
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Smart surface coating of drug nanoparticles with cross-linkable polyethylene glycol for bio-responsive and highly efficient drug delivery†
Weijia
Wei
a,
Xiujuan
Zhang
*a,
Xianfeng
Chen
b,
Mengjiao
Zhou
a,
Ruirui
Xu
a and
Xiaohong
Zhang
*a
aFunctional Nano & Soft Materials Laboratory (FUNSOM) and Technology Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou, Jiangsu 215123, China. E-mail: xjzhang@suda.edu.cn; xiaohong_zhang@suda.edu.cn; Tel: +86 512 65880955
bSchool of Chemistry and Forensic Sciences, Faculty of Life Sciences, University of Bradford, BD7 1DP, UK
First published on 15th March 2016
Abstract
Many drug molecules can be directly used as nanomedicine without the requirement of any inorganic or organic carriers such as silica and liposome nanostructures. This new type of carrier-free drug nanoparticles (NPs) has great potential in clinical treatment because of its ultra-high drug loading capacity and biodegradability. For practical applications, it is essential for such nanomedicine to possess robust stability and minimal premature release of therapeutic molecules during circulation in the blood stream. To meet this requirement, herein, we develop GSH-responsive and crosslinkable amphiphilic polyethylene glycol (PEG) molecules to modify carrier-free drug NPs. These PEG molecules can be cross-linked on the surface of the NPs to endow them with greater stability and the cross-link is sensitive to intracellular environment for bio-responsive drug release. With this elegant design, our experimental results show that the liberation of DOX from DOX-cross-linked PEG NPs is dramatically slower than that from DOX-non-cross-linked PEG NPs, and the DOX release profile can be controlled by tuning the concentration of the reducing agent to break the cross-link between PEG molecules. More importantly, in vivo studies reveal that the DOX-cross-linked PEG NPs exhibit favorable blood circulation half-life (>4 h) and intense accumulation in tumor areas, enabling effective anti-cancer therapy. We expect this work will provide a powerful strategy for stabilizing carrier-free nanomedicines and pave the way to their successful clinical applications in the future.
Introduction
Cancer is a leading cause of death in all countries and chemotherapy has been widely used in combination with other treatments like radiotherapy for curing the disease. However, many current anticancer drugs suffer from serious limitations such as poor water solubility, rapid blood clearance, widespread targeting, low accumulation in disease sites, and detrimental side effects for healthy tissues.1,2 In an attempt to address these limitations, during the past few decades, many nanovehicles have been developed as drug carriers such as liposomes,3–5 micelles,6–9 polymeric nanoparticles (NPs),10–13 and metal NPs.14–16 These nanovehicles are able to tremendously enhance the water solubility of drugs and increase their accumulation in tumors via an enhanced permeability and retention (EPR) effect,2 leading to enhanced efficacy and alleviated side effects.17 Although effective, in these strategies, the carriers do not have a therapeutic function and simply act as a platform to load and transport drug molecules. In this circumstance, these drug carriers are predominantly a major component in the drug system and account for a higher weight portion than actual therapeutics. It apparently leads to a rather low drug loading capacity (DLC). Even worse, many carriers may have potential systemic toxicity when used in the clinic.18 Recently, pure anticancer therapeutic NPs19–24 have been developed for a new generation of drug delivery systems, which generally involve the preparation of one or more pure hydrophobic drugs into the form of NPs, and then followed by surface modification with a small amount of surfactants to realize water dispersity and bioenvironmental stability for efficient drug delivery. Such a drug delivery system can substantially boost drug loading capacity and avoid the risk of any unnecessary burden to patients. Nevertheless, this type of drug NPs possess inadequate in vivo stability, since the surfactant is anchored to the surface of NPs only through weak hydrophobic interactions. The lack of stability may result in premature drug release following administration into the blood stream.Herein, in this work, we have synthesized two bio-responsive and crosslinkable amphipathic surfactants which can efficiently minimize any premature drug release from drug NPs during blood circulation and achieve bio-responsive release at the tumor site. Considering that the intracellular concentration of glutathione (GSH) is considerably greater than that in the extracellular environment (∼10 mM vs. ∼2 μM),25 we developed two types of GSH-responsive molecules including PEG5000-Lys-LA2 (PEGA) and PEG5000-Lys-(Lys-LA2)2 (PEGB). Then they were applied in the surface functionalization of doxorubicin (DOX) NPs for controlled intracellular drug release. The stability, in vitro drug release, cytotoxicity, cellular uptake, blood circulation, biodistribution, and antitumor activity were systematically studied for different types of NPs including bare DOX NPs, DOX NPs coated with PEGA and PEGB (individually termed as DOX-NCLPEGA NPs and DOX-NCLPEGB NPs), as well as DOX NPs coated with cross-linked surfactant molecules (termed DOX-CLPEGA NPs and DOX-CLPEGB NPs). Compared with other NPs, the cross-linked surfactant modified DOX NPs show much better bio-stability, longer blood circulation time, and promoted drug delivery to tumor sites as a result of the adequately inhibited premature release of payload molecules. This is the first report in which crosslinkable polymer molecules are applied to modify pure drug nanoparticles for improved stability and intracellular environment responsive release. This type of pure drug nanomedicine will have a great potential for clinical applications as it does not contain an extra drug carrier material and is less toxic. We conceive the experimental findings to be of great importance to guide the preparation and application of carrier-free nanomedicines in research and future clinical settings.
Results and discussion
Synthesis and characterization of PEG5000-Lys-(Lys-LA2)2 and PEG5000-Lys-LA2
To exhibit an excellent performance, the amphiphilic ligand molecules should include hydrophilic, hydrophobic and crosslinkable segments. The ratio of hydrophilic segments to hydrophobic and crosslinkable segments is a key factor. Herein, two types of novel ligand, PEG5000-Lys-(Lys-LA2)2 and PEG5000-Lys-LA2, containing hydrophobic and crosslinkable lipoic acid (LA) and hydrophilic PEG were synthesized. The synthetic route of PEG5000-Lys-(Lys-LA2)2 is depicted in Scheme 1. The 1H NMR spectrum of PEG5000-Lys-(Lys-LA2)2 (Fig. 1a) displays signals at 1.85–1.90, 3.51, and 3.69 ppm, attributed to the hydrogen protons in LA, PEG backbone and lysine (Lys), respectively.26,27 In addition, the molecular weight of PEG5000-Lys-(Lys-LA2)2 in MALDI-TOF Mass Spectrometry (Fig. S4†) was almost identical to the theoretical value comparing it to the starting PEG-NH2. | ||
| Scheme 1 Synthetic route of PEG5000-Lys-(Lys-LA2)2. | ||
 | ||
| Fig. 1 (a) 1H NMR spectrum of PEG5000-Lys-(Lys-LA2)2. (b) SEM and TEM (inset) images of bare DOX NPs. (c) The evolution of the sizes of different NPs over a period of 3 days. | ||
Preparation and characterization of DOX NPs
DOX NPs were synthesized by a solvent exchange method. As Scheme 2 shows, doxorubicin hydrochloride (DOX·HCl) is firstly converted to being hydrophobic. To achieve this, HCl is removed from the molecule by adding triethylamine (TEA) to DOX·HCl in dimethyl sulfoxide (DMSO) solution in a basic environment. Then the DOX/DMSO solution was injected dropwise into water under vigorous stirring to obtain DOX NPs. Subsequently, a solution of PEGB was injected into the bare DOX NPs suspension to obtain the NPs with non-cross-linked ligands on their surface. To improve the stability of NPs, the surfactant molecules were cross-linked on the surface of NPs by adding dithiothreitol (DTT) to the solution containing DOX NPs and PEGB under nitrogen. The formation of the cross-link between surfactant molecules can be demonstrated by the apparent change of the absorbance at 330 nm in the UV-Vis absorption spectra of the non-cross-linked PEG5000-Lys-(Lys-LA2)2 and cross-linked PEG5000-Lys-(Lys-LA2)2 (Fig. S5†).26 | ||
| Scheme 2 Schematic illustration of DOX NPs coated by PEG5000-Lys-(Lys-LA2)2. | ||
SEM and TEM images of bare DOX NPs are presented in Fig. 1b. These NPs are mostly spherical and the size is relatively uniform. Dynamic light scattering (DLS) measurement (Fig. S6†) reveals that the average size of these DOX NPs is about 100 nm. After surface modification with PEGB, the size of the NPs does not change much no matter whether the ligand molecules are cross-linked or not. The stability of these NPs in phosphate buffered saline (PBS) is shown in Fig. 1c. Apparently, PEGB modified NPs are very stable. During the observation period of 3 days, the size of the surface coated NPs increased only slightly. In great contrast, the bare DOX NPs have poor stability, demonstrated by the dramatic size increase during the same observation time window. Apparently, almost all of the uncoated NPs have aggregated. For the NPs coated with cross-linked PEGB on the surface, even when we add 10 mM DTT into the solution to break the cross-link, they still remain relatively stable.
Release profiles of DOX from various types of NPs
To understand the influence of the surface modification of DOX NPs on their release profiles, we performed a study by placing various types of NPs in dialysis containers and measuring the release of DOX. In the investigation, we studied three types of DOX NPs including DOX-C18PMH-PEG NPs, DOX-NCLPEGB NPs and DOX-CLPEGB NPs. The results are presented in Fig. 2. For the groups of DOX-NCLPEGA NPs and DOX-CLPEGA NPs, the results are displayed in Fig. S7.† For DOX-C18PMH-PEG NPs, DOX is rapidly released and about 70% of DOX is released within the first 10 h. For DOX-NCLPEGB NPs, although the release of DOX is slightly slower, the difference is rather small. Within the same period of the first 10 h, the release only slightly decreases from about 70% to 65%. In a stark contrast, the release of DOX from DOX-CLPEGB NPs is dramatically alleviated. For the same period of time, only about 15% of DOX has been liberated. Compared to DOX-CLPEGB NPs, the release of DOX from the DOX-CLPEGA NPs is slightly higher. About 23% of DOX is released within the same period (Fig. S7b†). The different release profiles between DOX-CLPEGA NPs and DOX-CLPEGB NPs may be ascribed to the amount of the LA group in the ligands. PEGB contains double number of the LA group in comparison with PEGA, which leads to double cross-linked disulfide bonds in the DOX-CLPEGB NPs. | ||
| Fig. 2 The release profiles of DOX from different types of NPs and under different conditions. (a) The release profiles of DOX from DOX-C18PMH-PEG NPs, DOX-NCLPEGB NPs and DOX-CLPEGB NPs. (b) The release profiles of DOX-CLPEGB NPs in the presence of different concentrations of DTT in PBS. (c) The release profiles of DOX-NCLPEGB NPs and DOX-CLPEGB NPs. For the DOX-CLPEGB NPs, the release was in PBS for the first 5 h. After 5 h, 10 mM DTT was added to the solution. | ||
To demonstrate that the improved retention of DOX within the DOX-CLPEGB and DOX-CLPEGA NPs is due to the cross-linked ligand molecules, we purposely added 10 mM DTT into the solution to break the cross-linking. This amount of DTT is similar to the level of intracellular GSH. Expectedly, the release of DOX from the NPs sharply increases and the profile is almost the same as that of the NPs without cross-linked PEG molecules on the surface (Fig. 2a). In consideration of the release profile of DOX from NPs, PEGB seems a very suitable surfactant and can be used to modify nanomedicines for improved blood circulation stability.
When the DTT concentration in dialyzed media is the same as the extracellular level of GSH (2 μM), the release profile from the DOX-CLPEGB NPs is similar to that in the release media without DTT (Fig. 2b). Notably, the DOX release is gradually promoted as the DTT concentration increases from 2 μM to 1 and 10 mM. This means that the release of drug molecules will only be boosted when the nanomedicine is internalized by cells. This characteristic is very useful in drug delivery.
To further demonstrate that the drug release can be conveniently controlled, we incubated DOX-NCLPEGB NPs and DOX-CLPEGB in PBS. For DOX-CLPEGB NPs, after 5 hours incubation, 10 mM DTT was added to the solution. The results are shown in Fig. 2c. It is obvious that there is a burst of drug release from the NPs with cross-linked PEG5000-Lys-(Lys-LA2)2 once 10 mM was added. After the burst release of DOX from the NPs with cross-linked surfactant molecules, the subsequent release profiles (after 9 h incubation) of both types of NPs are nearly identical. Overall, as shown in these release profiles, for NPs modified with cross-linked PEG molecules, the premature drug release during circulation can be efficiently inhibited. Also attractively, the release of therapeutic molecules from these NPs is potentially controllable by both intracellular and extracellular environments.
In vitro cellular uptake and cell imaging
The intracellular uptake of DOX-NCLPEGB NPs and DOX-CLPEGB NPs was investigated in human cervical cancer (HeLa) cells by using confocal microscopy. Free DOX·HCl molecules were used as a control group. As shown in Fig. 3a, the HeLa cells treated with DOX·HCl for 6 h exhibit more intense fluorescence signals than the two types of NPs. The variation is due to the different internalization mechanisms of free DOX and DOX NPs. Briefly, DOX·HCl molecules enter cells through diffusion and then diffuse further into the nuclei, while NPs are internalized by endocytosis.28–30 Moreover, we can see that the HeLa cells treated with DOX-CLPEGB NPs possess stronger fluorescence intensity than those incubated with DOX-NCLPEGB NPs.26 The finding is similar for the cells with incubation time of both 6 and 12 h with the NPs. The cellular uptake was also quantitatively studied by flow cytometry analysis and the results are in agreement with the above findings. As shown in Fig. 3f and g, a much greater amount of DOX·HCl is internalized in HeLa cells, when compared with both types of DOX NPs. Additionally, the cells incubated with DOX-CLPEGB NPs display more intense fluorescence signals than those treated with DOX-NCLPEGB NPs.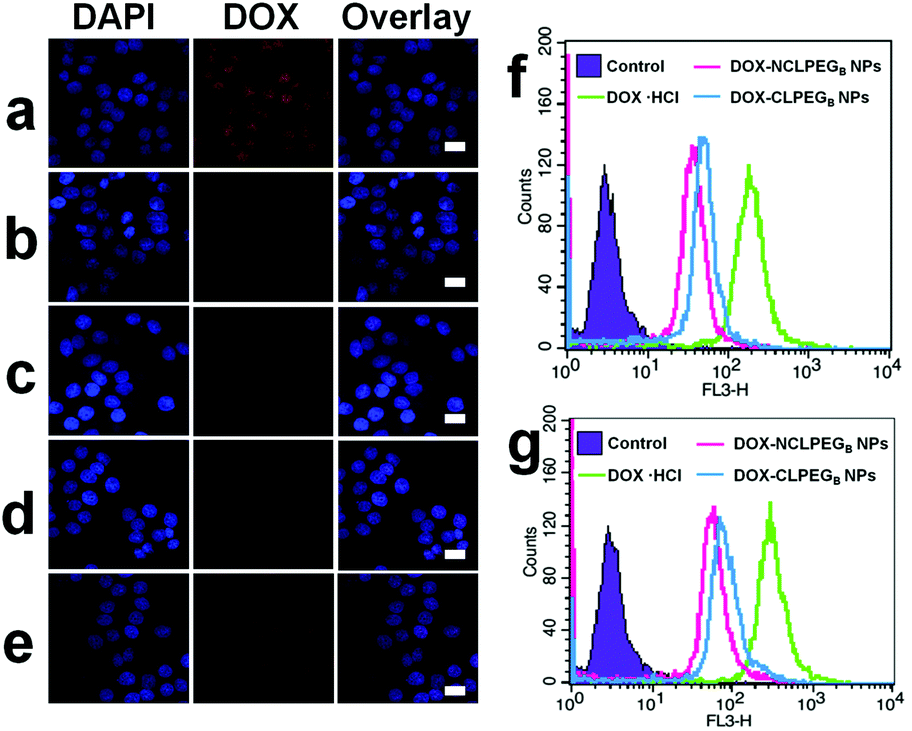 | ||
| Fig. 3 (a) Intracellular distribution of DOX·HCl in HeLa cells after 6 h incubation. (b, c) Intracellular distribution of DOX-NCLPEGB NPs and DOX-CLPEGB NPs in HeLa cells after 6 h incubation, (d, e) Intracellular distribution of DOX-NCLPEGB NPs and DOX-CLPEGB NPs in HeLa cells after 12 h incubation. Scale bars: 40 μm. (f, g) Flow cytometry analysis of DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs in HeLa cells after 6 h and 12 h incubation. | ||
In vitro cytotoxicity of DOX NPs
In vitro cytotoxicity of DOX NPs was assessed with two types of tumor cell lines including mouse metastatic breast cancer cell line (4T1), and HeLa cell line, as well as one type of normal cells, human hepatocytes (HL-7702). In the experiment, DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs were cultured with all cell lines at different concentrations ranging from 1.25 to 20 μg mL−1 (1.25, 2.5, 5, 10 and 20 μg mL−1) for 24 and 48 h. As presented in Fig. 4a and b, all samples show time and dose dependent cytotoxicity to 4T1 cells. At all concentrations, DOX NPs possess low toxicity than free DOX molecules. For example, after 24 h incubation, at the highest drug concentration of 20 μg mL−1, DOX·HCl exhibits higher cytotoxicity (viability of 23%) than both groups of DOX NPs (viabilities of about 30–40%). This is possibly due to the delayed release of DOX molecules from the NPs inside the cells, while free DOX molecules can easily diffuse through passive diffusion. Meanwhile, as illustrated in Fig. 4c and d, in normal cell line, two types of DOX NPs exhibit dramatically lower cytotoxicity than DOX·HCl. This may be attributed to the high pH and low intracellular GSH concentration in normal cell line in comparison with those in cancer cells. | ||
| Fig. 4 (a) and (b) Cell viabilities of 4T1 cell lines after being incubated with DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs for 24 and 48 h, respectively. (c) and (d) Cell viabilities of HL-7702 cell lines after being incubated with DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs for 24 and 48 h, respectively. | ||
In vivo blood circulation and biodistribution of DOX NPs
To study whether the cross-link strategy can be applied for inhibiting premature drug release during blood circulation, we intravenously injected DOX-NCLPEGB NPs and DOX-CLPEGB NPs into different groups of BALB/c mice. Blood samples were collected at different time points after injection. The mice administered with DOX-C18PMH-PEG NPs were adopted as a control. The fluorescence intensity of each blood sample was recorded and calibrated and the results are presented in Fig. 5a and these indicate that the blood circulation half-life of the DOX-CLPEGB NPs is much longer (>4 h) than those of the other two types of NPs (∼3 h). This indicates that cross-linking surfactant molecules is able to better encapsulate the nanomedicine and enable longer blood circulation time. In other words, the premature release of drug molecules from the nanomedicine can be effectively inhibited.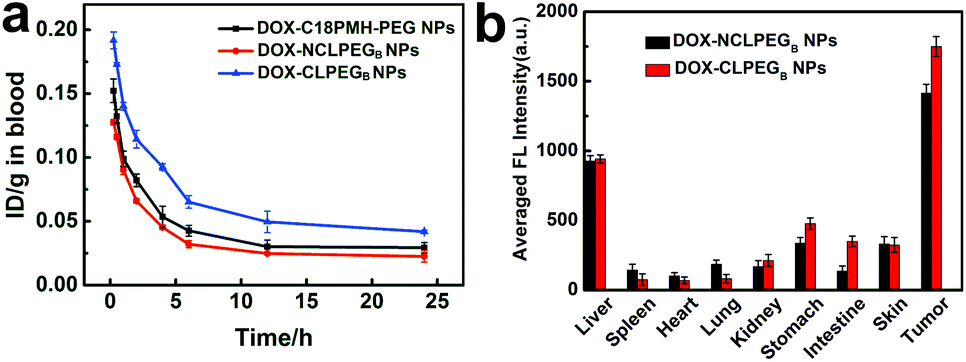 | ||
| Fig. 5 (a) Blood circulation curve of DOX-C18PMH-PEG NPs, DOX-NCLPEGB NPs and DOX-CLPEGB NPs, which was obtained through recording the fluorescence intensities of DOX in the blood at various times after drug administration. The unit is the percentage of the injected dose per gram tissue (% ID g−1). (b) Biodistribution of DOX-NCLPEGB NPs and DOX-CLPEGB NPs determined by measuring the fluorescence intensities of DOX in major organs and tumor tissues in vivo at 24 h after injection. | ||
In vivo biodistribution study was also carried out. Different DOX NPs were intravenously injected into 4T1 tumor bearing mice, and then the fluorescence intensities of DOX in various organs were monitored at different time intervals. Fig. 5b shows the biodistribution of DOX in major visceral organs and tumors of the mice treated with the DOX-NCLPEGB NPs or DOX-CLPEGB NPs at 24 h post injection. The results clearly demonstrate that the tumors of the mice treated with the DOX-CLPEGB NPs possess higher levels of DOX. Collectively, all of these outcomes suggest that the cross-link strategy works efficiently and have inhibited premature drug release during circulation. This leads to improved delivery of drug molecules to the tumor sites.
In vivo anticancer activities
To investigate the in vivo therapeutic efficacy of the DOX NPs, we chose 4T1 tumor bearing BALB/c mice as an animal model. For tumor treatment, PBS and DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs containing 1 mg ml−1 of DOX were intravenously administered in different groups of mice. The same treatment was repeated at 7 days after the primary injection. The tumor size and body weight of each mouse were recorded daily for two weeks to quantitatively evaluate the antitumor efficacy. As shown in Fig. 6a, the DOX NPs adequately inhibit the growth of tumor and the therapeutic efficacy is certainly better than the free DOX molecules. Particularly, the DOX-CLPEGB NPs realize a treatment efficacy (the tumor volume is 1.86 ± 0.41 fold of the initial value), which is clearly better than DOX-NCLPEGB NPs (2.90 ± 0.49 fold), DOX·HCl (4.92 ± 0.51 fold) and the PBS (7.35 ± 0.66 fold). | ||
| Fig. 6 In vivo anticancer effect of free DOX, DOX-NCLPEGB NPs and DOX-CLPEGB NPs. (a) The evolution of the tumor volume with time after the mice were administered with saline, free DOX and DOX-NCLPEGB NPs and DOX-CLPEGB NPs. Each formulation was injected on Day 0 and 7, and the injection concentration was 1 mg mL−1. (b) Body weight changes with time of 4T1 tumor bearing mice during the treatment period. | ||
Meanwhile, the mice treated with DOX NPs maintain a steady body weight to a certain extent (Fig. 6b). However, the mice which received injection of DOX·HCl experience a drop in body weight probably due to its serious side effect. Apparently, compared with DOX·HCl, the DOX-CLPEGB NPs can achieve greatly enhanced therapeutic efficiency with alleviated side effects.
Experimental
Materials
DOX·HCl (99%, Beijing ZhongShuo Pharmaceutical Technology Development Co., Ltd), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), di-Boc-lysine, DTT and LA (J&K Scientific Ltd) were used as received. Dimethyl sulfoxide-d6 (DMSO-d6), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), and 4-dimethylaminopyridine (DMAP) were from J&K Scientific Ltd TEA, DMSO, ethanol, ether, trifluoroacetic acid (TFA) and dichloromethane (DCM) were ordered from Sinopharm Chemical Reagent Co., Ltd. Monomethoxy poly(ethylene glycol) (MePEG-NH2, MW = 5 kDa, purity >95%) was from Shanghai Yare Biotech, Inc. All reagents and solvents were of analytical grade.Characterization
1H NMR spectra were obtained from a Unity Inova 400 spectrometer operating at 400 MHz using DMSO-d6 as a solvent. The chemical shifts were calibrated against residual solvent signals. The mass spectra were collected on a ABI 4700 MALDI-TOF/TOF mass spectrometer (linear mode) using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile as a matrix. UV-Vis absorption spectra were recorded on a Perkin-Elmer Lambda 750 UV/Vis/NIR Spectrophotometer. The fluorescence spectra were recorded with a FluoroMax 4 (Horiba Jobin Yvon) Spectrofluorometer. Scanning electron microscopic (SEM) images were collected on a FEI Quanta 200 FEG field emission scanning electron microscope. Transmission electron microscopy (TEM) images were observed using a Tecnai G220. DLS analysis measurements were recorded using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).Cell culture
HeLa cells were cultured in Dulbecco's Modified Eagle's medium (DMEM) and HL-7702 and 4T1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium. The culture media contain 10% of fetal bovine serum (FBS) and antibiotics including penicillin and streptomycin with 50 units per milliliter at 37 °C under a humidified atmosphere containing 5% CO2.Synthesis of PEG5000-Lys-LA2 and PEG5000-Lys-(Lys-LA2)2
Di-Boc-lysine (1.5 eq.) was dissolved in CH2Cl2, mixed with EDC·HCl (1.8 eq.), DMAP (1.8 eq.) and TEA (2 eq.) and activated at 37 °C for 4 h. This is followed by mixing with PEG5000-NH2 (1 eq.), and stirring at room temperature for 24 h. Then the solution was mixed with ice-cold ether. Subsequently, the precipitate was filtered and washed with cold ethanol and ether to obtain purified di-Boc-lysine-PEG5000. The obtained PEG derivative ester was dissolved in CH2Cl2/TFA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and stirred for 2 h at room temperature. Finally, the solvent was discarded and the product was precipitated in cold ether, and washed with cold ethanol and ether.
:1, v/v) and stirred for 2 h at room temperature. Finally, the solvent was discarded and the product was precipitated in cold ether, and washed with cold ethanol and ether.
LA (3 eq.), EDC·HCl (3.6 eq.), DMAP (3.6 eq.) and TEA (4 eq.) were dissolved in CH2Cl2 and activated at 37 °C for 4 h, followed by mixing with di-NH2-lysine-PEG5000 (1 eq.). The reaction lasted for 24 h at room temperature under rigorous stirring. The solution was then added into 10-fold volume of cold ethyl ether to precipitate the PEG5000-Lys-LA2, followed by three washes with cold ethanol and ether.
The NH2-terminated PEG5000-lysine (1 eq.) was conjugated with di-Boc-lysine (3 eq.) that was pre-activated with EDC·HCl (3.6 eq.), DMAP (3.6 eq.) and TEA (4 eq.) at 37 °C for 4 h. The conjugating reaction was stirred for 24 h at room temperature. After filtration and precipitation, the obtained PEG5000-Lys-(di-Boc-lysine)2 was treated with CH2Cl2/TFA (1:1, v/v) as described formerly. Then the PEG5000-Lys-(Lys-LA2)2 was synthesized with the same procedures as described above by conjugating NH2-terminated PEG5000-Lys-(Lys)2 (1 eq.) with pre-activated LA (6 eq.).
Preparation and modification of DOX NPs
First, 10 mL of 2 mg mL−1 DOX·HCl/DMSO solution was prepared and then 12 μL of TEA (1.5 eq.) was added under a moderate stirring at 25 °C. Through this step, hydrophilic DOX·HCl molecules were converted to being hydrophobic. Subsequently, DOX NPs were synthesized by a dropwise injection of 200 μL of the above prepared hydrophobic DOX in DMSO into 10 mL of water under robust stirring and then the stirring was continued for 5 min. Finally, 400 μL of 1.0 mg mL−1 PEGA and PEGB aqueous solutions were individually added to two solutions of 10 mL of bare DOX NPs. The mixtures were then ultrasonicated for 5 min followed by 1 h incubation at room temperature to obtain PEGA and PEGB modified DOX NPs.Preparation of DOX-CLPEG NPs
The pH of the above-prepared NPs solution was adjusted to be 8.5 using 0.7 M borate buffer (pH 9.0), and the dispersion was purged with nitrogen for 10 min. The cross-linking of the ligand molecules on the surface of NPs was achieved through the addition of 10 mol% DTT relative to the quantity of the lipoyl units in the surfactant. Subsequently, the solution was stirred under nitrogen for 22 h at room temperature.Stability of NPs in PBS
The stability study was performed to monitor the evolution of the size distribution of bare DOX NPs, DOX-NCLPEGA NPs, DOX-NCLPEGB NPs, DOX-CLPEGA NPs and DOX-CLPEGB NPs in PBS.In vitro release of DOX
The DOX NPs modified by C18PMH-PEG were used as a reference for comparison. The C18PMH-PEG was synthesized as described in our previously published paper.20 Solutions of 2 mL of DOX-C18PMH-PEG NPs, DOX-NCLPEGA NPs and DOX-CLPEGA NPs (2 samples) were injected into separate dialysis cartridges with a 14 kDa MWCO. These cartridges were individually dialyzed against 100 mL PBS at 37 °C except that one cartridge containing DOX-CLPEGA NPs was dialyzed in 100 mL PBS with 10 mM DTT. At desired time intervals, 1 mL solution was collected from each sample for fluorescence measurement and the dialysis medium was replenished with an equal volume of fresh one. In a separate experiment, cartridges containing DOX-CLPEGA NPs solution were individually dialyzed against PBS with various DTT concentrations (0, 2 μM and 1 mM) at 37 °C. In another experiment, DTT (10 mM) was added to the release medium at 5 h after starting dialysis. For the other group of DOX-NCLPEGB NPs and DOX-CLPEGB NPs, the experimental design was the same as the above.The quantity analysis of DOX release was performed through the measurement of its fluorescence intensity by a FluoroMax 4 (Horiba Jobin Yvon) spectrofluorimeter (excitation at 488 nm). The fluorescence spectra of DOX of different solutions were recorded from 520 to 650 nm. The experiments were performed in triplicate and the average was presented for analysis.
In vitro cytotoxicity of NPs
Eighty μL of complete media with HeLa, 4T1, HL-7702 cells were placed into different 96-well plates (∼60000 per well) followed by incubation of 24 h. Next, 20 μL of DOX NPs in PBS of different concentrations were added to each well for incubation (37 °C, 5% CO2). The concentrations of NPs were 1.25, 2.5, 5, 10 and 20 μg mL−1 in different groups. After the addition of DOX NPs, the cells were further incubated for 24 and 48 h, followed by being treated with 20 μL of MTT solution (5 mg mL−1 in PBS) for 4.5 h for a standard MTT assay measurement.
In vitro cellular uptake of DOX and DOX NPs
Flow cytometry (FCM) and confocal laser scanning microscopy (CLSM) were performed to study the cellular uptake of the DOX·HCl, DOX-NCLPEGB NPs and DOX-CLPEGB NPs. For analysis, HeLa cells were cultured with different DOX materials for 6 h and then washed with PBS three times. Following this, the cells were fixed with 4% formaldehyde for 5 min followed by another 3 times of washing in PBS. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, blue) for 10 min. Then the cells were washed with PBS three times for subsequent confocal fluorescence microscopy imaging using a Leica laser scanning confocal microscope. For FCM, the cells were seeded in 12-well plates (∼12000 cells per well). After incubation, the cells were collected by trypsin and centrifuged at 1000 rpm for 3 min. This process was repeated three times and the cells were washed with fresh PBS. Finally, the cells suspended in DMEM media were analyzed for fluorescence intensity with a flow cytometer (BD Biosciences, USA).
In vivo blood circulation and biodistribution of NPs
All in vivo experiments were performed in compliance with the relevant laws and institutional guidelines and also approved by Laboratory Animal Center of Soochow University. Different groups of BALB/c mice were administered with 200 μL of 1 mg mL−1 DOX-NCLPEGB NPs, DOX-CLPEGB NPs, and DOX-C18PMH-PEG NPs. The circulation of NPs was investigated by measuring the fluorescence intensity of DOX at 560 nm in each blood sample. For the measurement, approximately 10 μL of blood was collected from the tail vein of each mouse at desired time intervals (0.25, 0.5, 1, 2, 4, 6, 12 and 24 h). Each blood sample was dissolved in 1 mL of lysis buffer (1% SDS, 1% Triton X-100, 40 mM Tris acetate). The fluorescence intensities of DOX NPs with a series of concentrations were recorded for a standard calibration curve. This experiment was also run in triplicate and the average results were adopted for analysis.For the analysis of the biodistribution of NPs, 4T1 tumor bearing mice were injected with solution containing either DOX-NCLPEGB NPs or DOX-CLPEGB NPs via a tail vein. The dose was 1 mg mL−1. At specific time intervals including 2, 6, 12 and 24 h, the mice were sacrificed. The fluorescence intensities of a number of organs and tissues including liver, spleen, heart, lung, kidney, stomach, intestine, skin and tumors were recorded by the Maestro system. The results were calibrated by subtracting the autofluorescence of individual tissues and organs as well as the background reading using the untreated mouse as a reference group for a semi-quantitative biodistribution analysis.
In vivo antitumor activity
Five to six weeks old female BALB/c mice were raised to developed therapy models for solid tumor by implanting 4T1 cells into the right backside. After 6 days, the volumes of the tumors were estimated to be about 55–90 mm3. The value was calculated by the equation of volume = a × b2/2 in which a and b represent the longest and shortest diameters of a tumor. Subsequently, the mice were divided into 4 groups (n = 6) in a random manner. Subsequently, one mg mL−1 of DOX·HCl, DOX-NCLPEGB NPs, DOX-CLPEGB NPs or PBS solution was administered to the different groups of mice via a tail vein injection. The same treatment was repeated at 7 days after the primary injection. The tumor size and body weight of each individual mouse were recorded daily for two weeks.Conclusion
In summary, we designed and synthesized a GSH-responsive and crosslinkable amphipathic surfactant, PEG-Lys-(Lys-LA2)2, and applied it to modify carrier-free DOX NPs with a cross-linking structure. The release of DOX from NPs with cross-linked PEG-Lys-(Lys-LA2)2 molecules is significantly slower than that from the ones with non-cross-linked ligand coating. This indicates that the problem of premature drug release from carrier-free nanomedicine can be efficaciously solved with surface coating of cross-linked surfactant molecules. In the meantime, for the DOX NPs with cross-linked surface molecules, bio-responsive drug release and efficient intracellular uptake can be achieved. In vivo studies show that the DOX NPs coated with cross-linked ligands exhibit favorable blood circulation half-life (>4 h), intense accumulation in the tumor area and potent anticancer efficacy. This work further paves a way of using carrier-free nanomedicines for powerful drug delivery in clinical applications.Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB933500, 2012CB932400), National Natural Science Foundation of China (61422403), Natural Science Foundation of Jiangsu Province (BK20131162), QingLan Project, Collaborative Innovation Center of Suzhou Nano Science and a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- D. Le Garrec, M. Ranger and J.-C. Leroux, Am. J. Drug Delivery, 2004, 2, 15–42 CrossRef CAS.
- Y. P. Li, K. Xiao, W. Zhu, W. B. Deng and K. S. Lama, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS PubMed.
- F. F. Fu, Y. L. Wu, J. Y. Zhu, S. H. Wen, M. W. Shen and X. Y. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 16416–16425 CAS.
- K. M. Cai, X. He, Z. Y. Song, Q. Yin, Y. F. Zhang, F. M. Uckun, C. Jiang and J. J. Cheng, J. Am. Chem. Soc., 2015, 137, 3458–3461 CrossRef CAS PubMed.
- H. L. Chu, T. M. Cheng, H. W. Chen, F. H. Chou, Y. C. Chang, H. Y. Lin, S. Y. Liu, Y. C. Liang, M. H. Hsu, D. S. Wu, H. Y. Li, L. P. Ho, P. C. Wu, F. R. Chen, G. S. Chen, D. B. Shieh, C. S. Chang, C. H. Su, Z. M. Yao and C. C. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 7509–7516 CAS.
- J. Q. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790–792 CrossRef CAS.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- Z. Z. J. Lim, J. E. J. Li, C. T. Ng, L. Y. L. Yung and B. H. Bay, Acta Pharmacol. Sin., 2011, 32, 983–990 CrossRef CAS PubMed.
- L. Yan, J. F. Zhang, C. S. Lee and X. F. Chen, Small, 2014, 10, 4487–4504 CrossRef CAS PubMed.
- X. Zhou, P. Cao, Y. Zhu, W. G. Lu, N. Gu and C. B. Mao, Nat. Mater., 2015, 14, 1058–1064 CrossRef CAS PubMed.
- K. J. Cho, X. Wang, S. M. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- Y. Li, J. Y. Lin, Y. Huang, Y. X. Li, X. R. Yang, H. J. Wu, S. C. Wu, L. Y. Xie, L. Z. Dai and Z. Q. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 25553–25559 CAS.
- W. Li, Y. L. Yang, C. Wang, Z. Liu, X. J. Zhang, F. F. An, X. J. Diao and X. J. Hao, Chem. Commun., 2012, 48, 8120–8122 RSC.
- J. F. Zhang, Y. N. Li, F. F. An, X. H. Zhang, X. F. Chen and C. S. Lee, Nano Lett., 2015, 15, 313–318 CrossRef CAS PubMed.
- J. F. Zhang, Y. C. Liang, X. D. Lin, X. Y. Zhu, L. Yan, S. L. Li, X. Yang, G. Y. Zhu, A. L. Rogach, P. K. N. Yu, P. Shi, L. C. Tu, C. C. Chang, X. H. Zhang, X. F. Chen, W. J. Zhang and C. S. Lee, ACS Nano, 2015, 9, 9741–9756 CrossRef CAS PubMed.
- C. T. Yu, M. J. Zhou, X. J. Zhang, W. J. Wei, X. F. Chen and X. H. Zhang, Nanoscale, 2015, 7, 5683–5690 RSC.
- P. Huang, D. L. Wang, Y. Su, W. Huang, Y. F. Zhou, D. X. Cui, X. Y. Zhu and D. Y. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. L. Li, L. Zhu, Z. Z. Liu, R. Cheng, F. H. Meng, J. H. Cui, S. J. Ji and Z. Y. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS PubMed.
- P. Y. Sun, H. X. Lu, X. Yao, X. X. Tu, Z. Zheng and X. L. Wang, J. Mater. Chem., 2012, 22, 10035–10041 RSC.
- H. S. Yoo and T. G. Park, J. Controlled Release, 2001, 70, 63–70 CrossRef CAS PubMed.
- X. W. Dai, Z. L. Yue, M. E. Eccleston, J. Swartling, N. K. H. Slater and C. F. Kaminski, Nanomedicine, 2008, 4, 49–56 CAS.
- Y. Q. Li, Y. L. Tong, R. X. Cao, Z. M. Tian, B. S. Yang and P. Yang, Int. J. Nanomed., 2014, 9, 1065–1082 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr09167e |
| This journal is © The Royal Society of Chemistry 2016 |