
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInternalized compartments encapsulated nanogels for targeted drug delivery†
Jicheng
Yu
ab,
Yuqi
Zhang
ab,
Wujin
Sun
ab,
Chao
Wang
ab,
Davis
Ranson
a,
Yanqi
Ye
ab,
Yuyan
Weng
*c and
Zhen
Gu
*abd
aJoint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Raleigh, NC 27695, USA. E-mail: zgu@email.unc.edu
bMolecular Pharmaceutics Division, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
cCenter for Soft Condensed Matter Physics and Interdisciplinary Research & College of Physics, Optoelectronics and Energy, Soochow University, Suzhou, 215006, China. E-mail: wengyy@suda.edu.cn
dDepartment of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC 27599, USA
First published on 14th April 2016
Abstract
Drug delivery systems inspired by natural particulates hold great promise for targeted cancer therapy. An endosome formed by internalization of plasma membrane has a massive amount of membrane proteins and receptors on the surface, which is able to specifically target the homotypic cells. Herein, we describe a simple method to fabricate an internalized compartments encapsulated nanogel with endosome membrane components (EM-NG) from source cancer cells. Following intracellular uptake of methacrylated hyaluronic acid (m-HA) adsorbed SiO2/Fe3O4 nanoparticles encapsulating a crosslinker and a photoinitiator, EM-NG was readily prepared through in situ crosslinking initiated under UV irradiation after internalization. The resulting nanogels loaded with doxorubicin (DOX) displayed enhanced internalization efficiency to the source cells through a specific homotypic affinity in vitro. However, when treated with the non-source cells, the EM-NGs exhibited insignificant difference in therapeutic efficiency compared to a bare HA nanogel with DOX. This study illustrates the potential of utilizing an internalized compartments encapsulated formulation for targeted cancer therapy, and offers guidelines for developing a natural particulate-inspired drug delivery system.
Introduction
Drug delivery systems based on natural particulates have emerged as one of the most promising strategies for cancer therapy.1 Taking advantages of physical morphologies and biological functions of natural particulates, these biomimetic drug delivery carriers offer several significant advantages such as selective targeting, prolonged circulation time, and low immunogenicity.2–6 Among them, natural biological membrane-derived nanoparticles (NPs) have received extensive attention as they are simple and viable due to their capability of mimicking the natural membrane properties. For example, red blood cell membrane-coated PLGA nanoparticles have shown superior circulation half-life in vivo.7–10 In addition, it has been validated that cancer cell membrane-coated nanoparticles with a full array of tumor antigens can promote a tumor-specific immune response and target the source cancer cells via an inherent homotypic binding interaction.11 Our previously reported work demonstrated that the platelet membrane coated core–shell nanovehicle with overexpressed P-selectin on the membrane could specifically bind to CD44 receptors on the surface of cancer cells.12Endosomes play an important role in regulating fundamental processes in eukaryotic cells, such as nutrient uptake, signaling, immunity and adhesion.13 The massive amount of membrane is internalized into the endosome by several endocytic pathways, and the membrane lipids and proteins can be recycled back to the plasma membrane in an efficient manner.13–15 Although precise mechanisms of the recycling and fusion remain to be fully elucidated, several previous studies revealed that the endosome-to-plasma membrane transport is mediated by several proteins including Eps15p homology (EH) domains containing proteins.16,17 The proteins from the sorting nexin protein family and others can also promote the fusion of the endosome with the plasma membrane.18–21
In this study, we describe an internalized compartments encapsulated nanogel with endosome membrane components (denoted as EM-NG) which is easily extracted from the source cancer cells for targeting and specific delivery of small molecular drugs. As shown in Fig. 1, the EM-NG has an inner core composed of hyaluronic acid (HA) nanogel containing SiO2/Fe3O4 nanoparticles, and an outer shell with specific targeting ligands. HA is chosen since it is highly biocompatible and it can target the hyaluronan receptor CD44, which is overexpressed in a variety of tumor types.22–26 In order to allow HA nanogel to form intracellularly after endocytosis, we firstly use core–shell mesoporous silica NPs with Fe3O4 nanocrystals as the core (SiO2/Fe3O4 NPs) to encapsulate a crosslinker and a photoinitiator, and coat with methacrylated HA (m-HA) on the surface through electrostatic interactions. Following the incubation of resulting NPs with source cells, an in situ formed nanogel in the internalized compartment is obtained by the photo-polymerization upon UV light irradiation.27–32 The internalized compartments encapsulated HA nanogel is readily collected via the magnetic extraction due to the entrapped magnetic Fe3O4 nanocrystals. After loading with anticancer drug doxorubicin (DOX), these EM-NGs can actively target source cancer cells by taking the advantage of the specific interaction with their source cells, and subsequently internalize to release DOX.
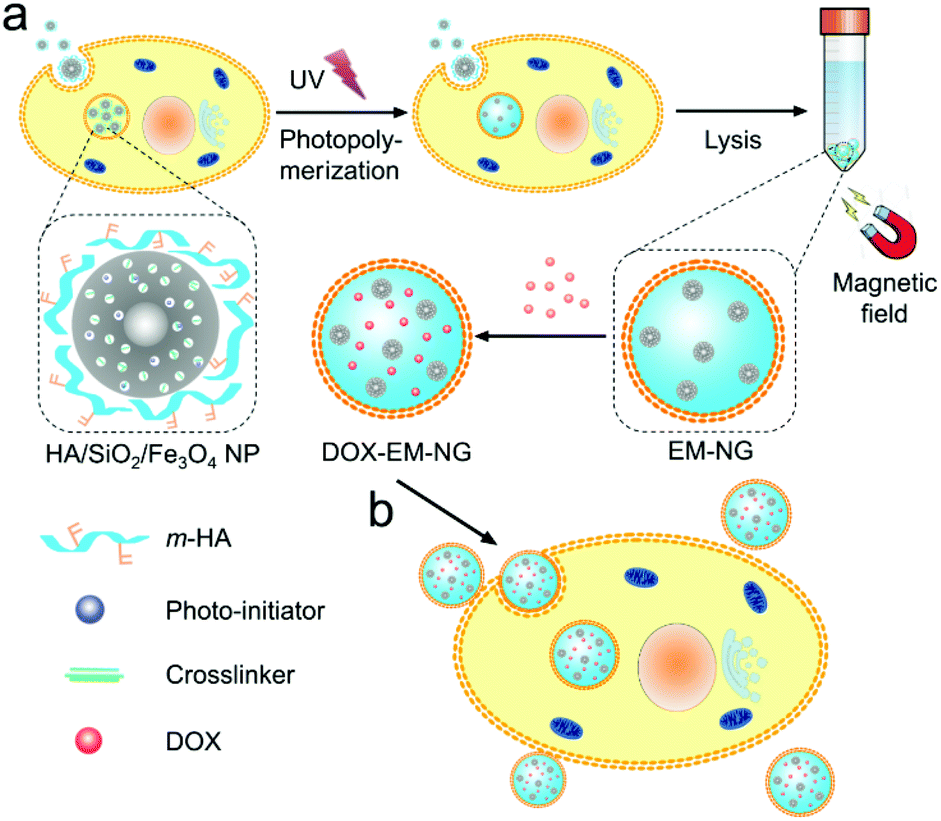 | ||
| Fig. 1 Schematic of EM-NG for targeted drug delivery. (a) Preparation of EM-NG from source cancer cells. (b) DOX-loaded EM-NGs for targeted drug delivery. | ||
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich and were used as received. Rhodamine-NHS was purchased from Life Technologies (Grand Island, NY, USA).Synthesis of Fe3O4 nanocrystals
A solution of 1 M iron(III) chloride hexahydrate and 0.5 M iron(II) chloride tetrahydrate in 25 mL DI water was added dropwise to a 0.5 M NaOH solution in 250 mL of DI water at 40 °C. The mixture was stirred for 1 h and the nanocrystals were subsequently washed with DI water until neutral pH. The resulting nanocrystals were dialyzed against DI water for 3 days. Finally, the Fe3O4 nanocrystals were stabilized with oleic acid and dispersed in chloroform with a concentration of 6.7 mg Fe per mL. The zeta-potential and size distribution were measured on a Zetasizer (Nano ZS; Malvern). The TEM images were obtained on a JEOL 2000FX TEM instrument.Synthesis of SiO2/Fe3O4 NPs functionalized with quaternary ammonium groups
1 mL of the Fe3O4 nanocrystals in chloroform was poured into 10 mL of 0.55 M aqueous cetyltrimethylammonium bromide (CTAB) solution and emulsified by sonication for 10 min. The resulting turbid brown solution was stirred and heated up to 65 °C for 10 min to evaporate the chloroform, resulting in a transparent black Fe3O4/CTAB solution. Then, the Fe3O4/CTAB solution was added to a mixture of 90 mL of water and 0.6 mL of 2 M NaOH solution, and the mixture was heated up to 70 °C under stirring. After adding 1 mL of tetraethylorthosilicate (TEOS) and 6 mL of ethylacetate, the solution was stirred at 70 °C for 3 h. Thereafter, the obtained SiO2/Fe3O4 NPs were washed 3 times with ethanol to remove the unreacted species and dispersed in 30 mL of ethanol. To extract CTAB, 80 μL of HCl was added to the solution and stirred for 3 h at 60 °C. After washing them with ethanol three times, the NPs were dispersed in 10 mL of chloroform, and 120 μL of 3-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride (TMAPS) (50%) in methanol was added. The reaction solution was stirred at room temperature for 24 h. The resulting SiO2/Fe3O4 NPs functionalized with quaternary ammonium groups were washed 3 times with ethanol and dried in a vacuum.Preparation of HA/SiO2/Fe3O4 NPs
3 mg of SiO2/Fe3O4 NPs was mixed with 0.5 mg N,N′-methylenebisacrylamide (MBA) and 0.1 mg photoinitiator (Irgacure 2959) in PBS buffer for 24 h. The unloaded crosslinker and photoinitiator were removed by filtering using a centrifugal filter (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da molecular mass cutoff, Millipore). Then, 1 mg m-HA or rhodamine-HA derivative was added and stirred for another 4 h to obtain HA/SiO2/Fe3O4 NPs.
000 Da molecular mass cutoff, Millipore). Then, 1 mg m-HA or rhodamine-HA derivative was added and stirred for another 4 h to obtain HA/SiO2/Fe3O4 NPs.
Cell culture
HeLa and A549 cells were obtained from Tissue Culture Facility of UNC Lineberger Comprehensive Cancer Center and cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 U mL−1) and streptomycin (100 μg mL−1) in a 37 °C incubator (Thermo Scientific) under 5% CO2 and 90% humidity. The cells were regularly sub-cultured with trypsin-EDTA (0.25%, w/w) and cell density was determined with a hemocytometer before each experiment.Preparation of EM-NG and HA NG
HeLa cells (1 × 105 cells per well) were seeded in 6-well plates. The cells were allowed to culture for 24 h before exposure to the HA/SiO2/Fe3O4 NP dispersions. The NP dispersions were prepared by diluting the concentrated NP solution into the FBS free medium. The cells were incubated with NPs for 4 h, and then the NP containing medium was discarded. After washing the cells with PBS twice, the cells were harvested with trypsin and centrifuged at 1000 rpm for 4 min. Then, the cells were resuspended in 1 mL PBS solution and exposed to UV irradiation (wavelength: 365 nm) for 1 min to form a solid HA nanogel by crosslinking polymerization. The cells were lysed with Pierce IP lysis buffer (Thermo Scientific). Then, EM-NGs were collected using a magnet from the cell lysate. In order to obtain the bare HA nanogel without membrane, the EM-NGs were further lysed with lysis buffer for a second time, and collected using a magnet.Intracellular distribution of HA/SiO2/Fe3O4 NPs
HeLa cells (1 × 105 cells per dish) were seeded in confocal dishes and cultured for 24 h. Then, the cells were incubated with rhodamine-HA/SiO2/Fe3O4 NPs for 1 h and 4 h. Afterward, the cells were washed with PBS twice and stained with LysoTracker green (50 nM) (Life Technologies) at 37 °C for 30 min. Then, the cells were washed with PBS twice and stained with Hoechst 33342 (1 μg mL−1) for 10 min. After washing with PBS twice, the cells were immediately observed using CLSM (LCM 710, Zeiss).Preparation of DOX-EM-NG
To prepare DOX-EM-NG, 0.03 mL of DOX solution (0.1 mg mL−1) was added into 0.27 mL of EM-NG solution, and mixed at room temperature for 24 h. Then, the excess DOX was removed by filtering using a centrifugal filter (10000 Da molecular mass cutoff, Millipore). The loading capacity (LC) of DOX-EM-NG was determined by measuring the amount of encapsulated DOX by analyzing the fluorescence intensity of DOX at 590 nm with an excitation wavelength of 480 nm using a microplate reader (Infinite M200 Pro, Tecan). LC was calculated as LC = encapsulated amount of DOX/total weight of DOX-EM-NGs.
Endosome membrane protein characterization
SDS-PAGE was used to separate the proteins contained in the endosome only, EM-NGs and NGs. Samples were diluted in protein loading buffer and incubated for 10 min at 100 °C. 10 μL of the sample was loaded into each well in a 10% polyacrylamide gel. After separation by SDS-PAGE, the proteins were transferred to a nitrocellulose membrane at 300 mA for 3 h using a wet transfer method. The membrane was incubated with a 3% BSA blocking solution in the PBS TWEEN solution (0.01%) for 30 min at room temperature, and then incubated with early endosome antigen 1 (EEA1) in blocking solution overnight at 4 °C. Afterwards, the membrane was washed 3 times with 0.01% PBS TWEEN and incubated with the anti-rabbit HRP in the blocking solution for 1 h. The membrane was then washed with 0.01% PBS TWEEN 3 times and visualized using a 1-StepTM TMB–Blotting solution (Pierce, USA).In vitro DOX release study
The release profile of DOX was determined using the dialysis method in PBS buffer. 400 μL of DOX-EM-NG solution was added into a dialysis tube (10000 Da molecular mass cutoff) (Slide-A-Lyzer, Thermo Scientific) against 1 mL of PBS buffer solution with different pH values (7.4 and 5.0). The dialysis tube was incubated at 37 °C. At predetermined time intervals, the total buffer solution was withdrawn, followed by replacing with fresh buffer solution with the same pH value. The amount of release DOX was measured through the same method mentioned above.
Determination of endocytosis pathways
HeLa cells (1 × 105 cells per well) were seeded in 6-well plates and cultured for 48 h. Afterwards, the cells were pre-incubated with different specific inhibitors for different endocytosis pathways, including chlorpromazine (CPZ, 10 μM) for the clathrin-mediated endocytosis, nystatin (NYS, 25 μg mL−1) for the caveolin-mediated endocytosis inhibition, amiloride (AMI, 1 mM) for the macropinocytosis inhibition, and methyl-β-cyclodextrin (MCD, 3 mM) for the lipid raft inhibition. Then, the cells were incubated with DOX-EM-NG at a DOX concentration of 1 μM in the presence of inhibitors for another 2 h. After washing the cells with PBS twice at 4 °C, the fluorescence intensity of DOX in the cells was measured by flow cytometry.Evaluation of DOX-EM-NG and DOX-NG uptake of HeLa cells
HeLa cells (1 × 105 cells per well) were seeded in 6-well plates and cultured for 48 h. Afterwards, the cells were added with DOX-EM-NGs, DOX-NGs, and free DOX with the same concentration, and incubated for 2 h. After washing the cells with PBS twice at 4 °C, the fluorescence intensity of DOX in the cells was measured by flow cytometry.In vitro cytotoxicity
HeLa or A549 cells (6 × 103 cells per well) were seeded in the 96-well plates. After 24 h culture, the cells were exposed to EM-NGs, free DOX solution, DOX-EM-NGs and DOX-NGs with different concentrations of DOX in FBS free medium for 24 h, respectively. Then, 20 μL per well of MTT solution (5 mg mL−1) was added and incubated for another 4 h. After removing the medium, 150 μL DMSO was added to each well. The absorbance was measured at a test wavelength of 570 nm with a reference wavelength of 630 nm by using a microplate reader (Infinite M200 PRO, Tecan).Statistical analysis
All results presented are mean ± SEM. Statistical analysis was performed using Student's t-test or ANOVA test. With a p value <0.05, the differences between experimental groups and control groups were considered statistically significant.Results and discussion
Preparation and characterization of HA/SiO2/Fe3O4 NPs
Magnetic Fe3O4 nanocrystals were synthesized by a traditional aqueous co-precipitation technique.33 The obtained Fe3O4 nanocrystals were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). As shown in Fig. 2a and b, the Fe3O4 nanocrystals were monodisperse with an average diameter of 22.5 nm. These nanocrystals were then coated with mesoporous silica using a sol–gel method.34 DLS results revealed that the size of SiO2/Fe3O4 NPs increased to 115.3 nm, and the wormhole-like mesopores with a size of about 2–3 nm were clearly observed in the silica shells in the TEM image (Fig. 2c and d). The zeta potential of silica nanoparticles was determined as −30.1 ± 3.3 mV due to surface hydroxyl groups.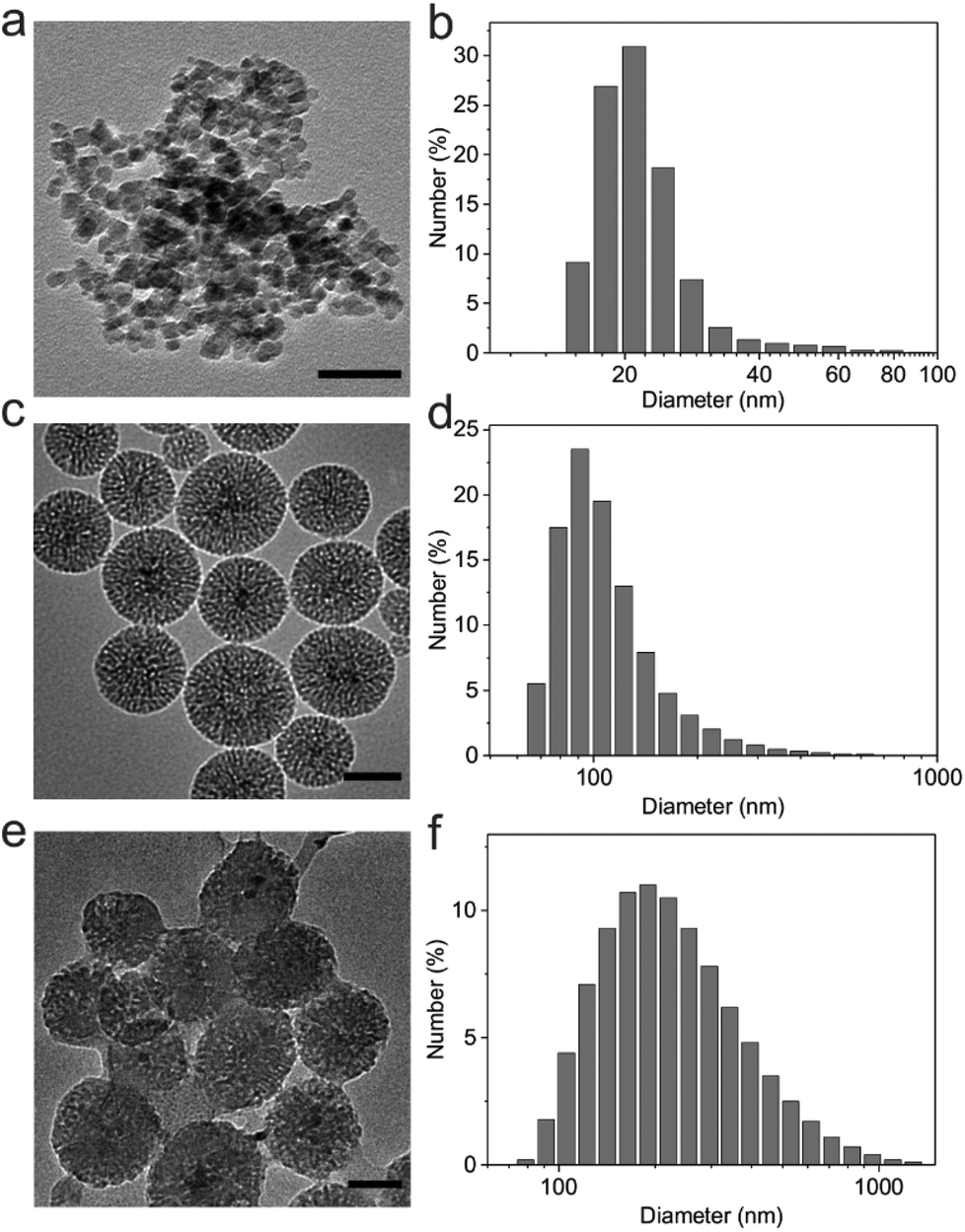 | ||
| Fig. 2 Characterization of Fe3O4 nanocrystals, SiO2/Fe3O4 NPs and HA/SiO2/Fe3O4 NPs. (a) The TEM image and (b) size distribution of Fe3O4 nanocrystals (scale bar: 50 nm). (c) The TEM image and (d) size distribution of SiO2/Fe3O4 NPs (scale bar: 50 nm). (e) The TEM image and (f) size distribution of HA/SiO2/Fe3O4 NPs (scale bar: 50 nm). | ||
To adsorb the negatively charged m-HA on the surface, 3-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride (TMAPS) was further functionalized with SiO2/Fe3O4 NPs to provide quaternary ammonium groups on the outer surface, which converted the zeta potential to 23.2 ± 0.4 mV. After being loaded with a crosslinker and a photoinitiator, m-HA was subsequently deposited onto the positively charged SiO2/Fe3O4 NPs by the electrostatic assembly. The TEM image clearly showed the HA shell on the surface of SiO2/Fe3O4 NPs (Fig. 2e). The negative zeta potential of the obtained HA/SiO2/Fe3O4 NPs (−36 ± 1.5 mV) and an apparent increase in diameter measured by DLS (Fig. 2f) further validated the successful coating of m-HA on the surface of SiO2/Fe3O4 NPs.
Preparation and characterization of EM-NGs
To achieve the internalized compartments encapsulated HA nanogels, HA/SiO2/Fe3O4 NPs were first required to be internalized. Human cervical carcinoma epithelial (HeLa) cells were chosen as a model cell line, and HA/SiO2/Fe3O4 NPs were incubated in the cell culture medium with cells. HA/SiO2/Fe3O4 NPs were allowed to be internalized via the endocytosis pathway. The successful uptake of HA/SiO2/Fe3O4 NPs was confirmed using confocal laser scanning microscopy (CLSM). The fluorescence signal of the rhodamine tagged HA/SiO2/Fe3O4 NPs was clearly observed in cells after 4 h of coincubation, and the fluorescence signals of rhodamine and LysoTracker Green showed high colocalization (Fig. 3a), suggesting that most of the HA/SiO2/Fe3O4 NPs entered the cells through endocytosis. After removing the free NPs, the cells were exposed to UV light to initiate the in situ photo-polymerization to form the crosslinked HA nanogel in the internalized compartments, namely EM-NG. The cells were then lysed, and EM-NG containing magnetic nanoparticles were collected via magnetic extraction (Fig. 3d). As determined by DLS, the EM-NG was 262.3 nm in hydrodynamic diameter (Fig. 3c). The TEM picture showed that the EM-NGs were round-oval in shape, and the membrane with a thickness of 10 nm and the multiple inclusions of silica NPs were clearly observed in each EM-NG (Fig. 3b). To further confirm the existence of endosome membrane components on the shell, western blotting analysis was performed against the early endosome antigen 1 (EEA1), the endosome membrane-specific marker. The result showed that a significant enrichment of EEA1 was present on the EM-NGs rather than the bare HA NGs (Fig. 3e). | ||
| Fig. 3 Characterization of EM-NGs. (a) Intracellular trafficking of rhodamine labelled HA/SiO2/Fe3O4 NPs on HeLa cell observed by CLSM. The late endo-lysosomes were stained by LysoTracker Green, and the nuclei were stained by Hoechst 33342 (scale bar: 20 μm). (b) The TEM image and (c) size distribution of EM-NG (scale bar: 200 nm). (d) The photograph of EM-NGs was collected under a magnetic field. (e) Western blotting analysis of endosomes, EM-NGs, and bare NGs against endosome membrane marker: the early endosome antigen 1 (EEA1). (f) DOX release profiles of DOX-EM-NGs at pH 7.4 and 5.0, respectively. | ||
Drug loading and release from EM-NGs
DOX, as a model hydrophilic anticancer drug, was loaded into EM-NG via dispersion.35,36 The loading capacity was determined as 7.3%. The in vitro drug release behavior was investigated under different pH conditions over time. The DOX-EM-NG exhibited a near zero-order release kinetics at pH 7.4 (Fig. 3f). In contrast, DOX released from nanogel was much faster under acidic conditions than physiological conditions. The accelerated drug release at acidic pH is mainly due to the weakening of the binding between the EM-NG and drug, and improved solubility of DOX at low pH.37,38 This pH-dependent drug release behavior can play a crucial role in tumor-targeted drug delivery via an endocytosis pathway.In vitro delivery of DOX by EM-NG
In order to determine the endocytosis pathway of DOX-EM-NG, HeLa cells were pre-incubated with several specific inhibitors of different kinds of endocytosis, including chlorpromazine (CPZ) for clathrin-mediated endocytosis, nystatin (NYS) for the caveolin-mediated endocytosis inhibition, amiloride (AMI) for the macropinocytosis inhibition, and methyl-β-cyclodextrin (MCD) for the lipid raft inhibition. As shown in Fig. 4b, CPZ, NYS and AMI all reduced the uptake of DOX-EM-NG significantly, suggesting that DOX-EM-NGs were taken up by HeLa cells through clathrin-mediated endocytosis, caveolin-mediated endocytosis, and lipid rafts. In contrast, there was insignificant inhibition of uptake efficiency in the cells pretreated with AMI for the macropinocytosis inhibition. These results indicated that several pathways were involved in the internalization of cell membrane with DOX-EM-NG, due to a large number of receptors and proteins on the surface of DOX-EM-NG.13,15 The fluorescence of DOX was clearly observed in cells after 1 h of incubation with DOX-EM-NG, visualized by CLSM (Fig. 4a and Fig. S1 in the ESI†), which validated the cellular internalization of DOX-EM-NG. When the incubation time was prolonged to 4 h, DOX was remarkably released and delivered into the nuclei of cells. To explore the targeting capability of DOX-EM-NG, EM-NG extracted from HeLa cells was further lysed to obtain the bare HA nanogel (NG) by removing the shell containing the endosome membrane components.39 By quantitative analysis using flow cytometry, it was demonstrated that incubation of DOX-EM-NG with HeLa cells in vitro led to significantly increased uptake as compared to the DOX loaded bare HA nanogel (DOX-NG) and free DOX (Fig. 4c).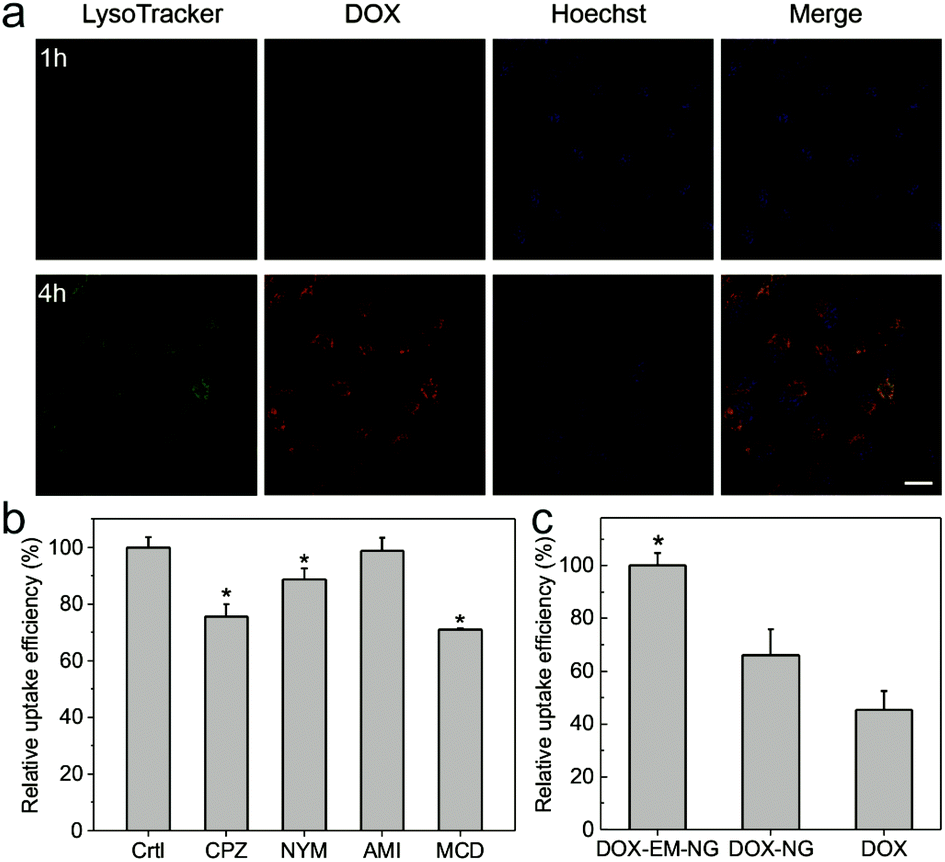 | ||
| Fig. 4 In vitro delivery of DOX by EM-NG. (a) Intracellular trafficking of DOX-EM-NG observed by CLSM. The late endo-lysosomes were stained by LysoTracker Green, and the nuclei were stained by Hoechst 33342 (scale bar: 20 μm). (b) Investigation of cellular uptake mechanism. Relative uptake efficiency of DOX-EM-NG in the presence of various endocytosis inhibitors. Inhibitor of clathrin-mediated endocytosis: chlorpromazine (CPZ, 10 μM); inhibitor of caveolin-mediated endocytosis: nystatin (NYS, 25 μg mL−1); inhibitor of macropinocytosis: amiloride (AMI, 1 mM); inhibitor of lipid raft: methyl-β-cyclodextrin (MCD, 3 mM). *p < 0.05 compared with the control group. (c) Relative uptake efficiency of DOX-EM-NG, DOX-NG and free DOX on HeLa cells. *p < 0.05 for treatment with DOX-EM-NG compared with DOX-NG and free DOX. | ||
In vitro cytotoxicity of DOX-EM-NGs
Next, the in vitro cytotoxicity of nanogel against HeLa cells was evaluated by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Both DOX-EM-NG and DOX-NG showed significantly enhanced cytotoxicity toward HeLa cells compared to free DOX solution upon 24-hour incubation (Fig. 5a). Notably, the cell viability of cells treated with DOX-EM-NG was much lower than that of those treated with DOX-NG, suggesting the better cell targeting ability of the endosome membrane components than the pure HA. The results were consistent with the cellular uptake studies mentioned above. Bare EM-NG did not exhibit significant cytotoxicity within the studied range of concentrations. In order to assess the capability of DOX-EM-NG to homotypically target cancer cells, the human lung adenocarcinoma epithelial (A549) cells, as a heterotypic cell line, were incubated with DOX-EM-NG or DOX-NG for 24 h. The results showed that there was insignificant difference in cell viability for DOX-EM-NG and DOX-NG (Fig. 5b), which further indicated that the enhanced binding effect was specifically associated with the membrane coating.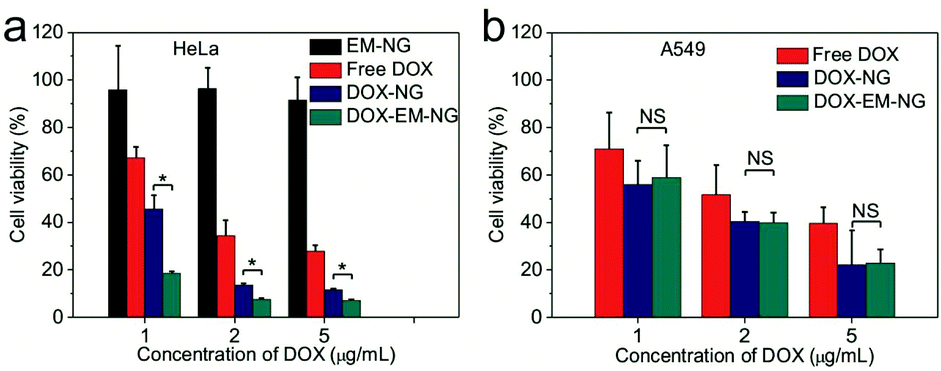 | ||
| Fig. 5 In vitro cytotoxicity of DOX-EM-NG. (a) In vitro cytotoxicity of EM-NG, free DOX, DOX-NG and DOX-EM-NG towards HeLa cells after incubation for 24 h. (b) In vitro cytotoxicity of free DOX, DOX-NG and DOX-EM-NG towards A549 cells after incubation for 24 h. *p < 0.05, NS: not significant. | ||
Conclusions
We have developed an innovative strategy utilizing internalized compartments encapsulated nanogels for enhanced delivery of anticancer drug. HA/SiO2/Fe3O4 NPs encapsulating a crosslinker and a photo-initiator were confirmed to efficiently be internalized by endocytosis by CLSM, and a subsequent in situ polymerization happened in the internalized compartments, resulting in the internalized compartments encapsulated HA nanogels with endosome membrane components. These EM-NGs containing magnetic nanocrystals were easily collected using a magnet after cell lysis. Through western blotting analysis against endosome membrane specific protein (EEA1), the existence of endosome membrane components on the surface was substantiated, and the shell with specific targeting ligands could be removed via further lysis for a long time. After loading with DOX, the resulting nanogel was demonstrated to efficiently target the source cancer cells through a specific homotypic affinity, which increased 1.5-fold in uptake efficiency compared to the bare HA NGs. Moreover, DOX-EM-NGs were able to deliver an anticancer drug to source cancer cells with enhanced efficacy, while there were insignificant differences compared to DOX-NGs when treated with non-source cancer cells.Furthermore, stimuli-responsive moieties can be integrated with these EM-NGs to achieve controllable drug release.40 We will also evaluate the in vivo targeting capability, antitumor efficacy and systemic toxicity of the nanogel. This strategy provides a guideline to develop internalized compartments encapsulated nanomedicine from primary tumors of patients for personalized anticancer treatments.
Acknowledgements
This work was supported by the grant from NC TraCS, NIH's Clinical and Translational Science Awards (CTSA, NIH grant 1UL1TR001111) at UNC-CH. We acknowledge the use of the Analytical Instrumentation Facility (AIF) at NC State, which is supported by the State of North Carolina and the National Science Foundation (NSF).Notes and references
- S. Mitragotri, D. G. Anderson, X. Y. Chen, E. K. Chow, D. Ho, A. V. Kabanov, J. M. Karp, K. Kataoka, C. A. Mirkin, S. H. Petrosko, J. J. Shi, M. M. Stevens, S. H. Sun, S. Teoh, S. S. Venkatraman, Y. N. Xia, S. T. Wang, Z. Gu and C. J. Xu, ACS Nano, 2015, 9, 6644–6654 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- W. Gao, R. H. Fang, S. Thamphiwatana, B. T. Luk, J. Li, P. Angsantikul, Q. Zhang, C.-M. J. Hu and L. Zhang, Nano Lett., 2015, 15, 1403–1409 CrossRef CAS PubMed.
- J.-W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, Curr. Opin. Biotechnol., 2013, 24, 1167–1173 CrossRef CAS PubMed.
- S. C. Balmert and S. R. Little, Adv. Mater., 2012, 24, 3757–3778 CrossRef CAS PubMed.
- J. A. Copp, R. H. Fang, B. T. Luk, C.-M. J. Hu, W. Gao, K. Zhang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13481–13486 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, B. T. Luk and L. Zhang, Nat. Nanotechnol., 2013, 8, 933–938 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, J. Copp, B. T. Luk and L. Zhang, Nat. Nanotechnol., 2013, 8, 336–340 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- R. H. Fang, C.-M. J. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- Q. Hu, C. Qian, Y. Ye, C. Wang and Z. Gu, Adv. Mater., 2015, 27, 7043–7050 CrossRef CAS PubMed.
- C. C. Scott, F. Vacca and J. Gruenberg, Semin. Cell Dev. Biol, 2014, 2–10 CrossRef CAS PubMed.
- M. Seaman, Cell. Mol. Life Sci., 2008, 65, 2842–2858 CrossRef CAS PubMed.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- S. Caplan, N. Naslavsky, L. M. Hartnell, R. Lodge, R. S. Polishchuk, J. G. Donaldson and J. S. Bonifacino, EMBO J., 2002, 21, 2557–2567 CrossRef CAS PubMed.
- N. Naslavsky, M. Boehm, P. S. Backlund and S. Caplan, Mol. Biol. Cell, 2004, 15, 2410–2422 CrossRef CAS PubMed.
- E. H. Hettema, M. J. Lewis, M. W. Black and H. R. Pelham, EMBO J., 2003, 22, 548–557 CrossRef CAS PubMed.
- M. N. Seaman, J. Cell Sci., 2012, 125, 4693–4702 CrossRef CAS PubMed.
- J. Gruenberg and K. E. Howell, Annu. Rev. Cell Biol., 1989, 5, 453–481 CrossRef CAS PubMed.
- L. Foret, J. E. Dawson, R. Villaseñor, C. Collinet, A. Deutsch, L. Brusch, M. Zerial, Y. Kalaidzidis and F. Jülicher, Curr. Biol., 2012, 22, 1381–1390 CrossRef CAS PubMed.
- V. P. Chauhan, J. D. Martin, H. Liu, D. A. Lacorre, S. R. Jain, S. V. Kozin, T. Stylianopoulos, A. S. Mousa, X. Han and P. Adstamongkonkul, Nat. Commun., 2013, 4 CAS , 2516.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS PubMed.
- M. Culty, M. Shizari, E. W. Thompson and C. B. Underhill, J. Cell. Physiol. Suppl., 1994, 160, 275–286 CrossRef CAS PubMed.
- M. Swierczewska, K. Y. Choi, E. L. Mertz, X. Huang, F. Zhang, L. Zhu, H. Y. Yoon, J. H. Park, A. Bhirde and S. Lee, Nano Lett., 2012, 12, 3613–3620 CrossRef CAS PubMed.
- M. Zhao, Y. Liu, R. S. Hsieh, N. Wang, W. Tai, K.-I. Joo, P. Wang, Z. Gu and Y. Tang, J. Am. Chem. Soc., 2014, 136, 15319–15325 CrossRef CAS PubMed.
- M. Yan, J. Du, Z. Gu, M. Liang, Y. Hu, W. Zhang, S. Priceman, L. Wu, Z. H. Zhou and Z. Liu, Nat. Nanotechnol., 2010, 5, 48–53 CrossRef CAS PubMed.
- R. Mo, T. Jiang, R. DiSanto, W. Tai and Z. Gu, Nat. Commun., 2014, 5 Search PubMed.
- J. Yu, Y. Zhang, Y. Ye, R. DiSanto, W. Sun, D. Ranson, F. S. Ligler, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8260–8265 CrossRef CAS PubMed.
- Z. Gu, M. Yan, B. Hu, K.-I. Joo, A. Biswas, Y. Huang, Y. Lu, P. Wang and Y. Tang, Nano Lett., 2009, 9, 4533–4538 CrossRef CAS PubMed.
- T. Jiang, R. Mo, A. Bellotti, J. Zhou and Z. Gu, Adv. Funct. Mater., 2014, 24, 2295–2304 CrossRef CAS.
- A. H. Lu, E. e. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- K. Qian, Y. Ma, J. Wan, S. Geng, H. Li, Q. Fu, X. Peng, X. Kan, G. Zhou and W. Liu, J. Controlled Release, 2015, 212, 41–49 CrossRef CAS PubMed.
- W. Wu, W. Yao, X. Wang, C. Xie, J. Zhang and X. Jiang, Biomaterials, 2015, 39, 260–268 CrossRef CAS PubMed.
- Y.-J. Jin, U. Termsarasab, S.-H. Ko, J.-S. Shim, S. Chong, S.-J. Chung, C.-K. Shim, H.-J. Cho and D.-D. Kim, Pharm. Res., 2012, 29, 3443–3454 CrossRef CAS PubMed.
- N. V. Nukolova, H. S. Oberoi, S. M. Cohen, A. V. Kabanov and T. K. Bronich, Biomaterials, 2011, 32, 5417–5426 CrossRef CAS PubMed.
- F. Bertoli, G. L. Davies, M. P. Monopoli, M. Moloney, Y. K. Gun'ko, A. Salvati and K. A. Dawson, Small, 2014, 10, 3307–3315 CrossRef CAS PubMed.
- Y. Lu, W. Sun and Z. Gu, J. Controlled Release, 2014, 194, 1–19 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of m-HA; synthesis of rhodamine-HA derivative; supplementary data on relative fluorescence intensity of DOX-EN-NGs on HeLa cells. See DOI: 10.1039/c5nr08895j |
| This journal is © The Royal Society of Chemistry 2016 |