
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophobic matrix-free graphene-oxide composites with isotropic and nematic states†
Martin
Wåhlander
a,
Fritjof
Nilsson
a,
Anna
Carlmark
a,
Ulf W.
Gedde
a,
Steve
Edmondson
b and
Eva
Malmström
*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Fibre and Polymer Technology, SE-100 44 Stockholm, Sweden. E-mail: mavem@kth.se
bUniversity of Manchester, School of Materials, Oxford Road, Manchester, M13 9PL, UK
First published on 18th May 2016
Abstract
We demonstrate a novel route to synthesise hydrophobic matrix-free composites of polymer-grafted graphene oxide (GO) showing isotropic or nematic alignment and shape-memory effects. For the first time, a cationic macroinitiator (MI) has been immobilised on anionic GO and subsequently grafted with hydrophobic polymer grafts. Dense grafts of PBA, PBMA and PMMA with a wide range of average graft lengths (MW: 1–440 kDa) were polymerised by surface-initiated controlled radical precipitation polymerisation from the statistical MI. The surface modification is designed similarly to bimodal graft systems, where the cationic MI generates nanoparticle repulsion, similar to dense short grafts, while the long grafts offer miscibility in non-polar environments and cohesion. The state-of-the-art dispersions of grafted GO were in the isotropic state. Transparent and translucent matrix-free GO-composites could be melt-processed directly using only grafted GO. After processing, birefringence due to nematic alignment of grafted GO was observed as a single giant Maltese cross, 3.4 cm across. Permeability models for composites containing aligned 2D-fillers were developed, which were compared with the experimental oxygen permeability data and found to be consistent with isotropic or nematic states. The storage modulus of the matrix-free GO-composites increased with GO content (50% increase at 0.67 wt%), while the significant increases in the thermal stability (up to 130 °C) and the glass transition temperature (up to 17 °C) were dependent on graft length. The tuneable matrix-free GO-composites with rapid thermo-responsive shape-memory effects are promising candidates for a vast range of applications, especially selective membranes and sensors.
Introduction
Graphene, a 2D carbon allotrope and the newest member in the nanocarbon family, has gained enormous attention world-wide since its discovery by Novoselov et al. in 2004.1 The extraordinary properties of graphene, graphene oxide (GO) and other 2D nanoparticles (NPs) make them unique candidates for a broad range of applications.2,3 The research field of 2D NPs is rapidly changing from basic science to applied technologies such as flexible electronics, corrosion protection, energy storage, ion and gas barriers, sensors and membranes, low friction coatings, composite materials and biomedical applications.2,4A major challenge, in order to obtain high-performance polymer nanocomposites, is the ability to control the dispersion of NPs in polymeric matrices. Graphene and GO spontaneously stack in the dry state to form aggregates including restacking through π–π stacking interactions, which suppress the unique properties of isolated NPs such as superior thermal and electrical conductivity of graphene. Hence, if NPs would be kept well-dispersed in a polymer matrix, the true properties of the isolated NPs can be exploited at low loadings.
GO is commonly produced by exfoliation of graphite by chemical oxidation via modified Hummer's method.5 The formation of oxygen-containing functional groups (epoxy-, carboxyl- and hydroxyl-groups) disrupts the aromaticity and imparts negative surface charges in aqueous suspension which causes repulsion.6 The disrupted aromaticity imparts modified separation properties, with applications such as gas-, salt- or ion-membranes, water purification and desalinisation, carbon capture etc.7–9 GO-“paper”, produced by solvent casting and filtration, has proven to be impermeable to inert gases (helium), but sensitive and very permeable to polar vapours (such as H2O).10 Therefore, covalent surface modifications to obtain hydrophobic GO have been conducted.3,4,11 However, the oxidation of GO is heterogeneous and forms carboxyl groups on the edges and epoxy and hydroxyl groups on the basal planes.12 Thus, it is challenging to obtain homogenously surface-modified GO, since any covalently attached reagent will be distributed to specific-reactive groups.
In this research, we aimed to attach initiator groups to GO for controlled polymerisation. To avoid inhomogeneity, it is attractive to avoid covalent modification routes and instead non-covalently decorate the GO with a cationic polyelectrolyte macroinitiator (MI). MIs of co-polyelectrolytes have previously been physisorbed by electrostatic interactions to a range of hydrophilic substrates (e.g. cellulose,13 alumina,14 silica15). Also GO, coated with a cationic polyelectrolyte, has recently been functionalised by an anionic block-MI.16 However, the direct immobilization of a cationic MI to GO has not yet been conducted. By the use of a statistical co-polymer as MI instead of a block co-polymer,17 the number of initiating sites can be tuned (i.e. the grafting density), thus more densely packed polymer brushes can subsequently be grafted from the substrate by surface-initiated polymerisation (SIP).11
Commonly, SIPs are conducted by controlled radical polymerisation techniques18 so called surface-initiated reversible-deactivation radical polymerisation techniques (e.g. atom transfer radical polymerisation (SI-ATRP),19 or reversible addition-fragment chain-transfer (SI-RAFT)).16,20 Well-dispersed inorganic NPs and highly filled transparent nanocomposites have been obtained by the synthesis of bimodal polymer grafts21 and multi-layered silanes with sparse polymer grafts,22 where the coatings of the dense short grafts or silanes screen the NP–NP attraction, whereas long sparse grafts offer either good miscibility with polymer matrices or cross-linking into matrix-free nanocomposites with high NP loadings.23
Previously, we reported dense grafting of hydrophilic methacrylates24 and pH-responsive brushes25 by SI-ATRP and SI-activators regenerated by electron transfer ATRP (SI-ARGET ATRP) from discrete SiO2 NPs decorated with statistical cationic MIs. These controlled polymerisation techniques can also be combined with precipitation polymerisation,26–28 where the solubility of the polymer in a homogeneous system decreases during the propagation and the product finally precipitates as droplets. For example, atom transfer radical precipitation polymerisation (ATRPP) combines the efficient precipitation polymerisation with ATRP and can therefore be used to obtain monodisperse polymer microspheres without the need of any surfactant and stabiliser.29 However, surface-initiated controlled radical precipitation polymerisation (SI-ARGET ATRPP) has not yet been widely used.
It is desirable to manufacture isotropic matrix-free GO-composites, due to the benefits of easier processing of well-dispersed, transparent, non-percolated NPs in combination with the elimination of miscibility issues between the NPs and the polymer matrix. However, to avoid poor mechanical performance of such materials, a prerequisite is long and densely grafted brushes with cohesive attraction, due to the high aspect ratio of GO, i.e. low percolation point. Cohesive grafts forming interconnections between nanofillers can induce novel and improved shape-memory or self-healing properties not possessed by the pure polymer.30 Further, densely grafted brushes are commonly associated with low-friction surfaces, which enhance the anisotropic orientation of GO along the direction of flow. Both anisotropic and isotropic materials are considered important for optical, barrier and mechanical applications.31
Our main objectives in this study were: to synthesise a cationic MI for homogeneous and non-covalent decoration of anionic GO; to graft hydrophobic polymers from the immobilised MI on well-dispersed GO using SI-ARGET ATRPP; to improve the dispersion and the interphase towards hydrophobic environments similar to a bimodal graft system; and to characterise the orientation and properties of grafted GO in suspensions and as matrix-free GO-composites.
Experimental section
Materials
N,N-Dimethylaminoethyl methacrylate (DMAEMA), 2-hydroxyethyl methacrylate (HEMA), methyl methacrylate (MMA), butyl methacrylate (BMA), butyl acrylate (BA), α-bromoisobutyryl bromide (α-BiB, 98%), ethyl-α-bromoisobutyryl bromide (E-BiB, 98%), 2,2′-bipyridine (bipy, ≥98%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), triethylamine (TEA, 99%), 4-dimethylaminopyridine (DMAP, ≥99%), copper(I) bromide (98% CuBr), copper(II) bromide (CuBr2, ≥99.999%), L-ascorbic acid (AA, ≥99%), methyl iodide (MeI, ≥99%), activated alumina powder (Al2O3, basic Brockmann I), 2-propanol, dichloromethane (DCM), diethyl ether, tetrahydrofuran (THF), toluene, acetone, ethanol (EtOH, 96%) and methanol (MeOH) were purchased from Sigma Aldrich and used without purification, if not otherwise stated. Graphene oxide N002-PS, denoted GO, was purchased from Angstron Materials® (Ohio, US) and was purified prior to use as described below. Dry dialysis tubing was purchased from Spectra/Por® (12–14 kDa MWCO). Linear high molar mass poly(methyl methacrylate)s (PMMAs, Mw: 410–550 kDa) with low dispersity were supplied by Alfa Aesar®.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 20 °C) and redispersion by ultrasonication in deionised water (15 MΩ cm at 25 °C) followed by dialysis in deionised water for 12 days using Spectra/Por® tubes. The pH was 3.6 after purification. The GO was characterised by confocal Raman, TGA, FT-IR, SEM, TEM, AFM, DLS and polyelectrolyte titration (Stabino®).
000g, 20 °C) and redispersion by ultrasonication in deionised water (15 MΩ cm at 25 °C) followed by dialysis in deionised water for 12 days using Spectra/Por® tubes. The pH was 3.6 after purification. The GO was characterised by confocal Raman, TGA, FT-IR, SEM, TEM, AFM, DLS and polyelectrolyte titration (Stabino®).
 | ||
| Scheme 1 Synthesis of the statistical cationic macroinitiator (3), through compound (1) and (2). | ||
Firstly, the statistical co-polymer of DMAEMA (4.4 ml, 26 mmol) and HEMA (3.2 ml, 26 mmol) was synthesised by ATRP from E-BiB (190 μl, 1.3 mmol) with bipy (410 mg, 2.6 mmol), CuBr (170 mg, 1.2 mmol) and CuBr2 (29 mg, 0.13 mmol) in 2-propanol (15 ml). The reagents were mixed in a round-bottomed flask equipped with a magnetic stirrer and a rubber septum, and thereafter degassed by four consecutive vacuum (5 min) and argon (5 min) cycles. The flask was placed in a preheated oil bath (30 °C) and the mixture was allowed to react for 2.5 h (>90% conversion) under argon atmosphere. The product was diluted by DCM, passed through a basic Al2O3-column to remove the yellow ligand/catalyst complex. The clear transparent product was precipitated into diethyl ether. The white powder was characterised by 1H-NMR (acetone-d6 or MeOD) and DMF-SEC. The final product contained 50 ± 1% DMAEMA and 50 ± 1% HEMA according to the 1H-NMR.
Secondly, the hydroxyl groups of the HEMA-side chains were esterified by the immobilization of the ATRP-initiator (α-BIB). The purified co-polymer (6.0 g, 21 mmol-residues HEMA), TEA (9.5 ml, 6.8 mmol) and catalytic amounts of DMAP (25 mg, 0.21 mmol) were dissolved in THF (25 ml) in an Erlenmeyer-flask at RT. The α-BiB (3.4 ml, 27 mmol) was separately mixed with cooled THF (25 ml at 0 °C) and added dropwise to the flask under weak argon flow and magnetic stirring. The flask was sealed with a rubber septum and aluminium foil and the white mixture was left on a shaking table for 16 h. The reaction was terminated by addition of EtOH and the white HBr-salt was removed by centrifugation (20000g, 10 min, 15 °C). The product was carefully concentrated using a rotary evaporator below 30 ○C (due to the risk of cross linking) and the product was precipitated in diethyl ether (cooled by dry ice) and filtered through a glass filter (P4). The beige product was redissolved in THF and the purification procedure was repeated once. The product (PDMAEMA-stat-PHEMA-I) was characterised by 1H-NMR in acetone-d6 and by 2D-NMR COSY90 in MeOD.
Finally, the tertiary amines of the DMAEMA-side chains were quaternised by MeI. PDMAEMA-stat-PHEMA-I (4.1 g, 9.4 mmol-residues DMAEMA) was dissolved in THF (200 ml) and degassed with argon. MeI (1.7 ml, 28 mmol) was added in excess at RT. The reaction mixture turned milky yellowish after ∼30 min as the solubility in THF decreased with increased quaternisation. The mixture was left to react overnight. Water (20 ml) was added to the reaction mixture to dissolve the quaternised polymer and the THF was removed using a rotary evaporator. The orange product was precipitated in THF (cooled by dry ice) and filtered, redissolved in water, concentrated in a rotary evaporator below 30 ○C, precipitated in cooled THF and filtered. The purified product was finally dissolved in water and freeze dried for 24 h. The yellow final MI of purified freeze-dried qPDMAEMA-stat-PHEMA-I was stored in the dark at 8 °C and was analysed by 1H-NMR in D2O, TGA, FT-IR and PET.
:1:1:0.25:1 for [BMA]:[I]:[CuBr]:[CuBr2]:[HMTETA]. n-BMA (20 ml, 8 °C) from which the inhibitor was removed by passing through a basic alumina column and was added to a round bottom flask equipped with rubber septum, containing E-BiB (14 μl, 97 μmol), HMTETA (26 μl, 97 μmol) and toluene (30 ml) cooled by an ice-bath. The reagents were stirred and the flask degassed by vacuum (4 min) and argon filled (4 min) before CuBr (14 mg, 97 μmol) and CuBr2 (5.4 mg, 24 μmol) were added. The flask was degassed by two additional vacuum-argon cycles (8 min each) before the flask was heated to 60 °C. The reaction kinetics were monitored by 1H-NMR and the reaction was terminated by exposure to oxygen at 73% conversion, the product dissolved in DCM purified through a basic alumina column, precipitated in MeOH and characterised by DMF-SEC (Mw: 182000 g mol−1, Đn: 1.2). The product was melt-processed by compression moulding and characterised by MOCON, TGA, DMA, and FT-IR.
:1:1:8:8 for [M]:[I]:[CuBr2]:[bipy]:[AA]. Fridge cool monomer (3.8–5.7 ml, 8 °C), from which the inhibitor was removed by passing through a basic alumina column, was introduced into an Erlenmeyer-flask equipped with a rubber septum and placed into an ice-bath and degassed by two vacuum/argon cycles. Typically, 20 mg of the MI (0.04 mmol Br) was dissolved in a round bottom flask under magnetic stirring. The flask was sealed with rubber septum and degassed by vacuum (3 min) and argon (3 min) where after ascorbic acid (51 mg, 0.29 mmol), bipy (45 mg, 0.29 mmol) and CuBr2 (8 mg, 0.04 mmol) were added. Finally, the flask was cooled in an ice-bath and cool degassed monomer was added by syringe under argon flow. The cooled reaction mixture was degassed by two final vacuum/argon cycles before the reagents were allowed to react for 3–24 h at 23–30 °C. Aliquots were collected regularly under argon flow in order to monitor the kinetics by 1H-NMR. The precipitated product, in the form of polymer particles, was swollen or dissolved in DCM and purified by solvent extraction with acetone, followed by precipitation in MeOH, filtration and drying in vacuum oven at 50 °C. The products were characterised by 1H-NMR and melt-processed films were characterised by DMA, TGA and FT-IR.
384g, 18 °C). After centrifugation, the MI-GO sheets were readily redispersed by ultrasonication in water and MeOH and were characterised by DLS, FT-IR and TGA.
:1:1:8:8 for [M]:[I]:[CuBr2]:[bipy]:[AA]. Typically, well-dispersed MI-GO (33 mg; 10 mg GO and 0.05 mmol Br), in a mixture of MeOH (40 ml) and water (2.5 ml), was added to a flask equipped with rubber septum and degassed (3 min) and argon filled (3 min) before ascorbic acid (72 mg, 0.41 mmol), bipy (64 mg, 0.41 mmol) and CuBr2 (11.4 mg, 0.05 mmol) were added. The monomers (3–20 ml) were passed through a basic alumina column to remove inhibitor, cooled in an ice-bath and degassed by vacuum-argon cycles in an Erlenmeyer-flask sealed with rubber septum. Finally, the degassed and argon-filled round-bottom flask was cooled and the cool monomer was added by syringe under argon flow. The reaction mixture was degassed by two vacuum/argon cycles before the temperature was increased to 23–30 °C and allowed to react for 0.5–24 h (conversions: 6.5–95.1%). Dispersible, low conversion PBA-MI-GO and PBMA-MI-GO samples were purified by two cycles of centrifugation (30000g, 10 min, 18 °C) and dispersion (ultrasonic bath for 3 min). High-conversion PMMA-MI-GO and PBMA-MI-GO were purified by solvent extraction followed by precipitation, filtration and drying in vacuum oven at 50 °C. The products were characterised by 1H-NMR, AFM, TGA, FT-IR, DLS, confocal Raman, TEM, SEM, POM, and melt-processed films characterised by DMA and MOCON.
Instrumentation
1H-NMR – Proton nuclear magnetic resonance spectroscopy data was obtained from a 400 MHz Bruker Aspect NMR, using D2O, acetone-d6, CDCl3 or MeOD as solvent.DMF SEC – Size exclusion chromatography was performed on a TOSOH EcoSEC HLC-8320 GPC system equipped with an EcoSEC refractive index detector and three columns (PSS PFG 5 mm; Microguard, 100 Å, and 300 Å) columns (MW resolving range: 0.1–300 kDa) from PSS GmbH, using DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was conducted using PMMA standards (MW: 0.8–1600 kDa). Corrections for flow rate fluctuations were made using toluene as internal standard. PSS WinGPC Unity software version 7.2 was used to process data.
UV-Vis spectrophotometry was performed by a Cary UV-Vis spectrophotometer with heating stage (CaryWinUV) to detect the lowest critical solution temperature (LCST) of PDMAEMA-stat-PHEMA. The instrument detected the absorbance/transmittance of a 3 mg per ml solution of the co-polymer at 670 nm while heating the sample from 20–50 °C with a heating rate of 1 °C min−1. The LCST temperature was recorded when the absorbance fell to 50% of the initial absorbance.
Stabino® polyelectrolyte titration technique was used to detect the isoelectric point of the GO using the Stabino® system (Microtrac Europe GmbH, Meerbusch Germany). In this instrument, shearing of the electric double layer produced by surface-immobilised particles produces a streaming potential. The magnitude and phase of the streaming potential is used to detect the isoelectric point. The titration steps are at intervals between 3 to 30 seconds. The amount of titration solution (PDADMAC: 0.275 μeq ml−1) added per step is between 10 and 100 μl, depending on the signal-change and the selected titration program.
PET – Polyelectrolyte titration was also used to detect the charge density of the cationic MI measured by a 716 DMS Titrino from Metrohm (Switzerland). Potassium polyvinyl sulphate (KPVS) was used as titrant at 0.319 μeq per ml concentration and orthotoluidine blue (OTB) was used as the indicator. The colour-change was recorded with a Fotoelektrischer Messkopf 2000 (BASF, Germany) and the MI samples were prepared according to Tera Yama to reach equilibrium around 1 ml of KPVS.34
DLS – dynamic light scattering was used to determine the size and charge of unmodified and modified GO at 20 °C using a Zetasizer Nano ZS particle characterisation system (Malvern Instruments Ltd, UK) with a 633 nm He–Ne laser collecting the scattered light at an angle of 173°. The particle size was calculated from the determined diffusion coefficient (1.333) and absorbance 0.1 assuming spherical symmetry. The size and zeta-potential of GO and MI-GO was determined in DI-water (RI: 1.330) and the PBA and PBMA grafted GO were characterised in THF (RI: 1.409) in glass cuvettes. The reported data represent an average of three measurements containing twelve scans each.
FT-IR spectroscopy was performed with a Perkin-Elmer Spectrum 2000 FT-IR instrument (Norwalk, CT, USA) equipped with a MKII Golden Gate, single reflection ATR system (Specac Ltd, London, UK). The ATR-crystal was a diamond 45° ATR Top plate. All samples were pressed with a force of 35 cNm onto the crystal and scanned between wavenumbers 4000–600 cm−1 at least 16 times.
Confocal Raman spectrometry was performed by a Renishaw 1000 Raman spectrometers with Olympus BH2-UMA microscopes system was used with a red laser (632 nm HeNe) with a spot size between 1–2 μm by the objective lens with ×50 magnification.
DSC – differential scanning calorimetry was performed by a DSC 1 from Mettler-Toledo. Samples of 5–10 mg were measured through a cycle of heating–cooling–heating with a rate of 5 °C min−1. Data was obtained from the second heating cycle: from −40–140 °C for PBMA-samples and from 30–180 °C for PMMA-samples.
TGA – thermogravimetric analysis was performed on a Mettler Toledo TGA/DSC1. STARe software was used to process the data. The samples (2 ± 0.2 mg) were placed in 70 μl ceramic crucibles and heated from 50–800 °C at a rate of 10 °C min−1 in N2 (flow rate = 50 mL min−1).
SEM – scanning electron microscopy (Hitachi S-4800 field emission SEM) was used to study grafted GOs on silicon wafers at an acceleration voltage of 1 kV. The samples were coated by a 5 nm thick Ag/Pt-coating by a sputter coater (Cressington 208HR) prior to imaging.
TEM – transmission electron microscopy (Hitachi HT7700) was used to study the unmodified and grafted GO. Suspensions of GO was deposited on 400 mesh copper grids of deionised water or THF (0.01 wt% GO). Samples were too difficult to microtome without prior moulding into epoxy. The slices were deposited onto 400 mesh copper grids and examined by the instrument operating at an acceleration voltage of 100 kV and high contrast mode.
AFM – atomic force microscopy images were obtained using a Bruker Multimode 8 using a Nanoscope 5 controller in ScanAsyst mode using a ScanAsyst Air cantilever (0.4 Nm−1 nominal spring constant). Scans were 256 × 256 pixel, 20 μm × 20 μm obtained at 0.977 Hz scan rate and cropped for display. Data was processed using Gwyddion V2.41 software (gwyddion.net). Data was levelled by matching scanline median heights and by subtraction of a 3rd order polynomial background, including only background regions in calculations (by manual selection).
DMTA – dynamical mechanical thermal analysis of the physical properties of the matrix-free GO composites was performed on a Q800 DMTA (TA instruments), equipped with a film fixture for tensile testing. The measurements of PMMA-samples and PBMA-samples were performed from 25 °C to 195 °C and from −20 °C to 135 °C, respectively and with a heating rate of 5 °C min−1. The tests were performed in controlled strain mode with a frequency of 1 Hz, oscillating amplitude of 0.1% of the sample length, preload of 0.02 N and force track of 125%.
MOCON Oxtran 2/20 (Modern Controls, Minneapolis, MN) equipped with a coulometric sensor was used to determine the oxygen permeability (OP) according to the ASTM standard D3985-02. Samples were conditioned at 50% or 80–90% RH and 23 °C for up to 30 h before the first measurement. All measurements were repeated three times. Sample films were mounted and sealed between two Al-foils with an open circular area of 5 cm2. The sample thickness was measured with a Mitutoyo micrometre and taken as the average value of five individual measurements.
Melt-processing by compression moulding of polymer films was performed by a TPB400 laboratory press (Fontijne Grotnes BV, Vlaardingen, The Netherlands) with a pressure of 140 kN for 20 min at 110 °C and 175 °C, respectively. The sample powders were pressed between two PET-films and shaped by a stainless steel mould (250 μm thick and 3.4 cm in diameter).
POM – polarized optical microscopy (Leitz Ortholux II POL-BK, Germany) was used to study the microscopic birefringence of matrix-free GO composites with 0° and 90° crossed-polarizers, equipped with a Leica DC300 camera, controlled by the Leica IM50 v1.20 software (Leica Microsystems AG, Switzerland).
Specific network density measurements, used to calculate a relative average molar mass between molecular interconnections were performed with a Mettler Toledo Precisa XR 20SSM-DR kit utilising the Archimedes principle. Each sample was measured at least three times in air and at least twelve times in ethanol, which was used as auxiliary liquid at 20 °C.
Methods and models
| P = DS | (1) |
A model for predicting composite permeability in composites containing sheet-shaped inclusions, the “smallest repeating box power model, (SRBP)”, was developed by Nilsson et al.35 An extension of that model, covering also composite diffusivity, is presented in the following:
An idealized geometric representation of a single sheet is a thin axis aligned brick with side lengths Lx, Ly and Lz (in our case 500 × 500 × 1). If the sheet is surrounded by a polymer matrix, the resulting representative composite domain also becomes brick-shaped and axis aligned with side lengths Cx, Cy, Cz. One corner of the domain is in common with one corner of the sheet. When the domain is cut along the faces of the brick, eight sub-domains occurs, one representing the matrix material (with diffusivity Dm) and seven representing the filler material (with diffusivity Df). The volume fraction of each sub-domain becomes ϕijk, where the three indexes represent the relative positions in the X- Y- and Z-directions, respectively. For (i): 1 = left and 2 = right, for (j): 1 = front, 2 = back and for (k): 1 = bottom, 2 = top. The volume fraction for the filler particle thus became ϕ111 = (LxLyLz)/(CxCyCz). When modelling the composite diffusivity, the eight geometry components can either be first coupled in series and then in parallel (Dα, eqn (2)), or the other way around (Dβ, eqn (3)).
 | (2) |
 | (3) |
These models correspond to Sarenis lamella formula36 and the brick layer model.37 It was however noticed that the geometrical sum of these two models (![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) , eqn (4)) was in even better agreement with corresponding high-accuracy finite element simulation data.35
, eqn (4)) was in even better agreement with corresponding high-accuracy finite element simulation data.35
| = Dmα·D1−mβ | (4) |
In GO-composites, the sheets are typically not perfectly positioned in axis-aligned parallel stacks without overlapping. The effect of overlapping can be accounted for by stretching the simulation domain in a systematic way that is described in detail by Nilsson et al.35 The effect of variable orientation of the sheets can be accounted for by introducing the 3 × 3 rotation matrix R = Rx(Ry·Rz), which describes the average orientation of the bricks. The effective composite diffusivity is finally obtained by multiplying the diffusivity tensor with R according to eqn (5):
| D = RRT | (5) |
![[M with combining tilde]](https://www.rsc.org/images/entities/i_char_004d_0303.gif) c) was calculated as
c) was calculated as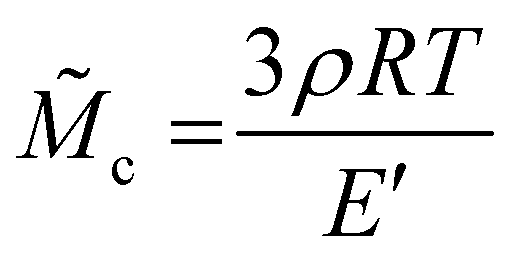 | (6) |
 | (7) |
Results and discussion
The desire to find efficient and controlled grafting routes of dense hydrophobic polymers from hydrophilic GO inspired us to synthesise a cationic MI for electrostatic anchoring to anionic GO. To our knowledge, this is the first time a cationic MI has been immobilised on GO. The procedure for adsorption of MI to GO was optimised and carefully controlled in order to avoid flocculation. Subsequently, a novel and surface-initiated controlled precipitation polymerisation was performed, utilising SI-ARGET ATRPP, to graft dense hydrophobic poly(meth)acrylates (PBA, PBMA and PMMA) from the physisorbed MI. The charge repulsion of MI-modified GO (MI-GO) prevented aggregation of GO and the hydrophobic grafts offered both homogenous distribution in organic solvents and cohesive attraction when dried. The design of the surface modification is reminiscent of a bimodal system of dense repellent short grafts and long cohesive grafts described by Benicewicz et al.21,38 The graft lengths were tuned from extra-short (XS) to extra-long (XL), which determined the properties and appearance of the grafted GO (from suspensions to matrix-free composites). Transparent or translucent black films of matrix-free GO-composites were melt-processed by compression moulding. The optical appearance, orientation of the GO-mesophase, thermo-mechanical properties and the barrier properties were characterised and compared to oxygen permeation models. Moreover, the matrix-free GO-composites exhibited rapid thermo-responsive shape-memory properties (Scheme 2). | ||
| Scheme 2 Immobilization of cationic MI to anionic GO followed by controlled surface grafting and manufacturing of isotropic materials of grafted GO, which transform into transparent matrix-free GO-composites in nematic state after melt-processing. The matrix-free GO-composites obtain shape-memory effects. | ||
Synthesis and characterisation of MI
The cationic MI was synthesised in three steps according to Scheme 1. Firstly, the random co-polymer of DMAEMA and HEMA was synthesised by ATRP using equimolar amounts of DMAEMA and HEMA (DPtarget = 40). According to 1H-NMR the monomer conversion exceeded 90% and the ratio of DMAEMA:HEMA was approximately 1:1 in the statistical co-polymer (Fig. 1-1). The molar mass (Mn) of the co-polymer was assessed to be 9400 g mol−1 at 92% conversion with a reasonably low dispersity (Đn = 1.3). The resulting PDMAEMA-stat-PHEMA (1) was thermally responsive, precipitating at a lower critical solution temperature (LCST) of 37 ± 0.6 °C upon heating and dissolving at 35 ± 0.7 °C upon cooling (Fig. S1†) which served as the upper temperature limit for further laboratory work. In a second step, the α-BiB was immobilised by esterification of the hydroxyl (OH) groups of HEMA-units to introduce a moiety capable of initiating ATRP (Fig. 1-2). The immobilization of α-BiB on PDMAEMA-stat-PHEMA-I (2) was efficient and proceeded to full conversion of the OH-groups according to 1H-NMR as both the e and g peaks shifted upwards (Fig. 1-1, 2). In the last step, the amine groups in the DMAEMA-units were quaternised by methyl iodide. Quaternised and protonated amines are readily distinguishable in 1H-NMR through peaks c and c′ and it was found that the degree of quaternisation was above 94% in the qPDMAEMA-stat-PHEMA-I (3) (Fig. 1-3). The synthesised compound 3 was subsequently used as the MI.
 | ||
| Fig. 1 1H-NMRs of (1) PDMAEMA-stat-HEMA in MeOD, (2) PDMAEMA-stat-PHEMA-I in acetone-d6, and (3) qPDMAEMA-stat-PHEMA-I in D2O. The synthesised compound (3) was subsequently used as cationic MI. | ||
Graft co-polymers
n-BA, n-BMA, and MMA were successfully polymerised from free MI utilising ARGET ATRPP in MeOH:H2O (10:1) at ambient temperatures (23–30 °C). As the side-chains propagated, the solubility of the graft co-polymer decreased in the monomer:solvent mixture, in accordance with free-radical precipitation polymerisations.26–28 The reaction kinetics was followed by 1H-NMR and found to be of first order with respect to monomer concentration up to a limited conversion (∼30%) (Fig. S2†) at which the critical molar mass of grafts had been reached. Above the critical molar mass, the graft co-polymer started to precipitate and the reaction mixture turned translucent and heterogeneous. The ARGET ATRPP gave rise to granular precipitation of graft co-polymers which could be extensively swollen and eventually dissolved in organic solvents, and thus purified by solvent extraction, precipitation and filtration. Dried granules of PBMA-MI and PMMA-MI were melt-processed into clear and transparent films and utilised as GO-free reference materials (Fig. S3†).
Immobilization of MI on GO
It was necessary to first determine the charge densities of GO (ρ−GO) and MI (ρ+MI) to avoid flocculation during the immobilization step of the cationic MI on the anionic GO to form MI-GO. The charge density of GO and MI was found to be ρ−GO: 540 ± 7 μeq g−1 and ρ+MI: 231 ± 6 μeq g−1, respectively. By keeping the cations (MI) in excess and slowly adding the dispersion of GO drop-wise into the solution of MI, the GO-sheets were individually coated and remained dispersed by cationic repulsion. We observed a slight colour-change from dark brown to amber and a significant change in Z-potential, from −49.4 ± 1.2 mV to +45.5 ± 1.1 mV. A small increase in particle size, from 256 to 285 nm, was observed along with a decreasing polydispersity (from 0.257 to 0.243), corroborating the successful immobilization without flocculation (Fig. S4†). Further, the birefringence of GO(aq) remained for MI-GO(aq), demonstrating the presence of lyotropic liquid crystals (LC).The FT-IR data of MI-GO (Fig. 2) demonstrate successful immobilisation of the MI to GO as the spectrum appeared as a combination of the individual MI and GO spectra; containing a large combined C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak (1730 cm−1) next to the overlapping CC peak of GO (1620 cm−1) and OH-bending peak of absorbed water in MI (1624 cm−1).
O peak (1730 cm−1) next to the overlapping CC peak of GO (1620 cm−1) and OH-bending peak of absorbed water in MI (1624 cm−1).
 | ||
| Fig. 2 (a) FT-IR spectra of GO, MI and MI-GO. Spectra have been vertically scaled and offset for clarity. (b) Thermograms of GO, MI-GO and MI. The decomposition slopes of MI-GO are highlighted by arrows relating to the mass-loss of GO and MI, respectively. | ||
To determine the amount of MI immobilised on GO, thermal analysis of GO, MI and MI-GO was conducted. According to the thermogram (Fig. 2), the main mass loss of GO under nitrogen occurs between 50–230 °C, where the first loss is due to removal of moisture and the second larger loss is due to decomposition of the oxygen-containing functional groups.39 Above 230 °C, minor decomposition of the graphitic backbone occurs. The major decomposition of the pure MI occurs between 210–600 °C. Based on this, the mass loss of MI-GO below 210 °C was interpreted as being dominated by the degradation of GO and the mass loss above 210 °C dominated by the degradation of MI. The mass ratio of MI:GO (2.3:1.0) was obtained from the ratio between the first and second mass loss of the MI-GO, according to the description in Fig. S5.† The MI:GO ratio is in close agreement with the mass ratio of MI:GO at the neutralisation point 2.32 ± 0.04 mg MI per mg GO.
Grafting from MI-GO
Polymer brushes of PBA, PBMA and PMMA were grafted from the MI-GO by SI-ARGET ATRPP in MeOH:H2O (10:1) at ambient temperature (23–30 °C). The colour of the GO-suspension changed from brown to grey during the grafting reaction, an effect of simultaneous reduction of GO by the reducing agent (ascorbic acid) present in excess. The precipitation was absent or delayed in the presence of GO in comparison to polymerisations conducted from free MI. For example, the synthesis of PBA-MI-GO (up to 15 h) never gave rise to large precipitated particles, in contrast to PBA-MI without GO. The decrease of the overall reaction rate is most likely an effect of the radical scavenging effect of GO,40 similar to the scavenging effect of carbon black.41
In order to assess the grafting density, the surface area of GO and the number of initiating sites are required. However, it is challenging to determine the specific surface area (SSA) of GO as it differs from the projected surface area (PSA of GO: 1000 m2 g−1) due to the heterogeneous shape and curvature of the sheets. Also, the immobilised MIs may roughen the surface topography by the formation of loops, enhancing the accessibility of initiators and further increase the SSA without affecting the PSA. The large total amount of Br-groups was assessed from the MI:GO-ratio. As most reactions reached high conversions a relatively high grafting efficiency was obtained from the literature.42,43 Further, the presence of GO, CuBr2, excess of ligand, low-swelling of solvent in combination with modest reaction temperatures will increase the grafting efficiency of the MI-GO due to reduced rate of propagation and increased rate of deactivation.44 By assuming a constant grafting efficiency of MI-GO of 50%, the theoretical grafting density of the PSA was determined to be 2.5 mmol grafts per g GO (∼1.5 grafts per nm2), or about 78 grafts per 1000 carbons (∼1 graft per 13 carbons), which is more dense than the dense grafts previously reported (∼1 graft per 49–73 carbons).45,46 As explained above, the high grafting density is likely overestimated as the SSA of GO might be significantly larger than the PSA. Despite that, the grafts on the basal plane are still assumed to be dense (with strong steric constraint) which will prevent sheet-stacking, while the grafts on the edges are considered less constrained, thus more able to entangle.
Further, the polymer graft length was tuned by reaction time (i.e. monomer conversion) or by the amount of monomer added to the reaction. The average molar mass per graft was assessed by thermogravimetry (Fig. 3, S6 and S7†) supported by the monomer conversion and yield which classified the samples as extra-short (XS), short (S), medium (M), long (L), and extra-long (XL) polymer grafts together with the assumed constant grafting density (Table 1). The appearance of the final product was significantly altered by the graft length. GO with the shorter polymer grafts (<4000 g mol−1), including all PBA-MI-GO and the PBMAS-MI-GO, resulted in transparent suspension in THF, which remained stable for weeks. The suspensions of grafted GO were characterised by DLS and the size of PBA-MI-GO increased with graft length indicating controlled grafting (Fig. 3). According to AFM, the surface morphology of PBAS-MI-GO and PBAM-MI-GO remained planar after grafting while the average thickness increased with graft length, from 2.6 ± 0.7 to 5.3 ± 0.3 nm and 17 ± 5.4 nm, respectively. The dimensions are in accordance with dense brushes from unmodified GO (Fig. S8†). The size of PBMAS-MI-GO after 0.5 h of reaction was comparable to the size of PBAM-MI-GO after 15 h of reaction, according to DLS, and was an effect of the much faster overall reaction rates of the methacrylates in comparison to the acrylate, as found during the solution polymerisation described above.
 | ||
| Fig. 3 (a) Thermograms of PMMA-MI-GO and linear PMMA. The thermal stabilisation of the PMMA-MI-GO is superior to the unfilled linear PMMA and the decomposition temperature increase with graft length, thus decreasing amount of GO and increasing time of reduction. The arrow indicates increasing graft length. (b) Size distribution and size increase by graft length of PBA-MI-GO and PBMAS-MI-GO, determined by DLS in THF. | ||
| Sample name | Reaction time (h) | GO contenta (wt%) | DPgraftb |
M
w, graftb (g mol−1) |
D
z
c (nm) |
Pdlc |
|---|---|---|---|---|---|---|
| a As determined by TGA at 600 °C (confirmed by the conversion of monomer and the yield of product). b As determined by TGA and the graft density. c As determined by DLS in THF. | ||||||
| PBAXS-MI-GO | 7 | 15.4 | 10 | 1300 | 543 | 0.163 |
| PBAS-MI-GO | 8 | 14.5 | 11 | 1400 | 576 | 0.161 |
| PBAM-MI-GO | 15 | 8.1 | 28 | 3600 | 763 | 0.242 |
| PBMAS-MI-GO | 0.5 | 8.4 | 24 | 3400 | 674 | 0.258 |
| PBMAM-MI-GO | 8 | 0.67 | 330 | 47000 |
— | — |
| PBMAL-MI-GO | 10 | 0.50 | 450 | 64000 |
— | — |
| PBMAXL-MI-GO | 24 | 0.07 | 3100 | 440000 |
— | — |
| PMMAM-MI-GO | 8 | 0.66 | 490 | 49000 |
— | — |
| PMMAL-MI-GO | 16 | 0.22 | 1500 | 150000 |
— | — |
| PMMAXL-MI-GO | 24 | 0.13 | 2830 | 283000 |
— | — |
After grafting, GO was no longer miscible with water, suggesting efficient shielding of the hydrophilic core of MI-GO by dense short grafts. Thin homogenously coated PBMAM-MI-GO was observed by SEM to have topography and rounded diffuse edges compared to the sharp edges of unmodified GO observed by TEM (Fig. 4). Glass-vials containing suspensions of unmodified GO, MI-GO and grafted GO (PBAM-MI-GO and PBMAS-MI-GO) show birefringence at low mass fractions (fm) of GO when placed between crossed polarizers (Fig. 4). The self-assembly of lyotropic LCs of modified GO-sheets is likely assisted by the short polymer brushes in THF.
 | ||
| Fig. 4 (a) TEM image of unmodified GO sheets. (b) SEM-images of PBMAS-MI-GO sheets. The MI and the grafts increase topography of the basal plane and smooth the edges of pristine GO. (c) The appearance and mass fractions (fm) of solids in suspension A (GO, fm: ∼0.01 wt%) and B (MI-GO, fm: ∼0.03 wt%) in water and C (PBMAS-MI-GO, fm: ∼0.03 wt%) and D (PBAM-MI-GO, fm: ∼0.10 wt%) in THF from left to right. The birefringence was observed in A–D between crossed polarizers (bottom row). | ||
The GO with longer polymer grafts (>40000 g mol−1), i.e. PBMA-MI-GO and PMMA-MI-GO with M–XL grafts, formed homogeneous solid materials at high or complete monomer conversions. PBMA-MI-GO and PMMA-MI-GO samples appeared as dark brown solids and were initially swollen, then eventually dispersed in DCM and subsequently precipitated into MeOH to result in white-grey powders or continuous fibres depending on the precipitation method (Fig. S9†). Microtomed slices of PMMAXL-MI-GO were studied by TEM in high-contrast mode. The TEM-images revealed “state of the art” dispersed GO in frozen randomised arrangements (Fig. 5). The presence of polymer grafts on MI-GO results in a statistical distribution of the sheets, where the separation is controlled by the graft length and appeared homogeneous across the entire slice (Fig. S10†). However, the projection of GO appears more or less wide, depending on the orientation of the sheet. The actual thickness of the edge of GO is estimated to be 1–1.5 nm as highlighted in the insert of Fig. 5. Polymer-grafted GO in the isotropic state corroborates the successful and controlled grafting of isolated MI-GO separated by dense film-forming polymer brushes.
 | ||
| Fig. 5 TEM image of well-dispersed, randomly oriented GO in PMMA-MI-GO. The insertion highlights the bright thin top edges of the GO-sheets in PMMA-MI-GO with arrows. | ||
The grafted GO with M, L and XL graft lengths (PBAM-MI-GO, PMMAL-MI-GO and PBMAXL-MI-GO) were characterised by FT-IR-spectroscopy and appeared similar to the spectra of the pure polymer analogues with the exception of an aromatic CC peak (1620 cm−1) originating from GO (Fig. 6). The absorbance of the CO peak (1730 cm−1) increased with increasing graft length, while the CC peak (1620 cm−1) decreased. The GO was further characterised by confocal Raman (Fig. 6). The D-band (1350 cm−1), G-band (1582 cm−1) and 2D-band (∼2700 cm−1) of GO were clearly detected by the instrument. The relationship between the aromatic (G-band) and the disrupted aromatic (D-band) seemed to be unaffected by the immobilization of MI and the subsequent polymer grafting, indicating non-destructive surface modifications. The 2D-band is related to the exfoliation of GO and is more pronounced for the grafted GO (PBAM-MI-GO, PBMAS-MI-GO and PMMAM-MI-GO) in comparison to pure GO as an effect of separation by the polymer grafts. Further, the C–H band (2900–3000 cm−1) and the CO band (1730 cm−1) were pronounced in the polymer grafted GO and therefore considered to arise from the polymer grafts.
 | ||
| Fig. 6 (a) Normalised FTIR-spectra of GO, PBAM-MI-GO, PMMAL-MI-GO and PBMAXL-MI-GO. Spectra have been vertically scaled and offset for clarity. (b) Confocal Raman spectra of GO, PMMAM-MI-GO, PBAM-MI-GO and PBMAS-MI-GO. Note the presence of D and G and 2D bands originating from the GO and the pronounced C–H peaks at 2900–3000 cm−1 and CO peaks 1730 cm−1 in the grafted samples. | ||
The thermal decomposition temperature of PBA-MI-GO, PBMA-MI-GO and PMMA-MI-GO increased with graft length, i.e. decreased GO-content and increased reduction time, according to the thermograms of grafted GO (Fig. 3, S6 and S7†). Further, the decomposition temperature (at 2 wt% mass loss) increased ∼130 °C for PMMAXL-MI-GO in comparison to linear PMMA. This relationship is most likely an effect of improved encapsulation of GO by the longer polymer grafts in combination with increased reduction time of GO by the reducing agent as well as the decreasing GO-content. It seems beneficial to have a small amount of well-dispersed GO present in order to obtain improved thermal stabilisation. Increased thermal stability of polymers is beneficial, as it determines the temperature at which the materials can be processed and utilised.4 Further, the thermograms of PMMA-MI-GO and PBMA-MI-GO demonstrate a slower decomposition of polymer grafts, with increasing GO content. Thus, the GO-sheets may improve the thermal conduction on the surface of the samples, and thereby shield the polymer grafts in the interior of the sample.
Matrix-free GO-composites
Transparent films without GO (linear PBMA, linear PMMA, PBMA-MI and PMMA-MI) and brown-transparent or black-translucent matrix-free GO-composites of PBMA-MI-GO and PMMA-MI-GO were melt-processed by compression moulding into 250 μm thick homogenous discs (3.4 cm in diameter). The appearance of the films strongly depended on the graft length, i.e. the GO content (Fig. 7). Importantly, the films of PBMA-MI (without GO) started to wrinkle at RT, around the Tg, and were therefore stored at 8 °C <Tg. Despite the better dimensional stability of PBMA-MI-GO films, they were stored under the same conditions. | ||
| Fig. 7 Photographs of flexible (a) transparent brown PBMAXL-MI-GO film and (b) translucent black PBMAM-MI-GO film. Birefringence of GO in the nematic state for (c) PMMAXL-MI-GO and (d) PBMAXL-MI-GO observed by POM. The inserts are images of whole film samples (3.4 cm in diameter) showing (c) a giant single Maltese cross caused by the anisotropic alignment of PMMAXL-MI-GO and (d) GO of PBMAXL-MI-GO in a random or nematic state observed between crossed polarizers. | ||
Isotropic and nematic states of GO
The macroscopic mesophase (cm-scale) of GO-composites was observed between crossed polarizers, and the microscopic mesophase (μm-scale) of GO by optical microscopy using crossed polarizers (POM). Birefringence from frozen LC was observed in all GO-composites. Surprisingly, the birefringence patterns of PMMAXL-MI-GO films appeared in the form of a giant single Maltese cross, 3.4 cm in diameter (Fig. 7 and S11†). Clockwise rotation of the plane of the polarizers gave rise to a clockwise rotation of the extinction cross which is consistent with circular optical symmetry and radially oriented crystals.47 It is suggested that the orientation of GO (nematic state) is induced by the directional flow during the melt-processing by compression moulding. Coloured radial stripes, in the thin outer sections of the films, confirm the direction of flow (Fig. S11†). However, the centre of the films, with limited flow of material could occasionally give rise to colourful light scattering due to isotropic or less ordered GO (Fig. 7 and S11†). The possibility to align grafted GO and manufacture anisotropic materials (such as fibres) is considered to be an important feature for both optical, mechanical and barrier applications.31Thermo-responsive shape-memory effect
The PBMA-MI-GO composites were flexible enough to fold, twist and deform at RT. The deformed shapes were permanent after cooling <Tg of PBMA. The cooled composites became much stiffer, but were still not brittle. The original shape of the composites was rapidly regained when immersed in hot water >Tg of PBMA or by using a heat gun set at 100 °C (Fig. 8). The speed of recovery increased with GO-content and PBMAM-MI-GO (0.67 wt% GO) fully regained its original shape in less than 2 s. Videos are available online to demonstrate the shape-memory effect‡ of the PBMA-MI-GO samples. This is likely an effect of the increased thermal conductivity and the reinforcement by the GO-sheets. We believe that the stable network shape-memory GO-composites consist of mainly molecular entanglements assisted by sparse chemical interconnections, and that the reversible switching transition is an anisotropic/isotropic transition. However, the precise mechanism needs to be further investigated. Tuneable and rapid shape-memory composites have a vast range of potential applications.30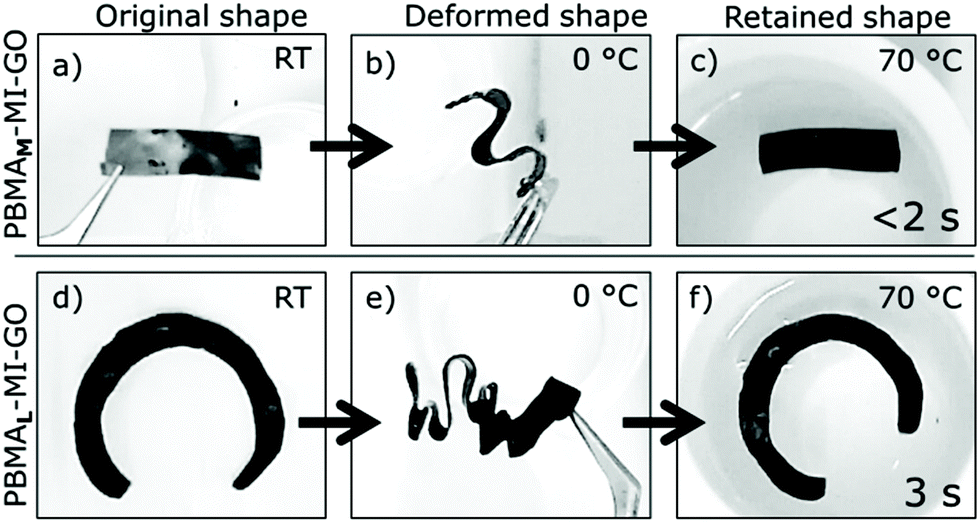 | ||
| Fig. 8 Thermo-responsive shape-memory effect of (a–c) PBMAM-MI-GO and (d–f) PBMAL-MI-GO. (b), (d) The deformed shapes are preserved after cooling in water (0 °C). (c), (f) Rapid shape transformations occur in hot water (70 °C) as the shape-memory composites regain their original shapes. Videos of these shape-memory effects are available online. | ||
The effect of GO orientation on oxygen barrier properties
The gas-barrier properties of the GO-composites were studied as a function of filler fraction in order to determine the 3D-orientation of the non-percolated GO and estimate the possibility to use grafted GO as membrane. The analytical permeability model was used to predict the oxygen permeability (OP) of composites with sheets aligned in the X-, Y-plane (along the direction of the permeate), in the Z-plane (perpendicular to the direction of the permeate) and in the isotropic state (random). The composite permeability was normalised by dividing by the permeability of the linear unfilled polymer (P/P0) and the model predictions were compared with the OP of PMMA-MI-GO and PBMA-MI-GO measured at 23 °C in 50% and 90% relative humidity (RH). The OPs of all GO-composites were successfully measured, including the large OP of 5700 ml μm m−2 24 h−1 kPa−1 of PBMAXL-MI-GO containing 0.04 vol% of GO. However, the OP of linear PBMA at the Tg was close to the limit of the instrument (6000 ml μm m−2 24 h−1 kPa−1).To conclude, the influence of non-percolated GO was much larger for permeable films (i.e. PBMA at Tg) in comparison to gas barriers (i.e. PMMA below Tg). The OP of PBMA-MI-GO decreased with increasing GO ratio and followed the prediction of the analytical permeability model for isotropic (random) GO over the entire range of filler fractions (Fig. 9). Previous OP-studies of solvent casted GO-composites48 demonstrates improved gas-barrier properties since the sheets in those studies are aligned perpendicular to the permeation direction, which is advantageous for barrier materials but not for membranes.
 | ||
| Fig. 9 Normalised permeability of (a) PMMA-MI-GO and (b) PBMA-MI-GO at 50% (squares) and 90% (circles) RH vs. filler fraction of GO. Experimental data is compared to permeation models (lines) of how the OP depend on the X, Y, Z and random orientation of GO. The GO-orientation and the direction of permeate is visualised by insert pictures and arrows, respectively. Due to symmetry the X and Y curves coincide. Storage modulus (E′) versus temperature of (c) linear PMMA, PMMA-MI and PMMA-MI-GO and (d) linear PBMA, PBMA-MI and PBMA-MI-GO. The vertical arrow marks the increase of E′ with GO ratio and the horizontal arrow marks the increase of Tg with graft length. | ||
According to literature, the OP for pure PMMA (Mw: 100000 g mol−1) ranges from 56 to 92 ml μm m−2 24 h−1 kPa−1.49,50 All of our PMMA-samples (filled or unfilled) outperform those reported by having OPs between 41–48 ml μm m−2 24 h−1 kPa−1 (Table S1†). The linear high molar mass PMMA forms strong chain entanglements, which improve the barrier properties in comparison to the shorter PMMA chains.49,50 Similarly, the PMMAXL-MI-GO was less permeable compared to samples having shorter grafts (PMMAL-MI-GO and PMMAM-MI-GO) despite its lower GO-content. The long PMMA grafts also improved the alignment of GO during melt processing due to improved lubrication, according to Fig. 7 and S11.† In the final film, long grafts of PMMA form more entanglements between GO-sheets which improve the film-forming properties. The OP of PMMAXL-MI-GO coincides with the permeation model predictions for randomly oriented GO (Fig. 9) at both 50% and 90% RH, while the OPs of PMMAL-MI-GO and PMMAM-MI-GO coincides with the prediction of GO-sheets oriented parallel to the direction of the permeate (X-, Y-direction). We interpret the improved permeability of the films with shorter PMMA grafts to be an effect of the limited orientation of GO along the Z-direction due to a more restricted flow during processing, forming nematic states including nanochannels for improved oxygen transport.
Previous studies of pure GO-films often report impressively low gas permeability with poor water stability.10 The water-moisture stability and high permeation through nanopores are important properties for new gas-, salt- or ion-selective membranes containing GO.51–53 The PBMA-MI-GO and PMMA-MI-GO were unaffected by changes in humidity, due to the dense hydrophobic grafts effectively shielding the hydrophilic core (MI, GO). The nanochannels between GO-sheets in PMMA-MI-GO and the disrupted aromatic structure of GO might function as selective membranes for larger molecules than oxygen. However, the specific selectivity of the matrix-free GO-composites needs to be further investigated.
Preliminary data (not presented here) of pervaporation studies with PBA-MI-GO as selective skin layers on PE-membranes, show promising tailoring of the pore size, flux and selectivity depending on the graft-length and GO-amount. The variation of inter-particle distance between individual GO sheets is a key parameter to predict and explain the permeation of GO membranes by various solvents and solutions.51 The ability to tune the separation of GO by graft length22 and the orientation of GO by the processing are desirable properties for the manufacturing of selective membranes. Also changing the nature of the grafts (e.g. by introducing stimulus-responsive functionality) might allow the production of molecular scale thermal, electrical and optical sensors.54
The effect of graft-length and GO content on thermo-mechanical properties
The mechanical properties of the PBMA-MI-GO and PMMA-MI-GO were analysed by DMTA and compared to both the linear analogues and to the corresponding graft co-polymer of either PBMA-MI or PMMA-MI. In order to better compare the DMTA data, the relative average molar mass between interconnections (c) of all samples were obtained using eqn (7) and the storage modulus (E′) above Tg at the rubber plateau (Table S2†). The films reinforcement by GO slightly increased the E′ in the rubbery plateau and obtained similar c in comparison to the linear analogues. Thus, the amount of interconnections (cross-links) is small between the grafted GO sheets. However, the MI-initiated polymers were more prone to form interconnections during precipitation polymerisation and therefore obtained a much shorter c. Since each MI hosts a large number of propagating chains, some biradical coupling terminations can easily occur despite the well-controlled polymerisations.
The E′ increased with increasing GO content for both PBMA-MI-GO and PMMA-MI-GO (Fig. 9). The PBMAM-MI-GO containing 0.67 wt% (0.38 vol%) of GO, showed an increase of about 50% in E′ (>600 MPa at −20 °C) compared to PBMA-MI, despite the lower c of PBMA-MI (Table S2†). The reinforcing effect of GO was less pronounced in PMMA-MI-GO, due to the high E′ of neat PMMA. However, the average E′ increased by up to 11% (∼300 MPa at 25 °C) as compared to linear PMMA, despite the low GO content (Fig. S12†). Also, significant increases in Tg were observed with increasing graft lengths (and so decreasing GO content) for both PBMA-MI-GO and PMMA-MI-GO. The Tg was determined in three ways: by the peak of loss modulus (E′′), by the peak of tanδ (E′′/E′) (Table S2†) and by DSC (Fig. S13†). The Tg at the peak of E′′ was in agreement with the DSC data and was therefore considered as most accurate. In comparison to the reference materials the Tg of PBMAXL-MI-GO and PMMAXL-MI-GO increased more than 12 °C to Tg = 40 °C and more than 17 °C to Tg = 127 °C, respectively.
Previous studies of PMMA-GO or PMMA-rGO composites often describe increasing thermal and mechanical properties with increasing GO content (up to 1 wt%).28,55,56 However, in our case the thermal properties of the matrix-free GO-composites dramatically improved with increasing molar mass of grafts i.e. decreasing GO content. Further, the matrix-free GO-composites outperform many of the previously reported ordinary GO-composites, due to dense graft entanglements, which might thermally protect the GO and mechanically stabilise the material at elevated temperatures. It is known that polymer brushes can obtain higher Tg than equivalent free polymers due to grafting,57 but seeing the same effect transferred to a bulk material has until now not been well reported.
The matrix-free GO-composites are tuneable materials, which possess a range of advantages compare to ordinary composites, such as tuning of the inter-particle distance, tuning of the GO-orientation, and the possibility to design highly filled non-percolated GO-composites. These advantages, together with thermal, mechanical and water stability, make them promising candidates for a range of applications such as durable low-friction coatings, flexible electronics, sensors and selective membranes for fuel-cells, batteries and gas/water-purification systems.
Conclusion
In this work, we have developed a novel route to graft hydrophobic polymers from hydrophilic GO sheets using a cationic MI, with the aim to obtain isotropic dispersions and matrix-free GO composites. The statistical cationic MI was synthesised by ATRP, physisorbed to GO and finally grafted with PBA, PBMA or PMMA brushes using SI-ARGET ATRPP. The surface modification of GO was reminiscent of a bimodal graft system, where the cationic MI creates repulsion, and the long grafts form entanglements and cohesion.The graft length of the polymer brushes was tuned from XS–XL, which determined the appearance of the grafted GO, as the GO content decreased with increasing graft length. Transparent and translucent black films of matrix-free GO-composites were melt-processed by compression moulding. TEM images show “state of the art” isotropic dispersions of GO prior to processing. However, after melt-processing the mesophase of matrix-free GO-composites show nematic alignment, seen as a single giant Maltese cross covering the entire film (3.4 cm in diameter). Rapid and tuneable thermo-responsive shape-memory effects were obtained, likely as an effect of reversible anisotropic/isotropic transitions. Permeability models for composites containing oriented 2D-fillers were developed and confirmed the isotropic or nematic state of the GO by comparing the experimental OP-data to the predicted OP of different GO-orientations.
The E′ of the matrix-free GO-composites increased with GO content, while the Tg increased with graft length. The increases in Tg observed for the matrix-free GO-composites are superior tostudies of ordinary composites filled with (r)GO and also show the opposite dependence on GO loading, as the Tg normally increase with GO content. The mechanical and thermal stability together with the tunability of matrix-free GO-composites make them promising materials for a range of applications, such as robust and selective membranes and sensors.
Acknowledgements
The authors gratefully acknowledge the financial support from ABB AB, European Institute of Innovation and Technology (EIT) – KIC InnoEnergy, and Swedish Centre for Smart Grids and Energy Storage (http://www.swegrids.se). Jason Hui (University of Manchester) is greatly acknowledged for the AFM imaging and thickness measurements of modified and unmodified GO. Oliver Andrén (KTH Royal Institute of Technology) is acknowledged for editing the SME-videos. Dr Yue Lin (University of Manchester) is acknowledged for the introduction to and discussion about confocal Raman.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo and N. Pugno, et al. , Nanoscale, 2015, 7, 4598–4810 RSC.
- M. Zhang, Y. Li, Z. Su and G. Wei, Polym. Chem., 2015, 6, 6107–6124 RSC.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Carbon, 2015, 95, 460–484 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- Y. You, V. Sahajwalla, M. Yoshimura and R. K. Joshi, Nanoscale, 2016, 8, 117–119 RSC.
- K. Huang, G. Liu, J. Shen, Z. Chu, H. Zhou, X. Gu, W. Jin and N. Xu, Adv. Funct. Mater., 2015, 25, 5809–5815 CrossRef CAS.
- J. Yang, C. Liu, L. Gao, J. Wang, Y. Xu and R. He, RSC Adv., 2015, 5, 101049–101054 RSC.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- N. Politakos, G. Liontos, A. Karanastasis, G. Zapsas, D. Moschovas and A. Avgeropoulos, Curr. Org. Chem., 2015, 19, 1757–1772 CrossRef CAS.
- M. Seifert, A. H. R. Koch, F. Deubel, T. Simmet, L. H. Hess, M. Stutzmann, R. Jordan, J. A. Garrido and I. D. Sharp, Chem. Mater., 2013, 25, 466–470 CrossRef CAS.
- F. L. Hatton, E. Malmström and A. Carlmark, Eur. Polym. J., 2015, 65, 325–339 CrossRef CAS.
- M. Moglianetti, J. R. P. Webster, S. Edmondson, S. P. Armes and S. Titmuss, Langmuir, 2011, 27, 4489–4496 CrossRef CAS PubMed.
- X. Y. Chen, S. P. Armes, S. J. Greaves and J. F. Watts, Langmuir, 2004, 20, 587–595 CrossRef CAS PubMed.
- V. T. Huynh, D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1413–1421 CrossRef CAS.
- Q. Wei, X. Wang and F. Zhou, Polym. Chem., 2012, 3, 2129–2137 RSC.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- C. M. Hui, J. Pietrasik, M. Schmitt, C. Mahoney, J. Choi, M. R. Bockstaller and K. Matyjaszewski, Chem. Mater., 2013, 26, 745–762 CrossRef.
- M. Baum and W. J. Brittain, Macromolecules, 2002, 35, 610–615 CrossRef CAS.
- Y. Li, P. Tao, A. Viswanath, B. C. Benicewicz and L. S. Schadler, Langmuir, 2012, 29, 1211–1220 CrossRef PubMed.
- M. Wåhlander, F. Nilsson, E. Larsson, W.-C. Tsai, H. Hillborg, A. Carlmark, U. W. Gedde and E. Malmström, Polymer, 2014, 55, 2125–2138 CrossRef.
- J. Yan, T. Kristufek, M. Schmitt, Z. Wang, G. Xie, A. Dang, C. M. Hui, J. Pietrasik, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2015, 48, 8208–8218 CrossRef CAS.
- L. A. Fielding, S. Edmondson and S. P. Armes, J. Mater. Chem., 2011, 21, 11773–11780 RSC.
- B. T. Cheesman, J. D. Willott, G. B. Webber, S. Edmondson and E. J. Wanless, ACS Macro Lett., 2012, 1, 1161–1165 CrossRef CAS.
- H. S. Mickley, A. S. Michaels and A. L. Moore, J. Polym. Sci., 1962, 60, 121–140 CrossRef CAS.
- W. I. Bengough and R. G. W. Norrish, Proc. R. Soc. London, Ser. A, 1950, 200, 301–320 CrossRef CAS.
- J.-M. Thomassin, M. Trifkovic, W. Alkarmo, C. Detrembleur, C. Jérôme and C. Macosko, Macromolecules, 2014, 47, 2149–2155 CrossRef CAS.
- H. Zhang, Eur. Polym. J., 2013, 49, 579–600 CrossRef CAS.
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef CAS.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef PubMed.
- Angstron_Materials, N002-PS-0.5 Graphene Oxide Solution, https://www.angstronmaterials.com/shop/n002-ps-graphene-oxide-solution-thickness-1-0-1-2nm-x-y/.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed.
- H. Terayama, J. Polym. Sci., 1952, 8, 243–253 CrossRef CAS.
- F. Nilsson, U. W. Gedde and M. S. Hedenqvist, Compos. Sci. Technol., 2011, 71, 216–221 CrossRef CAS.
- B. Sareni, L. Krähenbühl, A. Beroual and C. Brosseau, J. Appl. Phys., 1997, 81, 2375–2383 CrossRef CAS.
- J.-H. Hwang, D. S. McLachlan and T. O. Mason, J. Electroceram., 1998, 3, 7–16 CrossRef.
- A. Rungta, B. Natarajan, T. Neely, D. Dukes, L. S. Schadler and B. C. Benicewicz, Macromolecules, 2012, 45, 9303–9311 CrossRef CAS.
- Y. Qiu, F. Guo, R. Hurt and I. Külaots, Carbon, 2014, 72, 215–223 CrossRef CAS PubMed.
- C. Zhang, S. Chen, P. J. J. Alvarez and W. Chen, Carbon, 2015, 94, 531–538 CrossRef CAS.
- T. Liu, S. Jia, T. Kowalewski, K. Matyjaszewski, R. Casado-Portilla and J. Belmont, Langmuir, 2003, 19, 6342–6345 CrossRef CAS.
- P. Vana and E. Bourgeat-Lami, Controlled Radical Polymerization at and from Solid Surfaces, Springer, 2015 Search PubMed.
- A. El Harrak, G. Carrot, J. Oberdisse, C. Eychenne-Baron and F. Boué, Macromolecules, 2004, 37, 6376–6384 CrossRef CAS.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2010, 20, 1982–1992 RSC.
- H. Honda, H. Kimura and Y. Sanada, Carbon, 1971, 9, 695–697 CrossRef CAS.
- Y. Cui, S. I. Kundalwal and S. Kumar, Carbon, 2016, 98, 313–333 CrossRef CAS.
- K. E. Min and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 1021–1033 CrossRef CAS.
- S. Hess, M. M. Demir, V. Yakutkin, S. Baluschev and G. Wegner, Macromol. Rapid Commun., 2009, 30, 394–401 CrossRef CAS PubMed.
- A. Klechikov, J. Yu, D. Thomas, T. Sharifi and A. V. Talyzin, Nanoscale, 2015, 7, 15374–15384 RSC.
- Y. You, V. Sahajwalla, M. Yoshimura and R. K. Joshi, Nanoscale, 2016, 8, 117–119 RSC.
- P. Sun, M. Zhu, K. Wang, M. Zhong, J. Wei, D. Wu, Z. Xu and H. Zhu, ACS Nano, 2013, 7, 428–437 CrossRef CAS PubMed.
- H. H. Kung, J. Shen, M. C. Kung and C. M. Hayner, Polymer Functionalized Graphene Oxide and Thermally Responsive Ion Permeable Membranes Made Therefrom, US Pat., 2015/0318531 A1, 2015 Search PubMed.
- V. H. Pham, T. T. Dang, S. H. Hur, E. J. Kim and J. S. Chung, ACS Appl. Mater. Interfaces, 2012, 4, 2630–2636 CAS.
- Y. Yang, C.-E. He, W. Tang, C. P. Tsui, D. Shi, Z. Sun, T. Jiang and X. Xie, J. Mater. Chem. A, 2014, 2, 20038–20047 CAS.
- S. Yamamoto, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 6077–6079 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures of LCST, polymerization kinetics, melt-processed films, DLS, TGA, precipitated fiber and powder, TEM (of isotropic GO), birefringence, OP-data, DMTA-data and DSC. See DOI: 10.1039/c6nr01502f |
| ‡ Videos of the shape-memory effect are available online. See DOI: 10.1039/c6nr01502f |
| This journal is © The Royal Society of Chemistry 2016 |