
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlbumin-coated SPIONs: an experimental and theoretical evaluation of protein conformation, binding affinity and competition with serum proteins†
Siming
Yu
a,
Alex
Perálvarez-Marín
b,
Caterina
Minelli
c,
Jordi
Faraudo
a,
Anna
Roig
*a and
Anna
Laromaine
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain. E-mail: roig@icmab.es; alaromaine@icmab.es
bUnitat de Biofísica, Departament de Bioquímica i de Biologia Molecular, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
cAnalytical Science, National Physical Laboratory, Hampton Road, TW110LW Teddington, UK
First published on 24th May 2016
Abstract
The variety of nanoparticles (NPs) used in biological applications is increasing and the study of their interaction with biological media is becoming more important. Proteins are commonly the first biomolecules that NPs encounter when they interact with biological systems either in vitro or in vivo. Among NPs, super-paramagnetic iron oxide nanoparticles (SPIONs) show great promise for medicine. In this work, we study in detail the formation, composition, and structure of a monolayer of bovine serum albumin (BSA) on SPIONs. We determine, both by molecular simulations and experimentally, that ten molecules of BSA form a monolayer around the outside of the SPIONs and their binding strength to the SPIONs is about 3.5 × 10−4 M, ten times higher than the adsorption of fetal bovine serum (FBS) on the same SPIONs. We elucidate a strong electrostatic interaction between BSA and the SPIONs, although the secondary structure of the protein is not affected. We present data that supports the strong binding of the BSA monolayer on SPIONs and the properties of the BSA layer as a protein-resistant coating. We believe that a complete understanding of the behavior and morphology of BSA-SPIONs and how the protein interacts with SPIONs is crucial for improving NP surface design and expanding the potential applications of SPIONs in nanomedicine.
Introduction
The use of nanoparticles (NPs) for medicine is becoming a key technology. The development of NPs for a broad range of activities and with multifunctional properties is blooming. The successful commercialization of these NPs is limited due to a lack of control and prediction of the final NP properties, which decide the way NPs interact with the environment and their ultimate fate.1,2Proteins are commonly the first biomolecules that NPs encounter when they interact with biological systems either in vitro or in vivo. The adsorption of proteins on the surface of NPs leads to changes in the conformation and biological activity of the proteins, and modifies the designed properties of the NPs,3–5 which can subsequently affect in vitro or in vivo biological responses.6–9 The ability to develop NPs with protein-resistant coatings can impact the development of efficient drug-delivery systems with specific targeting strategies. Different strategies have been developed; for instance, using polyethylene glycol (PEG),10 which prevents protein adsorption, confers stability to the NPs, decreases the uptake of NPs by culture cells in vitro11,12 and by entire organisms in vivo,13 and increases their retention time in blood flow. However, some studies found that PEG can be oxidized by enzymes and some proteins still adsorb on to it.10,14,15
Natural products as bovine serum albumin (BSA) are also exploited to form a protective layer on the NPs to improve biocompatibility and transport of the NPs;16 for instance, novel commercial drugs, such as Abraxane, incorporate albumin in the drug formulation to efficiently accumulate the drug in the tumor due to an enhanced permeability and retention and to reduce the risk of hypersensitivity reactions. Drug release in albumin carriers can be triggered naturally by protease digestion or pH-responsive systems; the payload can be tuned by the amount of albumin on the NPs, and the unfolding of the protein on the NP can facilitate their clearance by phagocytes.17 Additionally, BSA maintains the colloidal stability of the NPs and it is a standard additive in the serum used in cell-culture experiments. Because of these properties, BSA seems a good coating candidate to obtain biocompatible and protein-resistant NPs.
The US Food and Drug Administration approved the use of super-paramagnetic iron oxide nanoparticles (SPIONs) in the 1990s as nuclear magnetic resonance imaging contrast agents, and they hold great promise for applications in magnetic separation, cell targeting, imaging, and in drug-delivery systems.18–20
The potential of SPIONs for biomedical applications, and the benefits of albumin coatings, lead us to investigate how BSA interacts with SPIONs (BSA-SPIONs; Scheme 1) and how it can improve SPIONs’ performance. Different strategies have been published using covalent or non-covalent immobilization of BSA on SPIONs. Strategies to covalently anchor BSA require the modification of the initial SPIONs with tedious reactions with organic solvents (toxic for biological applications), the exchange of solvents and they restrict and fix the conformation of the anchored protein.16,21 Non-covalent strategies require hydrophilic iron oxide surfaces, the common method of synthesis cannot offer it; and it adds an extra step.16,17 In our case, we synthesized SPIONs using microwave-assisted thermal decomposition and capped them with citrate, rendering hydrophilic SPIONs in a single step. Both strategies, covalent and non-covalent, afford more biocompatible and colloidally stable SPIONs systems and confirm the formation of a hard corona on the SPIONs surfaces. We chose non-covalent interaction for its practicality and efficiency. Here we study bovine albumin coated SPIONs (BSA-SPIONs) in detail by using a broad range of techniques; we also evaluate the binding strength of BSA on SPIONs and the potential of BSA coating to resist serum–protein adsorption by using isothermal titration calorimetry and gel electrophoresis. A complete understanding of the behavior and morphology of BSA-SPIONs and how the protein interacts with SPIONs are crucial for improving NPs surface design and expanding the potential applications of SPIONs in nanomedicine.
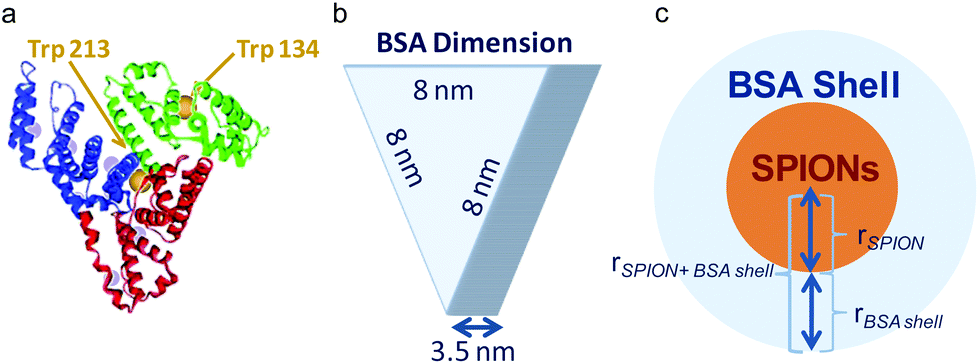 | ||
| Scheme 1 (a) Crystal structure of BSA where the position of the tyrosine (gray spheres) and tryptophan (labeled) residues are highlighted. (b) Scheme of the dimensions of the BSA as an equilateral triangular prism. (C) Schematic of the BSA-SPION complex. | ||
Methods
Chemicals and materials
Iron(III) acetylacetonate (Fe(acac)3, ≥97.0%), trisodium citrate dihydrate (citrate), and bovine serum albumin (BSA, ≥98%, Sigma, A7906-50G), phosphate-buffered saline (PBS, P5493-1L) 10× was diluted before use were purchased from Sigma-Aldrich and used as received. Fetal bovine Serum (FBS, 10270106) was bought from Life Technologies.Protocol for the preparation of bovine serum albumin coated SPIONs (BSA-SPIONs) are described previously in detail.22 In brief, SPIONs were synthesized by microwave-assisted thermal decomposition of the iron precursor and then coated with citrate in a single step, to obtain citrate-coated SPIONs (C-SPIONs). This approach produced monodisperse C-SPIONs in just 15 min. Transmission electron microscopy (TEM) revealed that C-SPIONs are spherical with a diameter of 6 ± 1 nm. Dynamic light scattering (DLS) indicated a hydrodynamic diameter (Dh) of 15 ± 1 nm in water, while the ζ-potential was −36 mV and a saturated magnetization of 62 emu g−1 was measured at room temperature by magnetometry.22 BSA was adsorbed on the C-SPIONs’ surface by using a pH-adjusted protocol, which significantly improved the stability of C-SPIONs in a wide range of biological media.16,22 C-SPIONs in water were adjusted to pH 11 with 0.1 mM NaOH and then BSA was incubated with them at this pH for 1 h. Subsequently, the system was neutralized to pH 7.4 with 0.6 mM HNO3 and the excess BSA was removed by centrifugation for all the experiments, (Fig. S1†). Hereafter, we will refer to this system as BSA-SPIONs.
BSA-SPIONs used in this work were all purified if not stated otherwise; purification followed the protocol: the obtained BSA-SPIONs dispersions were transferred to a centrifugal filter unit (Amicon, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MW, Millipore), centrifuged at 2548g for 25 min to remove the unbound BSA. The removed supernatant was collected and UV-vis spectra were recorded. The concentrated BSA-SPIONs were then redispersed in MilliQ water and filtered again under the same conditions. The filtration and centrifugation steps were repeated several times until the supernatant showed no UV-vis absorption at 280 nm. In our case, after three washing steps, UV-vis spectra of the isolated solution did not show any absorption at 280 nm (Fig. S2a†), which indicates the complete removal of free BSA from the BSA-SPIONs dispersion. The purified BSA-SPIONs were characterized further.
000 MW, Millipore), centrifuged at 2548g for 25 min to remove the unbound BSA. The removed supernatant was collected and UV-vis spectra were recorded. The concentrated BSA-SPIONs were then redispersed in MilliQ water and filtered again under the same conditions. The filtration and centrifugation steps were repeated several times until the supernatant showed no UV-vis absorption at 280 nm. In our case, after three washing steps, UV-vis spectra of the isolated solution did not show any absorption at 280 nm (Fig. S2a†), which indicates the complete removal of free BSA from the BSA-SPIONs dispersion. The purified BSA-SPIONs were characterized further.
Characterization
XPS spectra were acquired using an Axis-Ultra XPS instrument (Kratos Analytical, Manchester, UK) with monochromatic Al Kα X-rays (15 kV, 5 mA) in hybrid lens mode. Survey scans (single sweep) were acquired with a pass energy of 160 eV, step size 1000 meV, and a dwell time of 300 ms. Narrow scans were performed for the Fe 2p, C 1s, N 1s, O 1s, S 2p, and F 1s regions with a pass energy of 40 eV, step size 100 meV and a dwell time of 500 ms. Two sweeps were acquired for each of the narrow scan regions except for iron, carbon, and nitrogen, for which four sweeps were obtained. Charge neutralization was employed to reduce charging of the samples, and balance settings were determined on an individual basis. XPS spectra were analyzed by using CasaXPS software (Version 2.3.16) and intensities were calibrated with the NPL's (National Physical Laboratory) own transmission function and average matrix relative sensitivity factors. Tougaard backgrounds were used for peak quantification, except for a minority of cases where linear background was used instead.
Zeta potential (ζ-potential) was carried out on a Zetasizer Nano ZS (Malvern) apparatus with a zeta potential analyzer software. C-SPIONs (50 μg mL−1) solutions were placed in a disposable plastic cuvette, followed by collecting experimental data automatically using the built-in software. For each measurement, 20 cycles were collected, and at least 3 independent measurements were performed for each sample.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of BSA, while the one at 280 nm is due to aromatic amino acid residues such as tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe).4
O of BSA, while the one at 280 nm is due to aromatic amino acid residues such as tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe).4
Equation Fo/F vs. [SPIONs] were represented in logarithmic form, and a sigmoidal curve was fitted to the data, to give the equation:
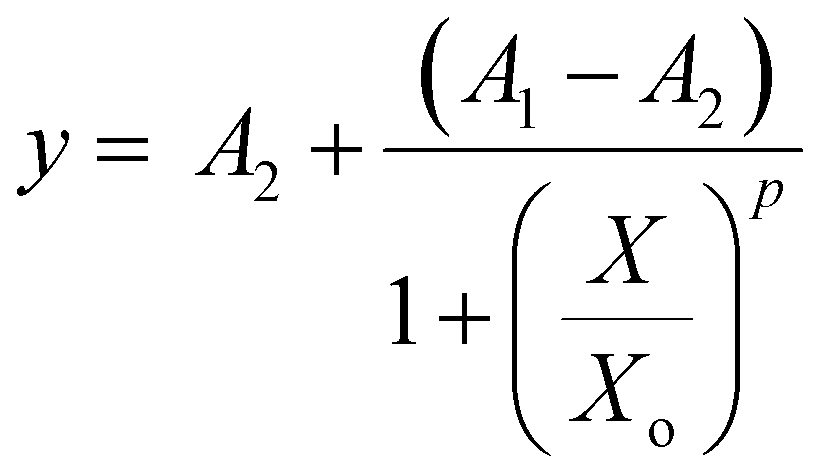
Ch2/DoF = 0.0056; r2 = 0.99745; A1 = 1.04 ± 0.045; A2 = 172 ± 188.2; Xo = 8.6 × 10−4 ± 6.52 × 10−3; p = 1.55 ± 0.24.
Data was fitted by using the software described in http://cbdm-01.zdv.uni-mainz.de/~andrade/k2d3/.23
Methodology for computer simulations
Results & discussion
Super-paramagnetic iron oxide NPs (SPIONs) were synthesized by microwaveassisted thermal decomposition of the iron precursor and then coated with citrate in a single step, to obtain citrate-coated SPIONs (C-SPIONs). BSA was adsorbed on the C-SPIONs’ surface by using a pH-adjusted protocol (Fig. S1†), detailed synthesis and characterization were provided previously.22 The excess BSA was removed by centrifugation for all the experiments. Hereafter, we will refer to this system as BSA-SPIONs.BSA shell formation
By using DLS, we monitored the increase of Dh of C-SPIONs upon BSA incubation at room temperature for 1 h for concentrations ranging from 0.1–100 μM BSA. DLS exhibits a slight increase in Dh at low BSA concentrations, and a sharp increase of Dh in the range of 1–20 μM BSA, which reaches a plateau at Dh = 23 nm for 20–50 μM BSA (Fig. 1a). Mean values and polydispersity are gathered in Table S1.† We infer that a concentration of 20 μM is sufficient to saturate the surface of the C-SPIONs with BSA. Additionally, the appearance of a peak in the DLS size distribution at 8 nm suggests the presence of free BSA in solution at 30 μM BSA, which supports the hypothesis of a saturated BSA coating on the NPs (Fig. 1b). Fig. S3† presents the DLS size distribution of only BSA. The difference of the hydrodynamic diameter value at the plateau (Dh_plateau) of BSA-SPIONs and the Dh value of C-SPIONs is 7 nm, which corresponds to a radius of 3.5 nm, and therefore a hydrodynamic-shell thickness of about 3.5 nm; indicative of a monolayer of BSA.17,22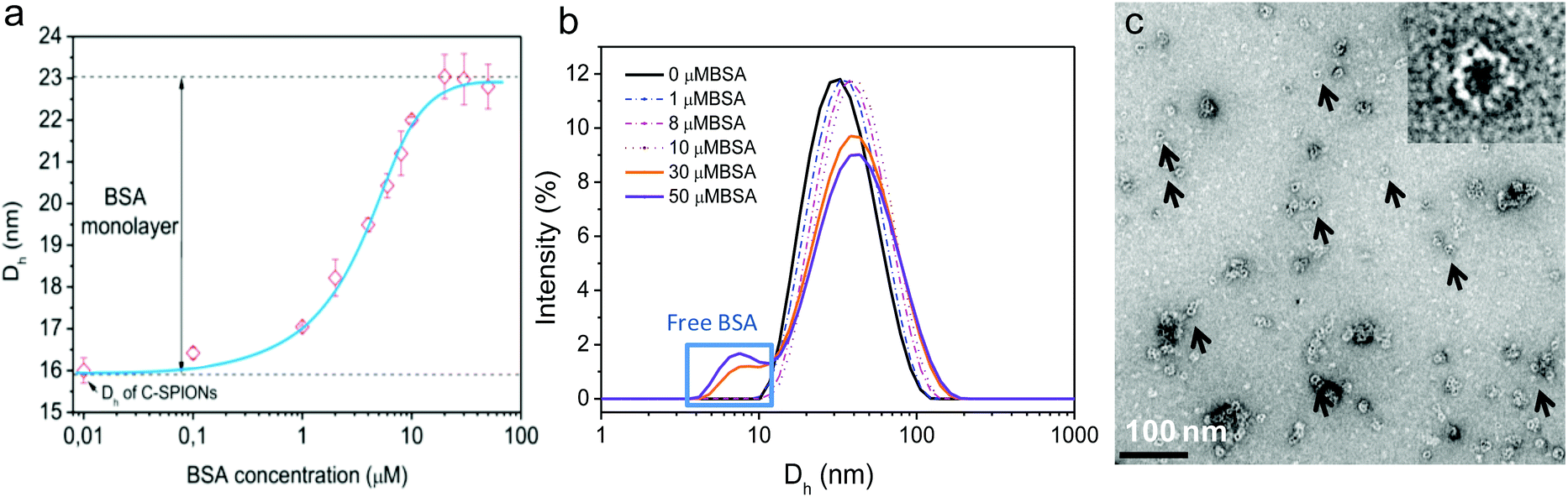 | ||
| Fig. 1 (a) Hydrodynamic diameter (Dh) change upon absorption of BSA, error bars are the standard deviation determined from 3 independent measurements of 15 scans each. (b) Intensity-weighted Dh distribution curves of BSA-SPIONs prepared at different BSA concentrations. The blue square indicates the presence of excess BSA in solution, (c) negative-staining TEM image of BSA-SPIONs. | ||
Negative-staining TEM of BSA-SPIONs was previously reported by us and it showed that the BSA shell on the SPIONs measured 3 ± 1 nm thick (Fig. 1c).22
BSA shell composition
To estimate the composition of the shell, we performed XPS of C-SPIONs and BSA-SPIONs and we identified all the expected elements in the system, Fig. 2 and S4.† The BSA-SPIONs C 1s spectrum exhibits a peak shoulder at 286 eV, which is characteristic of the N–C bond arising from BSA,30 and it is therefore present in the BSA-SPIONs and not in the C-SPIONs. Consistently, nitrogen was only detected in the BSA-SPIONs, as shown by the N 1s high resolution spectra in Fig. S4c.† Once we subtracted the contribution of the NP iron oxide core to the XPS signal, we computed the average relative elemental composition of the NP shell and compare it to that of pure BSA (Fig. 2c). Assuming that BSA is the only N-containing molecule in the shell, we estimated that 74% of the shell is covered by BSA. The higher level of C and lower level of N measured in the BSA-SPIONs’ coating compared to pure BSA suggest the presence of additional carbonaceous species such as citrate. By FT-IR we detected in BSA-SPIONs the typical bands of citrate (red color in Fig. 2d) and BSA (gray color in Fig. 2d); their slight red-shift indicates an interaction with the SPIONs. The detection of the citrate bands in the final BSA-SPIONs suggests the presence of sodium citrate as part of the shell, which indicates that BSA adsorbs on SPIONs’ surface through the interaction with citrate rather than replacing it.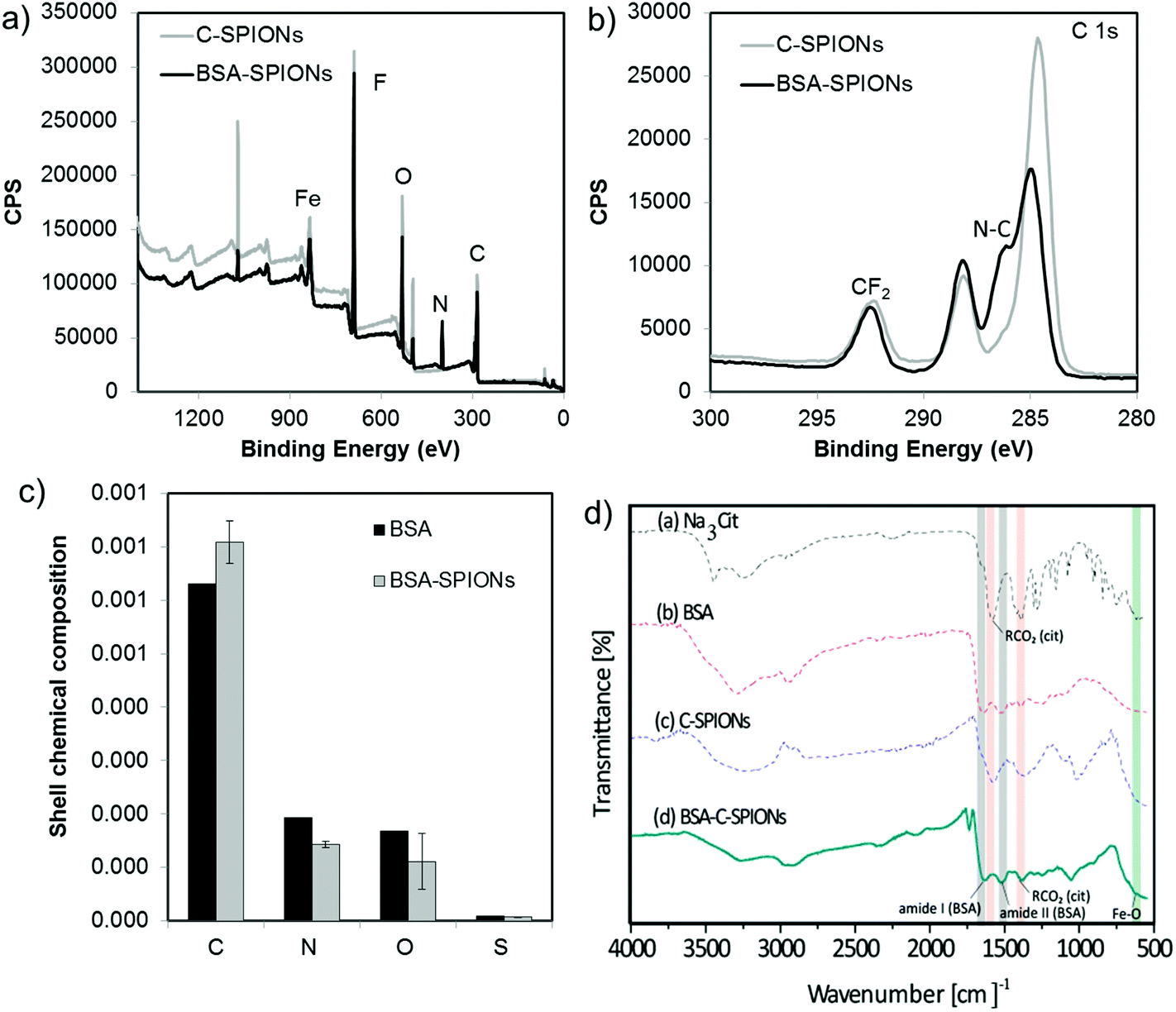 | ||
| Fig. 2 XPS (a) survey and (b) C 1s spectra of C-SPIONs and BSA-SPIONs. (c) Relative elemental composition of the coating on the BSA-SPIONs (gray) and, for comparison, the expected composition for pure BSA (black). XPS measurements were performed in triplicate and the declared uncertainty is the resulting standard deviation. (d) FTIR spectra of BSA-SPIONs, C-SPIONs, BSA, and citrate. | ||
Number of bovine serum albumin molecules in the coating shell
Considering the BSA dimensions,31 ≈8 × ≈8 × ≈3.5 nm3, (Scheme 1) the measured thickness of the observed BSA layer by TEM we estimated the number of molecules present in the shell of the C-SPIONs by using eqn (1).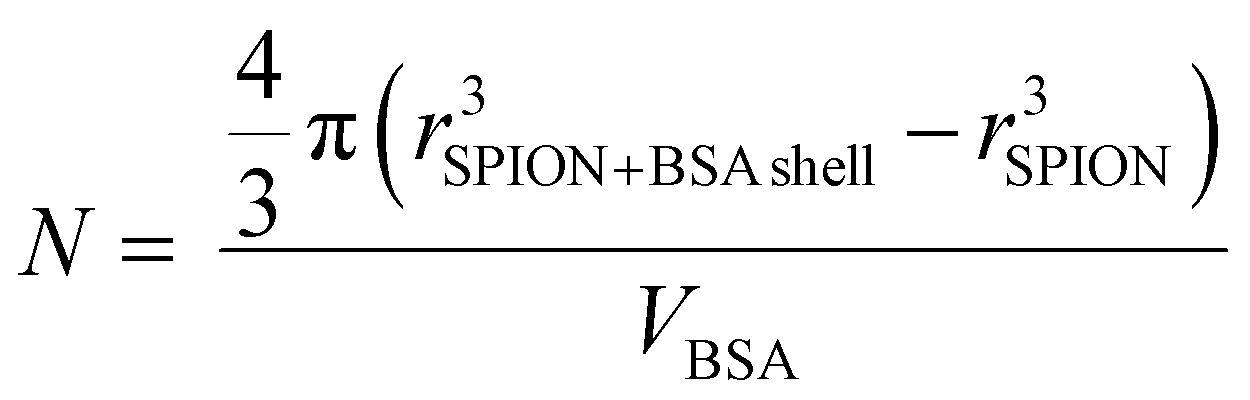 | (1) |
By taking the values obtained from the TEM, rSPION = 3.0 nm; rBSA shell = 3.0 nm; we conclude that approximately 10 ± 2 molecules form the saturated BSA layer on our SPIONs. We confirmed this number by using UV-vis spectroscopy, where we computed the number of proteins constructing the BSA monolayer from the optical absorption measured on the particle supernatant after incubation. We obtained a value of ten molecules of BSA per SPION at saturation, which is in good agreement with our previous estimate.
To confirm these results; we ran molecular dynamics (MD) simulations with atomistic resolution. We simulated the protein shell on a NP of 6 nm diameter to compute the maximum theoretical number of albumin molecules that can be adsorbed onto the NPs. The simulations were done by systematically adding two BSA proteins in solution, from 2 up to 12 BSA molecules, and allowing the proteins to structurally relax and adsorb to the surface (see Methods section for details). In Fig. 3, we show snapshots of configurations of adsorbed proteins obtained in these simulations, which indicate that the maximum value of BSA proteins that can be accommodated onto the 6 nm NP is ten. For the simulation with 12 BSA molecules, two of the proteins remained in solution. This result also indicates that, even in excess in solution; BSA does not form a multilayer over the NP, in accordance with the results from DLS.
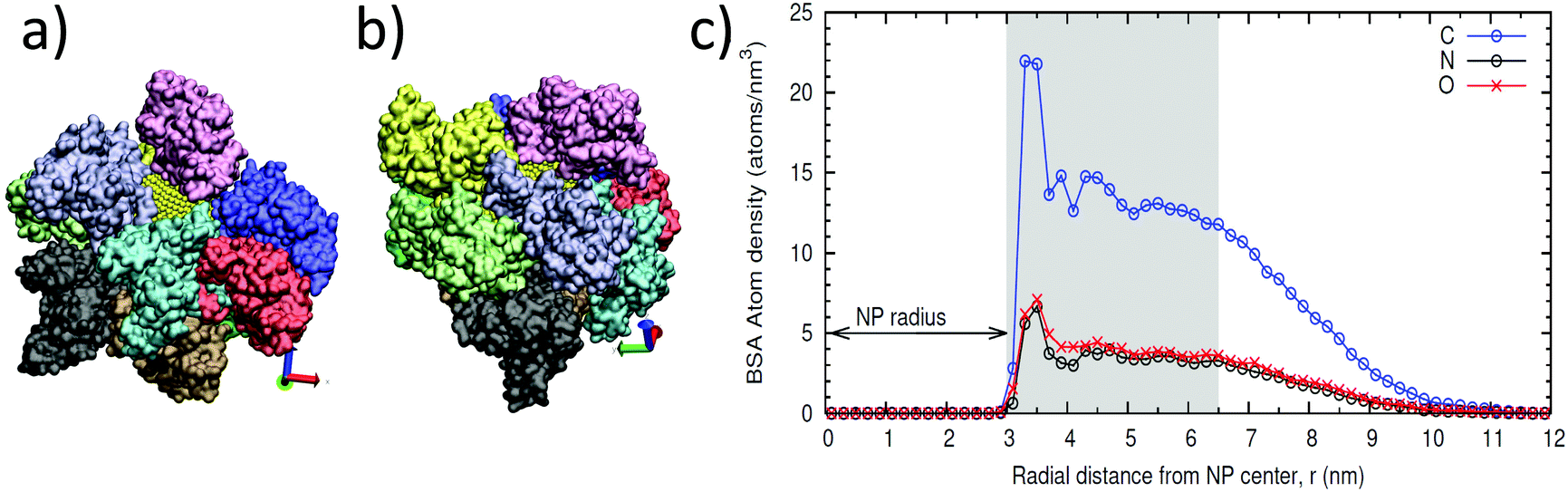 | ||
| Fig. 3 Results from MD simulations with ten BSA proteins adsorbed onto a NP. (a, b) Snapshots of the same configuration with different orientation. The NP is shown in yellow, although only small patches of it can be seen due to the adsorbed protein corona. Proteins are shown in surface representation. Each protein is shown in a different color. (c) Radial density profile of C, N, and O atoms of BSA proteins. The shaded region corresponds to the more compact layer. | ||
The structure of the protein adsorbed layer, containing ten BSA molecules, can be seen in Fig. 3a and b. The layer is relatively compact, with some of the adsorbed BSA proteins in close contact, but it has also small gaps between the proteins that fluctuate over time, and protrusions of protein groups at longer distances from the NP surface. This result can also be seen in the atomic density profile shown in Fig. 3c, which shows the time-averaged distribution of the C, N, and O atoms of the proteins in the adsorbed layer, measured from the center of the NP. There is a clear adsorption peak at r ≈ 3.5 nm, which corresponds to atoms in close contact with the surface of the NP (radius is ≈3 nm). After this peak, there is a region with an accumulation of BSA (with an approximately constant density of O and N atoms) which extends up to r ≈ 6.5 nm. After this region, the density profile decays. This behavior corresponds to a layer with only a partial and fluctuating presence of fragments of BSA proteins, which extends up to long distances, of about r ≈ 10 nm. Therefore, the thickness of the compact layer of adsorbed BSA can be estimated to be about 3 nm (from r = 3–3.5 to r = 6.5 nm), in agreement with the experimental estimate shown in Table 1.
| Method | r SPIONs+BSA (nm) | r SPIONs (nm) | Thickness of the BSA monolayer (nm) | N (saturation number of BSA molecule per SPION) |
|---|---|---|---|---|
| a BSA-SPIONs were negatively stained by using uranyl acetate before TEM measurement, for a better visual image of the BSA shell. b Thickness determined in a dry state. c Number determined according to the model described. | ||||
| TEMa | 6 | 3 | 3 ± 1b | 10 ± 2c |
| UV-vis | 10 | |||
| MD simulation | ≈6.5 | 3 | ca. 3–3.5 | 10 |
These experimental and theoretical results strongly support the formation of a BSA monolayer of about ten molecules. BSA molecules seems to adhere to the surface of C-SPIONs with a triangular base (“side on” adsorption) since adsorption on the “side up” of BSA would give us a protein thickness on the C-SPIONs of 8 nm rather than the detected value of 3 nm (Table 1).6,33 The “side on” adsorption of BSA expose positives patches of BSA, such as lysine and arginine amino acids and it could facilitate its tight binding.34,35 The adsorption of BSA in the “side on” fashion was previously reported for other NPs.36
Conformational change of BSA upon adsorption on C-SPIONs’ surface
By taking advantage of the optical properties of the aromatic residues (Phe, Tyr, Trp) of the BSA we evaluated any possible structural change that BSA underwent upon interaction with SPIONs, using UV-vis, fluorescence, synchronous fluorescence, and circular dichroism spectroscopy. Those results were also complemented with MD simulations at atomistic resolution.The UV-vis spectrum of BSA displays two characteristic adsorption peaks: at 210 nm (related to the polypeptide backbone of the BSA) and at 280 nm (due to the aromatic amino acid residues such as Trp, Tyr, and Phe). Changes in the UV-vis spectra are indicative of the interaction of BSA with the SPIONs.37 In our case the peak at 210 nm undergoes a red shift with increasing concentration of C-SPIONs (Fig. 4a), which reflects the modification of the polypeptide framework of the protein,38 while the peak at 280 nm blue-shifts, indicating that the aromatic residues of the protein are exposed to a more polar microenvironment.39,40
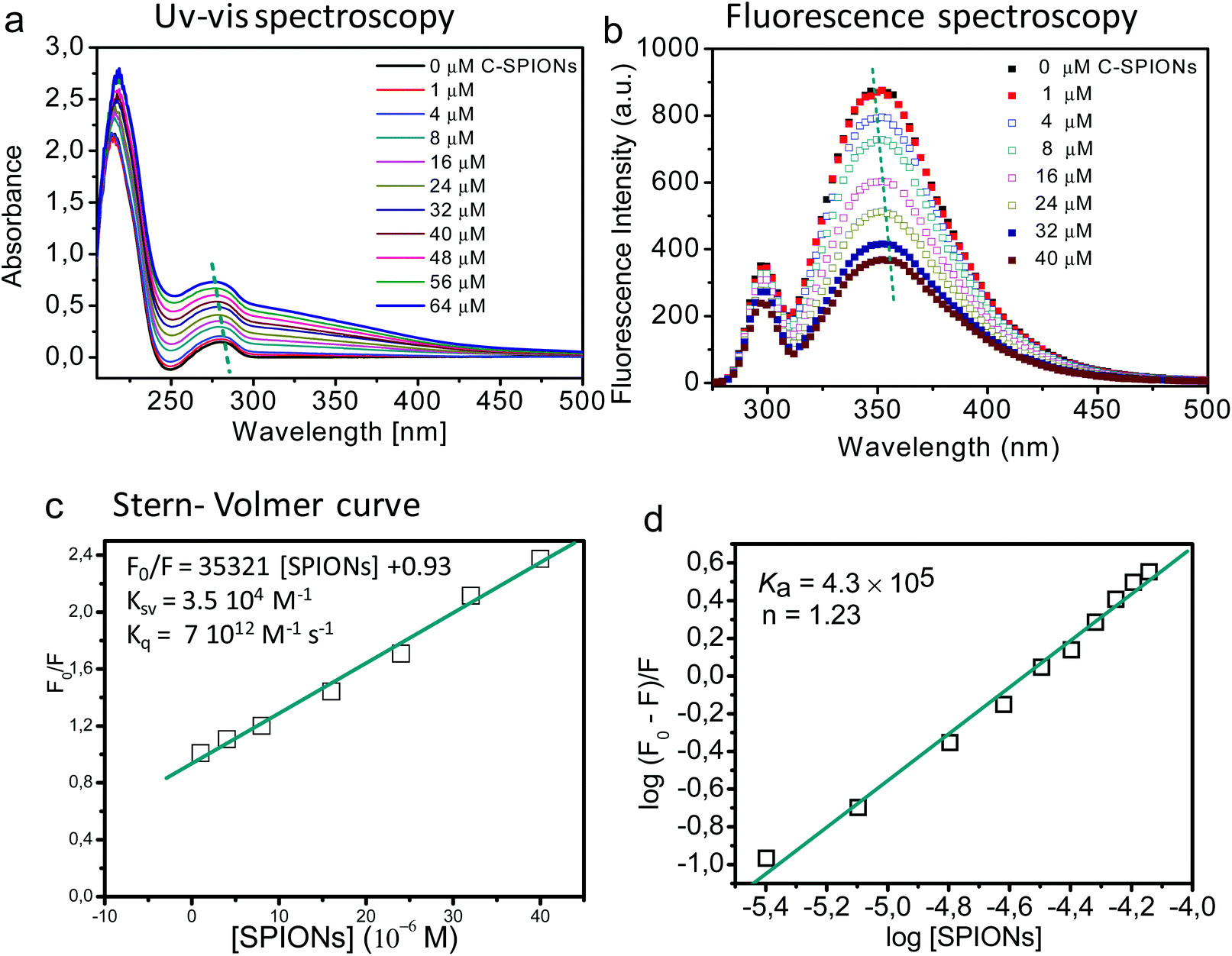 | ||
| Fig. 4 (a) UV-vis spectra of 4 × 10−6 M BSA with increasing concentration of C-SPIONs at pH 7.4. C-SPIONs concentration ranges from 0–64 μM. (b) Fluorescence spectra of 4 × 10−6 M BSA with increasing concentration of C-SPIONs at pH 7.4, upon excitation at 295 nm. (c) Linear Stern–Volmer plot. (d) Determination of the binding constant, Ka, from the fluorescence measurements. | ||
Trp, Tyr, and Phe are intrinsic fluorophores whose fluorescence is susceptible of quenching upon changes in the microenvironment of the residues;41 this allows us to investigate the interaction mechanism of BSA and SPIONs.37,42,43 Trp is the residue that contributes most to the intrinsic fluorescence of the protein since Phe has a low quantum yield and Tyr is quenched by neighboring chemical groups under an excitation wavelength of 295 nm. BSA has two Trp residues; Trp213 and Trp134, which are in the hydrophobic loop and hydrophilic region, respectively (Scheme 1). If BSA adsorption on the surface of C-SPIONs occurs in regions close to the Trp residues, perturbations in the fluorescence spectra intensity and maximum emission wavelength (λmax) appear (Fig. 4b).42,44
On selective excitation of the Trp residues at 295 nm, we observed a decrease of the fluorescence emission intensity at approximately 350 nm, which indicates quenching of C-SPIONs on BSA.37,39,45 The small red shift of λmax from 350.5 to 355.5 nm indicates some structural modification of BSA (Fig. 4b). C-SPIONs did not fluoresce when excited at 295 nm (Fig. S5†). By using the Stern–Volmer function (eqn (2)) we analyzed the quenching mechanism that C-SPIONs exert on the protein.
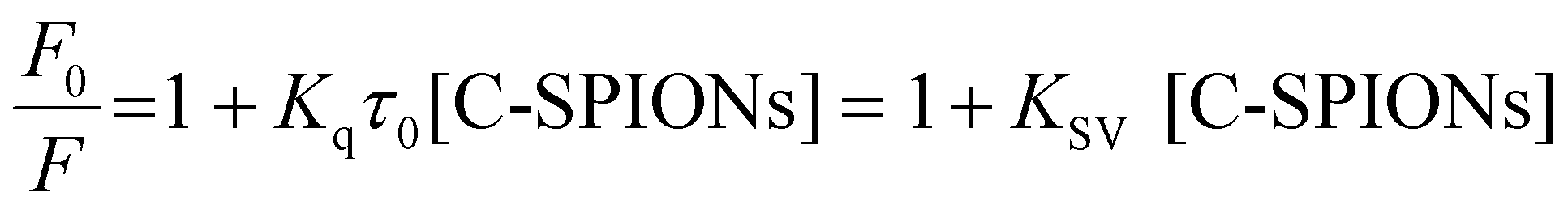 | (2) |
| Kq = KSV/τ0 | (3) |
Quenching mechanisms are classified in two types: static and dynamic, depending on the Kq value. The static mechanism (Kq > 2 × 1010 M−1 s−1) indicates the formation of a protein–quencher complex due to the strong interaction of proteins with the quencher, whereas dynamic quenching (Kq < 2 × 1010 M−1 s−1) implies a weak interaction between proteins and the quencher without stable complex formation.47 In our case, the value of Kq = 7 × 1012 M−1 s−1 indicates a static quenching mechanism, which confirms the formation of BSA-SPION complexes and a strong interaction between BSA and C-SPIONs. Additionally, the binding constant Ka and the number of binding sites n of the BSA-SPIONs complex can be determined by using eqn (4).48
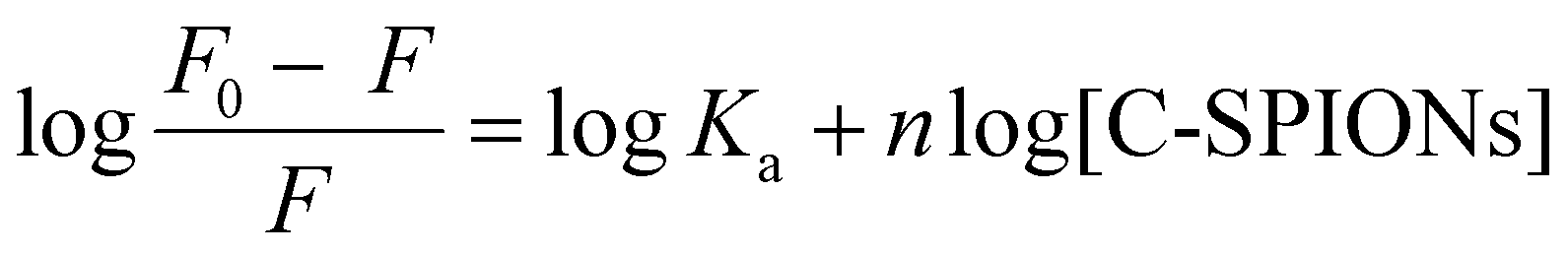 | (4) |
Synchronous fluorescence spectroscopy allows simultaneous scanning of the excitation and emission spectra of the BSA, while keeping a constant interval (Δλ) between them. This method provides detailed information on the microenvironment of specific aromatic amino acid residues such as Trp or Tyr. Those residues can be used to analyze the structure of the BSA due to its high content in the protein.50 When Δλ is set at 15 and 60 nm, the synchronous fluorescence spectra obtained trace characteristic information of Tyr and Trp residues, respectively.39Fig. 5 shows the exponential decrease in the synchronous fluorescence spectra of Tyr (287–290 nm) and Trp (279–282 nm) residues of BSA with increasing concentration of C-SPIONs. Synchronous fluorescence intensity of Trp is weaker than Tyr at Δλ = 60 nm, probably due to the lower content of Trp residues than Tyr within BSA (20 Tyr residues:2 Trp residues per BSA molecule).50 Red shifts in maximum emission wavelength (λmax) are related to the protein's conformational change51 and indicate that the residues’ microenvironment becomes less hydrophobic and residues are exposed to the polar solvent; it seems conformation's BSA changes expanding and decreasing its packed structure.
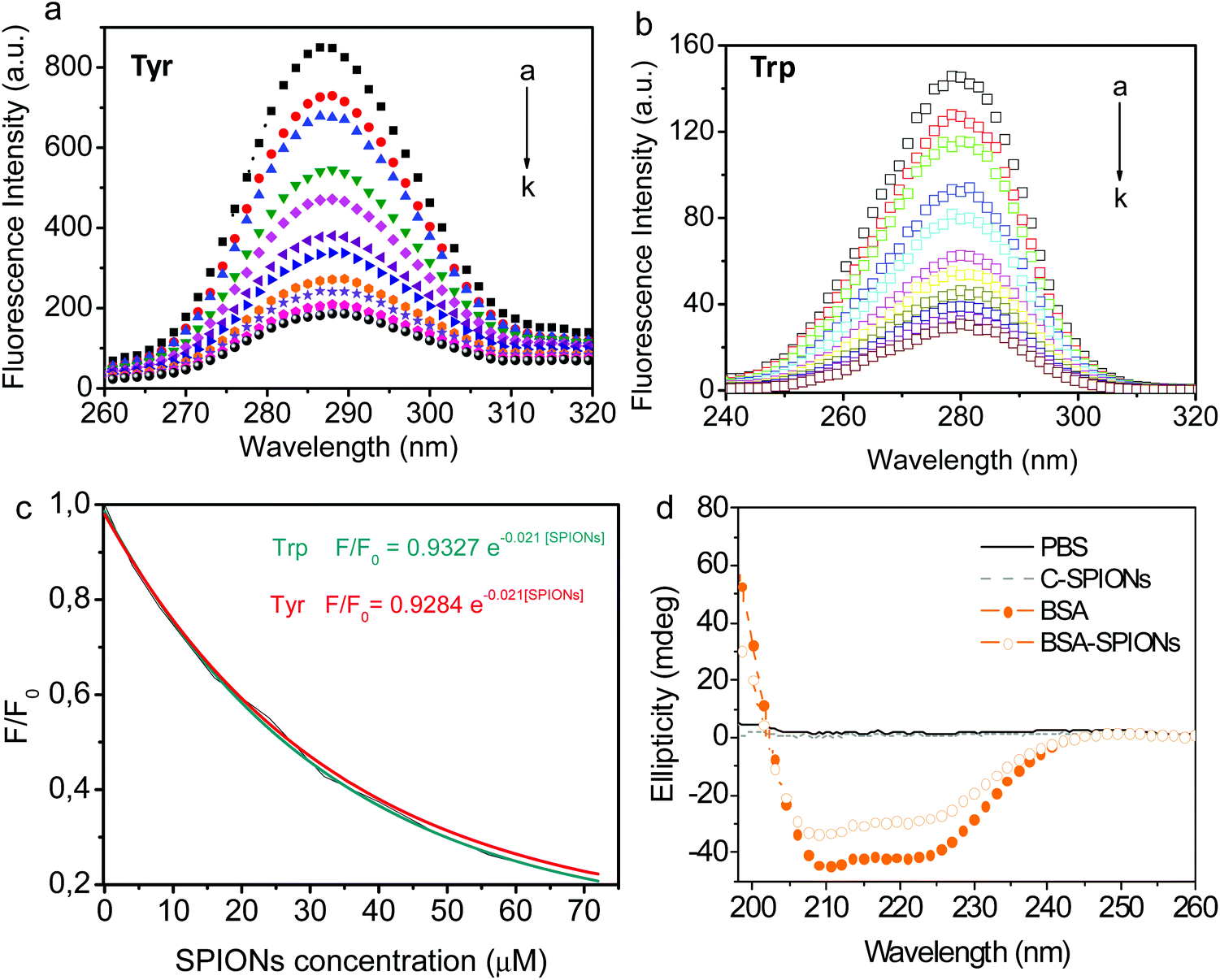 | ||
| Fig. 5 Synchronous fluorescence spectra of 4 × 10−6 M BSA for (a) Δλ = 15 nm (Tyr), (b) Δλ = 60 nm (Trp) upon increasing concentration of C-SPIONs. C-SPION concentration is from 0–72 μM (curves a–k). (c) Ratio of synchronous fluorescent intensity (F/F0) of 4 × 10−6 M BSA in the presence of increasing SPION concentrations for Δλ = 15 nm. (d) CD spectra of PBS, C-SPIONs, BSA, and BSA-SPIONs with a BSA concentration of 4 × 10−6 M. | ||
Finally, the secondary structure of the supported BSA was evaluated by using circular dichroism spectroscopy (CD) and indicated the presence of two main peaks at 208 and 222 nm that are characteristic of its α-helical content. There are no changes in the α-helical content of BSA and only a slight decrease in the β-strand is detected, Fig. 5d and Table S2.† This result indicates that BSA adheres to the SPIONs surface and modifies its hydrophobic/hydrophilic regions without significantly affecting the main secondary structure native features of BSA.
We studied the possible conformational changes during BSA adsorption in our MD simulations. The most significant changes occur during the adsorption of a BSA protein onto an initially empty NP. This process can be seen in movie S1.† Three snapshots of the process are also shown in Fig. 6. These snapshots represent three different stages in the BSA adsorption process. Initially, the BSA protein moves from bulk solution to touch the NP surface (Fig. 6a). Then, the protein spreads out over the NP, increasing the contact region between the protein and the NP (Fig. 6b). Finally, the protein relaxes its conformation into a more compact configuration (Fig. 6c). These changes were characterized by computing the solvent accessible surface area (SASA), the root-mean-square deviation of atomic positions (RMSD), and the α-helix content from the simulation trajectory, by using the VMD software.26 As the protein contacts the NP, the SASA increases from 325.1 nm2 in solution to 345 nm2 at the NP surface. After adsorption, the BSA relaxes to a more compact configuration with a SASA of 334.5 nm2 (a plot of SASA as a function of time is given in Fig. S7†). The RMSD between BSA adsorbed at the NP and BSA in solution is 5.8 Å2, so globally we obtained a relatively small change in atomic coordinates of the BSA. Conformational changes of BSA tend to be rather small for adsorption onto small NPs, such as in our case, Fig 5d and Table S2,† and substantial for particles with radii larger than around 100 nm.3
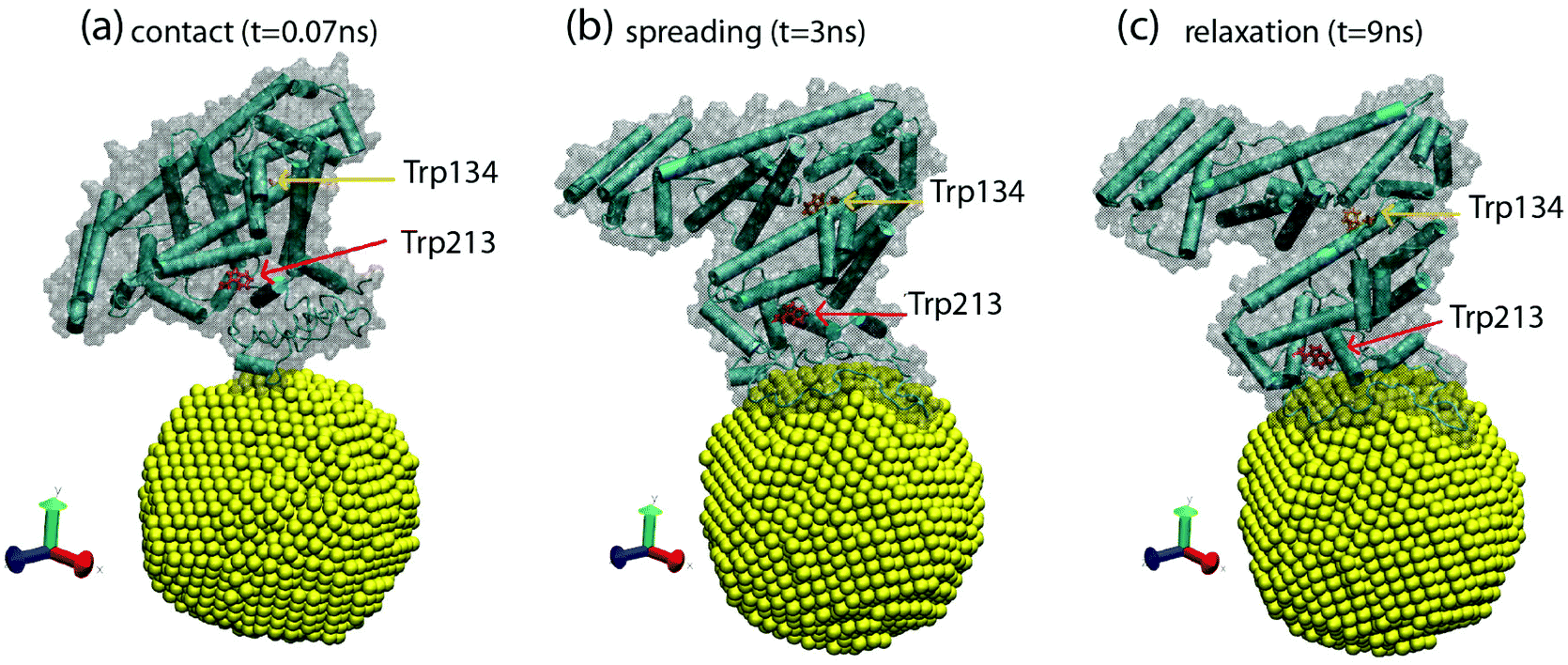 | ||
| Fig. 6 Snapshots from MD simulations obtained by using VMD software,26 corresponding to three different stages of protein adsorption. (a) Contact between BSA and the NP (t = 0.07 ns), (b) spreading of the BSA over the NP (t = 3 ns), and (c) relaxation of the adsorbed BSA protein (t = 9 ns). The NP is shown in yellow and the BSA is shown in cartoon representation, emphasizing the secondary structure. The surface of the protein is also shown as a shadow. The Trp134 and Trp213 residues are shown in bond representations in red and orange, respectively. | ||
We also monitored the residues Trp213 and Trp134, which are responsible for the changes in the Fluorescence spectra monitored in Fig. 4b. In Fig. 6, we also see that these changes affect the local environment and orientation of the Trp213 and Trp134 residues, which suggests a direct relation with the results shown in Fig. 4. Simulations indicate a significant change in the locations of the Trp213 and Trp134 residues. For these residues, we obtain a RMSD of 4 Å2, which mostly originates during the structural relaxation observed from Fig. 6b to c and movie S1.† This change is substantial, while the overall RMSD change in the BSA protein is 5.8 Å2, as mentioned above.
Protein resistant activity of BSA-SPIONs
The work described so far indicates the presence of a protein monolayer coating on the C-SPIONs, which in light of XPS analysis and MD simulations is quite compact. To further confirm these findings we computed the binding affinity of BSA to C-SPIONs by isothermal titration calorimetry (ITC), measuring the enthalpy, entropy, and Gibbs free energy of the adsorption process which is exothermic, therefore enthalpically favored (Fig. 7a). To quantify the thermodynamic parameters of the BSA–SPIONs interaction, the titration curve of BSA with C-SPIONs at room temperature was fitted using a single-binding model. The enthalpy (ΔH), binding stoichiometry (N) and constant (Ka), Gibbs energy changes (ΔG) and entropy (ΔS) are summarized in Table 2.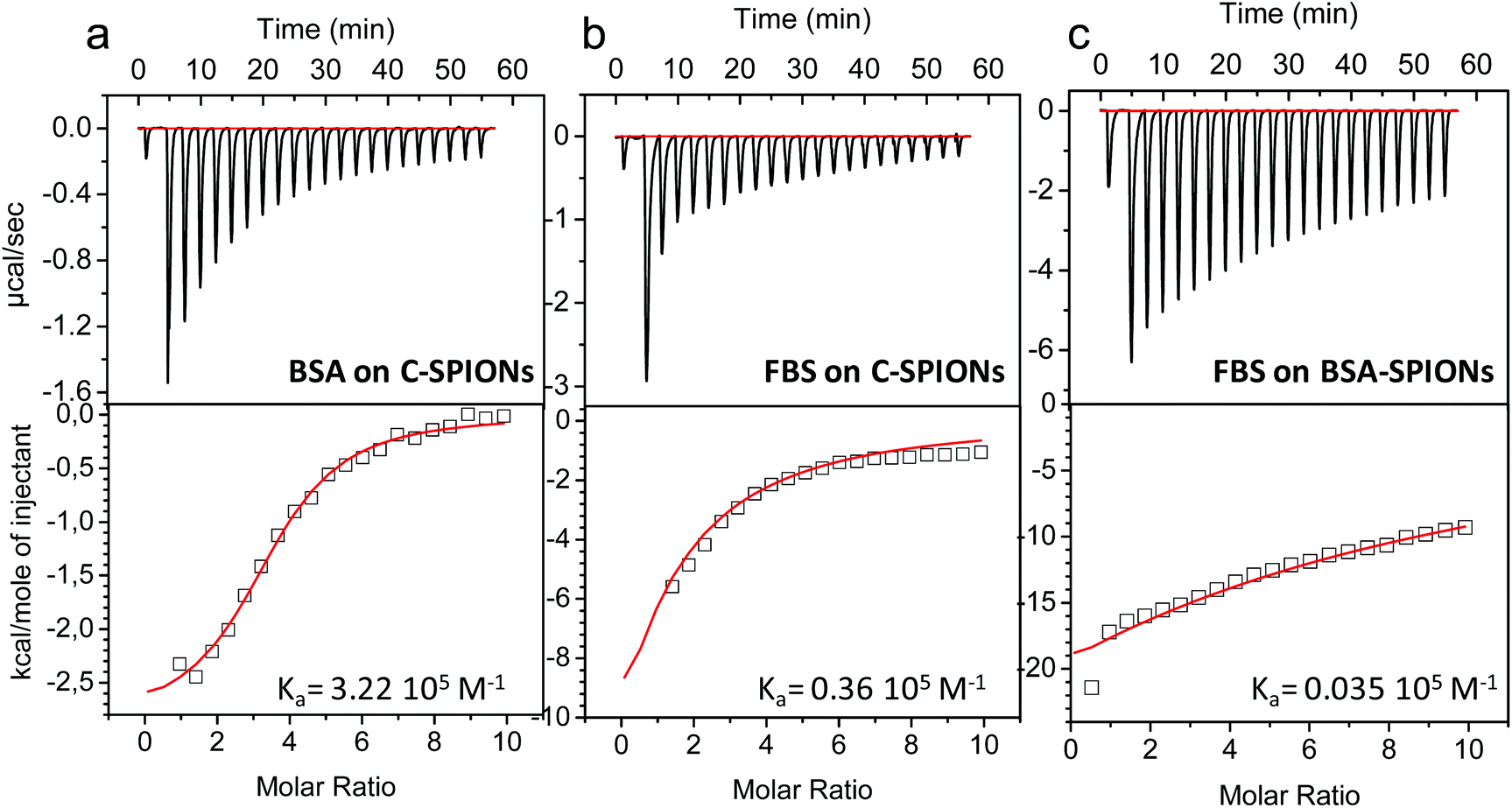 | ||
| Fig. 7 ITC data for the adsorption of BSA, (a) and FBS, (b) on C-SPIONs, and (c) adsorption of FBS on BSA-SPIONs. Upper graphs present the raw data obtained from the titrations and lower graphs the integrated heats of each peak with a corresponding fitting curve. | ||
| Sample | Protein | K a (105 M−1) | ΔG (Kcal mol−1) | ΔH (Kcal mol−1) | ΔS (cal mol−1 K−1) | N |
|---|---|---|---|---|---|---|
| C-SPIONs | BSA | 3.22 | –3.27 | –2.88 | 15.5 | 3.44 ± 0.08 |
| C-SPIONs | FBS | 0.36 | –66.3 | –71.8 | –220 | 0.48 ± 1.46 |
| BSA-SPIONs | FBS | 0.035 | –1327.7 | –1445.4 | –4710 | 0.48 ± 3.66 |
The values of ΔH < 0 and ΔS > 0 indicate that electrostatic interaction is the dominant force for the formation of the BSA-SPIONs complex.52 Although BSA and C-SPIONs are both negatively charged under the same conditions, BSA could bind by electrostatic attraction through its positive patches,34,35 which gives Ka = 3.22 × 105 M−1 in the corresponding titration. This value of Ka is remarkably similar to that measured from the fluorescence spectra, which supports the concept of a strong electrostatic interaction between BSA and the C-SPIONs.
ITC values indicate a strong interaction of BSA with the C-SPIONs, therefore we compared this interaction with that between fetal bovine serum (FBS) and C-SPIONs, which could mimic what our C-SPIONs would encounter in cell culture conditions or in intravenous injection if they are not protected with the BSA coating. We exposed C-SPIONs to FBS, and by using ITC (Fig. 7b and Table 2), we obtained the thermodynamic parameters ΔG < 0, ΔH < 0 and ΔS < 0, Ka = 0.36 × 105 M−1 and n = 0.48 ± 1.46. These results indicate that the interaction of C-SPIONs and FBS is mainly due to van der Waals’ or hydrogen bonding,53 with a Ka value ten times smaller than that of the Ka for BSA-SPIONs and ΔS > 0.
From these results, we wondered if BSA acts as a protein-resistant coating. Therefore, we exposed BSA-SPIONs to FBS. The hydrodynamic diameter measured by DLS after 1 h exposure of BSA-SPIONs to FBS did not change, which indicates non-specific adsorption (Fig. 8a). We titrated BSA-SPIONs with FBS and we obtained ΔG < 0, ΔH < 0 and ΔS < 0 and Ka = 0.035 × 105 M−1, 100 times smaller than Ka for BSA-SPIONs, corroborating the strong interaction of BSA with C-SPIONs and the protein-resistant activity of preformed BSA layer against FBS, Fig. 7c and Table 2.
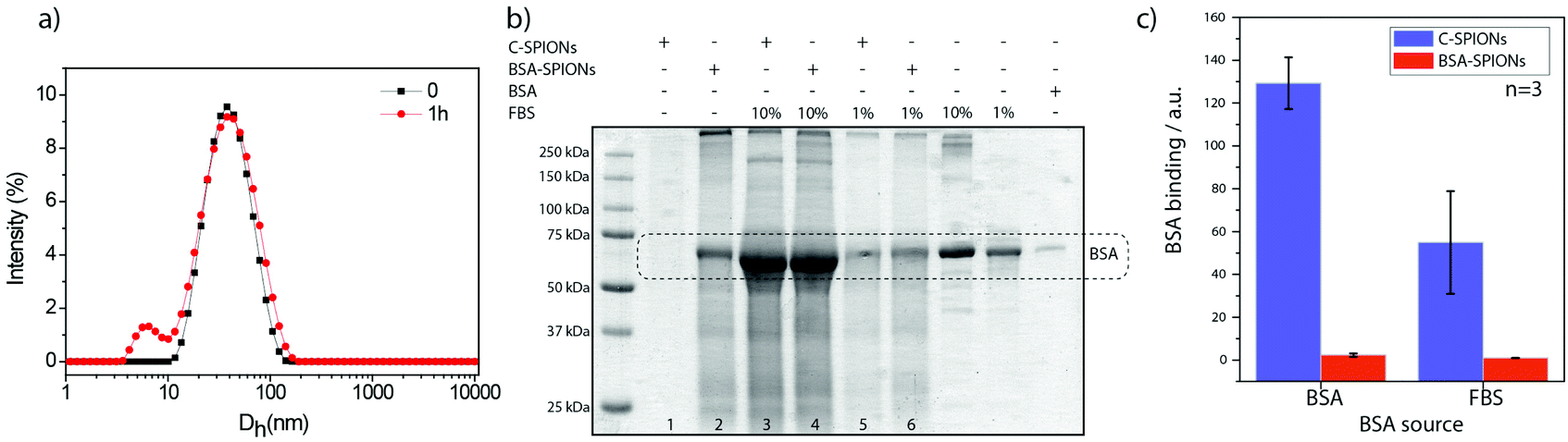 | ||
| Fig. 8 (a) DLS of BSA-SPIONs at time 0 (black line) and after 1 h (red line) exposure to FBS. (b) Representative SDS-PAGE gel of C-SPIONs and BSA-SPIONs isolated from PBS containing 10% FBS, after 1 h incubation. (c) BSA binding capacity of C-SPIONs and BSA-SPIONs mixed with pure BSA or FBS determined in three SDS-PAGE gels with three independent preparations, by the ratio of the area under the curve BSA band (ca. 67 kDa in (b)) for lanes 3:1 and 5:1 for C-SPIONs, and 4:2 and 6:2 for BSA-SPIONs, with BSA and FBS respectively. | ||
The protein repellency was further evaluated by means of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). We incubated C-SPIONs and BSA-SPIONs with BSA and FBS (pH 7.4) for 1 h, followed by purification with gel electrophoresis. C-SPIONs and BSA-SPIONs were used as the control. In the gel (Fig. 8b), we identified and quantified the amount of additional BSA bound to the samples. The amount of BSA bound from FBS or pure BSA to the BSA-SPIONs is negligible (Fig. 8c) compared to C-SPIONs, which bind a large amount of BSA from both pure BSA or FBS sources (Fig. 8c); this indicates that BSA shows a strong affinity to C-SPIONs’ surface. When the C-SPIONs’ surface is precoated by a BSA monolayer, it is unlikely that other proteins will adsorb on their surface due to the weak interaction of the BSA coating with FBS proteins. Molecular dynamics previously showed that extra molecules of BSA did not attach to the preformed monolayer of BSA, which validates the protein-resistant properties determined here.
Conclusions
Adsorption of proteins on the surface of NPs is a complex process, which depends on the physicochemical properties of the NPs, their native surface coating and the characteristics of the adsorbed proteins, as well as the nature of the medium exposed. These factors affect the amount and orientation of protein bound on the NPs’ surface. Our experiments indicate that the BSA electrostatically adsorbs on citrate-coated SPIONs’ surface to form a monolayer of 3 nm, which suggests the adsorption of BSA with a “side on” configuration on the C-SPIONs' surface. XPS and FTIR evidenced the presence of citrate on the BSA-SPIONs complexes. By using TEM, UV-vis, and molecular dynamics we determined that the monolayer of BSA is composed of ten molecules of BSA.By taking advantage of the intrinsic fluorescence of aromatic residues (Phe, Tyr, Trp) of the BSA we studied its conformational changes. UV-vis, fluorescence and synchronous fluorescence spectroscopies indicated that the regions close to Tyr and Trp are perturbed upon interaction with SPIONs and undergo a decrease in hydrophobicity that induces slight changes in the BSA conformation. CD also indicated a slight change in the BSA conformation which suggests that these polar changes could be localized close to the regions of Tyr and Trp and do not affect the overall secondary structure of the BSA. By using Stern–Volmer law we determined a single-binding-site binding mechanism, a static-quenching mechanism (Kq = 7 × 1012 M−1 s−1) and a value of Ka = 4.3 × 105 M−1 that confirms the formation of a strong BSA-SPIONs complex. By using molecular dynamics we visualized how the BSA proteins approximate the surface of the NPs and accommodate ten molecules of BSA to form a robust monolayer without significant modifications of the secondary structure of BSA.
Multiple forces such as van der Waals, hydrogen bonding, electrostatic and hydrophobic interactions, the desolvation of NPs and proteins, and solvation of newly formed complexes53 can be involved in the interaction of BSA with SPIONs. From ITC experiments, we observed that the interaction of BSA with C-SPIONs is enthalpically favored with a Ka value determined by ITC of 3.22 × 105 M−1; this is remarkably similar to the value measured from the fluorescence spectra, which supports the hypothesis of a strong electrostatic interaction between BSA and C-SPIONs.
Additionally, ITC allowed us to evaluate the competition of the BSA layer upon exposure to FBS; we observe that FBS attaches to the SPIONs surfaces through van der Waals rather than the electrostatic interactions observed for BSA, with a Ka value that is one order of magnitude smaller than for BSA-SPIONs. SDS-PAGE results also indicate that the amount of FBS or BSA adhered on BSA-SPIONs is negligible when the SPIONs are already decorated with BSA. We conclude that the protein shell of BSA is strongly adsorbed and does not change upon exposure to FBS and therefore acts as protein-resistant layer, similar to some PEGylated surfaces.
Our findings support the potential use of the BSA-SPIONs in medical applications, for example as an efficient carrier for targeted drug delivery in vivo. The preformed BSA monolayer serves as a protective shell for the SPIONs’ surface and the protein-resistant surface of BSA-SPIONs avoids the adsorption of additional serum proteins, hence potentially protecting and maintaining the targeting ability of the ligands adsorbed on the NP's surface.
Acknowledgements
The research leading to these results has received funding from; the People Program (Marie Curie Actions) of the EC 7th Framework Program under grant agreement no 303630 and co-funded by the European Social Fund, the Spanish Ministry of Economy MAT2015-64442-R, co-supported by FEDER funds, the Generalitat de Catalunya 2014SGR213, the COST Action MP1202, Severo Ochoa Program (SEV-2015-0496) co-funded by European Social Funds, Ramon y Cajal grant RYC-2010-06082 (AL) and the China Scholarship Council fellowship (SMY, 201206150053). CM thanks the Innovation R&D Programme of the UK National Measurement System. AP-M is a recipient of the Universitat Autònoma de Barcelona-Programa Banco de Santander Fellowship. JF acknowledges the CESGA Supercomputing Center (Finisterrae Supercomputer) for computing time and technical assistance and proyecto de excelencia CTS-6270 de la Junta de Andalucía. We acknowledge the support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- P. Satzer, F. Svec, G. Sekot and A. Jungbauer, Eng. Life Sci., 2016, 16(3), 238–246 CrossRef CAS.
- T. Sen, K. K. Haldar and A. Patra, J. Phys. Chem. C, 2008, 112, 17945–17951 CAS.
- R. Venerando, G. Miotto, M. Magro, M. Dallan, D. Baratella, E. Bonaiuto, R. Zboril and F. Vianello, J. Phys. Chem. C, 2013, 117, 20320–20331 CAS.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- Y. Zhang, N. Kohler and M. Q. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS PubMed.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–3166 CrossRef CAS.
- S. Hak, E. Helgesen, H. H. Hektoen, E. M. Huuse, P. A. Jarzyna, W. J. M. Mulder, O. Haraldseth and C. d. L. Davies, Langmuir, 2012, 6, 5648–5658 CAS.
- F. Kawai, T. Kimura, M. Fukaya, Y. Tani, K. Ogata, T. Ueno and H. Fukami, Appl. Environ. Microbiol., 1978, 35, 679–684 CAS.
- R. Gref, M. Luck, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Muller, Colloid Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- V. Kalidasan, X. L. Liu, T. S. Herng, Y. Yang and J. Ding, Nano-Micro Lett., 2016, 8, 80–93 CrossRef.
- P. Maffre, S. Brandholt, K. Nienhaus, L. Shang, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2014, 5, 2036–2047 CrossRef CAS PubMed.
- F. Canfarotta and S. A. Piletsky, Adv. Healthcare Mater., 2014, 3, 160–175 CrossRef CAS PubMed.
- E. Carenza, V. Barceló, A. Morancho, L. Levander, C. Boada, A. Laromaine, A. Roig, J. Montaner and A. Rosell, Nanomedicine, 2014, 10, 225–234 CAS.
- K. Hola, Z. Markova, G. Zoppellaro, J. Tucek and R. Zboril, Biotechnol. Adv., 2015, 33, 1162–1176 CrossRef CAS PubMed.
- A. Aires, S. M. Ocampo, D. Cabrera, L. de la Cueva, G. Salas, F. J. Teran and A. L. Cortajarena, J. Mater. Chem. B, 2015, 3, 6239–6247 RSC.
- S.-M. Yu, A. Laromaine and A. Roig, J. Nanopart. Res., 2014, 16, 1–15 Search PubMed.
- C. Louis-Jeune, M. A. Andrade-Navarro and C. Perez-Iratxeta, in Proteins: Structure, Function, and Bioinformatics, Wiley Subscription Services, Inc., A Wiley Company, 2012, vol. 80, ch. 374, pp. 374–381 Search PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wirkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- A. Bujacz, K. Zielinski and B. Sekula, in Proteins: Structure, Function, and Bioinformatics, 2014, vol. 82, pp. 2199–2208 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- D. E. Tanner, K.-Y. Chan, J. C. Phillips and K. Schulten, J. Chem. Theory Comput., 2011, 7, 3635–3642 CrossRef CAS PubMed.
- C. Calero, J. Faraudo and D. Bastos-González, J. Am. Chem. Soc., 2011, 133, 15025–15035 CrossRef CAS PubMed.
- N. A. Belsey, A. G. Shard and C. Minelli, Biointerphases, 2015, 10, 019012 CrossRef PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- K. M. Au and S. P. Armes, ACS Nano, 2012, 6, 8261–8279 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS PubMed.
- M. L. Quillin and B. W. Matthews, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 791–794 CrossRef CAS.
- P. Maffre, K. Nienhaus, F. Amin, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2011, 2, 374–383 CrossRef CAS PubMed.
- W. Hai-Dong, N. Catherine Hui, Y. Qiaoqin and B. Ildiko, Nanotechnology, 2011, 22, 145703 CrossRef PubMed.
- Y. Liu, F. Ji and R. Liu, Nanotoxicology, 2013, 7, 97–104 CrossRef CAS PubMed.
- B. K. Paul, K. Bhattacharjee, S. Bose and N. Guchhait, Phys. Chem. Chem. Phys., 2012, 14, 15482–15493 RSC.
- Y.-J. Hu, Y. Liu, Z.-B. Pi and S.-S. Qu, Bioorg. Med. Chem., 2005, 13, 6609–6614 CrossRef CAS PubMed.
- J. R. Albani, in Structure and Dynamics of Macromolecules: Absorption and Fluorescence Studies, ed. J. R. Albani, Elsevier Science, Amsterdam, 2004, pp. 141–192, DOI:10.1016/B978-044451449-3/50004-6.
- R. Huang, R. P. Carney, K. Ikuma, F. Stellacci and B. L. T. Lau, ACS Nano, 2014, 8, 5402–5412 CrossRef CAS PubMed.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- S. Chakraborti, P. Joshi, D. Chakravarty, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2012, 28, 11142–11152 CrossRef CAS PubMed.
- Q. Yang, J. Liang and H. Han, J. Phys. Chem. B, 2009, 113, 10454–10458 CrossRef CAS PubMed.
- Q. Xiao, S. Huang, Z.-D. Qi, B. Zhou, Z.-K. He and Y. Liu, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1020–1027 CrossRef CAS PubMed.
- B. K. Paul and N. Guchhait, Photochem. Photobiol. Sci., 2011, 10, 980–991 CAS.
- Z. Chi, R. Liu, Y. Teng, X. Fang and C. Gao, J. Agric. Food Chem., 2010, 58, 10262–10269 CrossRef CAS PubMed.
- N. Wang, L. Ye, F. Yan and R. Xu, Int. J. Pharm., 2008, 351, 55–60 CrossRef CAS PubMed.
- N. G. Zhdanova, E. A. Shirshin, E. G. Maksimov, I. M. Panchishin, A. M. Saletsky and V. V. Fadeev, Photochem. Photobiol. Sci., 2015, 14, 897–908 CAS.
- S. Gorinstein, I. Goshev, S. Moncheva, M. Zemser, M. Weisz, A. Caspi, I. Libman, H. Lerner, S. Trakhtenberg and O. Martín-Belloso, J. Protein Chem., 2000, 19, 637–642 CrossRef CAS PubMed.
- S. N. Khan, B. Islam, R. Yennamalli, A. Sultan, N. Subbarao and A. U. Khan, Eur. J. Pharm. Sci., 2008, 35, 371–382 CrossRef CAS PubMed.
- M. De, C.-C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr01732k |
| This journal is © The Royal Society of Chemistry 2016 |