
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent boronate-based polymer nanoparticles with reactive oxygen species (ROS)-triggered cargo release for drug-delivery applications†
Eliézer
Jäger
*,
Anita
Höcherl
*,
Olga
Janoušková
,
Alessandro
Jäger
,
Martin
Hrubý
,
Rafał
Konefał
,
Miloš
Netopilik
,
Jiří
Pánek
,
Miroslav
Šlouf
,
Karel
Ulbrich
and
Petr
Štěpánek
Institute of Macromolecular Chemistry v.v.i., Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic. E-mail: jager@imc.cas.cz; hocherl@imc.cas.cz
First published on 25th February 2016
Abstract
A new drug-delivery system of polymer nanoparticles (NPs) bearing pinacol-type boronic ester and alkyne moieties displaying triggered self-immolative polymer degradation in the presence of reactive oxygen species (ROS) with the capability of cellular imaging is presented. The NPs specifically release their drug cargo under concentrations of ROS that are commonly found in the intracellular environment of certain tumors and of inflamed tissues and exhibit significant cytotoxicity to cancer cells compared to their non-ROS-responsive counterparts.
The incorporation of selectively chemically degradable linkages into polymer-based nanoparticles (NPs) and microparticulate drug-delivery systems allows one to achieve external stimulus-triggered polymer degradation and triggered release.1–3 This is a very useful feature both to release the therapeutic cargo and to eliminate the biomaterial from the body after the cargo is released and the carrier is no longer needed. Such a stimulus may be an enzymatic removal of protecting groups, a pH change, light or more recently, the presence of reactive oxygen species (ROS) in the surrounding environment.4–6 The ROS plays a crucial role in human physiological and pathophysiological processes. An increasing amount of data indicates that ROS such as H2O2 are a component of cell signaling pathways that are necessary for the growth, development, and fitness of living organisms.7 On the other hand, imbalances in H2O2 production lead to oxidative stress and inflammation events, which damages the tissue and organ systems and are correlated with the onset and advancement of various diseases, including cancer, diabetes, cardiovascular and neurodegenerative diseases.8–11 Among the ROS species, H2O2 is the most expressed in tumors and at higher concentrations when compared to other ROS species, as well as, when compared with normal cells where intracellular concentration levels may span four orders of magnitude from 10−8 M in proliferation to 10−4 M in apoptosis.12,13 In line with this, several studies have detected elevated rates of ROS in almost all human cancer cells compared to their normal counterparts.14–18 Therefore, H2O2 has become a common marker for oxidative stress playing important roles in carcinogenesis and is also linked to apoptosis, cell proliferation and DNA mutations.19–21 Thus the involvement of ROS in cellular signaling and disease states has motivated the construction of clever chemical tools such as ROS-responsive micro- and NPs as drug carriers.22–24
The ability to generate a triggered NP carrier response (e.g., release of cargo or polymer degradation) in a ROS rich microenvironment is of particular interest, e.g., for the targeted drug delivery to tumors and sites of inflammation.4,5,23–26
Herein, a biocompatible and biodegradable ROS-sensitive polymer backbone with the capability of cellular imaging in a ROS-rich environment was synthesized by step-growth polymerization from monomers bearing a ROS-degradable pinacol-type boronic ester and an alkyne moiety suitable for click chemistry-based attachment of the active cargo (see Scheme 1).
 | ||
| Scheme 1 Synthetic route of the ROS-responsive polymer 1 (P1) bearing the alkyne group-containing monomeric unit 2 suitable for the click reaction. | ||
It is important to highlight that the pinacol-type boronic ester groups have been shown to be the most ROS selective and sensitive probes to detect H2O2 at physiological concentrations with high specificity.12,23,27,28
Initially, monomer 1 was synthesized according to the previously reported procedure23 (see the ESI† for the synthetic route). Monomer 2 (Scheme 1) was synthesized by the protection of 2,6-bis-(hydroxymethyl)-p-cresol with tert-butyldimethylsilyl chloride generating compound 2 that was then reacted with propargyl bromide to provide the protected alkyne compound 3 (Fig. S1, see the ESI† for the synthetic route). Monomer 2 (Scheme 1) was obtained in high yield (94%) after the removal of the protecting groups from compound 3 (ESI Fig. S2†). The synthesized monomers 1 and 2 were further successfully copolymerized with pimeloyl chloride generating the ROS-responsive polymer 1 (P1) (Scheme 1 and the ESI†).
Successful polymer synthesis was confirmed by 1H NMR (Fig. 1) and by size exclusion chromatography (SEC) analysis (Fig. 2a). Weight-average molecular weight (Mw) of polymer P1 was 21.5 kDa with a reasonable polydispersity index PDI = Mw/Mn = 1.49 (where Mn is the number-average molecular weight) as determined by SEC (Fig. 2a – black line). The 1H NMR spectrum of P1 shows characteristic signals for protons belonging to the repeating units of the monomers. The signals from the protons in monomer 1 and monomer 2 aromatic rings were detected at δ = 7.68 ppm (1 – see Fig. 1 for signal-structure assignment), δ = 7.41 ppm (2), and δ = 7.16 ppm (3). The methylene protons (4) of monomers 1 and 2 from the main chain of P1 were observed in the same position at δ = 5.08 ppm, whereas the signals attributed to the methylene groups of side chains of monomers 1 (5) and 2 (6) appear at δ = 4.88 and 4.57 ppm, respectively (Fig. 1). The signal of the proton of the terminal alkyne group (7) is at δ = 2.51 ppm (spectrum of P1 in d6-DMSO is also given, see ESI Fig. S3†). Furthermore, the spectrum displayed signals of methylene groups (10) from the pimeloyl chloride monomeric repeating unit at δ = 1.48 ppm, and the peaks of the methyl with methylene groups (8 + 9, 12 + 11) with chemical shifts at δ = 2.25 and 1.26 ppm, respectively. A ROS-insensitive counterpart to the P1 polymer (polymer 2; P2) was also synthesized to investigate the ROS response to the intracellular drug release efficiency.
 | ||
| Fig. 1 1H NMR spectra of the synthesized ROS-responsive polymer (P1) containing the monomer 2 units enabling the polymer modification by the click reaction. | ||
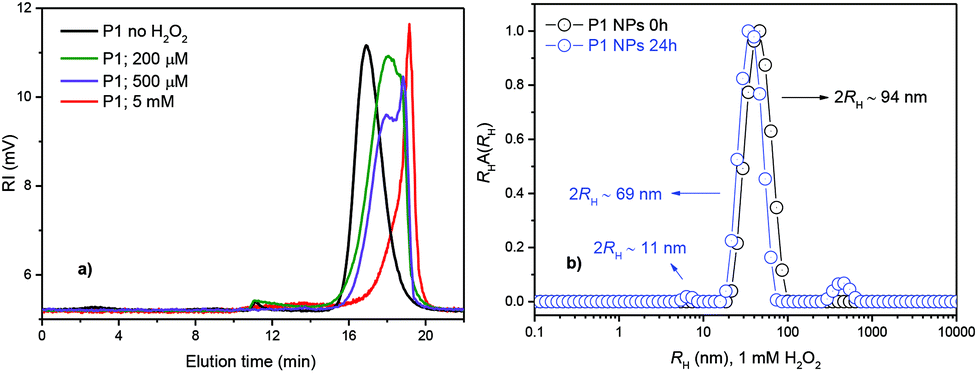 | ||
Fig. 2 SEC chromatograms of P1 prior to the addition of H2O2 (black line) and after degradation in 20% PBS/DMF solutions containing 200 μM and 500 μM (physiologically relevant levels) or 5 mM H2O2, respectively (incubation at 37 °C for 1 day) (a), and (b) intensity-weighted distributions of RH for P1 (○) prior to the H2O2 addition and ( ) after 24 h of incubation in 1 mM H2O2. ) after 24 h of incubation in 1 mM H2O2. | ||
Spectra of the ROS-insensitive counterpart polymer 2 in CDCl3 showed the characteristic peaks (Fig. S4, ESI†), which also indicated successful polymer synthesis.
The degradation of the P1 polymer in the presence of H2O2 was characterized by SEC analysis and 1H NMR following a modified methodology.23 Under exposure to H2O2 the aryl boronic ester groups are oxidized and subsequently hydrolyzed to display a phenol. The latter undergoes a quinone methide rearrangement to degrade the polymer in conformation with Scheme S1 (see the ESI†).
In a typical experiment, P1 was incubated in a 20% PBS/DMF (v/v) solution containing different H2O2 concentrations and at predetermined time intervals aliquots were examined by SEC (see the ESI† for methods).
The SEC chromatogram (Fig. 2) shows that P1 degraded into small molecules and oligomers in a time- and H2O2-dependent manner. Polymer degradation proceeds more extensively with an increasing incubation time and H2O2 concentration. P1 was shown to be responsive to physiologically relevant levels of H2O2 (≲1 mM)12,13,23,27,28 after 1 day of incubation (Fig. 2a) while the non-ROS-responsive counterpart polymer (P2) showed no degradation during the same time and under the same conditions (data not shown). When degradation of both the polymers was compared at higher H2O2 concentrations and for a longer time (5 mM, 4 days), the degradation of the P2 polymer was only partial (Fig. S5†). The degradation of polymer P1 evaluated with 1H NMR was complete after 5 days of incubation (see ESI Fig. S6†) as broad peaks in 1H NMR related to the polymer are replaced by sharp peaks of the low-molecular-weight degradation products (monomers) confirming the depolymerisation of P1 triggered by H2O2 (ESI, Fig. S6†).
The NPs from the P1 and P2 polymers were prepared by a nanoprecipitation protocol (see the ESI†) and their behavior under different ROS concentrations was evaluated in detail by dynamic light scattering (DLS), static light scattering (SLS), transmission electron microscopy (TEM) and by in vitro drug model release experiments.
Note that the NPs were prepared with a hydrodynamic radius (2RH = DH ∼ 94 nm), e.g., within a range known to be ideal for efficient tumor accumulation due to the enhanced permeability and retention (EPR) effect.29
The ROS-responsiveness capability of the P1 NPs was tested by DLS after 24 h of incubation with 1 mM of H2O2. Fig. 2b shows the distribution of RH for P1 NPs before and after 24 h of incubation as measured by DLS. The distribution of RH for P1 NPs appears as only one single distribution of RH relative to the presence of the single spherical polymer NP in PBS solution with an average diameter of 2RH ∼ 94 nm (Fig. 2b, black circles). Furthermore, the polydispersity of the NPs is very low as estimated through the cumulant analysis (μ/Γ2 = 0.08 ± 0.007) (ESI†). This is important for the homogeneous biological behavior of such NPs. However, after 24 h of H2O2 incubation, a trimodal distribution of RH was observed. In addition to the NP peak, the presence of molecularly dissolved copolymer chains as well as a peak of large aggregates could be noticed in the aqueous solution at 1 mM H2O2. Three well-defined peaks highlighting the three populations of the scattering polymer with average diameters of DH ∼ 11 nm, 69 nm and 1.9 μm were identified (Fig. 2b, blue circles). They can be attributed to free chains and their fragments, surface-eroded nanoparticles (decrease in DH of ∼25 nm) and polymer aggregates, respectively.3,30 The NP degradation can be clearly visualized in the volume-weighted distribution of RH (see Fig. S7, ESI†). Further the polymer degradation-triggered cargo release was studied using the release of the fluorescent model drug Nile Red (NR). Incorporation of the fluorescent marker also provided the means to study the cellular uptake of the NPs by microscopy and flow cytometry (FC). The NR-loaded ROS-responsive (from polymer P1) and non-ROS-responsive (from polymer P2) NPs were examined with fluorescence spectroscopy measurements over 24 h of incubation with 1 mM of H2O2 (Fig. S8–S10, ESI†). After 24 h the NR release from the ROS-responsive NPs was almost ∼6 times faster than the NR release from their non-ROS-responsive counterparts, thus confirming the potential of the P1 polymer NPs to release the model drug specifically in simulated ROS-rich microenvironments (Fig. S10, ESI†). The NP degradation was also investigated by TEM microscopy (Fig. 3). The TEM microscopy showed the particle size qualitatively comparable to that determined by DLS (Fig. 2b). Prior to incubation with H2O2, compact NPs of spherical morphology and narrow size distribution (DH ≤ 85 nm) were observed (Fig. 3a).
 | ||
| Fig. 3 TEM micrographs of polymer NPs from polymer P1 prior to incubation with H2O2 (a) and after 2 days of incubation with 1 mM of H2O2 (b). | ||
After incubation with 1 mM H2O2, the NPs showed diffused irregular shapes and a very broad size distribution, with the smallest NPs well below 10 nm and the largest NPs above ∼100 nm (Fig. 3b). This suggested that H2O2 caused decomposition of the NPs, and the decomposed parts were probably re-agglomerated due to their hydrophobicity. SLS data support the findings that the NPs underwent surface degradation as well as core decomposition, as the particles’ DH decreased (by 25 nm, see Fig. 2b) as well as the overall scattering intensity (Fig. S11a†) followed by the increase in particles RG (gyration radius), a characteristic of core hydration and swelling of the scattering particles in solution (Fig. S11b†).31–33 The decomposition was also in agreement with the observed low contrast for the incubated NPs (Fig. 3b). Compact NPs exhibited sharp edges and high contrast, whereas decomposed NPs, NP fragments and their agglomerates showed only a vague interface.
The cellular uptake of P1 and P2 NPs loaded with NR (dye loading ∼0.2 wt%, see ESI Fig. S8 and S9†) and the intracellular NR release were followed in vitro in human prostate cancer (PC-3) cells (see the ESI† for methods). The latter are known to produce high ROS levels.34 While NR is highly fluorescent when incorporated inside the NPs, after ROS-triggered NP degradation in the cells the dye is released and quenched outside the NPs (due to polarity changes in the micro-surrounding). After a short incubation of 4 h both the NPs displayed comparable fluorescence intensity in the cells under the microscope, however, after 20 h the fluorescence of P1 was much lower compared to P2 NPs (see Fig. S12†). This indicated a faster ROS-triggered degradation of P1 NPs after a prolonged exposure in ROS-producing cells. Based on a similar uptake rate of the NPs (as also confirmed by FC, see Fig. S13†), the ROS-mediated fluorescence decay and cargo release of P1 NPs was further pursued via fluorescence lifetime microscopy (FLIM) and FC. In a quantitative study via FC the NR quenching of the NPs was evaluated in PC-3 and human fibroblast (HF) cells (see the ESI† for methods). The latter is known for their low levels of ROS production contrary to PC-3.34–37 The cells were loaded with P1 NPs, washed and incubated for 4 h, thus exposing the internalized NPs to intracellular ROS insofar as present in the cells. After incubation the NR fluorescence was significantly reduced in PC-3 cells compared to the HF cells (Fig. 4).
 | ||
| Fig. 4 Nile Red fluorescence signals from NR-loaded P1 NPs in PC-3 and HF cells after 4 h of incubation. At t0 prior to incubation, the cells have been loaded with P1 NPs by a 2 h pre-incubation step, further the NPs were washed off (see the ESI† for methods). *0.01 level (ANOVA One-way). | ||
By the same experimental setup the Nile Red quenching of P1 and P2 NPs in PC-3 cells was compared. In line with the previous imaging data a lowered NR fluorescence of P1, factor 0.7, compared to P2 was observed. The NR release from the P1 and P2 NPs in PC-3 cells and in the presence of catalase (H2O2 scavenger agent) was also performed (see the ESI† for methods). A similar NR release from P2 NPs (non-responsive) was observed i.e., independent of catalase (Fig. S14b†), however, from the responsive P1 NPs the NR release is prevented in the presence of catalase (Fig. S14a†) demonstrating the specificity of the P1 NPs to release the drug in the presence of H2O2 (Fig. S14a and c†). In conclusion, the FC data acquired in PC-3 cancer cells (with and without catalase) and non-cancer HF cells indicated a ROS-induced degradation of P1, and demonstrated the polymer's potential to specifically trigger the cargo release in an ROS-rich intracellular environment. As the released NR inside the cells can interact with hydrophobic cell structures and partially recover fluorescence, released NR is never fully quenched and some residual fluorescence can be visualized by microscopy.
To show the NR release via co-localization in FLIM microscopy, a second dye Alexa Fluor® 647 (Alx647) azide was covalently bound to the clickable alkyne linker of P1via the click reaction (see ESI Fig. S15 and S16†). NR was physically entrapped into the Alx647-labeled NPs as described previously. With two fluorophores the intracellular fate of the cargo (NR) and the polymer (stained covalently with Alx647) could be tracked independently. The two dyes were visualized after separate excitation at 485 nm (NR) and 640 nm (Alx647). The co-localization of P1 NPs and the NR cargo after 8 h incubation in PC-3 (Fig. 5a and b) and HF cells (Fig. 5c and d) was compared.
 | ||
| Fig. 5 FLIM microscopy of dual-marked P1 NPs in PC-3 (a, b) and HF (c, d) cells after 8 h incubation, visualizing (released) NR and polymer-bound Alx647. Fluorescence was detected after separate excitation at 485 nm (NR, in a, c) and 640 nm (Alx647, in b, d). In a, b locations with high polymer content but little co-localizing NR are pointed out (circle), and vice-versa (square). In c, d the fluorescence patterns predominantly co-localize (squares). | ||
Analysis of the lifetime τ (see the ESI† for method) clearly differentiated the free NR (τ 4.2 ± 0.3 ns, exc. at 485 nm) from the NPs marked with Alx647 (τ 2.2 ± 0.1 ns, exc. at 640 nm).
After 1 h incubation of PC-3 cells with dual-marked P1 NPs, both NPs and NR were highly co-localizing and hardly any release of NR was observed (see ESI Fig. S17†). But after incubation in PC-3 cells for 8 hours, the cytoplasm was more homogeneously colored with the released NR (Fig. 5a), while the polymer was clustered up in few locations (Fig. 5b). Oppositely in HF cells only a little homogeneous NR fluorescence was visible outside the NPs after 8 h incubation (Fig. 5c), and the NR co-localized with the polymer to a high extent (Fig. 5d). However, after 8 h even in ROS-producing PC-3 cells the NPs likely were not fully degraded and the cells still contained NP-loaded NR as well (see the ESI†). In line with the findings after separate excitation, after simultaneous excitation at 485 nm and 640 nm the spread fluorescence of released NR was visible in PC-3 but barely in HF cells, and the co-localization of not-yet released NR with the particles was visible in HF but not in PC-3 cells (Fig. 6a and b).
 | ||
| Fig. 6 FLIM images of PC-3 (a) and HF (b) cells after 8 h incubation with dual-marked P1 NPs, color-coded by the averaged obtained lifetime per pixel. The localization of the polymer (covalently bound Alx647, tau ca. 2 ns, shown in blue) and of released Nile Red (spread throughout the cell, tau ca. 4 ns, shown in green), and local overlap of lifetimes (turquoise tones) was visualized after simultaneous excitation at 485 nm and 640 nm. | ||
This indicates that the P1 polymer NPs can be used for selective cargo release to PC-3 cancer cells, with the release rate in non-cancer HF cells being lower than that in the cancer cells with higher ROS levels.
Finally, to investigate the inhibitory effect on tumor cells, the ROS-responsive (P1) and non-responsive counterpart (P2) NPs were loaded with the antitumor drug paclitaxel (PTX) with an overall cargo content of ∼2.2 wt% and a loading efficiency of 94% (see the ESI†). Firstly, because stimuli-responsive NPs have been shown to have environment-dependent PTX release behavior,38,39 the in vitro PTX release from P1 and P2 NPs was evaluated in the presence of H2O2 (1 mM) and in PBS (pH 7.4) (see the ESI† for methods). The PTX release from the NPs was similar to the NR release (Fig. S10†) where after 24 h the PTX release from the ROS-responsive NPs was ∼2.5 times faster than the PTX release from their non-ROS-responsive counterparts (Fig. S19†).
Further, the alamarBlue® viability assay was used to evaluate the cytotoxicity of the PTX-loaded P1 and P2 NPs in cancer cell lines and in HF cells.
For this study various cancer cell lines, which are known for increased ROS production, such as human cervix carcinoma (HeLa),37 colorectal adenocarcinoma (DLD1)40 and prostate cancer (PC-3)34 cells were used and the NP cytotoxicity was compared with that found for the HF fibroblast cells as low ROS controls (see the ESI† for methods). The drug-loaded NPs were incubated with the ROS-producing cells and with HF cells for 24 up to 72 h. Both NPs were at all times more toxic than the free drug (Fig. S20 and S22, ESI†), which is generally attributed to the fact that the vast majority of the freely administered drug molecules are bound to serum proteins.41 Incubation of PC-3 and HF cells for 48 h showed a comparable toxic effect of the PTX-loaded P1 and P2 NPs (Fig. S20†). Note that the polymer material itself had no relevant effect on cell viability (Fig. S21, ESI†). As another test in HeLa cells had confirmed that NP toxicity steeply increases with the incubation time (raised from 12 h to 96 h in HeLa cells, see Fig. S22, ESI†), the difference in toxicity of P1 and P2 NPs could become more significant after longer incubation times (under conditions of extensive ROS-triggered NP degradation). Indeed after incubation for 72 h the higher toxicity of the PTX-loaded P1 NPs compared to P2 NPs was visible. The superior toxicity of the ROS-responsive NPs may be expected to be more prominent after a longer incubation time, because a longer incubation will increase the amount of internalized NPs, the timespan for ROS-triggered drug release and the exposure of cells to the drug. PC-3, HeLa and DLD1 cancer cells were incubated for 72 h with the NPs under standard cell culture conditions and in a second test under conditions of a low serum content of 2% in the medium (Fig. 7). Cell viability was not affected by the reduced serum content (data not shown). However, low serum content is known to enhance the overall uptake rate of the NPs,42 which in return might further enhance the superior toxicity of P1 NPs for cancer cells.
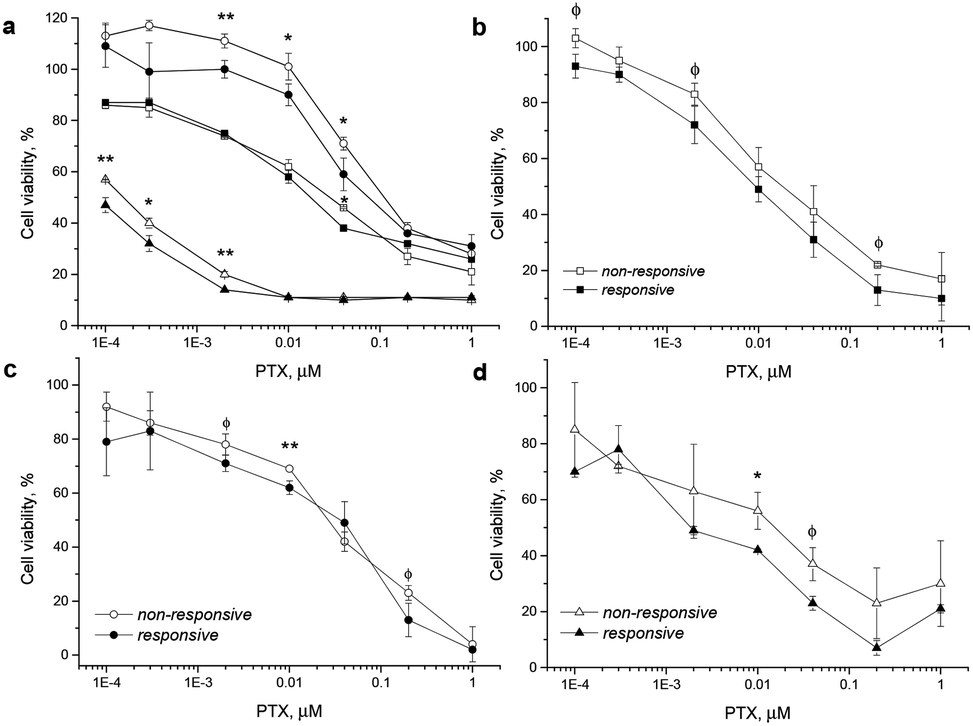 | ||
| Fig. 7 Incubation of PTX-loaded ROS-responsive P1 (filled marker) and non-responsive P2 (hollow marker) particles with DLD1 (circle), PC-3 (square), and HeLa (triangle) cells for 72 h in medium with 10% serum (a). Similar testing was done in medium with only 2% of serum, comparing again the cell lines PC-3 (b), DLD1 (c) and HeLa (d) cells after 72 h incubation. Significant differences: **<0.05; *<0.1; ϕ<0.2 (ANOVA). | ||
In all three cancer cell lines including the experiments in medium with 2% and with 10% serum content, the toxicity of the ROS-responsive P1 NPs was higher than that of non-responsive P2 NPs. The viability testing demonstrated herein that under the studied conditions the PTX-loaded ROS-responsive NPs appear more cytotoxic in tumor cells than their non-responsive counterpart NPs (Fig. 7).
To summarize, we have shown evidence that fluorescent polymer NPs, bearing pinacol-type boronic ester linkers trigger self-immolative polymer degradation and subsequently release the cargo drug in the presence of ROS concentrations typically present in an intracellular environment of certain tumor cells. Co-localization studies evidenced that the P1 polymer NPs can be used for selective cargo release to PC-3 cancer cells, with the release rate in non-cancer HF cells being lower. Finally the drug-loaded ROS-responsive NPs were shown to be more cytotoxic to tumor cells compared to their non-responsive counterparts making the presented polymer a promising candidate for applications as the delivery system and the imaging agent (theranostics) aimed at inflamed microenvironments and cancer tissue.
Acknowledgements
Financial support from Norwegian Grants (grant # 7F14009), the Ministry of Industry and Trade of the Czech Republic (grant # FR-TI4/625) and the Ministry of Education, Youth and sports (grant # LH14292; grant POLYMAT # LO1507) is gratefully appreciated. The electron microscopy at the Institute of Macromolecular Chemistry was supported through the Technology Agency of the Czech Republic (grant #TE01020118).Notes and references
- Y. Wu, D. Zhou, Y. Qi, Z. Xie, X. Chen, X. Jing and Y. Huang, RSC Adv., 2015, 5, 3523 Search PubMed.
- H. Cho, J. Bae, V. K. Garripelli, J. M. Anderson, H.-W. Jun and S. Jo, Chem. Commun., 2012, 48, 6043 RSC.
- S. Petrova, E. Jäger, R. Konefal, A. Jäger, C. G. Venturini, J. Špěváček, E. Pavlova and P. Štěpánek, Polym. Chem., 2014, 5, 3884 RSC.
- H. Chen, W. He and Z. Guo, Chem. Commun., 2014, 50, 9714 RSC.
- H.-L. Pu, W.-L. Chiang, B. Maiti, Z.-X. Liao, Y.-X. Ho, M. S. Shim, E. Y. Chuang, Y. Xia and H.-W. Sung, ACS Nano, 2014, 8, 1213 CrossRef CAS PubMed.
- D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murphy, Nat. Mater., 2007, 10, 765 CrossRef PubMed.
- B. D'Autréaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813 CrossRef PubMed.
- A. Mantovani, P. Allavena, A. Sica and F. Balkwill, Nature, 2008, 454, 436 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radicals Biol. Med., 2010, 49, 1603 CrossRef CAS PubMed.
- N. Houstis, E. D. Rosen and E. S. Lander, Nature, 2006, 440, 944 CrossRef CAS PubMed.
- B. Uttara, A. V. Singh, P. Zamboni and R. T. Mahajan, Curr. Neuropharmacol., 2009, 7, 65 CrossRef CAS PubMed.
- W. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3022 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722 CrossRef CAS PubMed.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS.
- G.-Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479 CrossRef CAS PubMed.
- G. Waris and H. Ahsan, J. Carcinog., 2006, 5, 14 CrossRef PubMed.
- P. T. Schumacker, Cancer Cell, 2006, 10, 175 CrossRef CAS PubMed.
- M. López-Lázaro, Cancer Lett., 2007, 252, 1 CrossRef PubMed.
- W. Dröge, Physiol. Rev., 2002, 82, 47 CrossRef PubMed.
- B. Kumar, S. Koul, L. Khandrika, R. B. Meacham and H. K. Koul, Cancer Res., 2008, 68, 1777 CrossRef CAS PubMed.
- E. Lallana and N. Tirelli, Macromol. Chem. Phys., 2013, 214, 143 CrossRef CAS.
- C. de G. Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758 CrossRef PubMed.
- M. S. Shim and Y. Xia, Angew. Chem., Int. Ed., 2013, 52, 6926 CrossRef CAS PubMed.
- E. A. Mahmoud, J. Sankaranarayanan, J. M. Morachis, G. Kim and A. Almutairi, Bioconjugate Chem., 2011, 22, 1416 CrossRef CAS PubMed.
- D. Jeanmaire, J. Laliturai, A. Almalik, P. Carampin, R. D'Arcy, E. Lallana, R. Evans, R. E. P. Winpeny and N. Tirelli, Polym. Chem., 2015, 5, 1393 RSC.
- A. Savina, A. Peres, I. Cebrian, N. Carmo, C. Moita, N. Hacohen, L. F. Moita and S. Amigorena, Immunity, 2009, 30, 544 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 9638 CrossRef CAS PubMed.
- E. Jäger and F. C. Giacomelli, Curr. Top. Med. Chem., 2015, 15, 328 CrossRef.
- F. C. Giacomelli, P. Štěpánek, C. Giacomelli, V. Schmidt, E. Jäger, A. Jäger and K. Ulbrich, Soft Matter, 2011, 7, 9316 RSC.
- A. Jäger, D. Gromadzki, E. Jäger, F. C. Giacomelli, A. Kozlowska, L. Kobera, J. Brus, B. Říhová, M. El Fray, K. Ulbrich and P. Štěpánek, Soft Matter, 2012, 8, 4343 RSC.
- T. Hu and C. Wu, Phys. Rev. Lett., 1996, 83, 4105 CrossRef.
- J. Fu and C. Wu, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 703 CrossRef CAS.
- B. Kumar, S. Koul, L. Khandrika, R. B. Meacham and H. K. Koul, Cancer Res., 2008, 68, 1777 CrossRef CAS PubMed.
- U. E. Martinez-Outschoorn, R. M. Balliet, Z. Lin, D. Whitaker-Menezes, A. Howell, F. Sotgia and M. P. Lisanti, Cell Cycle, 2012, 11, 4152 CrossRef CAS PubMed.
- M. P. Lisanti, U. E. Martinez-Outschoorn, Z. Lin, S. Pavlides, D. Whitaker-Menezes, R. G. Pestell, A. Howell and F. Sotgia, Cell Cycle, 2011, 10, 2440 CrossRef CAS PubMed.
- R. S. Bhimani, W. Troll, D. Grunberger and K. Frenkel, Cancer Res., 1993, 53, 4528 CAS.
- Y. Shi, C. F. van Nostrum and W. E. Hennink, ACS Biomater. Sci. Eng., 2015, 1, 393 CrossRef CAS.
- W. Chen, F. Meng, R. Cheng and Z. Zhong, J. Controlled Release, 2010, 142, 40 CrossRef CAS PubMed.
- J. M. Lluis, F. Buricchi, P. Chiarugi, A. Morales and J. C. Fernandez-Checa, Cancer Res., 2007, 67, 7368 CrossRef CAS PubMed.
- D. A. Smith, L. Di and E. H. Kerns, Nat. Rev. Drug Discovery, 2010, 9, 929 CrossRef CAS PubMed.
- G. Baier, C. Costa, A. Zeller, D. Baumann, C. Sayer and P. H. Araujo, Macromol. Biosci., 2011, 11, 628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr00791k |
| This journal is © The Royal Society of Chemistry 2016 |