
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imaging of transfection and intracellular release of intact, non-labeled DNA using fluorescent nanodiamonds†
V.
Petrakova‡
ab,
V.
Benson‡
ac,
M.
Buncek
d,
A.
Fiserova
ac,
M.
Ledvina
ae,
J.
Stursa
f,
P.
Cigler
*e and
M.
Nesladek
*agh
aFaculty of Biomedical Engineering, Czech Technical University in Prague, Sitna sq. 3105, 272 01 Kladno, Czech Republic
bInstitute of Physics AS CR, v.v.i, Na Slovance 1999/2, 182 21 Prague 8, Czech Republic
cInstitute of Microbiology AS CR, v.v.i, Videnska 1083, 142 20 Prague 4, Czech Republic
dGeneri Biotech Ltd., Machkova 587, 500 11 Hradec Kralove, Czech Republic
eInstitute of Organic Chemistry and Biochemistry AS CR, v.v.i., Flemingovo nam. 2, 166 10 Prague 6, Czech Republic. E-mail: cigler@uochb.cas.cz
fNuclear Physics Institute AS CR, v.v.i., 250 68, Rez near Prague, Czech Republic
gIMEC Division IMOMEC, Hasselt University, Wetenschapspark 1, B-3590, Diepenbeek, Belgium
hInstitute for Materials Research, Hasselt University, Wetenschapspark 1, B-3590 Diepenbeek, Belgium. E-mail: milos.nesladek@uhasselt.be
First published on 13th May 2016
Abstract
Efficient delivery of stabilized nucleic acids (NAs) into cells and release of the NA payload are crucial points in the transfection process. Here we report on the fabrication of a nanoscopic cellular delivery carrier that is additionally combined with a label-free intracellular sensor device, based on biocompatible fluorescent nanodiamond particles. The sensing function is engineered into nanodiamonds by using nitrogen-vacancy color centers, providing stable non-blinking luminescence. The device is used for monitoring NA transfection and the payload release in cells. The unpacking of NAs from a poly(ethyleneimine)-terminated nanodiamond surface is monitored using the color shift of nitrogen-vacancy centers in the diamond, which serve as a nanoscopic electric charge sensor. The proposed device innovates the strategies for NA imaging and delivery, by providing detection of the intracellular release of non-labeled NAs without affecting cellular processing of the NAs. Our system highlights the potential of nanodiamonds to act not merely as labels but also as non-toxic and non-photobleachable fluorescent biosensors reporting complex molecular events.
Introduction
Delivery of exogenous nucleic acids (NAs) into cells can provide highly specific treatments for many devastating diseases with unprecedented efficiency.1 In the past four decades, scientists have explored different approaches to gene therapy, including cellular delivery of mRNA, small interfering RNA, microRNA, DNA and plasmid DNA. However, these types of molecules are unstable in a biological environment, and vectors that enhance NA stability and targeting are under development to augment the therapeutic effect.2 Viruses, synthetic polymers, lipids and nanoparticles are currently considered among the most promising vectors. Among other delivery systems, detonation nanodiamonds and their complexes with poly(ethyleneimine) (PEI) have remarkably low toxicity and excellent cytocompatibility3–6 and recently were used as highly effective carriers for cell transfection with plasmid DNA,7,8 siRNA7–10 and miRNA.11 However, the detonation nanodiamond is non-fluorescent and does not allow a direct fluorescent tracking of the transfection. The use of so-called high-pressure high-temperature (HPHT) nanodiamonds has opened a possibility to track the transfection directly using engineered fluorescent nitrogen-vacancy (NV) centers.12,13Here we report on a NA nanocarrier device based on an oxygen terminated HPHT fluorescent nanodiamond containing NV centres. The device combines intracellular delivery with tracking and direct optical bio-molecular sensing of the delivery of an intact, non-labeled DNA. The NV color center operates here as a sensitive electric charge sensor which allows for monitoring of changes in a chemical environment close to the particle surface.14
A crucial point in transfection is an understanding of efficient unpacking and release mechanism of the NA payload.15 To study this process, considerable effort has been directed to the development of tools that allow monitoring of the intracellular fate of NAs. Most carriers described to date utilize fluorescent labels, which offer unprecedentedly high spatiotemporal resolution and a non-destructive approach. A number of NA conjugates with molecular fluorophores16–23 and fluorescent nanoparticles such as quantum dots,24 carbon dots,25 upconversion particles26 and lanthanoid-based polymer beacons27 have been prepared and examined. To gain a more detailed insight into NA polyplex dynamics and unpacking, researchers have used fluorescence resonance energy transfer (FRET)28–32 and lanthanide resonance energy transfer (LRET).33 These techniques have shown to be powerful for studying NA release dynamics, often at the single particle level.16,18,21,22,34
However, there are serious disadvantages in the use of NAs labeled with fluorescent probes. These unnatural modifications affect the cellular processing of NAs, and higher labeling densities can lead to drastic effects on expression levels and biased conclusions.35 The low stability of fluorescent labels is another significant issue. Whilst the labels quickly photobleach upon laser illumination, it does not allow monitoring of NA-cellular interactions for a prolonged period of time. Fluorescent nanoparticles such as quantum dots are blinking and typically contain a high concentration of cytotoxic elements. Moreover, such probes enable only passive labeling allowing for monitoring of position and motion. To our best knowledge, only two studies to date have successfully demonstrated imaging the intracellular release of non-labeled DNA. In one of these studies, the investigators reported that a complex ternary nano-assembly of poly(ethyleneimine) (PEI)-coated carbon dots, gold nanoparticles and plasmid DNA can detect the dissociation of DNA during transfection. The detection principle was based on quenching of carbon dot fluorescence by gold nanoparticles complexed with DNA and restoration of the fluorescence upon disassembly of the entire complex.36 In a different study, researchers used Gd(III)-based cationic polymer beacons to provide information on DNA dissociation based on the change in Gd relaxivity.27 The MRI readout of this process is valuable for in vivo imaging; however, it does not provide sufficient resolution for intracellular imaging. The development of alternative approaches is therefore urgently needed.
The nanoparticle carrier described here enables both effective transfection with non-labeled DNA and direct monitoring of payload detachment inside the cell. Our approach is based on the unique properties of biocompatible fluorescent nanodiamonds,37,38 which are broadly used as fluorescent labels, and expands their potential applications to fluorescent sensors reporting on a specific molecular event. We drew on two main principles for construction of the nanodevice.
First, upon oxidation, the HPHT FND surface is negatively charged and strongly binds the prototypical transfection reagent PEI.12,13
Second, the spectral distribution of FND fluorescence originating from NV color centers, is a powerful tool to monitor changes in the bonding environment close to the FND surface. NV centers in nanodiamonds are widely used for fluorescence-based detection techniques because they provide an exceptional combination of unlimited photostability, emission in the near-infrared region and lack of photoblinking.39 These properties have been utilized in a vast range of applications, from physics to biological applications such as single particle tracking inside cells,37 long-term in vivo particle tracking,40 tracing of neuronal processes,41 analysis of relationships between the particle shape and intracellular fate42 and fluorescence imaging in vitro43–45 and in vivo.40,46 Moreover, NV centers exhibit quantized electron and spin states, allowing optical manipulation even in a cellular environment.47,48
Recently, we discovered that fluorescent nanodiamonds are among the very few types of nanosensors that enable direct optical reading of noncovalent molecular events.14 The unique sensing mechanism is based on switching between the negatively charged and neutral states of NV centers which is induced by interaction of the FND surface with charged molecules. Notably, the detection is inherently insensitive to particle size polydispersity and operates as an intrinsic ratiometric system containing a natural non-photobleachable reference. In the presented approach, the non-covalent sensing capabilities of the NV color center,14 combined with the unique chemical properties of the diamond surface,49 led us to design a device that simultaneously enables effective vectoring of a DNA molecule to cells and serves as a nanoscopic electric charge sensor imaging complex molecular events, such as release of non-labeled DNA from an FND–PEI complex inside a cell during transfection.
Methods
Preparation of fluorescent nanodiamonds
Nanodiamond powder was supplied by Microdiamant, Switzerland (MSY 0–0.05; average diameter of nanoparticles ∼35 nm).50 Nanodiamonds were treated with a mixture of HNO3 and H2SO4 (85 °C, 3 days), washed with 0.1 M NaOH and 0.1 M HCl, washed five times with water and freeze-dried.51 Purified nanodiamond powder (160 mg), containing approximately 100–200 ppm of natural nitrogen impurities, was pressed in an aluminium target holder and irradiated with a 15.5 MeV proton beam extracted from the isochronous cyclotron U-120M for 70 min (fluence 6 × 1016 cm−2). The irradiated material was annealed at 900 °C for 1 h and subsequently oxidized in air for 6 h at 510 °C. The nanodiamond particles were then treated with a mixture of HNO3 and H2SO4 (85 °C, 3 days), washed with 0.1 M NaOH and 0.1 M HCl, washed five times with water and freeze-dried. Prior to use, the particles were dissolved in water (2 mg ml−1) and sonicated with a probe (Cole-Parmer, 750 W) for 30 min. The resulting transparent colloid was filtered using a 0.2 μm PVDF microfilter.Preparation of DNA
DNA used for preparation of the 137-bp fragment was isolated from the tail tissue of a DBA-2 mouse. Mouse tail was degraded in lysis buffer containing Nonidet P40, Tween 20 and proteinase K (Qiagen). DNA (7 μg per 100 μl PCR reaction) was amplified using HotStarTaqDNA Polymerase (Qiagen) and iCycler5 (BioRad). The following primers directed at beta-actin were used in the reaction: forward, 5′ agagggaaatcgtgcgtgac 3′; reverse, 5′ caatagtgatgacctggccgt 3′. The 137-bp PCR product was purified using a QIAquick PCR purification kit (Qiagen) and eluted into water. Its molecular weight was checked by agarose electrophoresis. The mouse beta-actin DNA fragment was then labeled and amplified by conventional PCR with the following primers: forward, 5′ agagggaaatcgtgcgtgac 3′; reverse, 5′ FAM-caatagtgatgacctggccgt 3′. For flow cytometry analysis, 5′ AlexaFluor 488-caatagtgatgacctggccgt 3′ was used.The pGFP expression plasmid used for verification of DNA transfection was obtained from Generi Biotech.
Coating of nanodiamonds with PEI and DNA
Poly(ethyleneimine) (Sigma Aldrich, MW 800, cat. no. 408719) was dissolved in water to prepare a stock solution (90 mg ml−1). Aqueous PEI was sonicated in a bath for 15 min. FND stock solution (2 mg ml−1) was added to the PEI solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol:vol) and sonicated with a probe (400 W, 15 min) under water cooling. The solution was washed with deionised water and centrifuged at 14500g for 15 min (3 times) to remove residual PEI. The pellet was then redispersed in deionised water to a final concentration of 0.48 mg per ml of FND–PEI complex (based on FND concentration).
:1 vol:vol) and sonicated with a probe (400 W, 15 min) under water cooling. The solution was washed with deionised water and centrifuged at 14500g for 15 min (3 times) to remove residual PEI. The pellet was then redispersed in deionised water to a final concentration of 0.48 mg per ml of FND–PEI complex (based on FND concentration).
DNA (160 ng) was dissolved in 5 μl water and sonicated in a bath cooled with ice for 1 h. This solution was then added to a 25 μl FND–PEI complex (0.48 mg ml−1, containing 12 μg FND) followed by sonication for 2 h. The FND–PEI–DNA complex was isolated by centrifugation at 14000g for 15 min, and the pellet was resuspended in an appropriate amount of DNAse-free water (Qiagen) to obtain a stock solution of 2 mg per ml FND–PEI–DNA.
Cell transfection and quantitative real-time PCR
IC-21 and HT-29 cell lines were maintained in RPMI1640 media supplemented with 20% and 10% fetal bovine sera, respectively. DNA (137-bp fragment or pGFP) was delivered into the cells using FND–PEI (25 μg per ml FNDs), PEI (3.3 μg ml−1, equivalent to the amount that covers FND surfaces) or the commercial transfection reagent X-tremeGENE HP DNA (Roche; 3:1 ratio, here, the manufacturers protocol has been followed). After 18 h incubation, we detected the level of transfected 137-bp fragment by real-time PCR with a TaqMan® probe using a commercial set of internal primers and probes for mouse beta-actin (TaqMan® Assay, Life Technologies). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene amplification was used as a reference, and quantitative levels were determined with Bio-Rad iQ5 2.0 software. All samples were performed in biological and technical triplicates. The 137-bp DNA fragment amplified and/or FAM-labeled was used in confocal analyses, PL measurements and toxicity assays.
Flow cytometry analysis
The equivalent of 105 cells IC-21 maintained in RPMI1640 media supplemented with 20% fetal bovine sera was seeded on a 12-well plate (In Vitro Scientific, USA). The AlexaFluor 488-modified oligonucleotide (0.25 μl, 1 mM solution, see Preparation of DNA) was incubated with a 12.5 μl FND–PEI complex (1 mg per ml of FND–PEI, sonicated for 30 minutes) for 60 minutes at room temperature. The FND–PEI–DNA complex was isolated by centrifugation at 14000g for 15 min, and the pellet was resuspended in an appropriate amount of DNAse-free water (Qiagen) to obtain a stock solution of FND–PEI–DNA (1 mg ml−1). IC-21 cells were incubated with the FND–PEI–DNA complex (final concentration 25 μg ml−1) for 30, 60, and 120 min. In parallel cell samples, an equal amount of the oligonucleotide was delivered into the cells using the commercial transfection reagent X-tremeGENE HP DNA (Roche; 3:1 ratio; the manufacturers protocol has been followed). At the end of the incubation period, cells were harvested, washed, and resuspended in PBS. Before the flow cytometry analysis, dead cells were stained with Hoechst 33258 (staining dead cells) for 5 minutes and the samples were measured with a flow cytometer BD LSR II and analyzed with a FlowJo 7.2.2 software.
Confocal microscopy
Colocalization of FND–PEI–DNA complexes with FAM-labeled DNA and expression of GFP delivered by the FND–PEI–DNA system were monitored in IC-21 and HT-29 cells by confocal microscopy (numerical aperture 1.2). The equivalent of 105 cells seeded on a glass bottomed 6-well plate (In Vitro Scientific, USA) was incubated either with FND–PEI or FND–PEI–DNA (containing the 137-bp DNA fragment or pGFP) complexes (final concentration in cell medium: 25 μg ml−1). At the end of the incubation period (15 min–2 h for FAM-labeled DNA and 48 h for pGFP), cell nuclei were stained with DAPI, and the images were recorded with an Olympus FV1000 SIM confocal microscope (objective 40×/0.95) and analyzed with Olympus FLUOVIEW 2.0a software. DAPI – excitation: 405 nm, emission: 461 nm; FNDs – excitation: 559 nm, emission: 655–755 nm; FAM – excitation: 473 nm, emission: 520 nm; GFP – excitation: 473 nm, emission: 520 nm. Photobleaching (with 405 nm laser) was used to distinguish the autofluorescence of the cells and verify the specificity of the FND signal.Photoluminescence measurements
If not stated otherwise, PL spectra were recorded using a Renishaw InVia Raman microscope; the excitation wavelengths were 514 nm and 488 nm with 15 mW laser power, using 50× long infinity corrected distance objective (numerical aperture 0.4). The measurements in aqueous solution (0.2 mg ml−1) and in cell medium (the same type as used for cell incubation) were performed in a Hellma fluorescence cuvette (type no. 105.252-QS). For cellular measurements, FND, FND–PEI or FND–PEI–DNA (final concentration 25 μg ml−1) were incubated with cells for 30, 60 and 120 min, washed with PBS (to remove efficiently the particles localized extracellularly), fixed in ethanol and stored at −20 °C before measurements. Measurements were performed from intracellular regions on 10 cells for each sample. Data show average of 10 measurements after background subtraction. All the measurements were performed using a 50× long distance objective.WST-1 assay
The biocompatibility of the FND systems was evaluated using a WST-1 assay according to the Roche on-line manual. Cells (5 × 103) were incubated with FND, FND–PEI or FND–PEI–DNA (final concentration 25 μg ml−1) for 24 h. The absorbance of formazan, a product of tetrazolium salt cleavage in proliferating cells, was measured at 450 nm. A reference absorbance was measured at 630 nm, and 1% SDS served as a negative control of proliferation. Experiments were performed in triplicate.Zeta potential measurements and dynamic light scattering
Dynamic light scattering and zeta potential measurements were recorded with a Zetasizer Nano ZS system (Malvern Instruments) at room temperature. Sample concentrations were 0.5 mg ml−1. For zeta potential measurements, the ‘dip cell’ was used.Colocalization analysis
To evaluate the statistical significance of the FND and FAM signal colocalization obtained with confocal microscopy, we performed colocalization analysis using Huygens Essential software (SVI, NL). The analysis included overlap, characterization of the degree of overlapping signals from red (FND) and green (FAM) channels and established colocalization coefficients. We analyzed colocalization coefficients (k1 and k2) that characterize the contribution of each signal (FND and FAM) to the overlap value. The overlap (ro) and colocalization coefficients (k1 and k2) were calculated by Huygens Essential colocalization analysis according to the following formulae (http://www.svi.nl):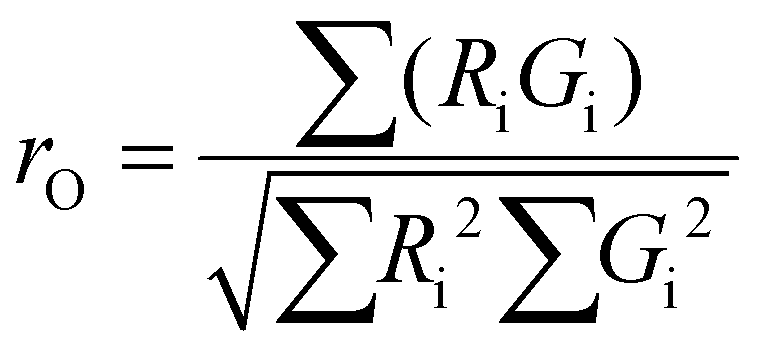 | (1) |
 | (2) |
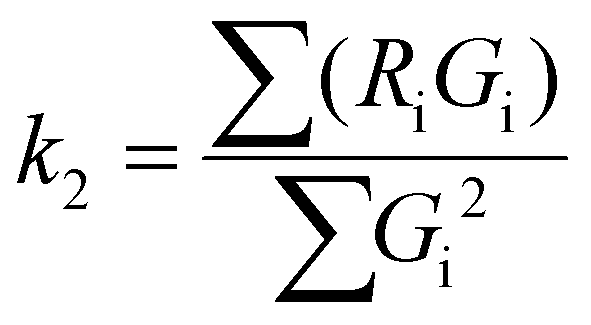 | (3) |
The value of ro is between 0 and 1. Ri and Gi are the intensities of the red and green channels, respectively.
Results and discussion
Principle of simultaneous vectoring and imaging
We constructed an FND device for intracellular NA delivery and monitoring using nanodiamond particles coated with low-molecular-weight branched PEI, which readily forms a complex with short DNA fragments and plasmids (Fig. 1a). The DNA binds electrostatically to the FND particle, forming a FND–PEI–DNA complex that penetrates the cell and liberates active DNA molecules. | ||
| Fig. 1 Principle of simultaneous vectoring, imaging and tracking of DNA payload release with an FND-based device. (a) Schematic of formation of the FND–PEI–DNA complex based on electrostatic interactions and the release of DNA from the complex after entering the cell. The color codes of the FND particles depict the expected changes in FND emission colors upon interaction with PEI and non-labeled DNA (this effect is not related to FAM fluorescence and can be observed for intact, fluorophore-free NA). (b) Schematics of electrical charge density in the proximity of an FND particle for FND–PEI (left) and FND–PEI–DNA (right) complexes and the corresponding band bending of energetic levels in the diamond. The alternations in occupancy of the NV− and NV0 energetic levels are related to Fermi level (EF). EVBM is the valence band maximum and EC is the conduction band minimum. (c) PL spectra of oxidized FNDs and FND–PEI and FND–PEI–DNA complexes recorded in aqueous solution (FND concentration: 0.2 mg ml−1) using an excitation wavelength of 514 nm. Formation of FND–PEI complex causes a significant decrease in NV− luminescence compared to oxidized FNDs. The level of NV− luminescence increases again upon binding of negatively charged DNA, which compensates for the positive charge of PEI. | ||
To monitor individual steps of construction and action of the FND cargo device, we use the ability of an NV center to serve as an electric charge sensor.52,53 Recently, we predicted54 and demonstrated14 that electrical charges carried by molecules such as NAs can, upon attachment to an FND, interact with a shallow NV center in the FND particle. Binding/release of charged molecules alters luminescence by changing the electron occupation of two charge states of the NV center, the so-called NV0 (neutral) and NV− (negatively charged) states. These two charge states exhibit different zero phonon lines (ZPLs). ZPL is an energy corresponding to the radiative transition of an electron from an excited state to its ground state.55 NV0 and NV− ZPL luminescent transitions occur at different wavelengths (NV0 ZPL = 575 nm, NV− ZPL = 636 nm). The binding/release of charged molecules manipulates the Fermi level position (EF) by the surface band bending (i.e., the upward or downward energy offset of a diamond band structure near the diamond surface, due to space charge effects; Fig. 1b). Such Fermi-level pinning has been achieved for FNDs with various covalent terminations.56–60 We took advantage of its manifestation for non-covalent molecular interactions. The relative position of the EF with respect to the energy level corresponding to the NV0 and NV− states determines the occupation of these states and consequently the strength of the luminescence of the NV0 and NV− ZPLs.
Covering the FND surface with the cationic polymer PEI and subsequent binding/release of negatively charged DNA are responsible for changes in surface charge density, which are directly reflected in NV−/NV0 electron occupation. By measuring the ZPL photoluminescence (PL) intensity corresponding to the particular charge state (NV− or NV0), we can quantitatively follow the occupation of the NV− and NV0 states. This allows us to identify changes in surface band bending upon DNA adsorption and release from the PEI–FND complex in cytoplasm. Our calculations, based on the Schrödinger–Poisson equation, show that for spherical nanoparticles the NV− and NV0 energy levels with respect to the Fermi level can be influenced up to a depth of approximately 20 nm from the FND surface, induced by band bending in the range of 1.5 eV.54
PL spectra recorded from aqueous solutions of FND–PEI and FND–PEI–DNA complexes and from oxidized FNDs using a confocal Raman setup with green laser excitation are shown in Fig. 1c. The wide PL bands observed at the higher wavelength side of ZPLs of NV− or NV0 centers are related to the phonon replicas of ZPLs.61 We built the FND sensor using oxidized FNDs62 in which the luminescence is dominated by the NV− ZPL. After attachment of polycationic PEI molecules, the NV− ZPL luminescence is dramatically reduced, while NV0 ZPL luminescence is slightly enhanced. Upon formation of the FND–PEI–DNA complex, the NV− ZPL luminescence is completely restored. Because of the non-covalent nature of these molecular events, the charge switching is reversible, and the sensing system responds dynamically to the association/dissociation of the corresponding complexes. The dynamic changes in the NV states caused by the interaction with non-labeled DNA can also be monitored by color changes of the luminescence emitted from FNDs. When the NV−/NV0 ratio changes the luminescence shows a color shift (see Fig. 1a), allowing us to optically visualize the molecular events occurring on the FND surface. This behavior corresponds to our previous findings on interaction of charged polymers with FNDs: NV− related luminescence intensity was substantially depleted upon non covalent binding of quaternary ammonium- or amine-containing polymers.14
To confirm the charge changes on FND particles, we measured zeta-potentials of the individual complexes. The negative zeta potential of uncoated oxidized FNDs (−33 mV) became positive after coating with PEI (+36 mV). Attachment of DNA led to restoration of negative zeta potential (−34 mV). These results justify our assumption that the PEI positive charge is compensated by DNA binding.
Optical monitoring of DNA transfection and release in macrophages
By confirming the successful construction of a FND NA carrier, we monitored the delivery of DNA molecules and their release into the cytoplasm in macrophages (IC-21). Macrophages represent an isolated cell population (circulating blood cells) that easily takes in a substantial amount of nanoparticles present in the surrounding media.For these model experiments, we prepared a random 137-bp double-stranded DNA fragment (see Methods for fragment synthesis details) that had no function in the transfected cells, consequently it was not amplified by any cellular mechanism, and was labeled at the 5′ end with fluorescein (FAM). We recorded fluorescence signals from confocal microscopy using two independent reading channels. The first monitors the fluorescence of NV centers (i.e. both NV− and NV0 charge states) and senses DNA release, while the second, or control channel, tracks the FAM label. FAM does not interfere with the NV fluorescence and enables an independent colocalization of the FND and DNA signals in this model experiment. We point out that we always set the focus depth on an intracellular structure (cell nuclei stained with DAPI). This enabled us to distinguish fluorescence originating in non-internalized and internalized FND–PEI–DNA complexes.
Before the FND–PEI–DNA complexes entered the cells, we confirmed that the fluorescence signal and specific signature of NV centers (i.e. combined NV− and NV0 fluorescence) were colocalized with the signal of FAM-labeled DNA, indicating the stability of the complex in the medium. Additionally, in the medium or cellular environment, nanoparticles are usually covered by a protein corona63 that might influence the observed NV PL changes. To rule out this possible phenomenon, we studied the PL spectra of uncoated FNDs and FND–PEI complexes in media and inside cells after particle internalization. However, no significant spectral changes in the NV luminescence were observed for FND in the media and after internalization (see ESI Fig. S1a and b†).
We added FND–PEI–DNA particles to macrophage cultures (see Methods) and observed the transfection process (Fig. 2). At the beginning of the experiment, the green FAM-labeled DNA signals and red luminescing NV centers (NV0 and NV− luminescence were integrated in a red detection channel) colocalized. The luminescence data indicate that both components stay together until they enter the cytoplasm (Fig. 2a). After the FND–PEI–DNA complexes were transfected into live cells, the red NV and green FAM signals quickly delocalized, indicating the release of the FAM-labeled DNA from the FND–PEI–DNA complex. Using an Olympus colocalization analytical tool, we visualized the colocalization of extracellular FND–PEI–DNA complexes as well as a separation of the FND signal that persisted in the cytoplasm after the delivery of FAM-labeled DNA. A representative graph of this analysis is shown in ESI Fig. S2.†
 | ||
| Fig. 2 Imaging of DNA release from the FND–PEI–DNA complex in IC-21 macrophages. (a) Time-lapse imaging of cells incubated with FND–PEI–DNA for 30, 60 and 120 min and measured using confocal microscopy. DNA was labeled with fluorescein (FAM) before formation of the complex with FND–PEI. The red luminescence corresponds to NV centers (FND). During the time-lapse, cells were maintained in a stabilized position in a tempered chamber without CO2. The chosen section of the plate was exposed to laser beams for 8 μs every 10 min. In all cells, FND luminescence was strictly localized in the cytoplasmic compartment, and the signal intensity did not change over a 2 h time period. (b) PL of NV centers measured upon 532 nm laser excitation, taken from FND–PEI–DNA complexes measured in water, medium and cells (fixed in ethanol) after 30, 60 and 120 min incubation with FND–PEI–DNA. Spectra are background-corrected and normalized to the luminescence intensity of NV0 ZPL (575 nm). The spectra show a clear decrease in the NV−/NV0 ZPL PL ratio as time elapses for FND–PEI–DNA located in cells. (c) Time development of [NV−/(NV0 + NV−)] intensity overlap coefficient, and colocalization coefficients k1 and k2 that describe the individual contributions of FAM and FND signals, respectively, to overlap. Values are an average of 10 measurements of each intracellular and extracellular signal. The parameters are compared for FND–PEI–DNA complexes inside the cells (red) and control FND–PEI–DNA complexes located extracellularly in cell medium (blue) measured after 30, 60 and 120 min incubation. TDI – transmission/bright field image. For a detailed report on confocal analysis, see ESI Fig. S3.† | ||
To describe the effect of DNA release quantitatively, we used colocalization analysis and determined colocalization coefficients k1 and k2 for individual time points (characterizing the contribution of FND and FAM channels to the total spectral overlap; see Methods) (Fig. 2c). Coefficient k1 corresponds to the colocalization of the FAM intensity weighted by the total FAM and FND signals. Coefficient k2 reflects the FND signal contribution and is reciprocal to k1. It could be confirmed that FAM-based fluorescence is much less photostable than FND fluorescence. The visualization of a combined color signal from FND and FAM-labeled DNA is therefore restricted to the extracellular environment and to the early phase of internalization. Correspondingly, we observed a partial dimming of the overall FAM signal during the experiment (apparently caused by FAM photobleaching). Nevertheless, the time evolution of the calculated colocalization coefficients clearly indicates that the release of the FAM-labeled DNA from the FND–PEI–DNA complex has occurred.
In parallel with the colocalization measurements (excitation with a 488 nm blue laser), we independently monitored the NV luminescence using confocal microscopy (excitation with a 514 nm green laser) and measured the characteristic spectral response of NV centers over the course of time. The PL spectra of NV centers at time points 30, 60 and 120 minutes after addition of FND–PEI–DNA complexes to the cells are plotted in Fig. 2b. We observed a significant decrease in the NV−/NV0 ratio over time that correlated very well with the decrease in overlap and k1 coefficient (which corresponds to DNA release; see Fig. 2c). These changes also agree well with the fluorescence changes observed in our model experiments in solution. Based on these data, we attributed the decrease in the NV−/NV0 PL ratio to the intracellular release of DNA from the FND–PEI–DNA complex. Notably, no FND photobleaching occurred during the experiment because of the extreme photostability of NV luminescence. The observed spectral changes in the NV−/NV0 luminescence therefore can be directly used for monitoring the changes in surface charges accompanying the process of DNA unpacking and release from FNDs.
Interestingly, we found that FNDs localized in the cytoplasmic region, but not in the nucleus, which is in agreement with other studies.12,43 Based on analogy with naked FNDs, we assume that FND–PEI–DNA particles enter the cell via endocytosis, and due to their sharp shape, escape the endosome by rupturing the endosomal membrane.42,64
For independent proof of the transfection efficiency of the FND–PEI–DNA complex we used the flow cytometry analyses (Fig. 3a and ESI Fig. S4†). We quantitatively compared the performance of FND–PEI and a commercial transfection reagent X-tremeGENE HP DNA for transfection of IC-21 cells with Alexa Fluor 488-labeled DNA oligonucleotide (see Methods). The percentage of cells positive for the Alexa Fluor 488 signal in 30, 60, and 120 min after transfection is shown in Fig. 3a. Two hours after transfection, we observed significantly higher Alexa Fluor 488-positive cell count in samples transfected with FND–PEI–DNA compared to the commercial reagent (81% of positive cells using the FND–PEI, 68% using the X-tremeGENE HP DNA).
 | ||
| Fig. 3 Flow cytometric analysis of (a) transfection efficiency of AlexaFluor 488-modified oligonucleotide to IC-21 cells and (b) the corresponding cytotoxicity estimated from a number of Hoechst-positive cells. FND–PEI (FND–PEI–DNA) and X-tremeGENE HP (HP-DNA) were used as transfection agents (see Methods for details). In graph (a), percentage of AlexaFluor 488-positive cells measured 30, 60, and 120 min after transfection is shown. The measurements are comparable to data shown in Fig. 2. For the source flow cytometric data see ESI Fig. S4.† The average of two measurements ± standard deviation is presented. Statistical significance of differences between tested groups was calculated by Student's t-test. Values of p ≤ 0.05 (*) and p ≤ 0.01 (**) were considered to be statistically significant. | ||
We also compared cytotoxicity estimated from a number of Hoechst-positive cells (Fig. 3b and ESI Fig. S4†). The cytotoxicity for FND–PEI–DNA was low in all cases, ranging from 6 to 17%. For shorter incubation times (30 and 60 min) the commercial reagent exhibited significantly higher cytotoxicity than the FND–PEI–DNA system (23 and 24%, respectively). After 120 min we found the cytotoxicity comparable for both reagents. We assume that the toxicity of the commercial reagent was triggered by the internalization process and later on those damaged cells were disintegrated and washed away during the preparation of samples for analysis. This resulted subsequently in apparently lower toxicity in 120 min (12%).
Optical monitoring of DNA internalization verified by PCR in human colon cancer cells
Our experiments with IC-21 macrophages provided proof-of-principle that our nanodiamond complexes can be used for monitoring of DNA delivery and release into the cytoplasm. As a next step, we tested DNA delivery and release in a more challenging system: HT-29 human colon cancer cells, which represent an established diagnostic and therapeutic target. First, we performed similar experiments with HT-29 as with macrophages and imaged the transfection and release of DNA from an FND–PEI–DNA complex.Confocal imaging confirmed that FND–PEI–DNA complexes serve as an efficient transfection system combined with tracking and intracellular sensing capabilities (Fig. 4a).
 | ||
| Fig. 4 Monitoring of FND–PEI–DNA complex intake by a human colon cancer cell line (HT-29). (a) Extracellular and internalized complexes incubated for 60 min with 25 μg per ml FND–PEI and 165 ng FAM-labeled DNA. Images with merged signals from FND (red) and FAM-labeled DNA (green) are shown. The colocalized signals from green channels appear in yellow (compared with IC-21 cells in Fig. 2). (b) Real-time PCR of the DNA fragment delivered into HT-29 cells by different transfection systems. NTC: negative control cells incubated in culture medium, PEI: incubation with PEI, FND: cells incubated with 25 μg per ml plain FNDs, FND–PEI: cells incubated with 25 μg per ml FND–PEI, FND–PEI–DNA: cells transfected with 25 μg per ml FND–PEI and 165 ng of FAM-labeled DNA, PEI–DNA: cells incubated with PEI combined with 165 ng of FAM-labeled DNA, HP-DNA: cells transfected with X-tremeGENE HP transfection reagent and 165 ng of the DNA. The experiment was performed in triplicate, and the level of target oligonucleotide sequence was normalized to an internal reference (glyceraldehyde-3-phosphate dehydrogenase, GAPDH). (c) Biocompatibility of FND-based systems transfected into HT-29 cells. Evaluation by WST-1 assay illustrating the mitochondrial activity of live cells. NTC: negative control cells incubated in culture media, FND: cells incubated with 25 μg per ml uncoated FNDs, FND–PEI: cells incubated with 25 μg per ml FND–PEI, FND–PEI–DNA: cells transfected with 25 μg per ml FND–PEI and 165 ng FAM-labeled DNA, PEI–DNA: cells incubated with PEI combined with 165 ng FAM-labeled DNA, SDS: cells incubated with 1% SDS. | ||
To verify that the DNA fragment had been successfully transported into the cells using the FND–PEI carrier, we performed quantitative real time PCR analysis. We observed an abundant level of the transfected 137-bp sequence in cells incubated with FND–PEI–DNA, indicating successful transfection of the DNA fragment (Fig. 4b).
Because PEI itself can serve as a transfection reagent, we performed a control transfection with free PEI. We obtained a similar level of transfected DNA using FND–PEI–DNA and PEI–DNA alone. This indicates that anchoring DNA via PEI to FNDs does not reduce the binding capacity of PEI to DNA or alter the transfection efficacy. As another control, we used the commercial transfection reagent X-tremeGENE HP DNA at a dose corresponding to the amount of DNA adsorbed to FND–PEI and under conditions recommended by the supplier. Cells transfected with X-tremeGENE HP DNA exhibited a lower level of the DNA fragment than cells transfected with FND–PEI–DNA or PEI–DNA (Fig. 4b).
We demonstrated in this way the high efficiency of both FND–PEI–DNA and PEI–DNA transfection systems for the DNA fragment used. However, FND–PEI as a transfection carrier showed a much lower impact on the cell mitochondrial activity than PEI alone, as determined by the WST-1 assay, which illustrates the mitochondrial activity of living cells (Fig. 4c). In comparison with cells transfected with PEI–DNA, cells transfected with FND–PEI–DNA showed mitochondrial activity comparable to control cells treated with medium alone. These results can be explained by the fact that PEI remains strongly bound to the FND surface after transfection and is not released into the cells, preventing its toxic impact.7,65
Verification of DNA transfection with a GFP expression plasmid
To confirm that DNA is successfully released to the cytoplasm in an active form after it gets separated from its FND–PEI carrier, we employed a control green fluorescent protein expression plasmid (pGFP) bound to the FND–PEI carrier. If released in the cell from the FND–PEI transfection carrier, pGFP would undergo transcription and translation, resulting in the formation of GFP protein, which can be easily visualized due to its inherent fluorescence. We transfected IC-21 and HT-29 cells with the FND–PEI–pGFP complex and with X-tremeGENE HP DNA complexed with pGFP. Two days after transfection, we observed a fluorescence characteristic for GFP expression. For IC-21 cells which engulfed a large amount of FND–PEI–pGFP and produced a large quantity of GFP protein, the signal was granular and showed a colocalization with NV center fluorescence (Fig. 5). For HT-29 cells with lower transfection efficacy we did not observe such signal overlap (see ESI Fig. S5†). | ||
| Fig. 5 Detection of green fluorescent protein 48 h after transfection of IC-21 cells with FND–PEI and X-tremeGENE HP transfection reagent, both complexed with the pGFP expression plasmid. FND–PEI: cells incubated with 25 μg per ml FND–PEI, GFP: cells transfected with X-tremeGENE HP transfection reagent and 2 μg pGFP, FND–PEI–GFP: cells transfected with 25 μg per ml FND–PEI and 2 μg pGFP. From the left: DAPI staining of nuclei; GFP signal; fluorescence of NV centers; merged fluorescence. Arrows point at cells positively transfected with nanodiamond particles and/or plasmid DNA. | ||
We also detected NV center fluorescence in the cytoplasm of cells transfected with FND–PEI–pGFP. Generally, once internalized, the pGFP produces a fluorescent GFP and they both persist in cytoplasm, next to FND particles, till the cell dies or divides enough to dilute them off. Our observation confirms that the pGFP is delivered into cells as a FND–PEI–pGFP complex, but soon after internalization, the DNA splits from the carrier and goes successfully through a translation process. Without this splitting from the FND–PEI carrier, the plasmid DNA cannot produce the fluorescent protein.
Interestingly, in Fig. 5 we can see a different morphology of IC-21 cells transfected with the GFP expressional plasmid. Here, a substantial amount of the pGFP with or without nanoparticles has been engulfed by phagocytizing cells and the subsequent expression of the GFP protein in a large quantity (see above) exhibited characteristic signs of toxicity.66 However, we have not experienced such a toxic effect of GFP in HT-29 cancer cells, which originate from colon tissue and secrete a protective layer of mucin-type glycans. This protective syncytium-like coat apparently obstructs the DNA delivery, so a good portion of transfection complexes persist glued on the cell surface without being internalized. This is shown in ESI Fig. S5,† where only a few colon cancer cells have internalized FNDs and/or pGFP (for comparison with internalization rate in macrophages see Fig. 5).
Conclusions
We have developed a non-toxic nanoscale diamond carrier for simultaneous transfection of cells and spatiotemporal fluorescence imaging of DNA payload release that does not require DNA labeling. The system is based on FND particles non-covalently coated with a cationic PEI polymer, which forms reversible complexes with DNA. The fluorescence of particles originates in NV color centers, which occur in neutral (NV0) or negative (NV−) charge states. Changes in the electrical charge environment in proximity to the FND surface correspond to complex molecular events such as binding and release of DNA. We harnessed this phenomenon, manifested by a color shift of the emitted luminescence, for monitoring of the course of transfection and intracellular release of non-labeled DNA. Our nanoprobe device thus reports directly on biomolecular interactions occurring at the cellular level with high spatial resolution, it is cytocompatible with different cell types and can operate in living cells for a prolonged period of time. Moreover, in comparison with a commercial transfection reagent, the transfection efficiency of FND–PEI is higher. Our results support recent findings regarding a great potential of fluorescent nanodiamonds in biological research and confirm them as excellent carriers/probes for biological macromolecules such as DNA. We believe that the detection mechanism we presented can be utilized for the construction of various types of nanoscale sensors monitoring directly the changes in charge or electrical field using NV centers in FND.Acknowledgements
This work was supported by Ministry of Health of the Czech Republic, grant no. 15e33094A and ESF European social fund in the Czech Republic – Investments in Education Development – Grant No. CZ.1.07/2.3.00/20.0306. The work of M. N. was supported by the FWO project (Flanders Diamond spin-magnetometers and nano-particle FRET sensors). Irradiation of NDs was carried out at the CANAM infrastructure of the NPI ASCR Rez supported through MŠMT project no. LM2011019. We thank Dr J. Micova, Dr I. Rehor, Mr J. Havlik and Mr M. Gulka for their very valuable help and Dr H. Hoffman for critical proofreading of the manuscript.Notes and references
- E. K.-H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv4 CrossRef PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- A. M. Schrand, L. Dai, J. J. Schlager, S. M. Hussain and E. Osawa, Diamond Relat. Mater., 2007, 16, 2118–2123 CrossRef CAS.
- A. M. Schrand, H. Huang, C. Carlson, J. J. Schlager, E. Osawa, S. M. Hussain and L. Dai, J. Phys. Chem. B, 2007, 111, 2–7 CrossRef CAS PubMed.
- V. Paget, J. A. Sergent, R. Grall, S. Altmeyer-Morel, H. A. Girard, T. Petit, C. Gesset, M. Mermoux, P. Bergonzo, J. C. Arnault and S. Chevillard, Nanotoxicology, 2014, 8, 46–56 CrossRef CAS PubMed.
- L. Moore, V. Grobárová, H. Shen, H. B. Man, J. Míčová, M. Ledvina, J. Štursa, M. Nesladek, A. Fišerová and D. Ho, Nanoscale, 2014, 2014, 11712–11721 RSC.
- X.-Q. Zhang, M. Chen, R. Lam, X. Xu, E. Osawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS PubMed.
- R. Kaur, J. M. Chitanda, D. Michel, J. Maley, F. Borondics, P. Yang, R. E. Verrall and I. Badea, Int. J. Nanomed., 2012, 3851 CAS.
- H. Kim, H. B. Man, B. Saha, A. M. Kopacz, O.-S. Lee, G. C. Schatz, D. Ho and W. K. Liu, J. Phys. Chem. Lett., 2012, 3, 3791–3797 CrossRef CAS PubMed.
- M. Chen, X.-Q. Zhang, H. B. Man, R. Lam, E. K. Chow and D. Ho, J. Phys. Chem. Lett., 2010, 1, 3167–3171 CrossRef CAS.
- M. Cao, X. Deng, S. Su, F. Zhang, X. Xiao, Q. Hu, Y. Fu, B. B. Yang, Y. Wu, W. Sheng and Y. Zeng, Nanoscale, 2013, 5, 12120–12125 RSC.
- A. Alhaddad, M.-P. Adam, J. Botsoa, G. Dantelle, S. Perruchas, T. Gacoin, C. Mansuy, S. Lavielle, C. Malvy, F. Treussart and J.-R. Bertrand, Small, 2011, 7, 3087–3095 CrossRef CAS PubMed.
- A. Alhaddad, C. Durieu, G. Dantelle, E. Le Cam, C. Malvy, F. Treussart and J.-R. Bertrand, PLoS One, 2012, 7, e52207 CAS.
- V. Petrakova, I. Rehor, J. Stursa, M. Ledvina, M. Nesladek and P. Cigler, Nanoscale, 2015, 7, 12307–12311 RSC.
- C. L. Grigsby and K. W. Leong, J. R. Soc., Interface, 2010, 7, S67–S82 CrossRef CAS PubMed.
- L. Liang, J. Li, Q. Li, Q. Huang, J. Shi, H. Yan and C. Fan, Angew. Chem., Int. Ed., 2014, 126, 7879–7884 CrossRef.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stöter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS PubMed.
- C. Rosazza, A. Buntz, T. Rieß, D. Wöll, A. Zumbusch and M.-P. Rols, Mol. Ther., 2013, 21, 2217–2226 CrossRef CAS PubMed.
- S. Coppola, L. C. Estrada, M. A. Digman, D. Pozzi, F. Cardarelli, E. Gratton and G. Caracciolo, Soft Matter, 2012, 8, 7919–7927 RSC.
- D. Vercauteren, H. Deschout, K. Remaut, J. F. J. Engbersen, A. T. Jones, J. Demeester, S. C. De Smedt and K. Braeckmans, ACS Nano, 2011, 5, 7874–7884 CrossRef CAS PubMed.
- A. M. Sauer, K. G. de Bruin, N. Ruthardt, O. Mykhaylyk, C. Plank and C. Bräuchle, J. Controlled Release, 2009, 137, 136–145 CrossRef CAS PubMed.
- S. Berezhna, S. Schaefer, R. Heintzmann, M. Jahnz, G. Boese, A. Deniz and P. Schwille, Biochim. Biophys. Acta, 2005, 1669, 193–207 CrossRef CAS PubMed.
- J. P. Clamme, J. Azoulay and Y. Mély, Biophys. J., 2003, 84, 1960–1968 CrossRef CAS PubMed.
- D. Shao, J. Li, X. Xiao, M. Zhang, Y. Pan, S. Li, Z. Wang, X. Zhang, H. Zheng, X. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 11082–11090 CAS.
- C. Liu, P. Zhang, X. Zhai, F. Tian, W. Li, J. Yang, Y. Liu, H. Wang, W. Wang and W. Liu, Biomaterials, 2012, 33, 3604–3613 CrossRef CAS PubMed.
- M. M. Rubner, D. E. Achatz, H. S. Mader, J. A. Stolwijk, J. Wegener, G. S. Harms, O. S. Wolfbeis and H.-A. Wagenknecht, ChemPlusChem, 2012, 77, 129–134 CrossRef CAS.
- J. M. Bryson, K. M. Fichter, W.-J. Chu, J.-H. Lee, J. Li, L. A. Madsen, P. M. McLendon and T. M. Reineke, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16913–16918 CrossRef CAS PubMed.
- H. Lee, I.-K. Kim and T. G. Park, Bioconjugate Chem., 2010, 21, 289–295 CrossRef CAS PubMed.
- H. H. Chen, Y.-P. Ho, X. Jiang, H.-Q. Mao, T.-H. Wang and K. W. Leong, Nano Today, 2009, 4, 125–134 CrossRef CAS PubMed.
- H. H. Chen, Y.-P. Ho, X. Jiang, H.-Q. Mao, T.-H. Wang and K. W. Leong, Mol. Ther., 2008, 16, 324–332 CrossRef CAS PubMed.
- V. Bagalkot, L. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065–3070 CrossRef CAS PubMed.
- Y.-P. Ho, H. H. Chen, K. W. Leong and T.-H. Wang, J. Controlled Release, 2006, 116, 83–89 CrossRef CAS PubMed.
- S. S. Kelkar, L. Xue, S. R. Turner and T. M. Reineke, Biomacromolecules, 2014, 15, 1612–1624 CrossRef CAS PubMed.
- E. Zagato, K. Forier, T. Martens, K. Neyts, J. Demeester, S. D. Smedt, K. Remaut and K. Braeckmans, Nanomed., 2014, 9, 913–927 CrossRef CAS PubMed.
- K. Rombouts, T. F. Martens, E. Zagato, J. Demeester, S. C. De Smedt, K. Braeckmans and K. Remaut, Mol. Pharm., 2014, 11, 1359–1368 CrossRef CAS PubMed.
- J. Kim, J. Park, H. Kim, K. Singha and W. J. Kim, Biomaterials, 2013, 34, 7168–7180 CrossRef CAS PubMed.
- Y.-R. Chang, H.-Y. Lee, K. Chen, C.-C. Chang, D.-S. Tsai, C.-C. Fu, T.-S. Lim, Y.-K. Tzeng, C.-Y. Fang, C.-C. Han, H.-C. Chang and W. Fann, Nat. Nanotechnol., 2008, 3, 284–288 CrossRef CAS PubMed.
- J. Stursa, J. Havlik, V. Petrakova, M. Gulka, J. Ralis, V. Zach, Z. Pulec, V. Stepan, S. A. Zargaleh, M. Ledvina, M. Nesladek, F. Treussart and P. Cigler, Carbon, 2016, 96, 812–818 CrossRef CAS.
- S.-J. Yu, M.-W. Kang, H.-C. Chang, K.-M. Chen and Y.-C. Yu, J. Am. Chem. Soc., 2005, 127, 17604–17605 CrossRef CAS PubMed.
- V. Vaijayanthimala, P.-Y. Cheng, S.-H. Yeh, K.-K. Liu, C.-H. Hsiao, J.-I. Chao and H.-C. Chang, Biomaterials, 2012, 33, 7794–7802 CrossRef CAS PubMed.
- T.-C. Hsu, K.-K. Liu, H.-C. Chang, E. Hwang and J.-I. Chao, Sci. Rep., 2014, 4, 5004 CAS.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C.-Y. Fang, I. Rehor, P. Cigler, H.-C. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 4495 Search PubMed.
- O. Faklaris, V. Joshi, T. Irinopoulou, P. Tauc, M. Sennour, H. Girard, C. Gesset, J.-C. Arnault, A. Thorel, J.-P. Boudou, P. A. Curmi and F. Treussart, ACS Nano, 2009, 3, 3955–3962 CrossRef CAS PubMed.
- J. Slegerova, M. Hajek, I. Rehor, F. Sedlak, J. Stursa, M. Hruby and P. Cigler, Nanoscale, 2015, 7, 415–420 RSC.
- I. Rehor, J. Slegerova, J. Kucka, V. Proks, V. Petrakova, M.-P. Adam, F. Treussart, S. Turner, S. Bals, P. Sacha, M. Ledvina, A. M. Wen, N. F. Steinmetz and P. Cigler, Small, 2014, 10, 1106–1115 CrossRef CAS PubMed.
- R. Igarashi, Y. Yoshinari, H. Yokota, T. Sugi, F. Sugihara, K. Ikeda, H. Sumiya, S. Tsuji, I. Mori, H. Tochio, Y. Harada and M. Shirakawa, Nano Lett., 2012, 12, 5726–5732 CrossRef CAS PubMed.
- A. Ermakova, G. Pramanik, J.-M. Cai, G. Algara-Siller, U. Kaiser, T. Weil, Y.-K. Tzeng, H. C. Chang, L. P. McGuinness, M. B. Plenio, B. Naydenov and F. Jelezko, Nano Lett., 2013, 13, 3305–3309 CrossRef CAS PubMed.
- L. P. McGuinness, Y. Yan, A. Stacey, D. A. Simpson, L. T. Hall, D. Maclaurin, S. Prawer, P. Mulvaney, J. Wrachtrup, F. Caruso, R. E. Scholten and L. C. L. Hollenberg, Nat. Nanotechnol., 2011, 6, 358–363 CrossRef CAS PubMed.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- I. Rehor and P. Cigler, Diamond Relat. Mater., 2014, 46, 21–24 CrossRef CAS.
- L.-C. L. Huang and H.-C. Chang, Langmuir, 2004, 20, 5879–5884 CrossRef CAS PubMed.
- F. Dolde, M. W. Doherty, J. Michl, I. Jakobi, B. Naydenov, S. Pezzagna, J. Meijer, P. Neumann, F. Jelezko, N. B. Manson and J. Wrachtrup, Phys. Rev. Lett., 2014, 112, 097603 CrossRef PubMed.
- S. Karaveli, O. Gaathon, A. Wolcott, R. Sakakibara, O. A. Shemesh, D. S. Peterka, E. S. Boyden, J. S. Owen, R. Yuste and D. Englund, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3938–3943 CrossRef CAS PubMed.
- V. Petrakova, A. Taylor, I. Kratochvilova, F. Fendrych, J. Vacik, J. Kucka, J. Stursa, P. Cigler, M. Ledvina, A. Fiserova, P. Kneppo and M. Nesladek, Adv. Funct. Mater., 2012, 22, 812–819 CrossRef CAS.
- F. Jelezko and J. Wrachtrup, Phys. Status Solidi A, 2006, 203, 3207–3225 CrossRef CAS.
- V. Petráková, A. Taylor, I. Kratochvílová, F. Fendrych, J. Vacík, J. Kučka, J. Štursa, P. Cígler, M. Ledvina, A. Fišerová, P. Kneppo and M. Nesládek, Adv. Funct. Mater., 2012, 22, 812–819 CrossRef.
- V. Petrakova, M. Nesladek, A. Taylor, F. Fendrych, P. Cigler, M. Ledvina, J. Vacik, J. Stursa and J. Kucka, Phys. Status Solidi A, 2011, 208, 2051–2056 CrossRef CAS.
- M. V. Hauf, B. Grotz, B. Naydenov, M. Dankerl, S. Pezzagna, J. Meijer, F. Jelezko, J. Wrachtrup, M. Stutzmann, F. Reinhard and J. A. Garrido, Phys. Rev. B: Condens. Matter, 2011, 83, 081304 CrossRef.
- K.-M. C. Fu, C. Santori, P. E. Barclay and R. G. Beausoleil, Appl. Phys. Lett., 2010, 96, 121907 CrossRef.
- J. Havlik, H. Raabova, M. Gulka, V. Petrakova, M. Krecmarova, V. Masek, P. Lousa, J. Stursa, H.-G. Boyen, M. Nesladek and P. Cigler, Adv. Funct. Mater., 2016 DOI:10.1002/adfm.201504857.
- K. Iakoubovskii, G. J. Adriaenssens and M. Nesladek, J. Phys.: Condens. Matter, 2000, 12, 189–199 CrossRef CAS.
- J. Havlik, V. Petrakova, I. Rehor, V. Petrak, M. Gulka, J. Stursa, J. Kucka, J. Ralis, T. Rendler, S.-Y. Lee, R. Reuter, J. Wrachtrup, M. Ledvina, M. Nesladek and P. Cigler, Nanoscale, 2013, 5, 3208–3211 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- Z. Chu, K. Miu, P. Lung, S. Zhang, S. Zhao, H.-C. Chang, G. Lin and Q. Li, Sci. Rep., 2015, 5, 11661 CrossRef PubMed.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- H.-S. Liu, M.-S. Jan, C.-K. Chou, P.-H. Chen and N.-J. Ke, Biochem. Biophys. Res. Commun., 1999, 260, 712–717 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional photoluminescence spectra, colocalization data and confocal analyses. See DOI: 10.1039/c6nr00610h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |