determined with the SNP tagging method described by Nikamo and Ståhle18 after the naked-eye colorimetric genotyping for the four SNPs rs10484554, rs3130457, rs6904246 and rs7745906.Direct sequencing
Each PCR was carried out using 15 ng of genomic DNA, the AmpliTaq Gold 360 Master Mix (Applied Biosystems, Foster City, CA) and 1 μM of each primer, in a total volume of 25 μL. The PCR conditions for TNFAIP3 were as follows: heat denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 62 °C for 30 s, 72 °C for 45 s, and final extension at 72 °C for 10 min. The PCR conditions for the HLA C*06:02 related SNPs were as follows: heat denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, 64 °C for 15 s, 72 °C for 40 s, and final extension at 72 °C for 10 min. The PCR primer sequences are listed in Table S2.† After amplification, PCR products were cleaned by using a USB ExoSAP-IT PCR Product Cleanup kit (Affymetrix, S. Clara, CA). The PCR products were then directly sequenced using an ABI 310 PRISM™ sequencer (Applied Biosystems, Foster City, CA), using the Big-Dye Terminator Cycle Sequencing Ready Reaction Kit™ (Applied Biosystems, Foster City, CA). The genotypes of the five SNPs were examined using the software packages Sequencing Analysis 5.1 and SeqScape Software Version 2.1 (Applied Biosystems, Foster City, CA).Reverse dot blotting
The HLA typing tests were performed by reverse hybridization with the INNO-LiPA HLA-C kit (Fujirebio, Gent, Belgium). The test was performed using the Auto-LiPA automated test system, and the results were analysed with the LiRAS software (Fujirebio, Gent, Belgium). Briefly, biotinylated amplified targets, were chemically denatured, and then hybridized with membrane-immobilized probes. Afterwards, stringent washing removed mismatched hybridization. Then, streptavidin-conjugated alkaline phosphatase was added, followed by a chromogenic substrate for this enzyme. A purple/brown coloration of a probe band indicated perfect match of that specific probe with the target.Automatic analysis of the dataset
Automatic analysis of the dataset was performed on a photograph taken with a smartphone, exploiting established image processing tools, in two steps. First the contour of each well was detected as a circle using the circular Hough transform33 and, subsequently, a clustering step based on the internal color of each circle was applied to automatically group the data. In the second step, the RGB pixel values were converted to the YUV color space, because it is more similar to the average human perception. Then, the k-means clustering algorithm34 was used to separate the wells, selecting the average YUV color within the wells as a descriptive feature. k-Means is an iterative data-partitioning algorithm that assigns n observations to exactly one of the k clusters, where k is chosen before the algorithm starts. In the case of this dataset, n is 300, equal to sample cardinality (i.e. the number of wells) and k is 2. A warning label (blue), which requires a human control, was used in the case of rare light red-orange points wrongly clustered as yellow (because the RGB orange value is nearer to the yellow level than the red one). The k-mean algorithm has the advantage of finding two groups without any prior knowledge of the RGB level of yellow and red (so it can work with any two colors under any light conditions).Acknowledgements
This work was partially supported by the Italian Flagship Project NanoMax and by Pfizer through “HLA chip” project.References
- T. I. S.NP. Map and W. Group, A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms, Nature, 2001, 409, 928–933 CrossRef PubMed.
- A. Maxmen, Pharmacogenomics: playing the odds, Nature, 2011, 474, S9–S10 CrossRef CAS PubMed.
- J. Wapner, Pharmacogenomics. Gene variants affect hepatitis C treatment, but link is elusive, Science, 2010, 330, 579–579 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Bionanotechnology. Colloidal nanoparticles as advanced biological sensors, Science, 2014, 346, 53 CrossRef CAS PubMed.
- G. K. Perera, P. Di Meglio and F. O. Nestle, Psoriasis, Annu. Rev. Pathol.: Mech. Dis., 2012, 7, 385–422 CrossRef CAS PubMed.
- J. L. Harden, J. G. Krueger and A. M. Bowcock, The immunogenetics of Psoriasis: A comprehensive review, J. Autoimmun., 2015, 64, 66–73 CrossRef CAS PubMed.
- R. C. Trembath, R. L. Clough, J. L. Rosbotham, A. B. Jones, R. D. Camp, A. Frodsham, J. Browne, R. Barber, J. Terwilliger, G. M. Lathrop and J. N. Barker, Identification of a major susceptibility locus on chromosome 6p and evidence for further disease loci revealed by a two stage genome-wide search in psoriasis, Hum. Mol. Genet., 1997, 6, 813–820 CrossRef CAS PubMed.
- E. Mallon, M. Bunce, F. Wojnarowska and K. Welsh, HLA-CW*0602 is a susceptibility factor in type I psoriasis, and evidence Ala-73 is increased in male type I psoriatics, J. Invest. Dermatol., 1997, 109, 183–186 CrossRef CAS PubMed.
- J. E. Gudjonsson, A. Karason, E. H. Runarsdottir, A. A. Antonsdottir, V. B. Hauksson, H. H. Jonsson, J. Gulcher, K. Stefansson and H. Valdimarsson, Distinct clinical differences between HLA-Cw*0602 positive and negative psoriasis patients–an analysis of 1019 HLA-C- and HLA-B-typed patients, J. Invest. Dermatol., 2006, 126, 740–745 CrossRef CAS PubMed.
- M. Talamonti, E. Botti, M. Galluzzo, M. Teoli, G. Spallone, M. Bavetta, S. Chimenti and A. Costanzo, Pharmacogenetics of psoriasis: HLA-Cw6 but not LCE3B/3C deletion nor TNFAIP3 polymorphism predisposes to clinical response to interleukin 12/23 blocker ustekinumab, Br. J. Dermatol., 2013, 169, 458–463 CrossRef CAS PubMed.
- W. H. Boehncke and M. P. Schon, Psoriasis, Lancet, 2015, 386, 983–994 CrossRef CAS.
- Genetic Analysis of Psoriasis Consortium, the Wellcome Trust Case Control Consortium, A. Strange, F. Capon, C. C. Spencer, J. Knight, M. E. Weale, M. H. Allen, A. Barton and G. Band, et al., A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1, Nat. Genet., 2010, 42, 985–990 CrossRef CAS PubMed.
- L. C. Tsoi, S. L. Spain, J. Knight, E. Ellinghaus, P. E. Stuart, F. Capon, J. Ding, Y. Li, T. Tejasvi and J. E. Gudjonsson, et al., Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity, Nat. Genet., 2012, 44, 1341–1348 CrossRef CAS PubMed.
- R. Prieto-Perez, T. Cabaleiro, E. Dauden and F. Abad-Santos, Gene polymorphisms that can predict response to anti-TNF therapy in patients with psoriasis and related autoimmune diseases, Pharmacogenomics J., 2013, 13, 297–305 CrossRef CAS PubMed.
- T. Tejasvi, P. E. Stuart, V. Chandran, J. J. Voor-hees, D. D. Gladman, P. Rahman, J. T. Elder and R. P. Nair, TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis, J. Invest. Dermatol., 2012, 132, 593–600 CrossRef CAS PubMed.
- R. P. Nair, K. C. Duffin, C. Helms, J. Ding, P. E. Stuart, D. Goldgar, J. E. Gudjonsson, Y. Li, T. Tejasvi and B. J. Feng, et al., Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways, Nat. Genet., 2009, 41, 199–204 CrossRef CAS PubMed.
- S. Leslie, P. Donnelly and G. McVean, A statistical method for predicting classical HLA alleles from SNP data, Am. J. Hum. Genet., 2008, 82, 48–56 CrossRef CAS PubMed.
- P. Nikamo and M. Ståhle, Cost-effective HLA-Cw06:02 typing in a Caucasian population, Exp. Dermatol., 2012, 21, 221–223 CrossRef CAS PubMed.
- P. Valentini, R. Fiammengo, S. Sabella, M. Gariboldi, G. Maiorano, R. Cingolani and P. P. Pompa, Gold-nanoparticle-based colorimetric discrimination of cancer-related point mutations with picomolar sensitivity, ACS Nano, 2013, 7, 5530–5538 CrossRef CAS PubMed.
- O. Olerup and H. Zetterquist, HLA-DRB1*01 subtyping by allele-specific PCR amplification: a sensitive, specific and rapid technique, Tissue Antigens, 1991, 37, 197–204 CrossRef CAS PubMed.
- O. Olerup and H. Zetterquist, HLA-DR typing by PCR amplification with sequence-specific primers (PCR-SSP) in 2 hours: an alternative to serological DR typing in clinical practice including donor-recipient matching in cadaveric transplantation, Tissue Antigens, 1992, 39, 225–235 CrossRef CAS PubMed.
- M. Bunce, C. M. O'Neill, M. C. Barnardo, P. Krausa, M. J. Browning, P. J. Morris and K. I. Welsh, Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP), Tissue Antigens, 1995, 46, 355–367 CrossRef CAS PubMed.
- S. G. Marsh, Nomenclature for factors of the HLA system, update June 2015, Tissue Antigens, 2015, 86, 234–237 CrossRef PubMed.
- P. Valentini and P. P. Pompa, Gold nanoparticles for naked-eye DNA detection: smart designs for sensitive assays, RSC Adv., 2013, 3, 19181–19190 RSC.
- Z. Yuan, C.-C. Hu, H.-T. Chang and C. Lu, Gold nanoparticles as sensitive optical probes, Analyst, 2016, 141, 1611–1626 RSC.
- Y. Akiyama, H. Shikagawa, N. Kanayama, T. Takarada and M. Maeda, DNA dangling-end-induced colloidal stabilization of gold nanoparticles for colorimetric single-nucleotide polymorphism genotyping, Chemistry, 2014, 20, 17420–17425 CrossRef CAS PubMed.
- P. Valentini and P. P. Pompa, A universal polymerase chain reaction developer, Angew. Chem., Int. Ed., 2016, 55, 2157–2160 CrossRef CAS PubMed.
- T. Yu, P.-P. Dai, J.-J. Xu and H.-Y. Chen, Highly Sensitive Colorimetric Cancer Cell Detection Based on Dual Signal Amplification, ACS Appl. Mater. Interfaces, 2016, 8, 4434–4441 CAS.
- G. Wang, Y. Akiyama, T. Takarada and M. Maeda, Rapid Non-Crosslinking Aggregation of DNA-Functionalized Gold Nanorods and Nanotriangles for Colorimetric Single-Nucleotide Discrimination, Chemistry, 2016, 22, 258–263 CrossRef CAS PubMed.
- D. Y. Zhang, Cooperative hybridization of oligonucleotides, J. Am. Chem. Soc., 2011, 133, 1077–1086 CrossRef CAS PubMed.
- R. Jin, G. Wu, Z. Li, C. A. Mirkin and G. C. Schatz, What controls the melting properties of DNA-linked gold nanoparticle assemblies?, J. Am. Chem. Soc., 2003, 125, 1643–1654 CrossRef CAS PubMed.
- P. P. Pompa, G. Vecchio, A. Galeone, V. Brunetti, G. Maiorano, S. Sabella and R. Cingolani, Physical assessment of toxicology at nanoscale: nano dose-metrics and toxicity factor, Nanoscale, 2011, 3, 2889–2897 RSC.
- D. H. Ballard, Generalizing the Hough transform to detect arbitrary shapes, Pattern Recognit., 1981, 13, 111–122 CrossRef.
- G. A. F. Seber, Multivariate Observations, John Wiley & Sons, Inc., Hoboken, NJ, 1984 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr02200f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |