
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical networks of redox-active reduced crumpled graphene oxide and functionalized few-walled carbon nanotubes for rapid electrochemical energy storage†
Byeongyong
Lee
a,
Chongmin
Lee
bc,
Tianyuan
Liu
a,
Kwangsup
Eom
d,
Zhongming
Chen
e,
Suguru
Noda
e,
Thomas F.
Fuller
d,
Hee Dong
Jang
*bc and
Seung Woo
Lee
 *a
*a
aGeorge W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: seung.lee@me.gatech.edu
bRare Metals Research Center, Korea Institute of Geoscience and Mineral Resources, Yuseong-gu, Deajeon 305-350, Korea. E-mail: hdjang@kigam.re.kr
cDepartment of Nanomaterials Science and Engineering, University of Science & Technology, Deajeon 305-350, Korea. E-mail: hdjang@kigam.re.kr
dSchool of Chemical & Biomolecular Engineering, Center for Innovative Fuel Cell and Battery Technologies, Georgia Institute of Technology, Atlanta, GA 30332, USA
eDepartment of Applied Chemistry, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 25th May 2016
Abstract
Crumpled graphene is known to have a strong aggregation-resistive property due to its unique 3D morphology, providing a promising solution to prevent the restacking issue of graphene based electrode materials. Here, we demonstrate the utilization of redox-active oxygen functional groups on the partially reduced crumpled graphene oxide (r-CGO) for electrochemical energy storage applications. To effectively utilize the surface redox reactions of the functional groups, hierarchical networks of electrodes including r-CGO and functionalized few-walled carbon nanotubes (f-FWNTs) are assembled via a vacuum-filtration process, resulting in a 3D porous structure. These composite electrodes are employed as positive electrodes in Li-cells, delivering high gravimetric capacities of up to ∼170 mA h g−1 with significantly enhanced rate-capability compared to the electrodes consisting of conventional 2D reduced graphene oxide and f-FWNTs. These results highlight the importance of microstructure design coupled with oxygen chemistry control, to maximize the surface redox reactions on functionalized graphene based electrodes.
Introduction
The rapid penetration of portable electronic devices and electric vehicles into our daily lives increasingly demands multi-functional electrochemical energy storage devices having high energy, high power and long-lasting lifetime.1 Rechargeable lithium-ion batteries (LIBs) meet these demands of high energy density and efficiency, but their power density and cycling life are limited.2,3 On the other hand, electrochemical capacitors (ECs) can provide significantly higher power and longer cycling life than LIBs, but their energy density is limited.4,5 Numerous nanostructured materials have been studied to improve the performance of electrodes in both LIBs and ECs.6–8 In particular, nanocarbons, including graphene and carbon nanotubes (CNTs), have been intensively investigated for high-performance electrode materials utilizing their high electrical conductivity and surface area.9–12Meanwhile, recent progress in the large scale synthesis of graphene oxide (GO) dispersion from graphite13,14 has stimulated the investigation of reduced graphene based electrodes.12,15–17 Reduced graphene films can be prepared through a vacuum-filtration (VF) process18 of a GO dispersion and subsequent reduction process,19,20 yielding mechanically stable free-standing electrodes. However, a critical hurdle of graphene based electrodes prepared from GO dispersions is the irreversible restacking of graphene sheets during the preparation of electrodes due to the strong π–π interaction, decreasing the electrochemically accessible surface area.17,21,22 Various methods have been investigated in order to prevent the restacking issues of graphene based electrodes.22–26 Nanocomposite electrodes with physical ‘spacers’, such as carbon black,27 nanodiamond,28 and CNT,29 were employed to separate the graphene sheets, thereby securing the active surface area. In addition, 3D graphene assembly was developed by hydrothermal reduction30 or freeze-drying31 processes of GO dispersions, providing larger accessible surface with an interconnected network structure.32,33
Recently introduced crumpled graphene has a strong aggregation-resistive characteristic owing to its unique 3D ball morphology, proposing a promising solution to prevent the restacking problem.22,34 The crumpled graphene was employed as electrode materials for the electrochemical double layer capacitor (EDLC), a type of ECs, demonstrating enhanced capacitance and rate-capability.35 However, the energy density of the crumpled graphene based EDLC is still significantly lower than those of pseudocapacitors or LIBs utilizing redox (Faradaic) reactions.35,36 A rising strategy to improve the energy density of carbon based electrodes is introducing redox-active functional groups to support pseudocapacitance (surface redox reaction) on top of double layer capacitance.36–45 Previous studies showed reversible redox reactions between Li ions and oxygen functional groups, such as carbonyl groups, on the various carbon supports, including CNTs,36,40–45 reduced graphene,46,47 and biomass derived carbon spheres,48 at high redox potentials of ∼3 V vs. Li/Li+. Thus, we envision that if these redox reactions are introduced onto the partially reduced crumpled graphene oxide (r-CGO), the energy density of the r-CGO based electrodes can be considerably improved while maintaining their high rate-capability.
In this study, we demonstrate the redox-active properties of the r-CGO for electrochemical energy storage applications. We assemble free-standing composite electrodes consisting of oxygen functionalized few-walled carbon nanotubes (f-FWNTs)41,49 and r-CGO (f-FWNT/r-CGO) via a VF process. We show that the f-FWNT/r-CGO has a hierarchical network structure confirmed by scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM). The composite electrodes exhibit high gravimetric capacities of up to ∼170 mA h g−1 in Li-cells with superior rate-capability compared to that of a compositionally similar electrode consisting of conventional 2D reduced graphene oxide (rGO) and f-FWNTs. By correlating the microstructure and surface chemistry of the electrodes with their electrochemical performance, we reveal that the enhanced performance of the f-FWNT/r-CGO electrode can be attributed to the 3D hierarchical network structure, facilitating ion and electron transport for the effective utilization of the surface redox reactions between copious oxygen functional groups on the carbon supports and Li ions. These results provide important insights on the microstructure design of the functionalized carbon electrodes for the effective utilization of their surface redox reactions.
Results and discussion
Previously, the crumpled graphene was prepared by an aerosol spray evaporation of aqueous GO droplets at a high temperature of 800 °C followed by a chemical reduction process,35 resulting in very low oxygen contents. In this study, the partially reduced crumpled graphene oxide particle was prepared at a mild temperature of 300 °C without further chemical reduction process to maintain the oxygen functional groups on the surface of the r-CGO. Scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM) images of the as-synthesized r-CGO displayed a wrinkled particle shape with the size range of 300–700 nm (Fig. S1a and S1b†). High-magnification HRTEM images of the r-CGO particle displayed distorted ridges with thicknesses of several tens of nanometers (Fig. S1c and S1d†).22 The f-FWNTs were also prepared by chemically oxidizing FWNTs via previously reported protocols.41,49,50 The HRTEM investigation clearly showed that the smooth outer surface of the pristine FWNTs (Fig. S1e†) has roughened after the oxidization process (Fig. S1f†). This surface structural change is ascribed to the introduction of the oxygen functional groups on the surface of the FWNTs.43 The composite, f-FWNT/r-CGO, films were prepared by filtering the mixture of the f-FWNTs and r-CGO dispersion with varied mass ratios, yielding free-standing, additive-free electrodes with a density of ∼0.4 g cm−3 (Scheme 1). It should be mentioned that the assembly of the r-CGO only free-standing electrode is impossible owing to its strong aggregation resistant property. Therefore, sub-millimeter long FWNTs49 were employed as a conductive binder to prepare a free-standing composite electrode. The f-FWNT/rGO composite electrode was also prepared using the mixture of the f-FWNTs and conventional GO via a VF process.44 In the mixture of the f-FWNTs and GO, same mass of the f-FWNTs and GO was used. The GOs within this electrode were further thermally reduced at 200 °C for 2 h under an Ar environment, finally producing the f-FWNTs/reduced GO (rGO) composite electrodes. The microstructures of the f-FWNT/r-CGO and f-FWNT/rGO composite electrodes were compared (Fig. 1). The f-FWNT/r-CGO electrode with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio between the f-FWNTs and r-CGO showed a 3D hierarchical porous structure, where the r-CGO was randomly entangled with f-FWNTs (Fig. 1a–d). We can envision fast ion and electron transport through this 3D porous network structure, enabling fast charge storage. In contrast, the f-FWNT/rGO electrode displayed a 2D alternating layered structure, where rGO sheets are separated by the inserted f-FWNTs (Fig. 1e and f). In this 2D structure, electrolyte ions can be readily transported along the in-plane direction, but their fast transport through the perpendicular direction can be restricted, potentially limiting the rate-capability of the electrode.
:1 mass ratio between the f-FWNTs and r-CGO showed a 3D hierarchical porous structure, where the r-CGO was randomly entangled with f-FWNTs (Fig. 1a–d). We can envision fast ion and electron transport through this 3D porous network structure, enabling fast charge storage. In contrast, the f-FWNT/rGO electrode displayed a 2D alternating layered structure, where rGO sheets are separated by the inserted f-FWNTs (Fig. 1e and f). In this 2D structure, electrolyte ions can be readily transported along the in-plane direction, but their fast transport through the perpendicular direction can be restricted, potentially limiting the rate-capability of the electrode.
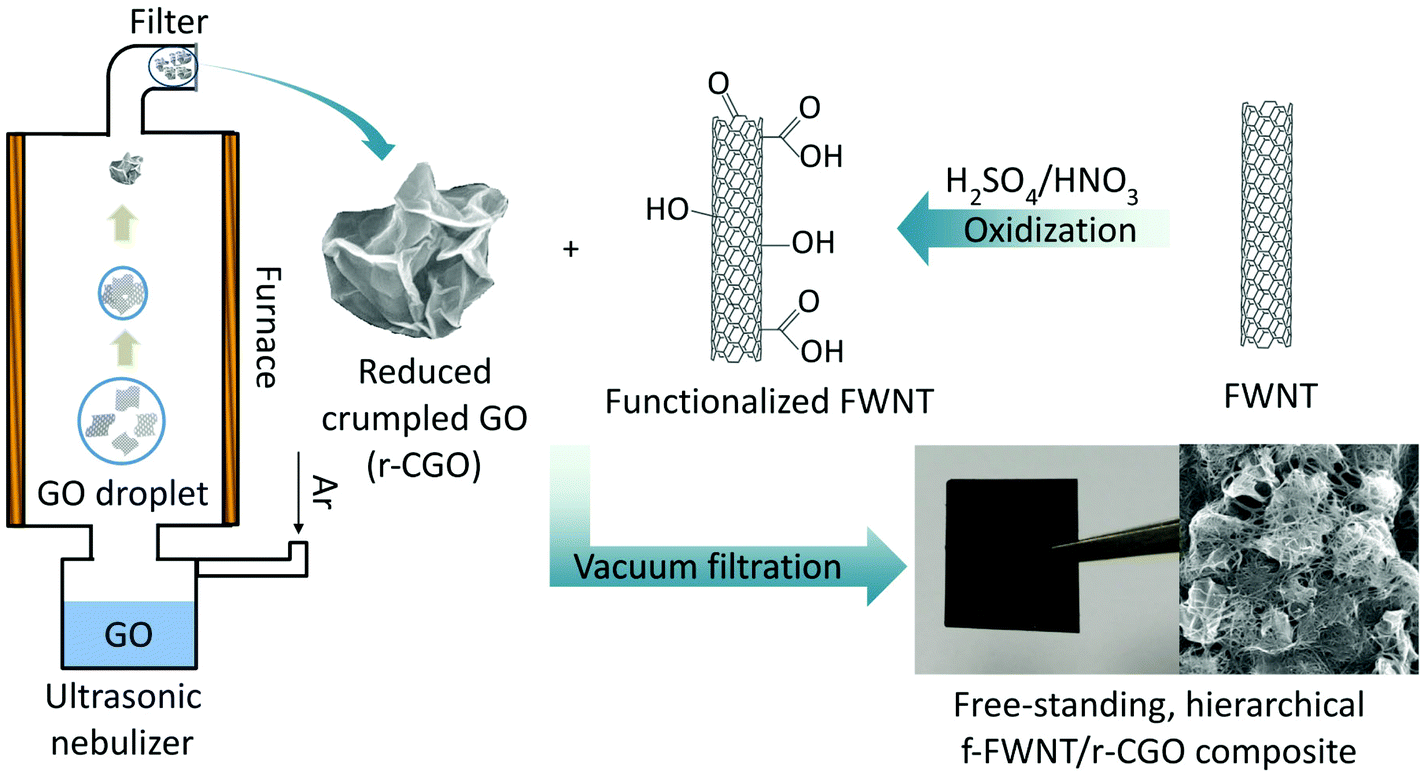 | ||
| Scheme 1 Fabrication process of the f-FWNT/r-CGO electrode; the nebulized GO dispersion was crumpled in a furnace at a mild temperature of 300 °C, producing partially reduced crumpled graphene oxide (r-CGO) particles. The f-FWNTs were prepared by oxidizing FWNTs in a mixture of acids, H2SO4/HNO3. The mixture of the r-CGO and f-FWNTs dispersion was vacuum-filtered, producing the free-standing f-FWNT/r-CGO composite electrode. | ||
 | ||
| Fig. 1 (a, b) Scanning electron microscopy (SEM) cross-section view images of the f-FWNT/r-CGO electrode. (c, d) High resolution transmission electron microscopy (HRTEM) images of the f-FWNT/r-CGO electrode. (e, f) SEM cross-section view images of the f-FWNT/rGO electrode. | ||
The f-FWNT/rGO and f-FWNT/r-CGO electrodes showed both a D band at ∼1370 cm−1 and a G band at ∼1600 cm−1 (Fig. S2†).51 The D band arises from disorder of sp2-hybridized carbon in graphene, while the G band originates from the in-plane stretching mode of the carbon–carbon bond in graphitic materials.52 The relative intensity of D to G band (ID/IG) of the f-FWNT/r-CGO electrode was slightly higher (ID/IG = 0.98) than that of the f-FWNT/rGO electrode (ID/IG = 0.82), indicative of the greater number of defect sites in the f-FWNT/r-CGO electrode. X-ray diffraction (XRD) analysis was used to further investigate the structure of the assembled electrodes (Fig. 2a and S3†). The f-FWNT/GO electrode showed an intense peak at 2θ = 11.6° (an interlayer distance of 0.76 nm), which is similar to that observed in stacked graphene oxide sheets (Fig. 2a).53 This indicates that some of the GO sheets are still locally restacked, as opposed to the fully separated structure resulting from the inserted f-FWNTs. A layered structure of graphitic materials can be characterized by a (002) peak at the 2θ = 26.2°.30 Previous study showed that graphite or restacked graphene sheets exhibit sharp (002) peaks at 2θ = 26.2° due to their 2D layered structures, whereas the 3D graphene hydrogels display broad and weak (002) peaks owing to the poor ordering along the stacking directions and their porous structure.54 After the thermal reduction process from the f-FWNT/GO to the f-FWNT/rGO, the intense peak at 2θ = 11.6° of the f-FWNT/GO electrode disappeared and the broad peak at 2θ = 25.7° (d-spacing of 0.35 nm) increased, which can be attributed to the removal of oxygen groups with the concomitant formation of the loosely packed rGO structure.54,55 In contrast, the f-FWNT/r-CGO electrode showed negligible peaks compared to those of other films, further illustrating its disordered structure (Fig. 1).
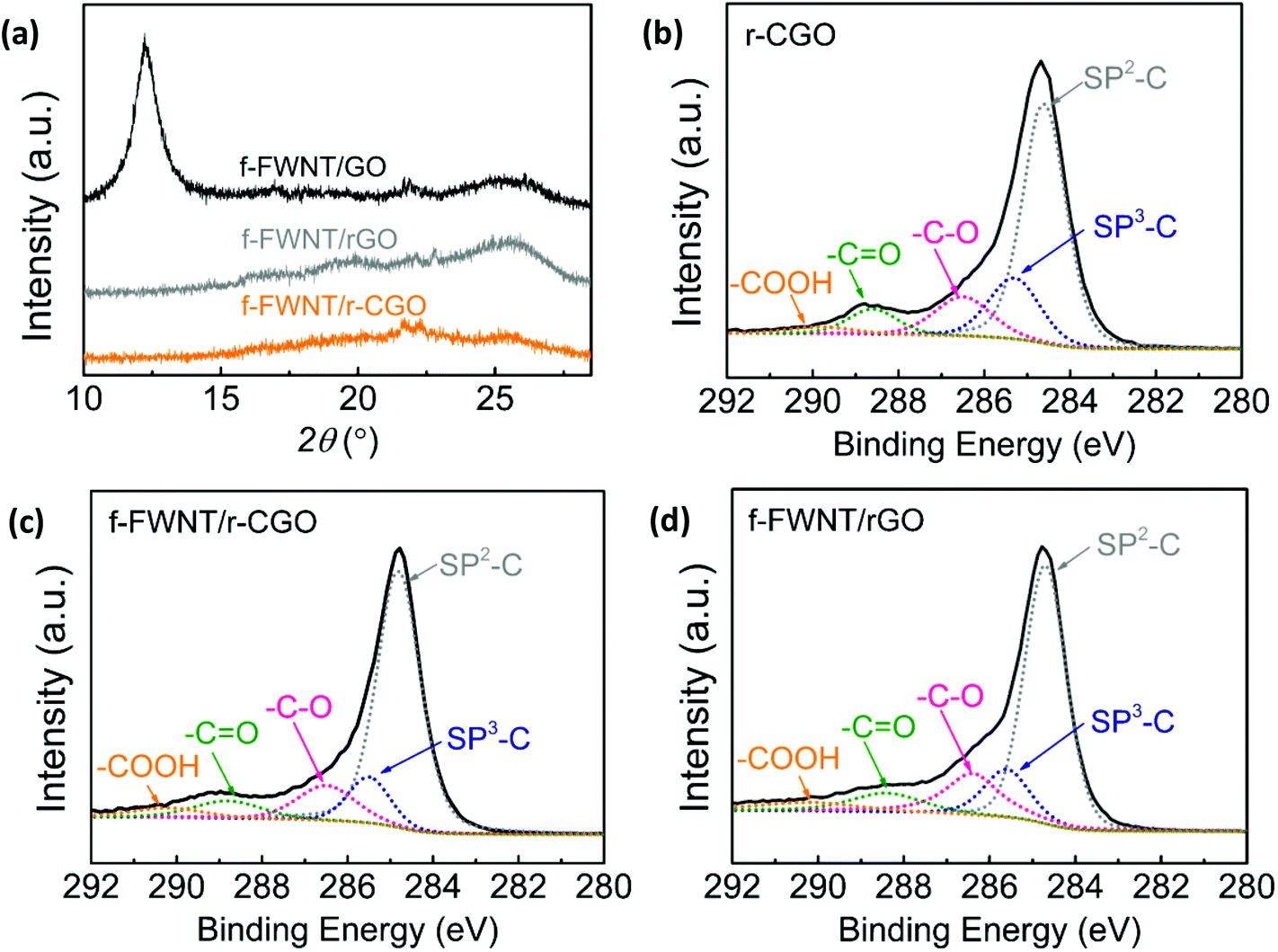 | ||
| Fig. 2 (a) X-ray diffraction (XRD) spectra of the composite electrodes. Surface chemistry investigation by high resolution C 1s spectra of the (b) partially reduced crumpled graphene oxide (r-CGO), (c) f-FWNT/r-CGO and (d) f-FWNT/rGO. | ||
The surface oxygen chemistry of the carbon materials was examined by XPS. Wide survey scan peaks with atomic ratios of oxygen to carbon (O/C) were compared for GO, r-CGO, f-FWNTs, and the assembled electrodes (Fig. S4a†). The GO had the highest O/C ratio of 0.41, while the r-CGO showed an O/C ratio of 0.26, indicative of its partially reduced structure. The O/C ratio of FWNTs (0.13) is lower than those of GO and r-CGO and thus the composite electrodes showed decreased O/C ratios for f-FWNT/GO (0.28) and for f-FWNT/rGO (0.17). The f-FWNT/rGO also showed an O/C ratio of 0.17, indicating compositional similarity of the oxygen functional groups to the f-FWNT/r-CGO. The density normalized electrical conductivities of the f-FWNT/GO, f-FWNT/rGO, and f-FWNT/r-CGO electrodes were found to be 22.7, 74.9 and 67.1 S cm2 g−1, respectively, showing a correlation with the O/C ratios of the electrodes.56,57 The main components of the oxygen functional groups were investigated by the high resolution C 1s spectra (Fig. 2b–d, S4b, S4c and S4d†). The peaks were fitted with sp3-hybridized carbon at 285.2 ± 0.2 eV, hydroxyl or epoxide (–C–O) at 286.5 ± 0.2 eV, carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 288.2 ± 0.2 eV, and carboxyl group (–COOH) at 290.2 ± 0.2 eV by setting the sp2-hybridized carbons at 284.5 eV as the basis.21,53,58 The high resolution C 1s spectra of the r-CGO (Fig. 2b) displayed considerably reduced hydroxyl or epoxide group (–C–O) peak intensity and negligible change of carbonyl (–CO) and carboxyl (–COOH) group peaks compared to those of the GO (Fig. S4b†) owing to the partial reduction process at a mild temperature.44 The high resolution C 1s spectra of the f-FWNTs electrode showed similar oxygen functional group peaks yet slightly decreased intensities compared to those of the r-CGO (Fig. S4c†). It is worth mentioning that the f-FWNT/r-CGO and FWNT/rGO are compositionally similar as supported by the same O/C ratio of 0.17 (Fig. 2c and d), providing good model systems to compare the electrochemical performance of the 2D and 3D nanostructured carbon electrodes.
O) at 288.2 ± 0.2 eV, and carboxyl group (–COOH) at 290.2 ± 0.2 eV by setting the sp2-hybridized carbons at 284.5 eV as the basis.21,53,58 The high resolution C 1s spectra of the r-CGO (Fig. 2b) displayed considerably reduced hydroxyl or epoxide group (–C–O) peak intensity and negligible change of carbonyl (–CO) and carboxyl (–COOH) group peaks compared to those of the GO (Fig. S4b†) owing to the partial reduction process at a mild temperature.44 The high resolution C 1s spectra of the f-FWNTs electrode showed similar oxygen functional group peaks yet slightly decreased intensities compared to those of the r-CGO (Fig. S4c†). It is worth mentioning that the f-FWNT/r-CGO and FWNT/rGO are compositionally similar as supported by the same O/C ratio of 0.17 (Fig. 2c and d), providing good model systems to compare the electrochemical performance of the 2D and 3D nanostructured carbon electrodes.
Electrochemical charge storage characteristics of the composite electrodes were investigated by cyclic voltammetry (CV) tests in the voltage range of 1.5–4.5 V vs. Li/Li+ in Li-cells (Fig. 3a, b and S5†). Comparison of CV scans at a slow scan rate of 1 mV s−1 showed that the f-FWNT/r-CGO electrode exhibited higher gravimetric current density compared to that of the f-FWNT/rGO electrode (Fig. 3a). The f-FWNT/GO electrode displayed negligible redox peaks despite its high oxygen content (Fig. S5a†), due to the poor electrical conductivity of the GO.12 After the reduction process, the f-FWNT/rGO electrode showed indistinct redox features in the broad voltage range of 2.5–4 V vs. Li/Li+. On the other hand, the f-FWNTs/r-CGO electrode displayed significantly enhanced redox peaks centered at ∼3 V vs. Li/Li+, that are ascribed to the redox reactions between oxygen functional groups and Li ions.36,41,44,47,59 The difference of the gravimetric current density between f-FWNT/r-CGO and other electrodes became prominent at a high scan rate of 5 mV s−1 (Fig. 3b). The f-FWNT/r-CGO electrode still maintained its redox peaks with the increased gap between oxidation and reduction peak potentials, while other f-FWNT/rGO and f-FWNT/GO electrodes only showed a skewed CV shape with negligible redox behaviour. Since the O/C ratios and electrical conductivities of the f-FWNT/r-CGO and f-FWNT/rGO electrodes are similar, the enhanced charge storage performance of the f-FWNT/r-CGO can be attributed to its unique microstructure facilitating the surface redox reactions of the oxygen functional groups. We postulate that the 3D hierarchical structure of the f-FWNT/r-CGO can provide a continuous ion transport pathway into the inner space of the electrode for effective utilization of the oxygen functional groups, while the 2D layered structure of the f-FWNT/rGO have limited ion accessibility through the rGO layers. Galvanostatic rate capability tests were carried out for the assembled electrodes, showing sloped discharge and charge curves (Fig. 3c, d, and S5b†). The sloped discharge and charge profiles of the functionalized carbon electrodes can be attributed to the coupling of the charge storage mechanisms: double-layer capacitance and the surface redox reactions of various oxygen functional groups, as reported in a previous study.47 At a slow discharge rate of 0.1 A g−1, the f-FWNT/r-CGO electrode exhibited a gravimetric capacity of ∼154 mA h g−1, which is higher than those of the f-FWNT/GO (∼95 mA h g−1, Fig. S5b†) and f-FWNT/rGO (∼126 mA h g−1) electrodes. As the current density increased to 1 A g−1, the capacity of the f-FWNT/rGO rapidly decreased to ∼40 mA h g−1, whereas the f-FWNT/r-CGO still retained a high capacity of ∼100 mA h g−1. The difference in gravimetric discharge capacities between the composite electrodes gradually increased with increasing current density (Fig. 3e). At a high current density of 10 A g−1, the f-FWNT/r-CGO exhibited ∼68 mA h g−1, while the f-FWNT/rGO showed negligible gravimetric capacity. Consistent with the CV results, the trend in the galvanostatic discharge further supports faster charge storage performance of the f-FWNT/r-CGO electrode compared to the f-FWNT/rGO electrode owing to its unique 3D structure than can facilitate ion transport.
 | ||
| Fig. 3 Cyclic voltammetry (CV) scans in the voltage range of 1.5–4.5 vs. Li/Li+ at scan rates of (a) 1 mV s−1 and (b) 5 mV s−1 of the composite electrodes. Galvanostatic rate-dependent discharge and charge profiles of (c) the f-FWNT/r-CGO and (d) f-FWNT/rGO at the varied current densities of 0.1–10 A g−1. (e) Gravimetric discharge capacity comparisons of the f-FWNT/r-CGO and f-FWNT/rGO as a function of current density. (f) Electrochemical impedance spectroscopy (EIS) investigation of the f-FWNT/r-CGO and f-FWNT/rGO electrodes. Nyquist plot of the composite electrodes was analysed by an equivalent electrical model, Randles circuit. −ZIm and ZRe were multiplied by electrode area. | ||
We employed electrochemical impedance spectroscopy (EIS) to further understand the difference of the rate-capability between the electrodes. Fig. 3f shows Nyquist plots and the Randles equivalent circuit of the f-FWNT/r-CGO and f-FWNT/rGO electrodes. Charge transfer resistance (Rct) typically gives information about the kinetics of the redox reactions occurring at the electrode surface.60 The values of Rct for the f-FWNT/r-CGO and f-FWNT/rGO are ∼1000 and ∼2350 Ω cm2, respectively, indicating much faster redox reactions of the f-FWNT/r-CGO than f-FWNT/rGO. At the low frequency region, the f-FWNT/r-CGO electrode exhibited a steeper slope compared to that of the f-FWNT/rGO, implying that the hierarchical porous structure can efficiently support fast ion diffusion to keep up with the change of frequency.28,61 It is worth mentioning that the tested f-FWNT/r-CGO had the optimum mass ratio (f-FWNT/r-CGO = 1:1) which showed the best electrochemical performance. We compared rate-dependent discharge and charge profiles of the f-FWNT/r-CGO electrodes with various mass ratios (1:0, 3:1, 2:1, 1:1, 1:2 and 1:3) (Fig. S6†). The f-FWNT/r-CGO electrode with the 1:1 mass ratio showed highest gravimetric capacity and rate-capability, indicating the optimal structure for maximizing the accessible active surface area and facilitating electron and ion transport to support the fast redox reactions. The assembled electrodes showed stable cycling stability for up to 3000 cycles with their coulombic efficiency values close to ∼100% (Fig. 4a). During the initial several hundred cycles, the specific capacities slightly increased, approaching a stable capacity of ∼170 mA h g−1. This capacity is comparable or higher than those of the reported carbon based free-standing positive electrodes (117–160 mA h g−1) (Table S1†).43–47,62 The increased capacity during the initial several hundred cycles may be attributed to the gradual electrolyte diffusion into the electrode surface and the activation of functional groups within the electrode. Comparison of the voltage profiles of the electrodes showed no dramatic change between the 100th and 3000th cycles (Fig. 4b).5,47,63 During the cycling process, the electrode maintained its initial hierarchical structure well (Fig. S7a†). In addition, the main oxygen functional groups on the cycled electrode showed negligible difference compared to the initial surface chemistry before cycling (Fig. S7b† and Fig. 2c). The cycling stability can be attributed to the surface limited charge storage mechanisms, including both double-layer capacitance and surface redox reactions.
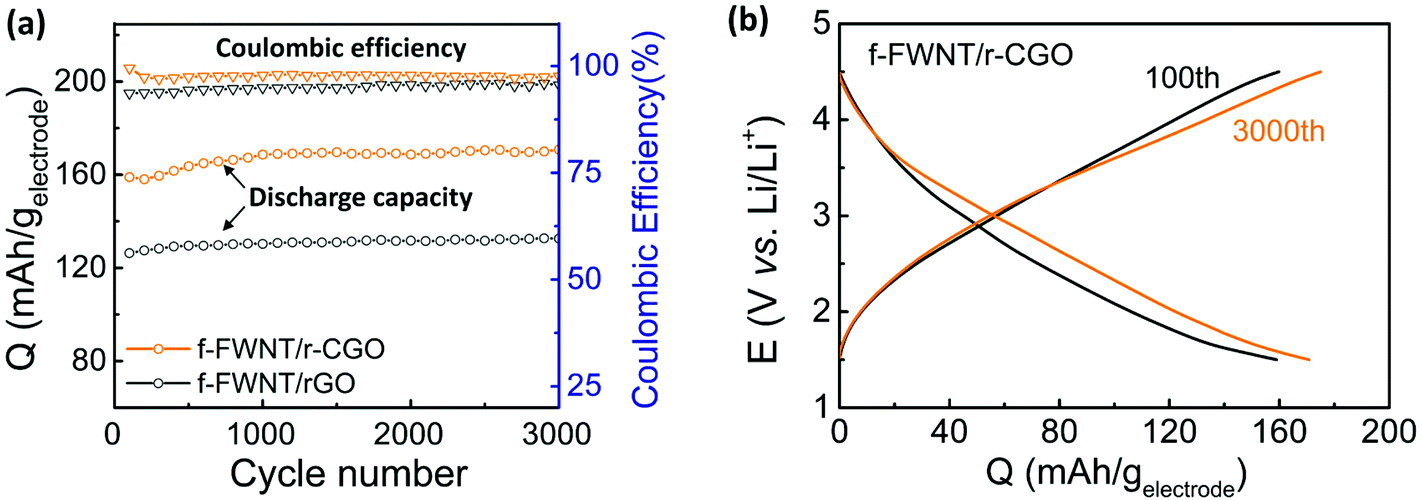 | ||
| Fig. 4 (a) Gravimetric discharge capacities and coulombic efficiencies of the electrodes (at a current density of 0.1 A g−1) as a function of cycle number. (b) Comparisons of the galvanostatic discharge and charge voltage profiles at the 100th and 3000th cycles of the f-FWNT/r-CGO electrode. | ||
Conclusions
In summary, we have demonstrated the effective utilization of the surface redox reactions on the r-CGO for enhanced electrochemical energy storage. The r-CGO was assembled as free-standing electrodes with the f-FWNTs via a VF process. The assembled f-FWNT/r-CGO electrode showed a 3D hierarchical porous structure, enabling fast ion and electron transport through the electrode. The f-FWNT/r-CGO electrode exhibited higher gravimetric capacity and significantly enhanced rate-capability compared to those of the compositionally similar, yet geometrically different f-FWNTs/rGO electrode. Thus, the superior electrochemical performances of the f-FWNT/r-CGO electrodes can be attributed to the 3D hierarchical nanostructures, which can provide not only efficient electron pathways but also facilitate fast ion diffusion to support fast surface redox reactions. These results provide meaningful insights on the microstructure design of functionalized carbon electrodes for the effective utilization of their surface redox reactions, which can be directly incorporated for the development of high-power Li-ion capacitors or LIBs.Experimental
Preparation of GO, r-CGO and f-FWNTs
Colloidal GO solution was prepared using the modified Hummers’ method.13,41 Partially reduced crumpled graphene oxide (r-CGO) was synthesized from sprayed droplets of the colloidal GO solution by aerosol spray pyrolysis.22,64 The 0.5 wt% of the GO solution was nebulized in an ultrasonic atomizer (Htech Co, model US-06) to generate sprayed water droplets containing GO nanosheets. The droplets were carried by Ar gas with a flow rate of 10 L min−1 into a pre-heated (∼300 °C) tubular furnace. The as-fabricated r-CGO via evaporation of water in the droplets and concomitant thermal reduction was collected from a filter located at the end part of the tubular furnace, and the gathered r-CGO was dispersed in deionized water and ethanol (volume ratio, 1:1) for 10 min in a sonicator bath. FWNTs were prepared by a chemical vapor deposition method.49,50 The FWNTs were oxidized by an acidic treatment.41 In brief, 100 mg of the pristine FWNTs were refluxed in a mixture of 150 mL of H2SO4 and 50 mL of HNO3 at 80 °C for 2 h. The f-FWNTs were obtained after rinsing the mixture with 5 vol% of HCl solution in deionized (DI) water followed by rinsing with DI water. Finally, the f-FWNTs dispersion was obtained by ultrasonication of f-FWNTs in a water and ethanol mixture (volume ratio, 1:1) for 40 min.
Preparation of free-standing, hierarchical composite electrode
The free-standing f-FWNT/r-CGO films were obtained by a vacuum filtration process.25 The f-FWNTs and r-CGO dispersions were mixed and agitated for 30 min. The mixture was vacuum-filtered with a polypropylene separator (Celgard 2500) as a filtration membrane. After an air-drying process for 1 h, the film was detached from the membrane followed by vacuum drying overnight at 70 °C. The f-FWNTs, GO and f-FWNT/GO electrodes were also obtained by the same preparation process of the f-FWNT/r-CGO. The f-FWNT/rGO was prepared by thermally reducing the f-FWNT/GO in a tube-furnace at 200 °C for 2 h under an Ar environment.Characterization
The morphology of the electrodes was investigated by cold field emission scanning electron microscope (SEM) (Hitachi SU8010, operated at 3 kV). Transmission electron microscopy (TEM) was carried out using a JEOL JEM-ARM200F. The elemental analysis of the films was examined by X-ray photoelectron microscopy (XPS, Thermal Scientific K-alpha XPS instrument) and C 1s peaks were fitted by XPSPEAKS 4.1.47 The relative intensity of D and G bands was examined by Raman Spectroscopy (Thermo Nicolet Almega XR Dispersive Raman Spectrometer). X-ray diffraction (XRD) was investigated using an X'Pert Pro Alpha-1. The electrical conductivity was measured by Lucas Lab Pro 4-point systems. Electrochemical impedance analysis was carried out using AutoLab Metrohm.Electrochemical measurements
The prepared electrodes were cut into a small piece with a razor blade and directly used as positive electrodes without binders or conductive additives in Swagelok-type cells. A piece of pure lithium foil was used as the negative electrode. Celgard 2500 and 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (volume ratio, 3:7, BASF) were adopted for separator and electrolyte, respectively. The Li-cells were assembled in a glove box (MBraun), maintaining O2 and H2O <0.1 ppm. The electrochemical performances of the Li-cells were investigated with a potentiostat/galvanostat (Bio-Logic VMP3) in the voltage range of 1.5–4.5 V vs. Li/Li+. The Li-cells were discharged or charged with varied galvanostatic current densities (from 0.1 A g−1 to 10 A g−1), and the cell voltage was held constant for 30 min, 1.5 V and 4.5 V at each end of discharge and charge, respectively. The cycling stability was examined by an accelerated cycling test.47,59 After completing 99 cycles at 10 A g−1 without holding the voltage, the Li-cells were discharged/charged at 0.1 A g−1 for one cycle holding the voltage for 30 min.
Acknowledgements
This study was supported by the R&D Center for Valuable Recycling (Global-Top Environmental Technology Development Program), funded by the Ministry of Environment, Korea. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation.Notes and references
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972–1985 CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- R. Mukherjee, R. Krishnan, T. M. Lu and N. Koratkar, Nano Energy, 2012, 1, 518–533 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- Z. S. Wu, W. C. Ren, L. Xu, F. Li and H. M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- W. F. Chen, L. F. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Y. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J.M. Gibson and J. X. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- J. H. Lee, N. Park, B. G. Kim, D. S. Jung, K. Im, J. Hur and J. W. Choi, ACS Nano, 2013, 7, 9366–9374 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- G. K. Wang, X. Sun, F. Y. Lu, H. T. Sun, M. P. Yu, W. L. Jiang, C. S. Liu and J. Lian, Small, 2012, 8, 452–459 CrossRef CAS PubMed.
- Y. Q. Sun, Q. O. Wu, Y. X. Xu, H. Bai, C. Li and G. Q. Shi, J. Mater. Chem., 2011, 21, 7154–7160 RSC.
- L. Qiu, X. W. Yang, X. L. Gou, W. R. Yang, Z. F. Ma, G. G. Wallace and D. Li, Chem. – Eur. J., 2010, 16, 10653–10658 CrossRef CAS PubMed.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- L. L. Jiang and Z. J. Fan, Nanoscale, 2014, 6, 1922–1945 RSC.
- C. Li and G. Q. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- Y. Zhao, C. G. Hu, Y. Hu, H. H. Cheng, G. Q. Shi and L. T. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371–11375 CrossRef CAS PubMed.
- W. N. Wang, Y. Jiang and P. Biswas, J. Phys. Chem. Lett., 2012, 3, 3228–3233 CrossRef CAS PubMed.
- J. Y. Luo, H. D. Jang and J. X. Huang, ACS Nano, 2013, 7, 1464–1471 CrossRef CAS PubMed.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- C. T. Hsieh and H. Teng, Carbon, 2002, 40, 667–674 CrossRef CAS.
- J. Han, L. L. Zhang, S. Lee, J. Oh, K. S. Lee, J. R. Potts, J. Y. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2013, 7, 19–26 CrossRef CAS PubMed.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. G. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- S. W. Lee, B. S. Kim, S. Chen, Y. Shao-Horn and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671–679 CrossRef CAS PubMed.
- H. R. Byon, S. W. Lee, S. Chen, P. T. Hammond and Y. Shao-Horn, Carbon, 2011, 49, 457–467 CrossRef CAS.
- M. N. Hyder, S. W. Lee, F. C. Cebeci, D. J. Schmidt, Y. Shao-Horn and P. T. Hammond, ACS Nano, 2011, 5, 8552–8561 CrossRef CAS PubMed.
- S. W. Lee, B. M. Gallant, Y. Lee, N. Yoshida, D. Y. Kim, Y. Yamada, S. Noda, A. Yamada and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 5437–5444 CAS.
- H. R. Byon, B. M. Gallant, S. W. Lee and Y. Shao-Horn, Adv. Funct. Mater., 2013, 23, 1037–1045 CrossRef CAS.
- S. Y. Kim, J. Hong, R. Kavian, S. W. Lee, M. N. Hyder, Y. Shao-Horn and P. T. Hammond, Energy Environ. Sci., 2013, 6, 888–897 CAS.
- S. H. Ha, Y. S. Jeong and Y. J. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 12295–12303 CAS.
- T. Y. Liu, K. C. Kim, R. Kavian, S. S. Jang and S. W. Lee, Chem. Mater., 2015, 27, 3291–3298 CrossRef CAS.
- T. Liu, R. Kavian, Z. Chen, S. S. Cruz, S. Noda and S. W. Lee, Nanoscale, 2016, 8, 3671–3677 RSC.
- D. Y. Kim, H. Sugime, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2011, 49, 1972–1979 CrossRef CAS.
- Z. M. Chen, D. Y. Kim, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2014, 80, 339–350 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio and R. Saito, Annu. Rev. Condens. Matter. Phys., 2010, 1, 89–108 CrossRef CAS.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- J. Ma, M. X. Yang, F. Yu and J. Zheng, Sci. Rep., 2015, 5, 13578, DOI:10.1038/srep13578.
- S. H. Huh, Physics and Applications of Graphene - Experiments, 2011, pp. 73–90, DOI:10.5772/590.
- O. C. Compton, B. Jain, D. A. Dikin, A. Abouimrane, K. Amine and S. T. Nguyen, ACS Nano, 2011, 5, 4380–4391 CrossRef CAS PubMed.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- S. Kundu, Y. M. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CAS.
- T. Y. Liu, R. Kavian, I. Kim and S. W. Lee, J. Phys. Chem. Lett., 2014, 5, 4324–4330 CrossRef CAS PubMed.
- A. P. Periasamy, J. F. Liu, H. M. Lin and H. T. Chang, J. Mater. Chem. A, 2013, 1, 5973–5981 CAS.
- X. H. Cao, Y. M. Shi, W. H. Shi, G. Lu, X. Huang, Q. Y. Yan, Q. C. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- T. Y. Liu, R. Kavian, Z. M. Chen, S. S. Cruz, S. Noda and S. W. Lee, Nanoscale, 2016, 8, 3671–3677 RSC.
- H. Kim, K. Y. Park, J. Hong and K. Kang, Sci. Rep., 2014, 4, 5278, DOI:10.1038/srep05278.
- J. Y. Luo, X. Zhao, J. S. Wu, H. D. Jang, H. H. Kung and J. X. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr02013e |
| This journal is © The Royal Society of Chemistry 2016 |